
Abstract
The hemagglutinin (HA) glycoprotein of influenza viruses is posttranslationally cleaved into the disulfide-linked subunits HA1 and HA2, and this proteolytic processing event is critical to the virus life cycle as it is required to activate membrane fusion potential and virus infectivity. High-resolution structures are available for the HA precursor (HA0), the cleaved neutral pH conformation of HA, and the low pH conformation that the HA assumes when triggered by acidification of endosomes to mediate fusion of viral and cellular membrane during virus entry. These structures have provided clues regarding the mechanisms by which proteolytic cleavage activates membrane fusion potential and how subsequent acidification drives the fusion process. It has been known for decades that influenza strains and subtypes can vary with regard to HA cleavage properties and that cleavage site sequences and the proteases that recognize them can represent a major determinant for virus pathogenicity. However, a number of questions remain with respect to the identity and characteristics of the proteases that activate HAs in the various natural hosts and complex ecosystems that constitute the realm of influenza viruses. The continuing study of HA cleavage properties and the proteases involved should illuminate our understanding not only of pathogenicity but other aspects of influenza biology including host range, transmission, and interplay with other microorganisms such as bacteria.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
There are three genera of influenza viruses in the Orthomyxoviridae family: the influenza A, influenza B, and influenza C. All are human pathogens; however, influenza A viruses are generally of greatest concern as they are responsible for most of the annual seasonal epidemics, and their vast natural reservoir in aquatic avian species provides a fertile source for the unpredictable emergence of novel pandemic strains. For the influenza A and B viruses, the initial stage of entry into host cells is mediated by receptor-binding and membrane fusion functions provided by the hemagglutinin (HA) glycoprotein . A separate viral surface protein, the neuraminidase (NA), is responsible for the receptor-destroying sialidase activity that allows for progeny virus dissemination at the end of the replication cycle. For influenza C viruses, both the receptor-binding function and receptor-destroying esterase functions are provided by a single glycoprotein, termed HEF, for hemagglutinin/esterase/fusion. The HA and HEF proteins of all influenza viruses share a common requirement for protease activation in order to potentiate membrane fusion and activate virus infectivity. This chapter will focus on the proteolytic activation of the HA glycoproteins of influenza A viruses, but many of the concepts outlined herein are equally relevant for the HA and HEF proteins of influenza B and C viruses.
The influenza A viruses are distinguished by their complex ecology and dynamic transmission cycles involving more than 100 avian species and a range of mammalian hosts (Munster et al. 2007; Slusher et al. 2014; Webster et al. 1992). In aquatic birds belonging to the order Anseriformes (ducks, geese, and swans) and Charadriiformes (gulls, terns, and shorebirds) that serve as the “natural” hosts of influenza A virus, 16 antigenically distinct HA subtypes and 9 NA subtypes are known to circulate, though they rarely cause symptomatic disease in these species. Under conducive conditions, viruses maintained within the natural reservoir may transmit to Galliformes (chickens, turkeys, and quail) and passerine birds or mammalian species such as swine, horses, dogs, and humans, causing a range of morbidity and mortality. The outbreaks resulting from cross-species transmission are often limited, but when influenza A virus strains become established in a new host, they can perpetuate for varying lengths of time and have devastating consequences to human health and global economies. Cross-species transmission events of influenza A virus are often characterized by the relatively rapid evolution of the virus to facilitate adaptation to the new host. These adaptive changes may be influenced by a variety of selective pressures, including, but not limited to, the immune response of the host; the availability and structure of host cell receptors; variations in host cell replication machinery; differences in the site of replication, which may involve pH and temperature; and environmental persistence to enable continued transmission. While these concepts are generally understood, we are only at the inception of understanding the complex interplay of viral and host factors involved in host range, transmission, and pathogenicity of these viruses. What we do know is that HA cleavage properties constitute one of the most critical determinants of pathogenicity and the environments and biological niches where activating proteases can be found in nature may play an important role in the complex ecology of influenza A viruses.
Proteolytic cleavage is a fundamental biochemical process that activates a multitude of functions for viruses as well as their hosts. A requirement for proteolytic activation of influenza virus infectivity was first discovered in studies examining influenza virus replication in cell culture. For many years after influenza viruses were first isolated in the 1930s (Shope 1931; Smith et al. 1933), embryonated chicken eggs were the substrate of choice for influenza virus propagation, and even as cell culture systems were developed, only a few strains such as A/WSN/33 (H1N1) were able to replicate and form plaques on cell monolayers. Decades passed before it was reported that virus replication and plaquing efficiency could be enhanced appreciably by the addition of proteases such as trypsin or pancreatin to the cell monolayers (Appleyard and Maber 1974; Came et al. 1968; Tobita and Kilbourne 1974) and that the extent of HA cleavage correlated with cytopathology following infection (Lazarowitz et al. 1973a). These studies were extended to show that HA cleavage activation was required for an early stage in the infection process, but was not required for virion assembly, hemagglutination, or virus adsorption functions (Klenk et al. 1975; Lazarowitz and Choppin 1975). Polyacrylamide gel electrophoresis (PAGE) analyses revealed that cleavage activation resulted in the digestion of the HA precursor protein (HA0) into faster migrating polypeptides known as the HA1 and HA2 subunits. N-terminal sequencing of the HA2 polypeptides derived from various infectious viruses revealed a highly conserved sequence, GLFGAIAGFIE (Skehel and Waterfield 1975), which is the N-terminal portion of the domain now universally referred to as the fusion peptide. Subsequent studies clearly demonstrated that the critical function activated by proteolytic cleavage is HA-mediated membrane fusion of the viral and endosomal membranes, facilitating transfer of the viral genome into host cells during virus entry for replication (Huang et al. 1980, 1981; Maeda and Ohnishi 1980; White and Helenius 1980).
1 Structural Basis for Activation of HA Fusion Potential
From a structural perspective , the mechanisms by which influenza HA becomes fusogenic upon protease cleavage and the subsequent acid-induced conformational rearrangements that drive the membrane fusion process in endosomes are quite well developed. For the HA of H3N2 subtype viruses that have circulated in humans since 1968, there is high-resolution structural information for the uncleaved HA0 precursor, the cleaved pre-fusion neutral pH HA present on the surface of infectious virions, and the low pH conformation adopted by HA following the acid-induced structural rearrangements required for fusion (Bizebard et al. 1995; Bullough et al. 1994; Chen et al. 1998; Wilson et al. 1981). Therefore, we base most of our structural discussions on the HA of the 1968 human virus A/Aichi/2/68 (H3N2).
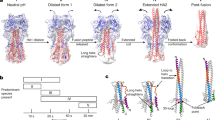
For discussion purposes, we generally assume that infection of a new host is initiated by virions that contain cleaved HAs on their surfaces and are therefore fully activated to facilitate membrane fusion; however, infectious virions can also contain mixed ratios of cleaved and uncleaved HAs on their surfaces, and there may be examples for which cleavage can also occur in endosomes during the early stages of entry. In any case, influenza viruses initiate infection by attaching to sialic acid-containing glycan receptors on host cell surfaces, foll owed by entry via the endocytic pathway. The trigger for fusion of the viral and endosomal membranes is the acidification of the vesicular compartments by cellular proton pumps. When the endosomal pH reaches a critical threshold, usually between pH 6.0 and 5.0 depending on the viral strain, a number of HA structural rearrangements are triggered that coordinately function to drive the fusion process (Bizebard et al. 1995; Bullough et al. 1994; Chen et al. 1999). The pre- and post-fusion structures of HA are shown in Fig. 1.1, and the conformational changes that take place include detrimerization of the membrane-distal head domains, extrusion of the HA2 N-terminal fusion peptide domains from the trimer interior, extension of the long HA2 coiled coil by recruitment of the short HA2 α-helix and connecting polypeptide to the N-terminal end of the long α-helix of each monomer, and 180° reorientation of the C-terminal domain of the long α-helices to “jackknife,” this domain against the central coiled coil. The extension of the central coiled coil directs the fusion peptides to the end of the low pH structure (the top, as shown in Fig. 1.1, right panel), and residues C-terminal to the “jackknifed” helices trace along a groove on the outside of the coiled coil, as can be viewed for the polypeptide chain of one monomer to the left of the coiled coil in Fig. 1.1, and this places the HA2 C-terminal transmembrane domains at the same end of the rod-shaped structure as the fusion peptides (Fig. 1.1, right panel). Based on these structural changes, a mechanism for initiating the membrane fusion process can be envisaged, in which hydrophobic fusion peptides are “harpooned” into the endosomal membrane to link the viral and cellular membranes via the HA2 subunit for influenza A virus, and the 180° “jackknife” of the long helices bring the viral and endosomal membranes into proximity as a prerequisite for the fusion process.

Structures of the neutral pH cleaved HA (left) and the low pH conformation (right) assumed when membrane fusion is triggered by acidification of endosomes. In the neutral pH structure (left), the HA1 subunit is shown in blue, the HA2 subunit is in gray, and the C-terminal residues of HA1 are highlighted in green, and HA2 N-terminal fusion peptide residues highlighted in red. Keep in mind that the neutral pH structure is derived from HA ectodomains solubilized by proteolytic treatment of virions, which removes the HA transmembrane domains located near the C-terminus of each HA2 subunit (the viral membrane would be at the bottom of the figure). The low pH rodlike structure (right) is composed entirely of HA2 residues (gray). Hatched lines indicate regions for which the structure is unknown as they are disordered in the crystal structure (linker region) or were removed by proteolysis to solubilize the protein for crystal preparation (fusion peptide). The rodlike structure illustrates that fusion peptide residues are relocated to the same end of the helical rod as the C-terminal end of the polypeptide chains (labeled C) that are proximal to the membrane anchor domains (removed by protease treatment); therefore, the viral and endosomal membranes are pulled into close proximity with one another by acid-induced conformational changes
Collectively, the acid-induced HA conformational changes that initiate membrane fusion are irreversible, and the observation that the rodlike low pH HA structure is considerably more stable than cleaved neutral pH HA (Carr et al. 1997; Chen et al. 1995, 1999; Ruigrok et al. 1988) indicates that the cleaved neutral pH HA is a metastable molecule that transitions to a more energetically stable form during the fusion process. In contrast, the uncleaved HA is relatively unresponsive to acidification with respect to structural rearrangements, suggesting that proteolytic cleavage primes the HA for fusion by allowing it to adopt the metastable conformation that can be triggered by acidification to induce membrane fusion. A comparison of the structures of the HA0 and cleaved neutral pH HA reveals that only 19 residues relocate upon protease cleavage (Chen et al. 1998; Wilson et al. 1981); however, two critical prerequisites for fusion transpire up on cleavage. First, protease cleavage liberates the hydrophobic fusion peptide from its internal position within the HA0 polypeptide chain, becoming the N-terminal domain of the newly generated HA2 subunit. Second, the N-terminal 10 residues of the HA2 fusion peptide relocate into a “cavity” present in the interior of the trimeric HA0 structure and form new contacts with a number of ionizable residues that line the cavity. These structural changes are depicted in Fig. 1.2, which shows a side view of the HA0 and cleaved HA ectodomains as well as a view down the threefold axes of symmetry. In the uncleaved HA0 structures (panels a and c), the cleavage loop extends out into solution; the conserved arginine residue that serves as the cleavage site of each monomer is indicated by the arrows. The residues colored in green and red are the ones that relocate upon protease cleavage; the relocation of HA2 N-terminal residues (indicated in red) into the trimer interior is best illustrated when viewed down the threefold axis of cleaved HA (panel d).

Structures of the HA ectodomains of A/Aichi/2/68 virus (H3N2 subtype). Panels a and c represent uncleaved HA0 viewed side-on (a) or side-down the threefold axis of symmetry (c), and panels b and d represent cleaved neutral pH HA from the same orientations. The residues that constitute the HA1 subunits are shown in blue and HA2 in gray, and these residues do not relocate following cleavage (they are superimposable in the two structures). The cleavage loops of HA0 are shown in red and green in panels a and c with arrows indicating the site of cleavage at residue Arg 329. These are the only residues that relocate following cleavage, with residues that constitute the N-terminus of HA2 (red) inserting into the trimer interior, as illustrated most clearly in panel d
A preponderance of evidence sugg ests that the new contacts formed when the HA2 N-terminal fusion peptide inserts into the interior of the HA trimer are critical for priming the metastable HA structure for subsequent acid-induced conformational changes. First, let us consider the fusion peptide itself. The N-terminal 11 residues of HA2 constitute the most highly conserved region of the HA, completely conserved in nearly all strains of the 16 influenza A HA subtype viruses and differing by only one residue in most influenza B strains (Cross et al. 2009; Nobusawa et al. 1991) (Table 1.1). Surprisingly, HAs having mutations in the fusion peptide domain that retain membrane fusion functionality can be selected for in the laboratory and generated easily by reverse genetics and have been documented at nearly every position (Cross et al. 2001, 2009; Daniels et al. 1985; Gething et al. 1986; Korte et al. 2001; Lin et al. 1997; Nobusawa et al. 1995; Orlich and Rott 1994; Qiao et al. 1999; Steinhauer et al. 1995; Yewdell et al. 1993). However, nea rly all HA fusion peptide mutants with substitutions in the N-terminal 10 residues mediate membrane fusion at elevated pH relative to wild-type (WT) HA (Cross et al. 2009). Furthermore, when infectious viruses containing such mutants are generated by reverse genetics, minimal passage frequently results in either reversion to the WT HA residue or pseudoreversion at the position that was altered (Cross et al. 2001). These observations suggest an evolutionary pressure on the HA beyond that which operates on functional interactions with target membranes and suggest that fusion peptide contacts in the metastable cleaved neutral pH HA serve as pH “sensors” that are reactive to acidification. As mentioned previously, in the HA0 structure, the cleavage loop is proximal to a cavity lined with ionizable residues, which are subsequently buried by the fusion peptide upon proteolytic cleavage (Chen et al. 1998). Since the relocation of fusion peptide residues constitutes the only structural change that takes place following cleavage, the newly formed contacts are likely to play a role in potentiating fusion activity. An analysis of this region of cleaved HA shows that HA2 ionizable residues A sp109 and Asp112 are invariantly conserved and form numerous hydrogen bonds with the fusion peptide. Other residues of interest in this region include conserved histidine residues that may serve as potential “trigger” residues for initiating acid-induced conformational changes. The pKa of the histidine side chain is around pH 6.0 in aqueous solution, and therefore, protonation of such residues would occur within the biologically relevant range during endosomal acidification, if accessible to solvent. Of particular interest are HA1 position 17 and HA2 positions 106 and 111. The 16 HA subtypes that circulate in aquatic birds can be phylogenetically and structurally segregated into two groups (Air 1981; Nobusawa et al. 1991; Russell et al. 2004), Group-1 (H1, H2, H5, H6, H8, H9, H11, H12, H13, H16) and Group-2 (H3, H4, H7, H10, H14, H15), and these three positions (HA1 17 and HA2 106 and 111) segregate along evolutionary lin es and are well conserved within groups. In Group-1, HA1 17 is a tyrosine, HA2 106 is an arginine, and HA2 111 is a histidine; in Group-2, HA1 17 is a histidine, HA2 106 is a histidine, and HA2 111 is a threonine. Extensive mutagenesis studies on these residues of an H3 subtype (Group-2) HA reveals that HA1 His17 may play a role in triggering structural changes upon acidification (Thoennes et al. 2008). In Group-1 HAs, HA1 17 is a tyrosine, but nearby HA2 residue His111 is invariantly conserved and may serve an equivalent role to Group-2 HA1 His17, and indeed, mutagenesis studies on a number of Group-1 HAs suggest that changes to His111 result in the inactivation of fusion activity (J. Trost, D.A.S., unpublished).
A number of additional studies support the idea that the contacts made between the fusion peptide and the cavity region may be critical in triggering acid-induced structural rearrangements. HA mutants that mediate membrane fusion at elevated pH relative to WT can be found throughout the HA trimer at domain interfaces that rearrange upon acidification (Byrd-Leotis et al. 2015; Cross et al. 2001; Daniels et al. 1985; Doms et al. 1986; Gething et al. 1986; Lin et al. 1997; Qiao et al. 1999; Steinhauer et al. 1995; Thoennes et al. 2008; Zaraket et al. 2013b), whereas stabilizing mutations, or those resulting in an HA that mediates membrane fusion at a lower pH compared to WT, have been identified at fewer positions to date (Byrd-Leotis et al. 2015; Steinhauer et al. 1991; Thoennes et al. 2008; Xu and Wilson 2011; Zaraket et al. 2013a). The identification of mutants that mediate membrane fusion at higher or lower pH relative to WT provided for the rational design of double mutant HAs, in which mutations known to confer high- and low-pH phenotypes were generated in various combinations to examine the cumulative effects. These studies revealed that when the structural locations of the mutations were proximal, the pH phenotype was additive, whereas when the mutations were distal to one another, the phenotype of one mutation was dominant, with changes in the fusion peptide regi on being most critical in determining the overall fusion phenotype (Steinhauer et al. 1996). These results supported data on the kinetics of conformational changes, as determined using a panel of conformation-specific monoclonal antibodies, which showed that structural alterations in the stem region preceded changes in the HA head domains (White and Wilson 1987). These interpretations were further supported by experiments on the fusion kinetics of single virions with planar lipid bilayers, which indicated that the “withdrawal” of the fusion peptide from the trimer interior is the rate-limiting step (Ivanovic et al. 2013). In addition, another study involving hydrogen-deuterium exchange and mass spectrometry approaches revealed that release of the fusion peptide occurs prior to the structural changes involving coiled-coil rearrangements (Garcia et al. 2015), and studies on the membrane fusion process by Cryo-EM indicate that extrusion of the fusion peptide and insertion into the target membrane as extended intermediates precedes the foldback into helical rods at fusion “dimples” where membrane merger occurs (Calder and Rosenthal 2016). Overall, the data strongly support a mechanism by which HA0 cleavage and coincident conformational changes prime the cleaved neutral pH structure for fusion by generating functional fusion peptides and critically relocating them to structural regions that can respond to acidification.
2 Structure of HA0 Cleavage Sites
Not only is cleavage acti vation required for infectivity, but the available evidence indicates that the sequence and structure of the HA cleavage loop may reveal clues about the ability of HAs to serve as substrates for specific activating proteases and dictate traits such as the pathogenicity of influenza strains and the ecology and host range of influenza viruses. To date, the HA0 precursor structure of three HA subtypes has been determined: the H3 subtype described above from a human 1968 pandemic H3N2 virus (Chen et al. 1998), the H1 subtype representing 1918 human pandemic H1N1 strains (Stevens et al. 2004), and the H16 subtype from an H16N3 avian virus isolated from a black-headed gull (Lu et al. 2012). Figure 1.3 depicts these three structures as a single monomer of the trimer, viewed from equivalent orientation. A visual comparison of these structures reveals major differences in the structural elements of the cleavage loop, including the location of the Arg329 cleavage site (green). Shown in yellow are the conserved electronegative residues discussed above, HA2 D109 and D112, HA2 His 17 for H3, and HA2 His 111 for H1 and H16. The H3 Arg329 cleavage site is located within a relatively standard loop structure that orients away from the trimer surface, leaving it easily accessible at a distal position in the loop. Unlike the H3 HA0, the loop structure of the H1 HA0 packs against the surface of the trimer, and the Arg329 residue is positioned such that it covers the electronegative cavity, though it orients out into solution. The H16 cleavage loop is somewhat similar to the H1 loop in that it packs against the trimer surface, but unlike the other two, it contains a five-residue α-helix that includes posi tions 325 through the Arg329 that is cleaved by activating proteases. Remarkably, this helix covers the electronegative cavity and orients the side chain of Arg329 into the cavity where it forms a salt bridge with highly conserved HA2 residue D112. The orientation of Arg329 in H16 HA0 is likely a factor in limiting its accessibility to proteases that can activate other subtypes, but not H16 (see below). It is possible that the differences in cleavage loop structures may exist, in part, due to the location of proximal glycosylation sites, which for H1 and H16 is at HA1 N20 (H3 numbering), whereas for H3 the glycosylation site is slightly farther away at HA1 N22 (Chen et al. 1998; Lu et al. 2012; Stevens et al. 2004). It should also be noted that the H3 HA0 structure was derived from protein expressed in mammalian cells, and the H1 and H16 structures were based on proteins expressed using baculovirus expression systems. It will be very interesting to extend our knowledge of HA0 precursor structures to additional subtypes and relate these to data th at is accumulating on the range of proteases capable of activating individual HA subtypes.

Cleavage site structures showing a single monomer of the HA0 trimer for H3 subtype (a), H1 subtype (b), and H16 subtype (c). (a) The cleavage domain of H3 (red) forms a loop structure with R329 (green) oriented out into solution. Shown in yellow are residues that line the electronegative cavity, residues HA2 D109, HA2 D112 (subscript denotes the HA2 subunit in the figure), and HA1 His 17 (between D2112 and R329). (b) The cleavage domain of H1 (magenta) packs more closely against the trimer, but R329 (green) remains oriented out into solution. HA2 D109, HA2 D112, and HA2 H111 are shown in yellow, though H111 is on the opposite face of the long helix and largely hidden in the depicted orientation. (c) The cleavage domain of H16 (orange) contains a small helical domain that orients the side chain of R329 (green) into the electronegative cavity, where it forms a salt bridge with HA2 D112 (again, ionizable residues shown in yellow). In all panels, residues that will constitute the HA1 and HA2 subunits following cleavage are shown in blue and gray, respectively
3 Activating Proteases
Whereas the residues of the cleavage loop that constitute the N-terminal portion of the fusion peptide are highly conserved, the upstream residues between the highly conserved Pro324 (H3 numbering) and Arg329 cleavage site are quite variable among subtypes and provide important clues regarding cleavage activation and the proteases involved (Tables 1.1 and 1.2). In particular, variations in the number and sequence of arginine and lysine residues at the cleavage site account for major differences in proteolytic activation and proved to be critical determinants of pathogenicity (Bosch et al. 1981; Garten and Klenk 1999; Klenk and Garten 1994; Steinhauer 1999) and host range (Galloway et al. 2013). Other factors that can influence the capacity for particular proteases to activate HAs include the presence or absence of nearby carbohydrates that can alter the accessibility of an activating protease (Deshpande et al. 1987; Kawaoka et al. 1984; Kawaoka and Webster 1989; Ohuchi et al. 1991). Studies on a wide range of influenza strains and subtypes have addressed functional activation of infectivity or fusion potential, and a v ariety of candidate proteases have been identified that activate low pathogenic avian influenza (LPAI) viruses and mammalian strains at monobasic cleavage sites. They include membrane-anchored serine proteases (MASP) (see Chap. 8) such as human airway tryptase (HAT) (Bottcher et al. 2006; Bertram et al. 2012), transmembrane protease, serine S1 member 2 (TMPRSS2) (Bottcher et al. 2006; Chaipan et al. 2009; Hatesuer et al. 2013; Sakai et al. 2014; Tarnow et al. 2014), TMPRSS4 (Bertram et al. 2010a; Kuhn et al. 2016), and matriptase (Baron et al. 2013; Hamilton et al. 2012), as well as soluble serine proteases (see Chap. 9) such as tryptase Clara (Kido et al. 1992), plasmin (Lazarowitz et al. 1973b), and kallikrein-related peptidases (Hamilton and Whittaker 2013). In considering the numerous studies published to date on an extended panel of proteases, it is apparent that n o single universal protease is involved in activating the membrane fusion potential of the HA, as most of the proteases examined activate subsets of viruses in nonoverlapping fashion and with extensive variation in cleavage efficiency. For example, Galloway et al. (2013) characterized cleavage activation from representatives of all 16 avian HA subtypes and several human strains for activation by trypsin, HAT, and TMPRSS2 and observed a variety of phenotypes. Trypsin displayed fairly broad cleavage activation, but showed low or no activity for H12, H13, and H16 subtype HAs, whereas HAT was more limited in its ability to cleave various subtypes, but was able to cleave H9, H11, and H12 HAs. On the other hand, TMPRSS2 was broadly effective but demonstrated no activity against H8 and H12 and was the only one of the three proteases examined in this study that showed specificity for the H13 and H16 HAs . Interestingly, H13 and H16 appear to have a limited host range, generally being isolated only from Charadriiformes, such as gulls and shorebirds, suggesting perhaps that cleavage activation can serve as a host range determinant for particular viruses. Furthermore, a study documenting a repository of Eurasian-lineage reverse genetics vectors and recombinant viruses found that the H13 and H16 recombinant viruses were only able to be recovered when transfected 293T cells were injected into embryonated chicken eggs for further propagation; embryonated chicken eggs are known to express a factor Xa-like serine protease that is capable of cleaving HA0, which may provide the rationale for this observation (Keawcharoen et al. 2010). As mentioned previously, the cleavage loop of the H16 HA appears to be rather structured with the critical Arg329 less exposed than observed in the loop structure of the H3 subtype HA. This may be indic ative of host-specific proteases found in Charadriiformes that have greater activity against H13 and H16 HA substrates, but this remains to be determined. Additionally, it was shown that human TMPRSS2 homologues in chicken and swine were capable of activating an influenza virus having a monobasic cleavage site in vitro (Bertram et al. 2012). In another example, studies using matriptase have shown that it can efficiently activate H1 and H9 subtype viruses, but is much less effective against H2 and H3 subtypes. Furthermore, while matriptase was able to cleave H1 subtype HAs, in general, it displayed selective activity against certain H1 subtype strains (Baron et al. 2013; Hamilton et al. 2012). Similarly, studies examining the cleavage activity of the thrombolytic proteases kallikrein (KLK) 5 and 12 with various HA strains belonging to H1, H2, and H3 subtypes found that KLK5 and KLK12 displayed differential activity against HAs within and across subtypes. For example, KLK12, but not KLK5, was able to cleave H2 HA; the opposite was observed with the H3 HAs examined. For the H1 HAs, KLK5 and KLK12 were able to efficiently cleave the HA from A/California/04/2009 (H1N1), b ut have substantially reduced activity against other H1 strains, such as A/New Caledonia/99, A/South Carolina/18, and the lab-adapted strains A/Puerto Rico/8/34 and A/WSN/33 (Hamilton and Whittaker 2013
Inextricably linked to the recognition of HA as a substrate by activating proteases is the requirement for the protease to generate specific cleavage products containing fusion-competent sequences at the HA2 N-terminus. As mentioned previously, the HA2 N-terminal fusion peptide domain that results from cleavage is the most conserved region in HA, with the sequence GLFGAIAGFIE being virtually invariant in natural isolates. Studies have shown that HA digestion by proteases such as thermolysin, which cleaves between the Gly1 and Leu2 of authentic HA2, renders the HA inactive for fusion and results in noninfectious virus (Garten et al. 1981; Lazarowitz and Choppin 1975; Orlich and Rott 1994; Steinhauer et al. 1995). Interestingly, the selection of influenza viruses capable of replicating in the presence of thermolysin revealed HA mutants having a single amino acid insertion just downstream of the Leu2, effectively restoring the length and spacing of critical residues of the fusion peptide (Orlich and Rott 1994), and fusion peptide length as well as composition has been demonstrated as a requirement for fusion function (Langley et al. 2009; Steinhauer et al. 1995). As mentioned previously, the HAT protease was able to generate HA1 and HA2 cleavage products with similar mobility as HAs digested with trypsin, but the HAT-cleaved H12 HA was not capable of mediating membrane fusion (Galloway et al. 2013). It was also shown that a recombinant virus expressing t he H12 HA replicated poorly in MDCK cells, but replicated quite well in embryonated chicken eggs (Keawcharoen et al. 2010), which express a factor Xa-like serine protease that is capable of activating HA0 (Gotoh et al. 1990).
An influenza HA cleavage anomaly is observed from studies on the H1N1 strain A/WSN/33 . This is a lab-adapted neurovirulent strain selected by passage of the first human influenza A isolate A/WS/33 (Smith et al. 1933) in mouse brain (Francis and Moore 1940) and is efficiently cleaved by the fibrinolytic protease plasmin (Lazarowitz et al. 1973b). The mechanism by which plasmin-mediated HA cleavage occurs was shown to be moderated by the ability of the NA of A/WSN/33 (H1N1) to sequester plasminogen, the zymogen precu rsor to plasmin, until it is converted to plasmin and able to cleave HA0 into HA1 and HA2. The ability of the NA to sequester plasminogen was shown to be dependent on the absence of a carbohydrate at position 146 (N2 numbering) and the presence of a carboxy-terminal lysine (Lys453) (Goto and Kawaoka 1998; Li et al. 1993). This feature has only been observed for the A/WSN/33 NA, but shows specificity for multiple HA subtypes (Goto and Kawaoka 1998). The HA cleavage site sequence of the A/WSN/33 H1 HA contains a serine to tyrosine substitution at the P2 position, resulting in the cleavage site IQY↓R rather than IQS↓R, as is found in most H1 HAs (Table 1.1) (Sun et al. 2010). In addition to the A/WSN/33 H1 HA, a subsequent study reported on the acquisition of a similar cleavage site sequence in a more contemporary seasonal H1N1 strain, A/Beijing/718/2009, that is preferentially cleaved by plasmin (Tse et al. 2013). These data are consistent with other studies showing that bulky hydrophobic amino acids, such as tyrosine, in the P2 position promote HA cleavage by plasmin. Unlike the A/WSN/33 HA, the ability of the HA to be cleaved by plasmin was found to be independent of the NA. Proteolytic conversion of plasminogen into plasmin is mediated by plasminogen activators. It has recently been shown that interferon stimulates the expression of the plasminogen activator inhibitor PAI-1 which prevents not only activation of plasmin but inhibits also other HA-activating proteases, such as TMPRSS2 and HAT. Inhibition of proteolytic activation of HA may therefore be an important mechanism in innate immunity (Dittmann et al. 2015).
Most studies evaluating the proteolytic activation of HA have been cell-based or in vitro studies; however, following the discovery of the ability of TMPRSS2 to cleave influenza A virus HA (Bottcher et al. 2006), several groups investigated this further in knockout and mutant mice by examining the ability of several different influenza A viruses to replicate and cause disease in Tmprss2-deficient mice. These studies showed that H1N1-, H3N2-, and H7N9-infected Tmprss2-deficient mice were generally protected from influenza-associate d morbidity and mortality (Hatesuer et al. 2013; Sakai et al. 2014; Tarnow et al. 2014). TMPRSS2-mediated influenza virus replication in vivo appears to be limited to influenza A viruses, as a recent study showed that influenza B viruses were able to efficiently replicate and cause disease in Tmprss2-deficient mice (Sakai et al. 2016).
Many of the proteases that have been implicated in activating cleavage are secreted and activate HA extracellularly; however, proteases such as TMPRSS2, TMPRSS4, and HAT have membrane-bound forms that may be active during HA transport or at the plasma membrane (Bottcher et al. 2009). In addition, a description of A/WSN/33 HA cleavage in endosomes during virus entry, distinct from the plasmin-directed cleavage described above, has been reported (Boycott et al. 1994). Though this was unique for MDBK cells and the protease involved was not identified, cell-associated proteases have the potential for HA activation during entry as well as during transport following de novo synthesis (Zhirnov et al. 2002). Cumulatively, it appears that among influenza strains, a broad range of proteases have the potential to activate infectivity and that activation can occur in cell-associated fashion at early or late stages of the replication cycle or extracellularly during virus spread in a host or during transmission (see Fig. 1.2 of Bertram et al. (2010b) and Fig. 1.1 of Bottcher-Friebertshauser et al. (2014) for useful illustrations). Proteolytic activation of HA is also an important pathogenic mechanism in viral-bacterial coinfection. It has long been known that various bacteria, including Staphylococcus aureus, secrete proteases that cleave HA and promote the development of pneumonia in virus-infected mice (Tashiro et al. 1987; Maeda 1996). Moreover, bacterial proteases may activate host proteases that cleave HA (Scheiblauer et al. 1992).
Perhaps less complicated are the HAs having cleavage site sequences that consist of a stretch of polybasic amino ac ids, with the general sequence motifs of R-X-(K/R)-R, which can be cleaved in the trans-Golgi by subtilisin-like proteases (see Chap. 10) such as furin and proprotein convertase 5/6 (PC5/6) (Horimoto et al. 1994; Klenk et al. 1984; Stieneke-Grober et al. 1992). The polybasic motif K-K-K-R that has only been observed on rare occasions with highly pathogenic avian influenza (HPAI) viruses (Kawaoka et al. 1984) is cleaved by TMPRSS13 belonging to the MASP group (Okumura et al. 2010). Notably, furin has been shown to proteolytically activate several other viral glycoproteins, including the respiratory syncytial virus fusion (F) protein, human immunodeficiency virus envelope glycoprotein GP120, Crimean-Congo hemorrhagic fever virus Gn prot ein, and the Ebola virus GP, among others (Klenk and Garten 1994; Chaps. 2, 5, 9 this volume). Polybasic cleavage sites are only observed with avian influenza viruses of subtypes H5 and H7 (Tables 1.1 and 1.2). Due to the ubiquitous expression of the proteases activating at polybasic cleavage sites, the viruses cause systemic infection with high lethality in poultry. The HPAI viruses evolve from LPAI variants by insertion of the polybasic motif (Garten and Klenk 1999; Klenk and Garten 1994; Steinhauer 1999). As a result of the insertion, the cleavage site loop is extended (Table 1.2), and this extension appears to be essential for high cleavability, since HAs that have acquired the polybasic motif by amino acid substitution are not cleaved by furin (Bottcher-Friebertshauser et al. 2014). However, sensitivity to furin is not always sufficient to confer a HP phenotyp e. Veits et al. (2012) assessed the capability of multiple HA subtypes to support a HP phenotype by genetically modifying the HA of H1, H2, H3, H4, H6, H8, H10, H11, H14, and H15 subtypes to contain a polybasic cleavage site, followed by the rescue of reassortants after co-transfection with the genes of either a low pathogenic H9N2 or a high pathogenic H5N1. Their results showed that only the reassortants consisting of the polybasic H2, H4, H8, or H14 HA and the remaining genes from the HP H5N1 were capable of supporting the HP phenotype of lethality in chickens. While this study does not rule out the capability of other subtype HAs supporting the HP phenotype, potentially with a different reassorting virus, it does highlight the fact that the polybasic cleavage site alone is not sufficient, and while H5 and H7 subtypes may be predisposed to acquiring a HP phenotype, other HA subtypes are also capable of supporting a HP phenotype.
There is no clear pathway for the emergence of highly pathogenic viruses. Aquatic birds generally display asymptomatic infection with both LPAI and HPAI viruses, despite the fact that they replicate equally well in aquatic birds and poultry, which makes surveillance for HPAI viruses difficult until they present in poultry. In some cases, the emergence of HPAI viruses is through direct transmission from aquatic birds, such as ducks and geese, to poultry, but there have been instances of transmission of HPAI viruses to poultry via aquatic or passerine birds, such as sparrows and starlings, which may display less severe pathogenicity than poultry to infection with HPAI viruses. In other cases, HPAI viruses have evolved from LPAI virus progenitors after introduction to poultry (Steinhauer 1999; Xu et al. 2017).
There are currently two primary mechanisms postulated to be responsible for the insertion of additional nucleotides that encode for basic amino acids. The first is believed to arise through accumulation of point mutations from polymerase slippage of the RNA-dependent RNA polymerase (RdRp) on purine-rich sequences, which are characteristic of the HA cleavage site coding sequences of many H5 and H7 viruses (Perdue et al. 1997). Notably, RdRp slippage on purine-rich or poly(A) sequences is a common feature of many RNA viruses. Briefly, during RNA synthesis of the (+)-sense template, the RdRp may encounter a region of secondary structure near the cleavage site sequence that causes the RdRp to pause and “slip,” or release and reprime, on these purine-rich or poly(A) sequences, resulting in the insertion of nucleotides. When these inserted nucleotides maintain the reading frame of the HA and display relative fitness in a given environment, these viruses may be selected for during replication, resulting in the emergence of a novel HPAI virus. The second mechanism is based on RNA recombination of viral or host RNA with that of the influenza A virus HA gene segment. To date, this has only been reported for H7 HPAI viruses. For example, sequence analysis of the viruses responsible for an outbreak of H7 HPAI in Mexico in 2012 revealed the presence of an eight-amino acid insertion (DRKSRHRRTR↓GLF) that resulted from RNA recombination with a host 28S rRNA (Maurer-Stroh et al. 2013); however, the first report of RNA recombination with a eukaryotic 28S rRNA was by Khatchikian et al. (1989) when the authors described the adaptation of A/turkey/Oregon/1971 (H7N3) to chicken embryo cells, which are not permissive for HA cleavage without the addition of exogenous trypsin. Upon sequencing, the authors discovered a 54-nucleotide (18 amino acids) insertion (SLSPLYPGRTTDLQVPTAR↓G) that resulted in an HA that could be cleaved in chicken embryo cells in the absence of exogenous trypsin as well as in MDCK, BHK, MDBK, and Vero cells. The recombination event was postulated to occur by polymerase jumping using the rRNA as a template during (+)-sense RNA synthesis of the (−)-sense RNA template. Notably, the (+)-sense sequence upstream of the insertion site, AAAGACUA, is identical to the 3′ border of the inserted 28S rRNA sequence, yielding a palindromic sequence (AAAGACU) in the adapted HA gene. Evidence for nonhomologous recombination in the laboratory has also been observed between viral HA and NP sequences with A/seal/Mass/1/80 virus (H7N7), resulting in cleavage loop insertions and accompanied by increased pathogenicity (Orlich et al. 1994). Similar recombination events between viral RNA sequences were subsequently observed in two separate outbreaks of H7 HPAI viruses reported in British Columbia in 2004 and Chile in 2002, both of which were the result of H7 LPAI acquiring insertions from an influenza gene segment that yielded a highly pathogenic variant (Pasick et al. 2005; Suarez et al. 2004). The strains isolated in British Columbia had a seven-amino acid insertion (QAYQKR/QMTR↓G) derived through RNA recombination with the M gene segment (M1), and the strains isolated in Chile had a ten-amino acid insertion (CSPLSRCRETR↓G) derived through RNA recombination with the NP gene segment. Analysis of several HPAI H7 HAs revealed the presence of palindromic sequences surrounding the insertions at the HA cleavage site (Maurer-Stroh et al. 2013). More recently, an outbreak of LPAI H7N9 in China is currently in its sixth epidemic wave and has been responsible for more than 1500 laboratory-confirmed human infections since its emergence in 2013 (WHO 2018). In December 2016, the emergence of an HPAI variant of the H7N9 virus was detected in two human patients as well as poultry that has a four-amino acid in sertion (KRKRTAR↓G) in the HA cleavage site postulated to have originated from a nonhomologous recombination event (Zhang et al. 2017; Zhu et al. 2017). These observations highlight the need to continue rigorous surveillance to monitor emerging strains for the presence of HA cleavage site characteristics that could lead to HP phenotypes.
Concluding Remarks
The link between HA cleavage properties and pathogenicity has been recognized for decades, beginning with early studies elucidating the effect of exogenous trypsin on influenza A virus replication in cell culture to the continual threat of emerging viruses with modified cleavage site sequences that confer a high pathogenic phenotype. Clearly, a variety of both cell-associated and secreted proteases are capable of cleavage activation of influenza A virus HA, in strain-specific fashion. Despite our existing body of knowledge, many gaps remain that relate to the identity and characteristics of proteases that activate most influenza viruses during replication and transmission in nature, to why certain subtypes are more prone to cleavage site modifications that yield high pathogenic phenotypes, and to what extent host-specific proteases restrict the transmission dynamics of certain influenza A virus HA subtypes. Continued investigation to endeavor to fill these gaps is important to our understanding of influenza virus biology and ecology and has critical implications for the development of novel cleavage-specific therapeutics to decrease influenza morbidity and mortality as well as a better understanding of the parameters contributing the emergence of novel pandemic and/or highly pathogenic influenza viruses. For influenza viruses that infect humans, further identification of candidate proteases as well as more detailed examination of those currently identified could lead to the development of antiviral strategies designed to inhibit cleavage activation. The inhibition of HA-activating proteases would present a novel therapeutic strategy for influenza A virus infection; however, as with any therapy that targets a host protein, there are substantial safety concerns. Interestingly, knockout mice generated for the TMPRSS2 studies discussed in this chapter were found to lack a discernable phenotype (Kim et al. 2006; Sales et al. 2011), providing conservative optimism for further pursuing this overall strategy. For all influenza strains, the virus must encounter an HA-activating protease at some stage of the replication or transmission cycle, be it during transport of newly synthesized HAs, at infected cell surfaces or during spread within infected hosts, or even at appropriate sites of infection in a new host, and this simple biochemical process can potentially have profound implications for a broad range of phenotypes related to virus stability, pathogenicity, host range, and natural ecology.
References
Air GM. Sequence relationships among the hemagglutinin genes of 12 subtypes of influenza A virus. Proc Natl Acad Sci U S A. 1981;78:7639–43.
Appleyard G, Maber HB. Plaque formation by influenza viruses in the presence of trypsin. J Gen Virol. 1974;25:351–7.
Baron J, Tarnow C, Mayoli-Nüssle D, Schilling E, Meyer D, Hammami M, Schwalm F, Steinmetzer T, Guan Y, Garten W, Klenk HD, Böttcher-Friebertshäuser E. Matriptase, HAT, and TMPRSS2 activate the hemagglutinin of H9N2 influenza A viruses. J Virol. 2013;87:1811–20.
Bertram S, Glowacka I, Blazejewska P, Soilleux E, Allen P, Danisch S, Steffen I, Choi SY, Park Y, Schneider H, Schughart K, Pohlmann S. TMPRSS2 and TMPRSS4 facilitate trypsin-independent spread of influenza virus in Caco-2 cells. J Virol. 2010a;84:10016–25.
Bertram S, Glowacka I, Steffen I, Kuhl A, Pohlmann S. Novel insights into proteolytic cleavage of influenza virus hemagglutinin. Rev Med Virol. 2010b;20:298–310.
Bertram S, Heurich A, Lavender H, Gierer S, Danisch S, Perin P, Lucas JM, Nelson PS, Pohlmann S, Soilleux EJ. Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS One. 2012;7:e35876.
Bizebard T, Gigant B, Rigolet P, Rasmussen B, Diat O, Bosecke P, Wharton SA, Skehel JJ, Knossow M. Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature. 1995;376:92–4.
Bosch FX, Garten W, Klenk HD, Rott R. Proteolytic cleavage of influenza virus hemagglutinins: primary structure of the connecting peptide between HA1 and HA2 determines proteolytic cleavability and pathogenicity of Avian influenza viruses. Virology. 1981;113:725–35.
Bottcher E, Freuer C, Steinmetzer T, Klenk HD, Garten W. MDCK cells that express proteases TMPRSS2 and HAT provide a cell system to propagate influenza viruses in the absence of trypsin and to study cleavage of HA and its inhibition. Vaccine. 2009;27:6324–9.
Bottcher E, Matrosovich T, Beyerle M, Klenk HD, Garten W, Matrosovich M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J Virol. 2006;80:9896–8.
Bottcher-Friebertshauser E, Garten W, Matrosovich M, Klenk HD. The hemagglutinin: a determinant of pathogenicity. Curr Top Microbiol Immunol. 2014;385:3–34.
Boycott R, Klenk HD, Ohuchi M. Cell tropism of influenza virus mediated by hemagglutinin activation at the stage of virus entry. Virology. 1994;203:313–9.
Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43.
Byrd-Leotis L, Galloway SE, Agbogu E, Steinhauer DA. Influenza hemagglutinin (HA) stem region mutations that stabilize or destabilize the structure of multiple HA subtypes. J Virol. 2015;89:4504–16.
Calder LJ, Rosenthal PB. Cryomicroscopy provides structural snapshots of influenza virus membrane fusion. Nat Struct Mol Biol. 2016;23:853–8.
Came PE, Pascale A, Shimonaski G. Effect of pancreatin on plaque formation by influenza viruses. Arch Gesamte Virusforsch. 1968;23:346–52.
Carr CM, Chaudhry C, Kim PS. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc Natl Acad Sci U S A. 1997;94:14306–13.
Chaipan C, Kobasa D, Bertram S, Glowacka I, Steffen I, Tsegaye TS, Takeda M, Bugge TH, Kim S, Park Y, Marzi A, Pohlmann S. Proteolytic activation of the 1918 influenza virus hemagglutinin. J Virol. 2009;83:3200–11.
Chen J, Lee KH, Steinhauer DA, Stevens DJ, Skehel JJ, Wiley DC. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell. 1998;95:409–17.
Chen J, Skehel JJ, Wiley DC. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA(2) subunit to form an N cap that terminates the triple-stranded coiled coil. Proc Natl Acad Sci U S A. 1999;96:8967–72.
Chen J, Wharton SA, Weissenhorn W, Calder LJ, Hughson FM, Skehel JJ, Wiley DC. A soluble domain of the membrane-anchoring chain of influenza virus hemagglutinin (HA2) folds in Escherichia coli into the low-pH-induced conformation. Proc Natl Acad Sci U S A. 1995;92:12205–9.
Cross KJ, Langley WA, Russell RJ, Skehel JJ, Steinhauer DA. Composition and functions of the influenza fusion peptide. Protein Pept Lett. 2009;16:766–78.
Cross KJ, Wharton SA, Skehel JJ, Wiley DC, Steinhauer DA. Studies on influenza haemagglutinin fusion peptide mutants generated by reverse genetics. EMBO J. 2001;20:4432–42.
Daniels RS, Downie JC, Hay AJ, Knossow M, Skehel JJ, Wang ML, Wiley DC. Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell. 1985;40:431–9.
Deshpande KL, Fried VA, Ando M, Webster RG. Glycosylation affects cleavage of an H5N2 influenza virus hemagglutinin and regulates virulence. Proc Natl Acad Sci U S A. 1987;84:36–40.
Dittmann M, Hoffmann HH, Scull MA, Gilmore RH, Bell KL, Ciancanelli M, Wilson SJ, Crotta S, Yu Y, Flatley B, Xiao JW, Casanova JL, Wack A, Bieniasz PD, Rice CM. A serpin shapes the extracellular environment to prevent influenza A virus maturation. Cell. 2015;160:631–43.
Doms RW, Gething MJ, Henneberry J, White J, Helenius A. Variant influenza virus hemagglutinin that induces fusion at elevated pH. J Virol. 1986;57:603–13.
Francis T, Moore AE. A Study of the Neurotropic Tendency in Strains of the Virus of Epidemic Influenza. J Exp Med. 1940;72:717–28.
Galloway SE, Reed ML, Russell CJ, Steinhauer DA. Influenza HA subtypes demonstrate divergent phenotypes for cleavage activation and pH of fusion: implications for host range and adaptation. PLoS Pathog. 2013;9:e1003151.
Garcia NK, Guttman M, Ebner JL, Lee KK. Dynamic changes during acid-induced activation of influenza hemagglutinin. Structure. 2015;23:665–76.
Garten W, Bosch FX, Linder D, Rott R, Klenk HD. Proteolytic activation of the influenza virus hemagglutinin: the structure of the cleavage site and the enzymes involved in cleavage. Virology. 1981;115:361–74.
Garten W, Klenk HD. Understanding influenza virus pathogenicity. Trends Microbiol. 1999;7:99–100.
Gething MJ, Doms RW, York D, White J. Studies on the mechanism of membrane fusion: site-specific mutagenesis of the hemagglutinin of influenza virus. J Cell Biol. 1986;102:11–23.
Goto H, Kawaoka Y. A novel mechanism for the acquisition of virulence by a human influenza A virus. Proc Natl Acad Sci U S A. 1998;95:10224–8.
Gotoh B, Ogasawara T, Toyoda T, Inocencio NM, Hamaguchi M, Nagai Y. An endoprotease homologous to the blood clotting factor X as a determinant of viral tropism in chick embryo. EMBO J. 1990;9:4189–95.
Hamilton BS, Gludish DW, Whittaker GR. Cleavage activation of the human-adapted influenza virus subtypes by matriptase reveals both subtype and strain specificities. J Virol. 2012;86:10579–86.
Hamilton BS, Whittaker GR. Cleavage activation of human-adapted influenza virus subtypes by kallikrein-related peptidases 5 and 12. J Biol Chem. 2013;288:17399–407.
Hatesuer B, Bertram S, Mehnert N, Bahgat MM, Nelson PS, Pohlmann S, Schughart K. Tmprss2 is essential for influenza H1N1 virus pathogenesis in mice. PLoS Pathog. 2013;9:e1003774.
Horimoto T, Nakayama K, Smeekens SP, Kawaoka Y. Proprotein-processing endoproteases PC6 and furin both activate hemagglutinin of virulent avian influenza viruses. J Virol. 1994;68:6074–8.
Huang RT, Wahn K, Klenk HD, Rott R. Fusion between cell membrane and liposomes containing the glycoproteins of influenza virus. Virology. 1980;104:294–302.
Huang RT, Rott R, Klenk HD. Influenza viruses cause hemolysis and fusion of cells. Virology. 1981;110:243–7.
Ivanovic T, Choi JL, Whelan SP, van Oijen AM, Harrison SC. Influenza-virus membrane fusion by cooperative fold-back of stochastically induced hemagglutinin intermediates. elife. 2013;2:e00333.
Kawaoka Y, Naeve CW, Webster RG. Is virulence of H5N2 influenza viruses in chickens associated with loss of carbohydrate from the hemagglutinin? Virology. 1984;139:303–16.
Kawaoka Y, Webster RG. Interplay between carbohydrate in the stalk and the length of the connecting peptide determines the cleavability of influenza virus hemagglutinin. J Virol. 1989;63:3296–300.
Keawcharoen J, Spronken MI, Vuong O, Bestebroer TM, Munster VJ, Osterhaus AD, Rimmelzwaan GF, Fouchier RA. Repository of Eurasian influenza A virus hemagglutinin and neuraminidase reverse genetics vectors and recombinant viruses. Vaccine. 2010;28:5803–9.
Khatchikian D, Orlich M, Rott R. Increased viral pathogenicity after insertion of a 28S ribosomal RNA sequence into the haemagglutinin gene of an influenza virus. Nature. 1989;340:156–7.
Kido H, Yokogoshi Y, Sakai K, Tashiro M, Kishino Y, Fukutomi A, Katunuma N. Isolation and characterization of a novel trypsin-like protease found in rat bronchiolar epithelial Clara cells. A possible activator of the viral fusion glycoprotein. J Biol Chem. 1992;267:13573–9.
Kim TS, Heinlein C, Hackman RC, Nelson PS. Phenotypic analysis of mice lacking the Tmprss2-encoded protease. Mol Cell Biol. 2006;26:965–75.
Klenk HD, Garten W. Host cell proteases controlling virus pathogenicity. Trends Microbiol. 1994;2:39–43.
Klenk HD, Garten W, Rott R. Inhibition of proteolytic cleavage of the hemagglutinin of influenza virus by the calcium-specific ionophore A23187. EMBO J. 1984;3:2911–5.
Klenk HD, Rott R, Orlich M, Blodorn J. Activation of influenza A viruses by trypsin treatment. Virology. 1975;68:426–39.
Korte T, Epand RF, Epand RM, Blumenthal R. Role of the Glu residues of the influenza hemagglutinin fusion peptide in the pH dependence of fusion activity. Virology. 2001;289:353–61.
Kuhn N, Bergmann S, Kosterke N, Lambertz RL, Keppner A, van den Brand JM, Pohlmann S, Weiss S, Hummler E, Hatesuer B, Schughart K. The proteolytic activation of (H3N2) influenza A virus hemagglutinin is facilitated by different type II transmembrane serine proteases. J Virol. 2016;90:4298–307.
Langley WA, Thoennes S, Bradley KC, Galloway SE, Talekar GR, Cummings SF, Vareckova E, Russell RJ, Steinhauer DA. Single residue deletions along the length of the influenza HA fusion peptide lead to inhibition of membrane fusion function. Virology. 2009;394:321–30.
Lazarowitz SG, Choppin PW. Enhancement of the infectivity of influenza A and B viruses by proteolytic cleavage of the hemagglutinin polypeptide. Virology. 1975;68:440–54.
Lazarowitz SG, Compans RW, Choppin PW. Proteolytic cleavage of the hemagglutinin polypeptide of influenza virus. Function of the uncleaved polypeptide HA. Virology. 1973a;52:199–212.
Lazarowitz SG, Goldberg AR, Choppin PW. Proteolytic cleavage by plasmin of the HA polypeptide of influenza virus: host cell activation of serum plasminogen. Virology. 1973b;56:172–80.
Li S, Schulman J, Itamura S, Palese P. Glycosylation of neuraminidase determines the neurovirulence of influenza A/WSN/33 virus. J Virol. 1993;67:6667–73.
Lin YP, Wharton SA, Martin J, Skehel JJ, Wiley DC, Steinhauer DA. Adaptation of egg-grown and transfectant influenza viruses for growth in mammalian cells: selection of hemagglutinin mutants with elevated pH of membrane fusion. Virology. 1997;233:402–10.
Lu X, Shi Y, Gao F, Xiao H, Wang M, Qi J, Gao GF. Insights into avian influenza virus pathogenicity: the hemagglutinin precursor HA0 of subtype H16 has an alpha-helix structure in its cleavage site with inefficient HA1/HA2 cleavage. J Virol. 2012;86:12861–70.
Maeda H. Role of microbial proteases in pathogenesis. Microbiol Immunol. 1996;40:685–99.
Maeda T, Ohnishi S. Activation of influenza virus by acidic media causes hemolysis and fusion of erythrocytes. FEBS Lett. 1980;122:283–7.
Maurer-Stroh S, Lee RT, Gunalan V, Eisenhaber F. The highly pathogenic H7N3 avian influenza strain from July 2012 in Mexico acquired an extended cleavage site through recombination with host 28S rRNA. Virol J. 2013;10:139.
Munster VJ, Baas C, Lexmond P, Waldenstrom J, Wallensten A, Fransson T, Rimmelzwaan GF, Beyer WE, Schutten M, Olsen B, Osterhaus AD, Fouchier RA. Spatial, temporal, and species variation in prevalence of influenza A viruses in wild migratory birds. PLoS Pathog. 2007;3:e61.
Nobusawa E, Aoyama T, Kato H, Suzuki Y, Tateno Y, Nakajima K. Comparison of complete amino acid sequences and receptor-binding properties among 13 serotypes of hemagglutinins of influenza A viruses. Virology. 1991;182:475–85.
Nobusawa E, Hishida R, Murata M, Kawasaki K, Ohnishi S, Nakajima K. The role of acidic residues in the “fusion segment” of influenza A virus hemagglutinin in low-pH-dependent membrane fusion. Arch Virol. 1995;140:865–75.
Ohuchi R, Ohuchi M, Garten W, Klenk HD. Human influenza virus hemagglutinin with high sensitivity to proteolytic activation. J Virol. 1991;65:3530–7.
Okumura Y, Takahashi E, Yano M, Ohuchi M, Daidoji T, Nakaya T, Bottcher E, Garten W, Klenk HD, Kido H. Novel type II transmembrane serine proteases, MSPL and TMPRSS13, proteolytically activate membrane fusion activity of the hemagglutinin of highly pathogenic avian influenza viruses and induce their multicycle replication. J Virol. 2010;84:5089–96.
Orlich M, Gottwald H, Rott R. Nonhomologous recombination between the hemagglutinin gene and the nucleoprotein gene of an influenza virus. Virology. 1994;204:462–5.
Orlich M, Rott R. Thermolysin activation mutants with changes in the fusogenic region of an influenza virus hemagglutinin. J Virol. 1994;68:7537–9.
Pasick J, Handel K, Robinson J, Copps J, Ridd D, Hills K, Kehler H, Cottam-Birt C, Neufeld J, Berhane Y, Czub S. Intersegmental recombination between the haemagglutinin and matrix genes was responsible for the emergence of a highly pathogenic H7N3 avian influenza virus in British Columbia. J Gen Virol. 2005;86:727–31.
Perdue ML, Garcia M, Senne D, Fraire M. Virulence-associated sequence duplication at the hemagglutinin cleavage site of avian influenza viruses. Virus Res. 1997;49:173–86.
Qiao H, Armstrong RT, Melikyan GB, Cohen FS, White JM. A specific point mutant at position 1 of the influenza hemagglutinin fusion peptide displays a hemifusion phenotype. Mol Biol Cell. 1999;10:2759–69.
Ruigrok RW, Aitken A, Calder LJ, Martin SR, Skehel JJ, Wharton SA, Weis W, Wiley DC. Studies on the structure of the influenza virus haemagglutinin at the pH of membrane fusion. J Gen Virol. 1988;69(Pt 11):2785–95.
Russell RJ, Gamblin SJ, Haire LF, Stevens DJ, Xiao B, Ha Y, Skehel JJ. H1 and H7 influenza haemagglutinin structures extend a structural classification of haemagglutinin subtypes. Virology. 2004;325:287–96.
Sakai K, Ami Y, Nakajima N, Nakajima K, Kitazawa M, Anraku M, Takayama I, Sangsriratanakul N, Komura M, Sato Y, Asanuma H, Takashita E, Komase K, Takehara K, Tashiro M, Hasegawa H, Odagiri T, Takeda M. TMPRSS2 Independency for Haemagglutinin Cleavage In Vivo Differentiates Influenza B Virus from Influenza A Virus. Sci Rep. 2016;6:29430.
Sakai K, Ami Y, Tahara M, Kubota T, Anraku M, Abe M, Nakajima N, Sekizuka T, Shirato K, Suzaki Y, Ainai A, Nakatsu Y, Kanou K, Nakamura K, Suzuki T, Komase K, Nobusawa E, Maenaka K, Kuroda M, Hasegawa H, Kawaoka Y, Tashiro M, Takeda M. The host protease TMPRSS2 plays a major role in in vivo replication of emerging H7N9 and seasonal influenza viruses. J Virol. 2014;88:5608–16.
Sales KU, Hobson JP, Wagenaar-Miller R, Szabo R, Rasmussen AL, Bey A, Shah MF, Molinolo AA, Bugge TH. Expression and genetic loss of function analysis of the HAT/DESC cluster proteases TMPRSS11A and HAT. PLoS One. 2011;6:e23261.
Scheiblauer H, Reinacher M, Tashiro M, Rott R. Interactions between bacteria and influenza A virus in the development of influenza pneumonia. J Infect Dis. 1992;166:783–91.
Shope RE. The etiology of swine influenza. Science. 1931;73:214–5.
Skehel JJ, Waterfield MD. Studies on the primary structure of the influenza virus hemagglutinin. Proc Natl Acad Sci U S A. 1975;72:93–7.
Slusher MJ, Wilcox BR, Lutrell MP, Poulson RL, Brown JD, Yabsley MJ, Stallknecht DE. Are passerine birds reservoirs for influenza A viruses? J Wildl Dis. 2014;50:792–809.
Smith W, Andrewes CH, Laidlaw PP. A virus obtained from influenza patients. Lancet. 1933;2:66–8.
Steinhauer DA. Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology. 1999;258:1–20.
Steinhauer DA, Martin J, Lin YP, Wharton SA, Oldstone MB, Skehel JJ, Wiley DC. Studies using double mutants of the conformational transitions in influenza hemagglutinin required for its membrane fusion activity. Proc Natl Acad Sci U S A. 1996;93:12873–8.
Steinhauer DA, Wharton SA, Skehel JJ, Wiley DC. Studies of the membrane fusion activities of fusion peptide mutants of influenza virus hemagglutinin. J Virol. 1995;69:6643–51.
Steinhauer DA, Wharton SA, Skehel JJ, Wiley DC, Hay AJ. Amantadine selection of a mutant influenza virus containing an acid-stable hemagglutinin glycoprotein: evidence for virus-specific regulation of the pH of glycoprotein transport vesicles. Proc Natl Acad Sci U S A. 1991;88:11525–9.
Stevens J, Corper AL, Basler CF, Taubenberger JK, Palese P, Wilson IA. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science. 2004;303:1866–70.
Stieneke-Grober A, Vey M, Angliker H, Shaw E, Thomas G, Roberts C, Klenk HD, Garten W. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease. EMBO J. 1992;11:2407–14.
Suarez DL, Senne DA, Banks J, Brown IH, Essen SC, Lee CW, Manvell RJ, Mathieu-Benson C, Moreno V, Pedersen JC, Panigrahy B, Rojas H, Spackman E, Alexander DJ. Recombination resulting in virulence shift in avian influenza outbreak, Chile. Emerg Infect Dis. 2004;10:693–9.
Sun X, Tse LV, Ferguson AD, Whittaker GR. Modifications to the hemagglutinin cleavage site control the virulence of a neurotropic H1N1 influenza virus. J Virol. 2010;84:8683–90.
Tarnow C, Engels G, Arendt A, Schwalm F, Sediri H, Preuss A, Nelson PS, Garten W, Klenk HD, Gabriel G, Bottcher-Friebertshauser E. TMPRSS2 is a host factor that is essential for pneumotropism and pathogenicity of H7N9 influenza A virus in mice. J Virol. 2014;88:4744–51.
Tashiro M, Ciborowski P, Klenk HD, Pulverer G, Rott R. Role of Staphylococcus protease in the development of influenza pneumonia. Nature. 1987;325(6104):536–7.
Thoennes S, Li ZN, Lee BJ, Langley WA, Skehel JJ, Russell RJ, Steinhauer DA. Analysis of residues near the fusion peptide in the influenza hemagglutinin structure for roles in triggering membrane fusion. Virology. 2008;370:403–14.
Tobita K, Kilbourne ED. Genetic recombination for antigenic markers of antigenically different strains of influenza B virus. J Virol. 1974;13:347–52.
Tse LV, Marcano VC, Huang W, Pocwierz MS, Whittaker GR. Plasmin-mediated activation of pandemic H1N1 influenza virus hemagglutinin is independent of the viral neuraminidase. J Virol. 2013;87:5161–9.
Veits J, Weber S, Stech O, Breithaupt A, Graber M, Gohrbandt S, Bogs J, Hundt J, Teifke JP, Mettenleiter TC, Stech J. Avian influenza virus hemagglutinins H2, H4, H8, and H14 support a highly pathogenic phenotype. Proc Natl Acad Sci U S A. 2012;109:2579–84.
Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. Evolution and ecology of influenza A viruses. Microbiol Rev. 1992;56:152–79.
White J, Helenius A. pH-dependent fusion between the Semliki Forest virus membrane and liposomes. Proc Natl Acad Sci U S A. 1980;77:3273–7.
White JM, Wilson IA. Anti-peptide antibodies detect steps in a protein conformational change: low-pH activation of the influenza virus hemagglutinin. J Cell Biol. 1987;105:2887–96.
Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature. 1981;289:366–73.
World Health Organization. (2018). Influenza at the human-animal interface [PDF file]. Retrieved from http://www.who.int/influenza/human_animal_interface/Influenza_Summary_IRA_HA_interface_25_01_2018_FINAL.pdf?ua=1
Xu R, Wilson IA. Structural characterization of an early fusion intermediate of influenza virus hemagglutinin. J Virol. 2011;85:5172–82.
Xu Y, Ramey AM, Bowman AS, DeLiberto TJ, Killian ML, Krauss S, Nolting JM, Torchetti MK, Reeves AB, Webby RJ, Stallknecht DE, Wan XF. Low-pathogenic influenza A viruses in North American Diving Ducks contribute to the emergence of a novel highly pathogenic influenza A(H7N8) virus. J Virol. 2017;91:pii: e02208-16.
Yewdell JW, Taylor A, Yellen A, Caton A, Gerhard W, Bachi T. Mutations in or near the fusion peptide of the influenza virus hemagglutinin affect an antigenic site in the globular region. J Virol. 1993;67:933–42.
Zaraket H, Bridges OA, Duan S, Baranovich T, Yoon SW, Reed ML, Salomon R, Webby RJ, Webster RG, Russell CJ. Increased acid stability of the hemagglutinin protein enhances H5N1 influenza virus growth in the upper respiratory tract but is insufficient for transmission in ferrets. J Virol. 2013a;87:9911–22.
Zaraket H, Bridges OA, Russell CJ. The pH of activation of the hemagglutinin protein regulates H5N1 influenza virus replication and pathogenesis in mice. J Virol. 2013b;87:4826–34.
Zhang F, Bi Y, Wang J, Wong G, Shi W, Hu F, Yang Y, Yang L, Deng X, Jiang S, He X, Liu Y, Yin C, Zhong N, Gao GF. Human infections with recently-emerging highly pathogenic H7N9 avian influenza virus in China. J Infect. 2017;75:71–5.
Zhirnov OP, Ikizler MR, Wright PF. Cleavage of influenza a virus hemagglutinin in human respiratory epithelium is cell associated and sensitive to exogenous antiproteases. J Virol. 2002;76:8682–9.
Zhu W, Zhou J, Li Z, Yang L, Li X, Huang W, Zou S, Chen W, Wei H, Tang J, Liu L, Dong J, Wang D, Shu Y. Biological characterisation of the emerged highly pathogenic avian influenza (HPAI) A(H7N9) viruses in humans, in mainland China, 2016 to 2017. Euro Surveill. 2017;22(19):pii: 30533.
Acknowledgments
The authors acknowledge the support by the US Department of Health and Human Services contract HHSN272201400004C (NIAID Centers of Excellence for Influenza Research and Surveillance).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Galloway, S.E., Liang, B., Steinhauer, D.A. (2018). Activation of the Hemagglutinin of Influenza Viruses. In: Böttcher-Friebertshäuser, E., Garten, W., Klenk, H. (eds) Activation of Viruses by Host Proteases. Springer, Cham. https://doi.org/10.1007/978-3-319-75474-1_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-75474-1_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-75473-4
Online ISBN: 978-3-319-75474-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)