
Abstract
Polycrystalline materials’ mechanical properties and failure modes depend on many factors that include segregation of different alloying elements as well as its grain boundaries (GBs) structure. Understanding the parameters affecting the diffusion and binding of alloying elements within GBs will allow enhancing the mechanical properties of the commercial engineering materials and developing interface dominant materials. In practice, the coincidence site lattice (CSL) GBs are experiencing deviations from their ideal configurations. Consequently, this will change the atomic structural integrity by superposition of sub-boundary dislocation networks on the ideal CSL interfaces. For this study, ideal ∑3 GB structures and their angular deviations in BCC iron within the range of Brandon criterion will be studied comprehensively using molecular statics (MS) simulations. GB segregation energy and free surface segregation energies are calculated for carbon atoms. Rice-Wang model will be used to assess the embrittlement impact variation over the deviation angles.
Access provided by CONRICYT-eBooks. Download conference paper PDF
Similar content being viewed by others


Keywords
Introduction
Mechanical properties and failure modes of steels depend on many factors that include segregation of different alloying elements as well as its grain boundaries (GBs) structure [1]. Understanding the parameters affecting the diffusion and binding of alloying elements within GBs will allow enhancing the mechanical properties of the commercial engineering steels and developing interface dominant materials [2]. Some of the elements dissolved in iron lead to enhancing the strength of steel, on the other hand others can degrade the toughness of steel significantly [3]. Being the main alloying element in steel, carbon segregation to different GBs is of ultimate importance.
Overall strength of steel can be greatly controlled through GB cohesion and strengthening. Several studies have been performed in this area. Many numerical studies indicated that carbon increases the cohesion at GB [4, 5]. It is of great importance to mention that the local structure within GB affects the segregation energy. First principle calculations have indicated that strong covalent bond is created between carbon and iron atoms at GB. Experiments showed that segregation of carbon to GBs of steel hinders grain coarsening and interface sliding leading to a steel alloy with ultrahigh strength of 7 GPa [6]. This is due to the role of carbon in decreasing GB energy that results in reducing the driving force for grain coarsening. Carbon segregation at nanocrystalline ferrite GBs, gives the material high thermal stability upon annealing.
Advancements in the field of grain boundary segregation engineering (GBSE) proved that GB segregation can be used as a microstructure design method since solutes affect the structure, phase state and atomic bonds within the steel interface [7]. Experiments in the field of GBSE indicated that deviated structures from coherency are common, even the most coherent GBs can contain deviated portions, where the alloying elements and impurities segregate and alter the GB mechanical properties. As a consequence, considering GB deviated and defected structures is of great importance since many cases ideal CSL GBs not formed; rather a deviation from the misorientation angle exists. Exponential property changes can occur if the GB plane is misaligned by only a few degrees from its most coherent position [7]. Deviant GBs are still considered CSL if the deviation angle is lower than a certain value for each ∑ indicated by Brandon [8]. The deviations introduce dislocation superimposing the GB and are associated with partial dislocations or elastic strain. Different deviation angle alters the trapping sites for impurities by changing the structural integrity of the grain boundary . This will affect segregation tendency of the impurity atoms to the grain boundary . It was showed experimentally using atom probe tomography that changing the deviation angle affects the carbon percentage segregated to the grain boundaries. It has been shown that the segregation values for Σ3 (112) is sensitive to the angular deviations [6]. No prior atomistic study examined the effect of carbon segregation to deviated GB structures within Brandon criteria. This study aims to investigate the effect of deviation angles from the ideal CSL structure for three different common symmetrical tilt grain boundaries (STGB) ∑3 (111), ∑3 (112) and ∑5 (310) on carbon segregation and the cohesive energy changes due to carbon segregation to these interfaces.
Methodology
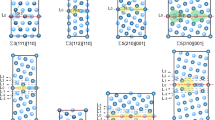
The concept of deviation from ideal CSL is shown in Fig. 1. Angle (ϴ) represents the misorientation angle between the two grains. If the one of the two grains, forming the bi-crystal, is rotated relative to the other by a deviation angle (ϕ) around the Z-axis, the GB will still be regarded as CSL structure. Nevertheless, increasing ϕ above a certain limit causes the GB to lose its special character. In order to determine the deviation angles to be studied, it should be mentioned that Brandon criterion sets the maximum deviation angle from the ideal CSL configuration above which the structure no longer preserve the CSL structure. The maximum deviation angle can be calculated according to Brandon criterion through Eq. (1):

Left: Bi-crystal simulation cell. Right: GB structure of ideal CSL sigma 3 (111) where ϴ represents the misorientation angle between the two grains and ϕ is the deviation angle
where \( \theta_{max} \) is Brandon limit, \( \theta_{ \circ } \) is Brandon limit constant approximately equals to 15° and ∑ is Reciprocal value of the CSL density, therefore the equation is function of ∑ only. For ∑3 grain boundaries the Brandon limit is 8.66°. Hence the chosen deviation for ∑3 grain boundaries are 5°, 9°, where 9° represents the extreme case of Brandon criteria.
Molecular statics (MS) simulations are performed using LAMMPS [9] with embedded atom method (EAM) potential developed by Veiga et al. [10]. The interatomic potential was developed to study of low concentrations of carbon in ferritic solid solution.. Bi-crystal cubic simulation cells are modeled with dimensions of 20 × 20 × 20 nm as shown in Fig. 1. Free surface boundary conditions were employed along the three-directions since the periodicity cannot be preserved for the deviated structures. For the current MS simulations, the simulation cells dimensions are sufficient to eliminate the free surface effect while computing the values of GB segregation energy. In order to define the two crystals geometrically, the first grain is defined with its GB plane normal and axis of rotation, then the second grain is rotated around this axis with angle equals to the sum of the misorientation angle (ϴ) and the deviation angle (ϕ) as shown in Fig. 1. For ideal GB structures, deviation angle is 0°, while for the deviated structures, the deviation angle has values higher than 0° up to Brandon limit. In the cuurent simulations, GB plane normal is taken to be in the Y-direction, while the rotational axis in the Z-direction.
Carbon is inserted at distance 15 Angstrom from GB plane at the octahedral site, which is the most preferable interstitial site for carbon accommodation. At each certain distance from GB, the simulation is done many times with carbon at different position on X and Z directions to reduce the experimental error. The aim of MS technique is to minimize the potential energy of the system, though a numerical iterative process. In order to reach the minimum energy configuration, the coordinates of each atom are iteratively adjusted. The iterations are completed, when satisfying one of the ending criteria. The ending criteria chosen are either the change of the system energy is lower than \( 10^{ - 6} \) or it can be terminated after 10000 iterations. The minimization is done at 0 K using the conjugate gradient (CG) algorithm (Polak-Ribiere version).
As a result of the different orientation of the two grains intersecting at GB, many atoms overlap resulting in an unphysical system with high energy. In order to reach the optimum configuration of the system with global minimum energy, the overlapping atoms should be eliminated. Hence a certain cut off radius under which the close atoms are removed should be determined. A certain criterion [11] is to examine all the cut off radii between 0.0275α and −0.7α (where a is the lattice constant) with increment of 0.05 until reaching the most relaxed structure with minimum energy.
For each GB structure carbon segregation to GB and the surface corresponding to each GB are calculated. GB and surface segregation are required to be quantified to compare the preference of carbon segregation to grain boundary or surface. This will allow to determine the enhancing effect of carbon regarding each GB according to Rice-Wang model as it will discussed later. Eq. 2 is used to calculate GB segregation energy.
where \( E_{seg GB}^{\alpha } \) is the GB segregation energy, \( E_{ GB }^{\alpha } \) is the total energy of the simulation cell with GB and carbon atom allocated at an interstitial octahedral site of the GB, \( E_{GB} \) without carbon , while the \( E_{ bulk}^{\alpha } \) and \( E_{bulk} \) are the total energy of the corresponding single crystal system with and without carbon respectively. The term \( (E_{ bulk}^{\alpha } - E_{bulk} ) \) represents the bulk segregation of carbon . It is put in the equation of grain boundary and surface segregation in order to measure all the segregation energy relative to the bulk segregation to allow for better comparison.
Carbon segregation to the free surface for each GB structure is also calculated. It is important to calculate the surface segregation as well as the GB segregation to establish the relation between the deviation angle and the change in the GB cohesive energy. Carbon tendency to bind with these free surfaces are calculated through determining the carbon free surface segregation energy using Eq. 3:
\( E_{seg FS}^{\alpha } \) is the carbon free surface segregation energy, \( E_{ FS }^{\alpha } \) is the total energy of the free surface single crystal system with carbon atom allocated at an interstitial octahedral site and \( E_{FS} \) is the corresponding system without carbon addition.
Rice-Wang model will be adopted in order to compare quantitatively the effect of GB segregation of carbon to the different deviated structures from ideal CSL. Moreover, this model will allow predicting the change in cohesive energy for each GB structure due to carbon segregation. A positive change in the cohesive energy means a strengthening effect for carbon at this GB structure. Rice and Wang model describes intergranular embrittlement mechanism through the contest between brittle boundary separation and plastic crack blunting. According to Rice and Wang model [12], the ability of an impurity atom to reduce the Griffith work of a grain boundary is a linear function of the difference in the binding energies at the grain boundary and free surface of this impurity atom. Generally, if the impurity atom tends to segregate to free surface rather than the GB, the impurity atom will cause the embrittlement of the material and enhance the intergranular fracture. On the other hand, if the impurity atom tends to segregate to the grain boundary rather than the free surface, the impurity atom will enhance the cohesion of the grain boundary and as a consequence the strength of the material and the creep resistance will be enhanced. According to this model, The change in cohesive energy or embrittlement potency can be calculated as indicated by Eq. (4).
where \( {\text{E}}_{\text{SE}}^{\alpha } \) is the change in cohesive energy or the strengthening energy due to segregation of one carbon atom to the GB and it is equal to the difference between average GB segregation and average free surface segregation for carbon . If the value of \( {\text{E}}_{\text{SE}}^{\alpha } \) is positive then the impurity atom enhance the cohesive strength of the grain boundary . This model will provide a powerful tool to compare the cohesive enhancement of each GB structure due to carbon segregation. Also, the effect of the deviation angles on the change in cohesive energy of those interfaces can be constructed easily.
Results
The GB segregation energy of carbon is calculated within the ideal and deviated GB structures using MS simulations. Extensive simulations have been made for these GB models over different distances from the GB plane. The results are summarized in Fig. (2) and statistical analysis has been made for segregation energy results at each distance level from the GB plane, where the mean of each data population is estimated at 95% confidence level. For \( \sum 3 \left( {111} \right) \) GB, All the segregation energy results among the tested deviation angles showed that GB is preferable location for segregation and binding since there is significant reduction in segregation energy as carbon atom position approaches a bulk site. In addition, the segregation behavior is affected by the angular deviations from the (111) ideal symmetry plane. For \( \sum 3 \left( {112} \right) \) GB, ideal and 5° deviated structure showed less preference for carbon , while 9° showed more binding sites.

GB segregation energy (ev) versus distance along GB plane (Angrstom) for the studied ideal and deviant structure showing less preference sites for carbon (higher segregation energy) for a \( \varvec{ }\sum 3\varvec{ }\left( {112} \right) \) GB compared to b \( \sum 3\varvec{ }\left( {111} \right) \) GB
In order to compare GB segregation of the different deviated structures from ideal CSL, average GB segregation energy is calculated by considering the values of segregation energy between −2 to + 2 Angstrom along the GB plane. The results show that angular deviation from the ideal symmetry plane has an observable effect on the segregation energy. The average GB segregation energy for the studied GBs and their deviations are shown in Fig. (3). For \( \sum 3 \left( {111} \right) \) GB, It is clear that the average GB segregation energy values increase as the deviation angle increases from 0° to 9° indicating less binding of carbon to GB region compared to the ideal one. Increasing the deviation angle to 9°, the average GB segregation energy decreases again with a value slightly lower than 5° structure. On the contrary, for \( \sum 3 \left( {112} \right) \) GB, it is observed that the average GB segregation energy value decrease as the deviation angle increases indicating a higher tendency for carbon to segregate to GB as the deviation increase. The most significant increase in GB segregation energy is for the 9° deviation configuration (slightly higher than Brandon limit) to reach −0.6 eV. GB segregation energy value at 9° is six times higher than the value for the ideal structure indicating that for \( \sum 3 \left( {112} \right) \) GB, exceeding Brandon limit alters the segregation properties considerably. Comparing the GB segregation values for \( \sum 3 \left( {112} \right) \) and \( \sum 3 \left( {111} \right) \) GBs, it is obvious that generally \( \sum 3 \left( {111} \right) \) GB is more preferred for carbon segregation than \( \sum 3 \left( {112} \right) \) GB within GB structures ranging from 0° to 5°. Deviated 9° is the only structure that GB segregation for \( \sum 3 \left( {112} \right) \) is lower than \( \sum 3 \left( {111} \right) \).

Average GB segregation energy (ev) versus deviation angle showing decrease in avg. segregation energy for ∑3 (111) and increase for ∑3 (112) compared to ideal
Due to the difference in the deviation angle for each GB structure, the orientation of the free surface laying in the orthogonal direction to GB plane varies over each GB structure. Hence, the carbon free surface segregation energy is calculated for ideal CSL structure as well as the other deviation angle structures. A single carbon atom at the octahedral site is placed near the free surface to calculate the free surface segregation energy. The positions are varied in different simulations to eliminate the statistical error. The results of the free surface segregation energy are shown in Fig. (4). For the free surface corresponding to \( \sum 3 \left( {111} \right) \) GB, It can be observed that the highest free surface segregation preference (lowest segregation energy) for carbon is to segregate to 9° deviation free surface, followed by the ideal free surface. For the free surface corresponding to \( \sum 3 \left( {112} \right) \) GB, the value of the average free surface segregation corresponding to ideal is the highest. Upon increasing the deviation angle, free surface segregation energy decrease. The highest free surface segregation preference is observed for the free surface of 9° deviation GB similar to \( \sum 3 \left( {111} \right) \) GB. The results of the change in cohesive energy vs deviation angle is shown in Fig. (5). For ∑3 (111), increasing the deviation angle showed a lower cohesive energy enhancement compared to the ideal, however still have a beneficial effect of GB structures. For ∑3 (112) GB, all the deviant structure have higher strengthening energy compared to the ideal one. 9° deviant structure showed the highest beneficial effect.

Average surface segregation energy (ev) versus deviation angle showing decrease in avg. segregation energy for ∑3 (112) compared to ideal, for ∑3 (111) it shows increase at 5° then decrease at 9°

Change in cohesive energy (strengthening energy) (eV) versus deviation angle showing decrease in strengthening energy for ∑3 (111) with increasing deviation angle, for ∑3 (112) strengthening energy increase with increasing deviation angle compared to ideal
Conclusion
Small deviation angle from the ideal misorientation angle of CSL configuration for \( \sum 3 \left( {111} \right) \) and \( \sum 3 \left( {112} \right) \) altered the segregation and binding of carbon to those GBs and their corresponding free surface. With the aid of Rice-Wang model, it was shown that carbon segregation to all the studied ideal and deviant structures has a beneficial effect on the cohesive energy of the GBs. Deviation angle has an observable effect on the strengthening energy of the GBs studied. \( \sum 3 \left( {111} \right) \) and \( \sum 3 \left( {112} \right) \) GB showed different responses to increasing the deviation angle. For \( \sum 3 \left( {111} \right) \), increasing the deviation angle resulted in decreased strengthening energy compared to the ideal structure. An opposite behavior is observed for \( \sum 3 \left( {112} \right) \) that increasing the deviation angle leaded to increased strengthening energy relative to the ideal structure.
References
Wachowicz E, Kiejna A (2008) Effect of impurities on grain boundary cohesion in bcc iron. Comput Mater Sci 43:736–743
Hatem TM, Zikry MA (2009) Dislocation density crystalline plasticity modeling of lath martensitic microstructures in steel alloys. Philos Mag 89:3087–3109
Krasko GL, Olson GB (1990) Effect of boron, carbon, phosphorus and sulphur on intergranular cohesion in iron. Solid State Commun 76:247–251
Hatem TM, Zikry MA (2009) Shear pipe effects and dynamic shear-strain localization in martensitic steels. Acta Mater 57:4558–4567
Kontis P et al (2016) On the effect of boron on grain boundary character in a new polycrystalline superalloy. Acta Mater 103:688–699
Kuzmina M, Ponge D, Raabe D (2015) Grain boundary segregation engineering and austenite reversion turn embrittlement into toughness: example of a 9 wt.% medium Mn steel. Acta Mater 86:182–192
Raabe D et al (2014) Grain boundary segregation engineering in metallic alloys: a pathway to the design of interfaces. Curr Opin Solid State Mater Sci 18:253–261
Brandon DG (1969) The structure of high angle grain boundaries. Phys Status Solidi 31:193–201
Plimpton S (1995) Fast parallel algorithms for short-range molecular dynamics. J Comput Phys 117:1–19
Veiga RGA, Becquart CS, Perez M (2014) Comments on atomistic modeling of an Fe system with a small concentration of C. Comput Mater Sci 82:118
Solanki KN, Tschopp MA, Bhatia MA, Rhodes NR (2013) Atomistic investigation of the role of grain boundary structure on hydrogen segregation and embrittlement in α-Fe. Metall. Mater Trans A Phys Metall Mate Sci 44:1365–1375
Rice JR, Wang JS (1989) Embrittlement of interfaces by solute segregation. Mater Sci Eng, A 107:23–40
Acknowledgment
This project is funded by the Academy of Scientific Research and Technology (ASRT) under joint collaborative Efforts of Egyptians Expatriates & Scientific Organizations towards Tackling R&D Challenges (JESOR), Contract No. 17.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 The Minerals, Metals & Materials Society
About this paper

Cite this paper
Hendy, M., Hatem, T.M., El-Awady, J.A. (2018). Atomistic Simulations of Carbon Diffusion and Segregation in α-Iron Grain Boundaries. In: & Materials Society, T. (eds) TMS 2018 147th Annual Meeting & Exhibition Supplemental Proceedings. TMS 2018. The Minerals, Metals & Materials Series. Springer, Cham. https://doi.org/10.1007/978-3-319-72526-0_30
Download citation
DOI: https://doi.org/10.1007/978-3-319-72526-0_30
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-72525-3
Online ISBN: 978-3-319-72526-0
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)