
Abstract
The secretion of antimicrobial proteins termed killer toxins is common among yeast species from diverse phylogenetic origin. Killer toxin-encoding genes may be localized either in the nucleus or on cytoplasmic double-stranded DNA or RNA molecules of viral ancestry. A number of distinct strategies are utilized by killer toxins to inhibit or kill competitors in a process typically initiated by binding of the toxin to a cell wall receptor. Some toxins target the cell wall itself, whereas others utilize a number of subsequent steps to reach the membrane or intracellular compartments. Finally, killer toxins interfere with vital cellular processes, such as cell wall and membrane integrity, DNA replication, or mRNA translation. In some cases, specific immunity mechanisms are required to prevent suicidal effects of toxin production. Killer strains, their toxins, or peptides derived from killer toxins have proven potential for different applications in human therapy or as antimicrobial agents in food and feed industries.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
I. Introduction
A large variety of antimicrobial substances are produced by pro- and eukaryotic microorganisms in order to improve their ability to dominate a certain environmental niche by killing or inhibiting competing microorganisms. Such substances include the classic antibiotics, either low-molecular-weight secondary metabolites or peptides that are routinely synthesized non-ribosomally and contain a limited number of amino acids. Larger antimicrobial proteins synthesized by the ribosome are also produced by pro- and eukaryotic microorganisms and include the bacteriocins and yeast killer toxins. Several of these natural microbial products are of considerable interest for applied aspects in medicine, agriculture, and food industries. In this review, we focus on the large group of ribosomally synthesized antimicrobial proteins termed killer toxins from different yeast species and summarize current knowledge of toxin diversity in terms of mechanism and structure. We further present selected examples highlighting their application potential. Reviews focusing on yeast killer toxins generally or more specifically addressing specific subtypes have been published (Stark et al. 1990; Bussey 1991; Magliani et al. 1997; Schaffrath and Meinhardt 2005; Golubev 2006; Schmitt and Breinig 2002, 2006; Klassen and Meinhardt 2007; Jablonowski and Schaffrath 2007). However, continuous progress in the identification of toxin modes of action and immunity mechanisms and in the demonstration of new application potential has been made recently.
The discovery of yeast killer toxins dates back to 1963, when Bevan and Makover (1963) described the secretion of molecules by a certain isolate of brewer’s yeast Saccharomyces cerevisiae that inhibited growth of other yeast strains. Later the secreted molecule was identified as a protein (Woods and Bevan 1968) which was named killer factor or killer toxin and the producing strain termed killer yeast. Until today, production of killer toxins was identified in over 100 ascomycetous and over 50 basidiomycetous yeast species (reviewed in Klassen et al. 2017). Several of these toxins were studied extensively, either with a focus on structure and mechanisms of cell killing or to investigate their applied aspects.
Even though killer toxin production is quite common among yeasts, the toxins are as heterogenous as is the phylogenetically diverse group of fungi regarded as “yeast.” Many of the yeast killer toxins are small basic proteins (<20 kDa), but other examples include multimeric protein complexes of over 100 kDa. Apart from the killer proteins which exhibit an antibiotic activity against other microorganisms, there are also other antimicrobial substances, such as toxic glycolipids that can be produced by different yeast species and confer a killer-like phenotype to the producer strain (reviewed in Golubev 2006). However, these non-proteinaceous yeast antibiotics are not considered “killer toxins” in the common sense and will not be focused on in this review.
The cellular localization of killer toxin-encoding genes varies for distinct killer types. Whereas the majority of yeast killer toxins are encoded in the nucleus, some are encoded by selfish genetic elements consisting of viral or viruslike dsDNA or dsRNA molecules that persist in the killer strain’s cytoplasm. In the following parts, we will separately address the diverse group of killer toxins based on the distinct localization of the encoding genes and summarize current knowledge on killer toxins of different genera and toxic mechanisms, focusing on well-characterized killer toxins.
II. Chromosomally Encoded Killer Toxins
A. Cyberlindnera
The genus Cyberlindnera (formerly Williopsis, Pichia, and Hansenula) comprises several species with well-characterized chromosomally encoded killer toxins (reviewed in Klassen et al. 2017). Two extensively studied toxins from this genus are HM-1 and WmKT from C. mrakii (formerly known as Williopsis saturnus var. mrakii and Hansenula mrakii) (Ashida et al. 1983; Kasahara et al. 1994; Guyard et al. 2002a).
HM-1 (also known as HMK) is a 10.7 kDa basic protein, which is encoded by the HMK gene and exhibits remarkable thermo- and pH-stability (Yamamoto et al. 1986a, b) that is probably achieved by five intramolecular disulfide bonds (Yamamoto et al. 1986a, b; Ashida et al. 1983; Lowes et al. 2000). HM-1 resists treatment at 100 °C for 10 min and remains active in between pH 2 and 11 (Ashida et al. 1983; Lowes et al. 2000).
As for several other toxins discussed below, HM-1 is initially translated as a preprotoxin that is subsequently processed by several cleavage events during maturation (Fig. 1). Specifically, the mature, 88 amino acid (aa) spanning HM-1 toxin is formed by signal peptidase-mediated cleavage of the N-terminal signal peptide and Kex2 endopeptidase-mediated removal of a propeptide region from the 125 aa preprotoxin during secretion (Fig. 1; Kimura et al. 1993). The three-dimensional structure of HM-1 has been solved (Antuch et al. 1996; Fig. 1). The structure revealed a surprising folding similarity to γB-crystallin, a major constituent of vertebrate eye lenses, a finding that was unexpected due to the lack of detectable homology between HM-1 and γB-crystallin at the amino acid level (Antuch et al. 1996). It is assumed that eye lens crystallins and HM-1 evolved from an ancestral single-domain precursor. As for HM-1, crystallins similarily exhibit outstanding stability, likely a necessity of their structural role in vertebrate eye lenses, where almost no protein turnover takes place (Wistow and Piatigorsky 1988).

Processing of toxin precursors. Toxin precursors are schematically depicted along with their mature toxins below the arrow. HM-1 and WmKT structures based on protein database entries 1KVE and 1WKT, respectively, are shown in cartoon (ribbon/coil) and surface views. Parts constituting mature toxins are given in gray, and parts being removed during processing are in white. Distinct subunits are indicated by shades of gray. Subunit sizes are given in kDa; processing sites by Kex1/Kex2 peptidases as well as signal peptidase (sp) are indicated as such. –S–S–: disulfide bridge in mature toxins; K1 contains multiple disulfide bridges
Earlier work indicated that HM-1 binds to and inhibits β-1,3-glucan synthase, a key enzyme involved in cell wall synthesis (Yamamoto et al. 1986a, b; Komiyama et al. 1996; Takasuka et al. 1995). This inhibitory action toward β-1,3-glucan synthesis was thought to impair cell wall resynthesis in zones of bud formation and to cause subsequent pore formation and cell lysis (Takasuka et al. 1995; Komiyama et al. 1996). It was further demonstrated that osmotic stabilization suppresses toxic effects of HM-1 (Komiyama et al. 1996).
Several toxins (see below) are known to initially bind to a certain cell wall receptor, followed by binding to a secondary membrane receptor. Thus, killer toxins may engage in stepwise cell wall and membrane receptor interactions preceding the actual cell-killing mechanism. HM-1, for example, is assumed to initially bind to β-1,6- and β-1,3-glucan in the cell wall since exogenously supplied β-1,6-/β-1,3-glucan can antagonize HM-1 toxicity, and Saccharomyces cerevisiae kre6 mutants with lowered β-glucan content as well as spheroplasts are toxin resistant (Kasahara et al. 1994; Komiyama et al. 2002). In addition, HM-1 was shown to bind to an unidentified protein in the membrane fraction of yeast cells, possibly representing the secondary membrane receptor (Miyamoto et al. 2006). When using S. cerevisiae alg3 mutants which lack the α-1,3-mannosyltransferase involved in protein glycosylation or other mutants with defects in protein glycosylation, a significantly increased HM-1 resistance was observed (Kimura et al. 1999; Miyamoto et al. 2011). At the same time, HM-1 binding efficiency to the membrane fraction is strongly reduced in the alg3 mutant, suggesting that HM-1 first binds to a glycosylated receptor protein before inhibition of beta-glucan synthase takes place (Miyamoto et al. 2011).
Screening of the genome-wide deletion library of S. cerevisiae identified a number of genes that impact HM-1 sensitivity. Mutants lacking HOG1 or SLT2 genes are defective in signaling pathways responding to high osmotic pressure (high osmolarity glycerol pathway, HOG) or disturbance in cell integrity (cell wall integrity, CWI), respectively, and the mutants are hypersensitive to HM-1 (Miyamoto et al. 2011, 2012). Such mutant responses are consistent with an effect of HM-1 on osmoregulation and induction of cell wall stress, and HM-1 indeed activates both stress response pathways (Miyamoto et al. 2011, 2012). While cell wall integrity could be affected by the action of HM-1 as an inhibitor of β-1,3-glucan synthase, the role of turgor regulation in the cellular response to HM-1 involves an additional, not previously recognized aspect. In fact, the screening of the genome-wide deletion mutant collection identified the genomic FPS1 locus of S. cerevisiae as being crucial for HM-1 toxicity (Miyamoto et al. 2011). Mutants lacking the gene are devoid of a porin membrane channel protein that mediates glycerol export under conditions of low osmotic pressure in order to reduce turgor (Tamás et al. 1999). In further support of a key role of Fps1 in HM-1 toxicity, it was noted that HM-1 sensitivity in distinct yeast species correlates with the presence of an Fps1 orthologue (Yamamoto et al. 1988; Miyamoto et al. 2012). Despite a defect in osmoregulation and proven induction HOG signaling by HM-1, fps1 mutants were shown to exhibit a 17-fold increase in the IC50 dose of HM-1, suggesting that high turgor caused by the mutation is HM-1 protective. Surprisingly, however, the high turgor of fps1 deletion mutants increases sensitivity to inhibition of β-1,3-glucan synthesis by other agents, such as echinocandins, since higher turgor likely increases the tendency of cells with injured wall to rupture (Miyamoto et al. 2012). Therefore, the HM-1-resistant response of fps1 mutant cells was interpreted to indicate that induction of cell wall stress, rather than inhibition of β-1,3-glucan synthesis, might play a major role in the toxic effect of HM-1. Thus, additional research is needed to characterize the role of Fps1 in the HM-1 response and further define the contribution of the described inhibitory effect on β-1,3-glucan synthesis to the toxic mechanism.
The C. mrakii WmKT of strain MUCL4198 is, even though the producing species are identical, rather different from HM-1. The size of 85 kDa (Table 1) is significantly larger than that of HM-1, and, compared to the latter, WmKT has a substantially reduced tolerance to pH and temperature variation (Guyard et al. 2002a). Optimal activity of WmKT was observed at a pH 4.6 and temperatures between 26 and 28 °C. Such acidic pH optimum applies for a number of other killer toxins (see below) and might represent an adaptation to the environmental setting in which killer toxins serve a benefit to the producing cell by inhibiting the growth of competitors. It is assumed that the toxic principle of WmKT is mediated by hydrolysis of cell wall β-glucans (Guyard et al. 2002a). This assumption is based on the suppression of toxic effects of WmKT by application of glucosidase inhibitors and on the detection of in vitro glucosidase activity of the killer toxin (Guyard et al. 2002a). Further, S. cerevisiae kre1 and kre4 mutants, defective in β-1,3- or β-1,6-glucan synthesis, display WmKT resistance (Guyard et al. 2002a, b). In addition to HM-1 and WmKT, strain NCYC500 of C. mrakii (formerly W. mrakii) produces a very small killer toxin (K500, 1.8-5 kDa) that does not exhibit the broad pH and temperature stability characteristic of HM-1 and based on its small size might rather unlikely exhibit an enzymatic activity similar to that of WmKT (Hodgson et al. 1995). Thus, different strains of the same species can produce killer toxins that may have little in common except for the overall purpose of competitor inhibition.
Another species of Cyberlindnera known to form a number of different toxins is C. saturnus (formerly W. saturnus). Strain IFO0117 produces a toxin termed HSK, which is similar to the above described HM-1 (Kimura et al. 1993). Strains IFO0117 and DBVPG4561 produce toxins designated HYI and KT4561 (Table 1; Komiyama et al. 1995, 1998; Buzzini et al. 2004). Of these, HYI might be similar to HM-1, both with respect to size and toxic principle as an inhibitor of the β-1,3-glucan synthase (Komiyama et al. 1995, 1998). The toxic mechanism of KT4561 is currently unknown, but similar to HM-1 it exhibits relatively good pH and temperature stability. However, with an experimentally determined size of 62 kDa, it is a much larger protein than HM-1.
B. Pichia
Several species of the genus Pichia are recognized as toxin producers. Some well-characterized Pichia killer species have been moved to different genera such as Millerozyma or Wickerhamomyces and will be discussed in a separate chapter. Pichia kluyveri (strain 1002) produces a 19 kDa killer toxin which induces toxic effects by forming ion-permeable channels (Middelbeek et al. 1979; Kagan 1983). Such ion channels were shown to result in cell shrinkage accompanied by leakage of ions, adenosine 5′-triphosphate and decrease of intracellular pH (Middelbeek et al. 1980a, b). A similar toxic principle was afterwards assigned to a number of additional toxins from diverse sources (see below). The P. kluyveri toxin is active at acidic conditions (pH 2.5 and 4.7) and at temperatures below 40 °C (Middelbeek et al. 1979, 1980a).
Another species of the genus, the halotolerant yeast P. membranifaciens, secretes a toxin termed PMKT (P. membranifaciens killer toxin), which exhibits a similar toxic principle as for the P. kluyveri toxin (Santos and Marquina 2004a). However, PMKT activity is enhanced by the presence of salt (Marquina et al. 1992; Lorente et al., 1997). PMKT is similar to the P. kluyveri toxin in size (18 kDa) and was found to be active against sensitive yeast cells at temperatures below 20 °C and at acidic pH (below 4.8; Santos et al. 2000). It is assumed that PMKT first binds to β-1,6-glucan as the primary receptor and subsequently interacts with Cwp2, a cell wall mannoprotein (Santos et al. 2007). Interestingly, the mature form of Cwp2 is covalently linked to β-1,6-glucan, while its precursor is attached to the plasma membrane via a glycosylphosphatidylinositol (GPI) anchor. Thus, it is assumed that interactions between PMKT and Cwp2 may assist transport of the toxin from its primary cell wall receptor to the cell membrane, where lethal ion channel formation occurs (Santos et al. 2007; Belda et al. 2017).
Transcriptional profiling of S. cerevisiae cells exposed to PMKT revealed the induction of genes of the high glycerol (HOG) pathway (Santos et al. 2005), resembling later observations described above for the mechanistically unrelated HM-1 (Miyamoto et al. 2011, 2012). In addition, mutants defective in Hog1 are hypersensitive to both HM-1 and PMKT (Santos et al. 2005; Miyamoto et al. 2011, 2012). Hence, PMKT and HM-1 both induce a coordinated transcriptional response in target cells resembling the response to osmotic stress which apparently counteracts the toxic effects of both toxins (Santos et al. 2005; Rep et al. 2000). Further studies are required to investigate whether both toxins have additional mechanistic similarities not yet recognized.
Another strain of the same species (P. membranifaciens CYC1086) is known to produce a toxin (PMKT2) with diverse properties (Santos et al. 2009, 2013). PMKT2 is larger than PMKT (Table 1) and exhibits a different mode of action. Instead of using β-glucan as the primary receptor, PMKT2 binds to mannoproteins and stops growth of target cells by inducing an early S-phase cell cycle arrest. At low doses, PMKT2 induces apoptotic cell death, similar to a number of mechanistically unrelated killer toxins (see below) (Santos et al. 2013). For a detailed comparison of PMKT and PMKT2, we refer to a recent review (Belda et al. 2017).
C. Wickerhamomyces and Millerozyma
A variety of killer toxins have been described in different strains of Wickerhamomyces anomalus (formerly Pichia anomala) (Table 1), several of which were isolated from agricultural or food sources (Comitini et al. 2004b; Izgü and Altinbay 2004; Wang et al. 2007a; Muccilli et al. 2013). Production of killer toxins or other growth inhibitory compounds is quite common in this species as a systematic screening revealed antagonistic activities in more than 70% of W. anomalus strains tested from the Russian Collection of Microorganisms (VKM) (Golubev 2015). For some of the Wickerhamomyces killer toxins, information about their killing mechanism is available. Similar to WmKT from C. mrakii, several of these toxins bind to β-glucan in the target cell wall and induce toxic effects by hydrolysis of this major cell wall constituent (Table 1). In particular the glucanase killer toxins from W. anomalus are known to exhibit a broad antimicrobial antimicrobial activity, not restricted to yeast species. Some W. anomalus toxins also inhibit pathogenic bacteria or mycelial fungi and even protozoans which have raised interest in industrial applications (see Chapter IV) (Sawant et al. 1989; Walker et al. 1995; Jijakli and Lepoivre 1998; Izgü et al. 2007a, b; Wang et al. 2007a; Muccilli et al. 2013; Valzano et al. 2016).
The halotolerant yeast Millerozyma farinosa (formerly Pichia farinosa) produces the SMKT (salt-mediated killer toxin), which is expressed as a preprotoxin of 222 aa (Suzuki and Nikkuni 1994). As for HM-1, SMKT preprotoxin is processed by the signal peptidase and the Kex protease during secretion, resulting in mature α- and β-subunits of 6.6 and 7.9 kDa, respectively. In this instance, Kex processing liberates the interstitial γ-polypeptide, forming the mature αβ dimer (Fig. 1; Suzuki and Nikkuni 1994). As for many other killer toxins, SMKT is active at acidic pH (below 5). Higher pH-values induce dissociation of the toxin subunits, resulting in loss of activity (Suzuki et al. 1997). The crystal structure of SMKT has been determined (Kashiwagi et al. 1997), but the nature of its primary receptor remains unknown. Interestingly, SMKT exhibits a remarkable folding similarity to a dsRNA-encoded toxin (KP4) from a phylogenetically distinct origin (Ustilago maydis) (see also Sect. III). In vitro studies with purified liposomes suggest that SMKT results in membrane permeabilization, similar to PMKT and P. kluyveri toxins (Suzuki et al. 2001).
D. Kluyveromyces, Lachancea, and Tetrapisispora
Several Kluyveromyces species secrete toxins with different characteristics. K. lactis produces the well-characterized toxin zymocin, which is encoded by a cytoplasmic plasmid system described in detail in part (Sect. II.B). Chromosomally encoded toxins are known in K. wickerhamii (KwKt) and K. marxianus (K6) (Izgü et al. 1999; Comitini et al. 2004a, b; Comitini and Ciani 2011). The species K. waltii and K. phaffii are also toxin producers and were reclassified as Lachancea waltii and Tetrapisispora phaffii, respectively (Table 1) (Young and Yagiu 1978; Kono and Himeno 1997; Ciani and Fatichenti 2001).
The T. phaffii toxin known as KpKt (Kluyveromyves phaffii killer toxin) is a 33 kDa protein encoded by the TpBGL2 gene and exhibits glucanase activity, similar to several other toxins from W. anomalus (Comitini et al. 2009; Oro et al. 2014). KwKt and K6 were purified as proteins of 72 and 42 kDa; however, their mode of action remains unknown so far.
III. Extrachromosomally Encoded Toxins
A. dsRNA Virus Toxins
A number of well-characterized yeast killer toxins are encoded by killer genes with unusual cellular localization. In fact, the firstly discovered S. cerevisiae killer strains (Woods and Bevan 1968) were found to harbor dsRNA viruses which carry the genetic information for toxin production (Bevan et al. 1973). These S. cerevisiae viruses of the Totiviridae family exist in pairs of separately encapsulated viruslike particles in the cytoplasm (for review, see Wickner 1992, 1996; Schmitt and Breinig 2002, 2006). Strictly required for the system is the presence of the 4.6 kb L-A helper virus, which encodes the major capsid protein (Gag) and a RNA-dependent RNA polymerase (Pol). The Gag-encoding ORF1 and Pol-encoding ORF2 of L-A overlap in the -1 reading frame, and a programmed -1 ribosomal frameshift results in the formation of a Gag-Pol fusion protein, which is required for the replicative cycle of the virus (Icho and Wickner 1989; Dinman et al. 1991). The L-A type Totivirus may occur with or without satellite dsRNAs with the prefix “M.” The M viruses termed M1, M2, M28, and Mlus encode different toxin types (K1, K2, K28, Klus) (Bevan et al. 1973; Schmitt and Breinig 2006; Rodríguez-Cousiño et al. 2011). These satellite viruses depend on the presence of an L-A-type virus, since they utilize the L-A-encoded Gag and Gag-Pol for encapsidation and replication. The capsid encoded by L-A contains 60 Gag-dimers and one or two Gag-Pol fusion proteins. The structure of the capsid contains pores to allow the exit of (+)ssRNA transcribed from the viral genome within the capsid. In addition, they allow acquisition of host metabolites but retain the dsRNA copy of the virus and exclude degradative enzymes (Castón et al. 1997). For the replicative cycle and virus gene expression, a (+)ssRNA copy is generated in the capsid by the RNA-dependent RNA polymerase activity of Gag-Pol. After release from the capsid, such (+)ssRNA copy of the virus is translated by the host ribosome into preprotoxin (M virus) or Gag and Gag-Pol fusion proteins (L-A virus). In addition, the (+)ssRNA is encapsidated into newly formed capsids, in which synthesis of the complementary (–)RNA strand occurs. These yeasts Totiviridae lack an extracellular route of transmission and are therefore termed viruslike particles (to distinguish from viruses with an infectious cycle). Well-characterized and functionally distinct S. cerevisiae toxins encoded by dsRNA viruses are K1 (encoded by M1 virus) and K28 (encoded by M28 virus) (see also Schmitt and Breinig 2002, 2006). More recently, a novel dsRNA-encoded toxin (Klus) was identified and significant progress made in the characterization of the Klus-encoding M and associated helper viruses (Rodríguez-Cousiño et al. 2011, 2013; Rodríguez-Cousiño and Esteban 2017). Also, more recent work established K2 as a toxin type with significant differences to K1 (Servienė et al. 2012; Lukša et al. 2015; Orentaite et al. 2016). Interestingly, the different types of dsRNA-encoded toxins are equally dependent on the presence of an L-A-type helper virus but generally lack conserved toxin sequences (de la Peña et al. 1981; Dignard et al. 1991; Schmitt and Tipper 1995; Suzuki and Nikkuni 1994; Rodríguez-Cousiño et al. 2011).
1. K1 and K2
As assumed for other yeast killer toxins, K1-induced target cell killing occurs in several discrete steps, involving initial contact to a primary cell wall receptor followed by binding of a distinct membrane receptor. K1 uses β-1,6-glucan in the cell wall as the primary receptor and the GPI-anchored cell wall glycoprotein Kre1 as the membrane receptor to reach the plasma membrane and ultimately forms cation-selective ion channels (Hutchins and Bussey 1983; de la Peña et al. 1981; Martinac et al. 1990; Breinig et al. 2002, 2004). The mode of action resembles the above chromosomal PMKT which also causes membrane permeabilization, and it utilizes the same primary but a distinct membrane receptor (Cwp1). Interestingly, however, both membrane receptor proteins (Kre1 and Cwp1) are GPI-anchored and appear in a mature glucan-bound and additionally in a membrane-bound (GPI-anchored) form. Thus, the subsequent interactions of the toxins with cell wall glucan and with both mature and GPI-anchored forms of a membrane receptor may represent a common strategy to mediate transport of the toxin from the initial binding site to the actual cellular target.
The K1 toxin is a dimer with subunit sizes of 9.5 (α) and 9 kDa (β), which are covalently linked by three disulfide bridges (Bostian et al. 1984). Site-directed mutagenesis indicated that both of the subunits are involved in binding to the primary receptor, while the α-subunit alone is required for membrane interaction (Bussey 1991; Zhu and Bussey 1991). Maturation of K1 is well characterized and involves common processing steps of a preprotoxin precursor identified in other toxins. The two subunits of K1 are formed by signal peptidase and Kex1-/Kex2-dependent processing and involve the release of the γ-peptide region (Bostian et al. 1984; Zhu et al. 1992). The processing of K1 proprotoxin is strikingly similar to the above described chromosomally encoded SMKT and the viral K28 toxin, even though the toxins are diverse at the sequence level (de la Peña et al. 1981; Schmitt and Tipper 1995; Suzuki and Nikkuni 1994).
More similarities between PMKT and K1 include the significance of a functional HOG signaling pathway for toxin resistance. S. cerevisiae cells with a defect in HOG signaling—due to the loss of Hog1—display strong sensitivity not only to PMKT but also to K1 (Pagé et al. 2003), suggesting a transcriptional response similar to other osmotic stresses being effective in suppressing K1 toxicity.
Other than the chromosomally encoded toxins, which typically target distinct species, dsRNA-encoded toxins are usually active against S. cerevisiae strains that do not carry the L-A- and M-type Totiviridae, while toxin producers are immune against their own, but not other K-type toxins (Schmitt and Breinig 2006). For K1, the immunity mechanism is known to be mediated by the K1 toxin precursor (preprotox). It was demonstrated, that expression of a cDNA copy of M1 in mutants lacking Kex2 confers K1 immunity in the absence of toxin production. Further, expression of the α-subunit together with 31 N-terminal residues of γ is sufficient for K1 immunity, indicating that the presence of a part of the preprotoxin confers K1 protection (Zhu et al. 1993).
The K2 toxin exhibits similarities to K1 in terms of the cell killing strategy causing membrane permeabilization (Orentaite et al. 2016). Like K1, K2 binds to β-1,6-glucan and apparently also uses Kre1 as the plasma membrane receptor; however, the primary K2 sequence is unrelated to K1, and K2 displays a more acidic pH optimum (Young and Yagiu 1978; Pfeiffer and Radler 1984; Dignard et al. 1991; Schmitt and Breinig 2002; Novotná et al. 2004; Lukša et al. 2015). In addition, K2 killers remain susceptible to K1 (and vice versa), and screening of the S. cerevisiaegenome-wide deletion collection revealed 332 genes changing susceptibility to K2, the majority of which not influencing K1 susceptibility (Dignard et al. 1991; Meskauskas and Citavicius 1992; Servienė et al. 2012). Thus, even though K1 and K2 may utilize similar cell wall and membrane receptors and target the plasma membrane to induce cell killing, functional differences exist between these toxins with respect to target cell interaction and immunity mechanism (Dignard et al. 1991; Meskauskas and Citavicius 1992; Novotná et al. 2004; Servienė et al. 2012).
2. K28
Another well-characterized toxin encoded by a S. cerevisiae dsRNA is K28. Similar to K1, it consists of two small subunits (α, β) of around 10 kDa (Table 2), which are covalently linked by a disulfide bridge (Schmitt and Tipper 1995). K28 is encoded by a single ORF and initially translated as preprotoxin which is processed during secretion by signal peptidase and Kex1/Kex2 to form two toxin subunits from a single polypeptide (Schmitt and Tipper 1990, 1995; Riffer et al. 2002; Schmitt and Breinig 2006). As for K1, the protoxin contains a γ-peptide region intervening the α-β sequences. This γ-peptide is deleted by processing with the Kex2 endopeptidase (Schmitt and Tipper 1995). Despite this striking similarity in toxin maturation between K1 and K28, target cell interaction and killing mechanisms are completely different for both of the toxins.
In fact, K28 is so far the only example of a yeast killer toxin gaining access to the target cell by endocytosis and subsequent retrograde passage of the secretory pathway, followed by exit from the endoplasmic reticulum (ER), a strategy followed by several bacterial toxins of the so-called A/B type. For K28, the toxic α-subunit subsequently enters the nucleus, where it blocks DNA replication (Fig. 2; Schmitt et al. 1996; Eisfeld et al. 2000; Heiligenstein et al. 2006).

Schematic representation of killer toxin mode of action. (a) Toxins targeting cell wall or membrane. β-1,6-glucan is the primary cell wall receptor for chromosomally encoded PMKT and dsRNA-encoded K1. PMKT and K1 utilize GPI-anchored Kre1 and Cwp2 as secondary membrane receptors and induce membrane channel formation. WmKT, W. anomalus toxin, and KpKt act as glucanases. (b) Toxins with intracellular targets. Mannoprotein represents the cell wall receptor for dimeric K28. Chitin is the proposed cell wall receptor for zymocin, PaT, and PiT. Erd2 acts as the secondary membrane receptor for K28, which gains access to the target cell’s cytoplasm by retrograde transport and finally inhibits DNA synthesis in the nucleus. The secondary membrane receptor for zymocin and related toxins PaT and PiT is unknown; however, Ipt1-synthesized sphingolipid is required for cellular uptake of the zymocins’ γ-subunit which cleaves cellular tRNAGlu. In analogy to zymocin γ, toxic subunits of PaT and PiT also enter the target cell to cleave tRNAGln or rRNA, respectively. See text for details and references
The K28 toxin uses cell wall mannoprotein as the primary receptor and subsequently interacts with Erd2 as the membrane receptor (Schmitt and Radler 1987; Schmitt and Breinig 2006; Eisfeld et al. 2000; Becker et al. 2016). Erd2 is a H/KDEL receptor protein which localizes mainly to the Golgi/endoplasmic reticulum and mediates retention of ER-resident proteins, but a minor fraction is present in the cytoplasmic membrane (Semenza et al. 1990; Becker et al. 2016). This minor fraction of Erd2 was shown to bind to the HDEL motif present in the K28 β-subunit (Schmitt and Breinig 2006; Becker et al. 2016), followed by endocytosis and retrograde transport of K28 to the ER (Eisfeld et al. 2000; Becker et al. 2016). The HDEL motif recognized by Erd2 is uncovered upon Kex1 processing of K28 β at the C-terminus (HDELR) and is a strict requirement for toxin function, due to its crucial role in mediating the reentry of the toxin into the secretory pathway of the target cell (Eisfeld et al. 2000).
The exit of the toxin from the ER to the cytoplasm occurs via the the Sec61 translocon (Eisfeld et al. 2000; Heiligenstein et al. 2006). The Sec61 complex mediates bidirectional translocation of protein across the ER membrane. This includes secretory proteins entering the ER and misfolded proteins, which are removed from the secretory pathway for subsequent degradation via the ERAD (ER-associated protein degradation) pathway (reviewed in Nakatsukasa and Brodsky 2008). K28 apparently mimics an ERAD substrate, and the ER chaperones Kar2, Pdi1, Scj1, Jem1, and Pmr1, which normally mediate the ER-specific unfolded protein response, assist exit of partially unfolded but covalently linked α-β subunits from the ER (Heiligenstein et al. 2006). Once in the cytosol, the α-β dimer is split by a toxin-intrinsic mechanism to reduce the disulfide bond between the two subunits, releasing the toxic α-subunit (Suzuki et al. 2017). The free β-subunit gets ubiquitinated and is subsequently degraded by the proteasome, while the α-subunit enters the nucleus and ultimatively inhibits DNA synthesis as the final growth inhibitory event (Schmitt et al. 1996; Heiligenstein et al. 2006). Screening collections of over 5000 mutants with deletions in nonessential or temperature-sensitive alleles of essential genes revealed a number of processes protecting the cells from K28, such as Hog1, vacuolar proteins, and the proteasome (Carroll et al. 2009). Also, the same study identified the AP2 clathrin adaptor subunits as crucial for K28 toxicity, by performing an important function in endocytosis of this toxin (Carroll et al. 2009).
Similar to K1, the preprotoxin of K28 also mediates toxin immunity specifically against K28, and again K28 immunity is also established in a kex2 mutant, which is unable to release active toxin (Schmitt and Tipper 1992; Zhu et al. 1992). While the precise mechanism of K1 immunity is still unknown, details for K28 are established (Breinig et al. 2006). K28 killer cells were shown to reinternalize mature K28 toxin, and an interaction with unprocessed K28 preprotoxin in the cytoplasm is the key step for immunity. Mechanistically, this involves ubiquitination and selective proteasomal degradation of the mature re-internalized K28 (Breinig et al. 2006). Notably, partial immunity is already conferred by the α-subunit alone when present in the cytoplasm and full immunity required only a nonspecific sequence extension to the α-subunit, which strikingly resembles the situation in K1 immunity (Breinig et al. 2006).
3. Other dsRNA Virus Toxins
Similar to S. cerevisiae, the yeasts Hanseniaspora uvarum and Zygosaccharomyces bailii are also known to carry L-A- and M-type dsRNA viruses of the Totiviridae family (Radler et al. 1990; Zorg et al. 1988; Schmitt and Neuhausen 1994; Schmitt et al. 1997; Weiler et al. 2002). The H. uvarum toxin is a 18 kDa monomer which utilizes cell wall β-1,6-glucan as the primary receptor and displays a relatively broad killing spectrum as compared to the S. cerevisiae K1 or K28 toxins (Radler et al. 1990; Schmitt and Neuhausen 1994; Schmitt et al. 1997). The 10 kDa Z. bailii toxin zygocin also exhibits a broad spectrum of sensitive yeasts, which includes human and phytopathogenic fungi (Weiler and Schmitt 2003). While the lethal mechanism of the H. uvarum toxin remains unknown, zygocin was shown to induce cell permeabilization, possibly due to membrane channel formation (Weiler and Schmitt 2003; Schmitt and Breinig 2006). In contrast to the other dsRNA-encoded toxins, the zygocin preprotoxin is not required for immunity of the producer cell. It was demonstrated that Z. bailii whole cells and protoplasts are naturally resistant to zygocin (Weiler et al. 2002), a situation that was observed for chromosomally encoded toxins as well (Weiler et al. 2002).
dsRNA viruses encoding killer toxins are not even restricted to the ascomycetous yeast species. They were also identified in the dimorphic fungus Ustilago maydis (Puhalla 1968), a basidiomycete which has budding yeast like and filamentous growth stages (reviewed in Steinberg and Perez-Martin 2008). Three different immunity specificities (P1, P4, and P6) have been found in this species (Koltin and Day 1976; Tipper and Bostian 1984). In contrast to the S. cerevisiaedsRNA-encoded toxins, which are routinely small dimers, KP1 and KP4 are monomeric proteins of 13.4 and 13.6 kDa, respectively (Park et al. 1994, 1996a); KP6, however, is a dimer with subunits of 8.6 and 9.1 kDa (Tao et al. 1990). The structure of KP4 and KP6 were determined (Gu et al. 1995; Li et al. 1999; Allen et al. 2013). KP4 was found to exhibit structural similarities to scorpion toxins, which are known to act on Na+ channels (Gu et al. 1995). Since KP4’s toxic effects could be suppressed by exogenous Ca2+, it was suggested that KP4 may act by inhibition of Ca2+ channels (Gu et al. 1995). This was further substantiated by demonstrating KP4-mediated inhibition of voltage-gated Ca2+ channels in mammalian neuronal cells (Gu et al. 1995). However, KP4 was also realized to be a close structural homologue of the chromosomally encoded toxin SMKT from M. farinosa (Kashiwagi et al. 1997), which is thought to directly induce membrane permeabilization (Suzuki et al. 2001). KP6 is structurally distinct from KP4; its two subunits exhibit remarkable structural similarity to each other but not to other known toxins (Allen et al. 2013). Again, α- and β-subunits of the toxin are formed by Kex processing of the protoxin and removal of a γ-peptide from the center of the protoxin, located between the α- and β-regions (Tao et al. 1990; Allen et al. 2013). Although it was suggested that KP6 may induce target cell killing by pore formation leading to leakage of cell contents (Peery et al. 1987; Li et al. 1999; Steinlauf et al. 1988), the presence of intramolecular disulfide bonds in the α- and β-subunits of KP6 were interpreted to be more consistent with an indirect induction of cell lysis by the toxin, for example, by activation of membrane channels (Allen et al. 2013).
A number of additional basidiomyceteous yeasts were identified to carry viruslike particles with dsRNA genomes, which are associated with killer phenotypes (Table 2) (Golubev et al. 2002, 2003; Pfeiffer et al. 2004; Fuentefria et al. 2008). Some characteristics of the encoded toxins are described, but their modes of action and structural details remain to be investigated.
B. Toxins Encoded by Viruslike Elements (VLEs)
A second type of extranuclear genetic information associated with killer toxin production is represented by the dsDNA elements previously termed linear plasmids, killer plasmids, but more recently, due to evidences for viral ancestry viruslike elements (VLE) (reviewed in Satwika et al. 2012a; Meinhardt and Schaffrath 2001; Schaffrath and Meinhardt 2005; Klassen and Meinhardt 2007). Reminiscent of the S. cerevisiaeL-A Totivirus being associated with satellite M viruses that confer the killer phenotype, dsDNA killer elements (Fig. 3) can be distinguished into autonomous and nonautonomous traits as well. They occur in pairs or triplets in a number of different ascomycetous genera such as Debaryomyces, Millerozyma, Babjeviella, Saccharomycopsis, Schwanniomyces, and Botryoascus but also in the basidiomycete Tausonia (Trichosporon) pullulans (Kitada and Hishinuma 1987; Ligon et al. 1989; Worsham and Bolen 1990; Hayman and Bolen 1991; Bolen et al. 1992; Cong et al. 1994; Fukuhara 1995; Chen et al. 2000).

Schematic representation of viruslike dsDNA plasmids from yeasts encoding killer toxins. dsDNA plasmids are grouped according to the target molecules. Arrows indicate ORFs and their transcriptional direction; terminal proteins are depicted as filled circles; terminal inverted repeats correspond to filled triangles. Known or proposed functions of encoded proteins are indicated. Toxin, intracellular toxic subunit; imm, immunity proteins. The predicted toxin uptake protein is homologous in all systems
As for the Totiviridae, most of these dsDNA molecules reside in the cytoplasm of the host, and the autonomous element provides key functions to facilitate cytoplasmic DNA replication and transcription, which the associated nonautonomous elements depend on (reviewed in Jeske et al. 2006; Klassen and Meinhardt 2007).
Cytoplasmic replication is initiated using a free –OH group of the VLE-encoded terminal protein (TP) (protein priming). The TP is expressed as a fusion protein with a viral B-type DNA polymerase, and after completion of replication, it remains covalently bound to the 5′ ends of the plasmids (Tommasino et al. 1988; Hishinuma and Hirai 1991; Hishinuma et al. 1984; Stark et al. 1984; Sor and Fukuhara 1985; Klassen et al. 2001; Klassen and Meinhardt 2003; Jeske and Meinhardt 2006). Related B-type DNA polymerases involved in a similar mode of replication are also found in adenoviruses and certain bacteriophages (reviewed in Klassen and Meinhardt 2007).
For cytoplasmic transcription, a uniquely structured VLE-encoded RNA polymerase and a mRNA capping enzyme are employed which are both encoded by the autonomous element as well (Wilson and Meacock 1988; Larsen et al. 1998; Schaffrath et al. 2000). The capping enzyme is most closely related to the one from cytoplasmic vaccinia virus (Larsen et al. 1998; Tiggemann et al. 2001; Klassen and Meinhardt 2007). The genes on the killer elements are equipped with unique cytoplasmic promoters characterized by a short 6 nt consensus sequence that is recognized by the plasmid-encoded RNA polymerase (Kämper et al. 1989a, b; Kämper et al. 1991; Romanos and Boyd 1988; Schaffrath et al. 1996; Schickel et al. 1996). Due to the unique promoter structure of cytoplasmic genes, nuclear genes cannot be expressed on the cytoplasmic dsDNA elements unless their promoter is exchanged, and—vice versa—plasmid genes cannot be expressed in the nucleus without modification (Romanos and Boyd 1988; Schaffrath and Meacock 1996; Stark et al. 1990; Meinhardt et al. 1994; Schaffrath et al. 1995; Schründer and Meinhardt 1995; Schickel et al. 1996). As for the satellite M viruses, killer toxin production is in all cases known exclusively associated with the nonautonomous elements (Fig. 3), but only some of the described nonautonomous elements are in fact associated with a killer phenotype. A number of nonautonomous elements are apparently cryptic, but several of them show remnants of killer toxin genes, suggesting loss of functional toxin genes during evolution (Klassen et al. 2002; Klassen and Meinhardt 2007; Satwika et al. 2012b).
The currently recognized dsDNA-encoded killer toxins are found in Kluyveromyces lactis (zymocin), Millerozyma (Pichia) acaciae (PaT), Debaryomyces (Wingea) robertsiae (DrT), and Babjevia (Pichia) inositovora (PiT). They were found to target distinct tRNA species in target cells, such as tRNA or rRNA.
1. tRNA-Targeting Toxins
a) Zymocin
The best studied instance of the tRNA-targeting toxins is zymocin from K. lactis, which is encoded by the dsDNA element pGKL1 that is associated with the autonomous pGKL2 (Stark and Boyd 1986; Stark et al. 1990). The toxin is a heterotrimer (αβγ) with subunit sizes of 99, 30, and 28 kDa, which are encoded by 2 separate genes on pGKL1 (Jablonowski and Schaffrath 2007). As for various other toxins (see above), the polypeptide encoded by the larger ORF (ORF2) is processed by signal peptidase and the K. lactis Kex1 (S. cerevisiae Kex2 homologue) endopeptidase during secretion to form α- and β-subunits (Hishinuma et al. 1984; Stark et al. 1984, 1990; Sor and Fukuhara 1985; Stark and Boyd 1986; Tokunaga et al. 1987). The γ-subunit is encoded by a separate gene and becomes covalently linked to β via a disulfide bond (Stark and Boyd 1986; Stark et al. 1990; Wemhoff et al. 2014). Zymocin utilizes target cell wall chitin as the primary receptor for target cell binding. This step is mediated by the α-subunit, which is characterized by the presence of a chitin-binding and chitinase domain (Stark et al. 1990; Butler et al. 1991a; Jablonowski et al. 2001). Unlike other known killer toxins not targeting the cell wall itself, the receptor-binding part of the toxin can also hydrolyze the receptor, and the ability to do so appears to be essential for toxin function (Butler et al. 1991a; Wemhoff et al. 2014). Following binding and possibly hydrolysis of chitin, the γ-subunit is imported into the target cell in a poorly understood process. This is thought to involve the aid of the hydrophobic β-subunit and depends on a particular membrane sphingolipid (M(IP)2C) as well as a proton gradient generated by the plasma membrane ATPase Pma1 (Mehlgarten and Schaffrath 2004; Zink et al. 2005).
In addition to the chitinase activity of the α-subunit, also the disulfide bond between β and γ is essential for the killing activity of zymocin (Butler et al. 1991a; Wemhoff et al. 2014). However, toxicity of the complex absolutely requires the presence of the γ-subunit and this protein alone, when conditionally expressed inside the cell mimics toxic effects of the trimeric complex (Tokunaga et al. 1989; Stark et al. 1990; Butler et al. 1991b; Frohloff et al. 2001; Wemhoff et al. 2014). The actual mechanism of cell killing is the selective enzymatic cleavage of tRNAGlu UUC by hydrolyzing the phosphodiester bond between the wobble nucleoside (U34) and the 3′ nucleoside (U35) (Lu et al. 2005; Jablonowski et al. 2006). Cleavage of this tRNA is dependent on the presence of the eukaryotic form of the conserved xm5U modification 5-methoxy-carbonyl-methyl-2-thiouridine (mcm5s2U) at the wobble position (Butler et al. 1994; Frohloff et al. 2001; Huang et al. 2005; Lu et al. 2005). The six-subunit Elongator complex (Elp1–Elp6, for a recent review see Schaffrath and Leidel 2017) and the tRNA methyltransferase Trm9 are required for the synthesis of the methoxy-carbonyl-methyl side chain (mcm5U), and a separate sulfur transfer pathway facilitates the thiolation at position 2 (s2U) of the uracil base (Kalhor and Clarke 2003; Huang et al. 2005, 2008; Lu et al. 2005; Noma et al. 2009; Leidel et al. 2009). Consistent with the importance of mcm5s2U for cell killing by the tRNase, loss of ELP1-ELP6 or TRM9 prevents, and loss of any member of the thiolation pathway genes reduces zymocin toxicity (Frohloff et al. 2001; Fichtner et al. 2003; Lu et al. 2005; Jablonowski et al. 2006). This dependency of the toxin on the presence of the complex tRNA modification has been utilized to identify further loci with a previously unknown role in tRNA mcm5s2U modification (Fichtner and Schaffrath 2002; Mehlgarten and Schaffrath 2003; Jablonowski and Schaffrath 2007; Fichtner et al. 2002; Huang et al. 2005; Lu et al. 2005; Bär et al. 2008; Zabel et al. 2008; Studte et al. 2008).
When the γ-subunit was purified and its tRNAse activity was analyzed in vitro, two other mcm5s2U-modified tRNAs (tRNAGln UUG and tRNALys UUU) were cleaved, although the efficiency was much lower compared to tRNAGlu UUC (Lu et al. 2005). Since combined overexpression of tRNAGlu, tRNAGln, and tRNALys increased zymocin resistance compared to overexpression of tRNAGlu alone, all three tRNAs were assumed to represent in vivo targets, but tRNAGlu is the preferred one (Lu et al. 2005; Jablonowski et al. 2006). Indeed, intracellular induction of the tRNAse subunit resulted in depletion of tRNAGlu, but no reduction in the abundance of tRNALys or tRNAGln was observed (Lu et al. 2005). In fact, it could be demonstrated that the anticodon sequence U34U35C36 and an adenosine in position 37 and a cytidine in position 38 are required for efficient cleavage by γ-toxin in vitro, a requirement that is met by tRNAGlu but not tRNAGln or tRNALys (Lu et al. 2008). Consistent with differential zymocin resistance phenotypes of Elongator/tRNA thiolation mutants, it was shown that the presence of the mcm5 side chain provides a strong stimulatory effect, while the presence of the s2U group revealed a weak positive effect on the cleavage efficiency (Lu et al. 2008). Interestingly, it was further demonstrated that the presence of a chemically slightly distinct form of the xm5 modification, the bacterial 5-methylaminomethyl (mnm5) group, is a negative determinant for cleavage (Lu et al. 2008).
Strinkingly, γ lacks sequence similarity to other ribonucleases. Site-directed mutagenesis identified Glu9, Arg151, and His209 of the γ-subunit as the probable catalytic residues (Keppetipola et al. 2009; Jain et al. 2011). Despite the absence of primary sequence similarity to other known ribonucleases, it is assumed that γ-toxin cleaves its target by an RNase A-like chemical mechanism of transesterification involving His209 and Glu9 as general acid-base catalysts with a stabilization of the transition state by Arg151 (Keppetipola et al. 2009; Jain et al. 2011). As for other RNases using such transesterification mechanism, γ also produces 2′3′cyclic phosphate and 5′OH ends that require specific end-healing enzymatic activities before repair by ligation is possible (Lu et al. 2005; Nandakumar et al. 2008). Eukaryotic cells, such as S. cerevisiae, carry a tRNA ligase enzyme (Trl1) that is capable of tRNA end healing and sealing and normally operates in the process of tRNA splicing. Therefore, it was somewhat surprising that the zymocin-induced tRNA cleavage products are evidently not efficiently repaired by Trl1, even though this enzyme can fix a very similar tRNA damage in the splicing reaction (Nandakumar et al. 2008). It was demonstrated that specifically the yeast tRNA ligase is inhibited by the presence of the mcm5s2U modification at the cleavage site, whereas other RNA ligases (plant and phage) are capable of repairing zymocin-induced tRNA damage and confer toxin resistance (Nandakumar et al. 2008).
b) PaT and DrT
The VLE-encoded killer toxins from Millerozyma acaciae (Pichia acaciae) and D. robertsiae (formerly Wingea robertsiae) display similarities but also differences to zymocin (Worsham and Bolen 1990; Meinhardt and Schaffrath 2001; Klassen and Meinhardt 2002; Klassen et al. 2004, 2008, 2014). In both cases, nonautonomous elements associated with autonomous elements were found to carry genes with similarity to the pGKL1 gene encoding the zymocin αβ precursor protein, while at the same time a gene encoding a γ-subunit homologue is absent (Klassen et al. 2004). PaT and DrT are thought to share initial steps of target cell interaction with zymocin: They both bind cell wall chitin and subsequently import a toxic subunit in a common process involving the conserved αβ-like protein with chitin-binding (and likely chitinase) activity (Klassen et al. 2004). However, the intracellularly active toxin subunits of PaT and DrT target a distinct tRNA species (tRNAGln UUG) for cleavage compared to zymocin (Klassen et al. 2008, 2014). PaT was shown to cleave tRNAGln UUG at position 34 as does zymocin but, unlike the latter, does not require the presence of mcm5s2U (Klassen et al. 2008). In vitro cleavage experiments using total yeast tRNA with or without the modification suggest that PaT may utilize an additional cleavage site upstream of U34, likely U32 (Klassen et al. 2008; Meineke et al. 2012). This assumption is based on the detection of two closely migrating cleavage protducts with fully modified tRNAGln and detection of only the faster migrating one in the absence of mcm5s2U (Klassen et al. 2008). It is assumed that in the presence of mcm5s2U, both sites may be cleaved, which could lead to the excision of a dinucleotide. This interpretation is further supported by the analysis of RNA repair enzymes on in vivo. Other than for zymocin (see above), plant and phage tRNA ligases were unable to suppress PaT toxicity in the presence of endogenous yeast TRL1 and mcm5s2U modification (Nandakumar et al. 2008; Meineke et al. 2012). Interestingly, however, plant and phage tRNA ligase were capable of suppressing toxic effects in the absence of mcm5s2U, a condition where available evidence suggests that only the U32 site is cleaved. A likely explanation for the differential rescue of toxic effects of the two distinct killer endonucleases is that PaT, but not zymocin can cleave two sites in its target tRNA, which may result in the excision of a dinucleotide and hence, may damage the target in a non-repairable fashion (Meineke et al. 2012). It should be noted, however, that dinucleotide excision could not yet directly be demonstrated in fully modified tRNAGln UUG and was undetectable when using synthetic unmodified substrates in in vitro cleavage studies with purified PaT PaOrf2 (Chakravarty et al. 2014). Thus, a discrepancy between results with unmodified stem-loop substrates and fully modified tRNA exists, and it could also be possible that other reasons than dual cleavage sites account for the detection of duplet bands in in vitro cleavage reactions with PaT. The crystal structure of the toxic subunit of PaT has been determined and indicated a novel type of folding pattern and active site arrangement distinct from any other ribonuclease (Chakravarty et al. 2014).
Apart from direct inhibition of translation, PaT was found to induce cellular effects resembling those induced by DNA-damaging agents (Klassen et al. 2004, 2008, 2011; Wemhoff et al. 2016a, b). PaT induces S-phase cell cycle arrest and S. cerevisiae mutants defective in DNA repair pathways base excision repair and homologous recombination exhibit strongly enhanced toxin susceptibility, suggesting a link between translational integrity and genome surveillance. Ribonucleotide reductase (RNR) was recently identified as a potential mediator of the DNA-damaging effect of PaT. RNR is periodically expressed and induced in early S-phase to satisfy the massively increased demand for ribonucleotide to desoxyribonucleotide conversion when genome replication is initiated. In the presence of sublethal doses of PaT, the induction of RNR in early S-phase is impaired, which likely accounts for the observed stalling of replication forks in the toxin mediated S-phase arrest. Impaired RNR formation and subsequent dNTP pool depletion will also impair repair of endogenous DNA lesions via pathways requiring dNTP and could indirectly increase DNA damage by inhibiting endogenous repair. In support of a general connection between inhibition of translation and DNA damage, specific DNA repair pathways were demonstrated to protect cells not only from PaT but also from zymocin and the ribosome inhibitor hygromycin B (Klassen et al. 2011; Wemhoff et al. 2016a, b).
c) PiT
A third dsDNA-encoded toxin related to zymocin, PaT, and DrT is produced in B. inositovora (formerly P. inositovora, Yamadazyma inositovora) (Hayman and Bolen 1991; Klassen and Meinhardt 2003; Kast et al. 2014). Again, a precursor protein similar to zymocin αβ is encoded by a nonautonomous plasmid, and a separate gene encodes a subunit that is imported into the target cell and induces the actual toxic effects (Klassen and Meinhardt 2003; Kast et al. 2014). As for the other dsDNA-encoded toxins, conditional expression of the toxic subunit devoid of its signal peptide mimics toxic effects of the holotoxin. In contrast to zymocin, PaT, and DrT, however, PiT apparently targets ribosomal RNA (rRNA) instead of tRNA, as the toxic subunit was shown to induce fragmentation of the 18S and 25S rRNAs (Kast et al. 2014). Positions of PiT-induced cleavage sites were approximately mapped using Northern hybridizations, and multiple positions were identified that are cleaved after induction of the toxic subunit. One of the cleavage sites in 18S rRNA was mapped at the nucleotide level and found to reside in a small loop of the 18S rRNA that exhibits some sequence similarity to the anticodon loop of tRNAGlu UUC (Kast et al. 2014). Hence, PiT might exhibit a distant relationship to zymocin, which specifically cleaves this tRNA. It remains to be determined whether the toxic subunit indeed exhibits rRNA-specific ribonuclease activity and whether cleavage occurs in the context of assembled ribosomes. In marked contrast to zymocin, however, loss of tRNA modification mcm5s2U only modestly increases toxin resistance (Kast et al. 2014). It is not known if RNA modifications, which occur in rRNA as well as in tRNA act as modulators of PiT-induced cleavage.
d) Immunity Against dsDNA-Encoded Toxins
All killer toxin-producing yeasts utilize a strategy to exclude themselves from the effects of their own toxin. One strategy is the production of toxins that utilize receptors not present in the producer strain. However, both the dsRNA- and dsDNA-encoded toxins are routinely active against strains of the same species devoid of the killer virus/viruslike element. It is assumed that this toxin specificity for other strains of the same species creates a strong positive selection to maintain the toxin-encoding genetic element. For the dsRNA viruses, the preprotoxin is often associated with immunity as well (see above). In contrast, there are separate immunity genes in case of the nonautonomus elements encoding zymocin, PaT, and DrT (Tokunaga et al. 1987; Paluszynski et al. 2007; Kast et al. 2015). An immunity gene is apparently lacking in the nonautonomous plasmid-encoding PiT (Hayman and Bolen 1991; Klassen and Meinhardt 2003). The immunity genes of PaT and DrT display detectable sequence similarity as do the corresponding tRNAse subunits of the toxins (Klassen et al. 2004, 2014; Paluszynski et al. 2007), and while each mediates full protection against the cognate toxin, at least the PaT immunity factor can provide detectable cross protection against DrT as well (Klassen et al. 2014). Since DrT and PaT toxic subunits are not detectably similar to zymocin γ either at the sequence level or with respect to the target tRNA, no cross protection between PaT/DrT and zymocin was observable (Kast et al. 2015). Based on these observations, it was concluded that these immunity proteins directly recognize the cognate toxin and protect against its toxic RNA-cleaving activity (Klassen et al. 2014; Kast et al. 2015). Interestingly, all three immunity factors entirely prevent toxic action of intrcellularly expressed tRNAse subunits (Paluszynski et al. 2007; Klassen et al. 2014; Chakravarty et al. 2014; Kast et al. 2015), indicating that immunity factors neutralize the reimported toxin subunit in the producer cell, rather than blocking its uptake. For PaT, direct inhibition of the in vitro tRNAse activity by the immunity protein was demonstrated (Chakravarty et al. 2014). A unusually high A/T content of PaT, DrT, and zymocin immunity genes was recently demonstrated to ensure exclusive gene expression in the cytoplasm (Kast et al. 2015). Even when equipped with a nuclear promoter, these genes cannot be functionally expressed in the nucleus due to recognition of A/T rich motifs within the immunity gene transcripts by the nuclear polyadenylation machinery. As a result, such transcripts become internally cleaved and polyadenylated. This mechanism is thought to prevent successful nuclear capture of immunity genes, which would undermine the autoselection principle imposed by VLE-encoded toxin and immunity gene combinations (Kast et al. 2015).
IV. Applications
A. Antifungals for Human Therapy
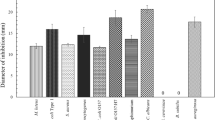
Some of the yeast killer toxins exhibit activity against human pathogens causing severe systemic infections, such as Candida albicans or Cryptococcus neoformans that are difficult to treat with conventional antimycotics. Based on this activity, they have been suggested to be potentially useful for therapy of human infections (Yamamoto et al. 1988; Walker et al. 1995; Weiler and Schmitt 2003; Buzzini et al. 2004; Theisen et al. 2000; Magliani et al. 1997; Izgü et al. 2007a). However, direct application of killer toxins is of limited practical importance because many of these proteins are unstable or inactive at temperatures around 37 °C or neutral pH. In addition, antigenicity and toxicity may prohibit application in the human bloodstream and therefore cannot directly be considered as a therapeutic option to treat severe systemic mycoses (Magliani et al. 2004). However, some specific killer toxins display a broad temperature stability which might facilitate their use as topical applications on superficial skin lesions (Buzzini et al. 2004). For example, W. anomalus K5 toxin, which exhibits stability at 37 °C was studied against dermatophytes and several pathogenic Candida species. All clinical isolates tested as well as type strains belonging to the genera Trichosporon, Microsporum, and Candida were found to be susceptible to K5, suggesting this toxin might indeed be applicable as a topical antifungal agent (Izgü et al. 2007a, b).
To overcome the abovementioned problems associated with the direct application of killer toxins, a strategy of using antibodies with killer activity has been initiated by Polonelli and Morace (1988). A monoclonal antibody (mAbKT4) which neutralized the in vitro activity of W. anomalus UCSC 25F (=ATCC 96603; Table 1) PaKT (Polonelli and Morace 1987) was used to raise anti-idiotypic antibodies, which display an internal image of the toxin’s active site. Strikingly, such natural polyclonal and subsequently developed monoclonal antibodies or single-chain variable fragments (scFv) derived from a phage display library were able to interact with the cell wall and kill yeast cells susceptible to the original W. anomala toxin (Polonelli and Morace 1988; Polonelli et al. 1990, 1997; Magliani et al. 1997, 2004). Vaccination with mAbKT4 in the mouse model resulted in the production of killer toxin-like antibodies, which conferred significant protection against experimental candidiasis (Polnelli et al. 1993, 1994). These antibodies displaying the activity of a killer toxin were termed antibiobodies (antibiotic-like antibodies) and are considered a significant addition to the repertoire of antifungals for the treatment of invasive fungal infections (reviewed in Magliani et al. 2012).
A further improvement in the field was achieved by development of fungicidal killer peptides (KPs) that are derived from antibiobodies displaying W. anomalustoxin-like activity. Such KPs are decapeptides, the sequence of which was originally derived from the active antibiobodies and was further optimized by alanine scanning, resulting in further improved stability and broad target spectrum (Polonelli et al. 2003; Magliani et al. 2012). Such W. anomalus killer toxin-derived KPs are active against pathogenic microorganisms which are known to induce severe systemic mycoses that are difficult to treat with conventional antimycotics (Cenci et al. 2004; Travassos et al. 2004). In addition, they are active against a variety of pathogenic prokaryotic microorganisms, such as Mycobacteria, Staphylococcus, or Streptococcus species, and plant pathogenic Pseudomonas strains (reviewed in Magliani et al. 2004). It is assumed that glucan or glucan-like molecules in the cell wall of susceptible pro- and eukaryotic microogranisms constitute the basis for the broad spectrum of toxin activity observed for idiotypic antibodies and decapeptides derived thereof (Magliani et al. 2004).
In addition to W. anomalus toxin, HM-1 was also used to produce toxin-neutralizing antibodies that were subsequently employed in idiotypic vaccination and production of killer toxin-like antibodies, which display an internal image of HM-1’s active site and inhibit target cell’s glucan synthase activity (Selvakumar et al. 2006a, b, c). As for the W. anomalus toxin, killer peptides could be derived from such killer activity bearing antibodies and may have application potential in the treatment of human fungal infections (Kabir et al. 2011). Small peptides derived from antibiobodies are of special interest since they can be produced much more economically when compared to the antibiobodies.
B. Antifungals in Agriculture, Food, and Feed Industry
The ability of preventing growth of competing microorganisms by secreting inhibitory killer toxins has raised interest in application of such strains as bioprotective agents in agriculture and food industries. In particular, fermented food and beverage products are often at risk to lose product quality due to the development of spoilage yeasts, fungi, or bacteria. For example, wine fermentations and post-fermentative aging processes can get spoiled by Dekkera/Brettanomyces or Kloeckera/Hanseniaspora yeast species, which results in loss of sensory product quality due to unpleasant odor and taste development (Comitini et al. 2004a; Wedral et al. 2010). Hence, there is an application potential for yeast killer toxins capable of inhibiting growth of spoilage yeasts and the specific conditions of wine fermentation and aging (low pH of ~3.5 and low temperatures). For example, KpKt from Tetrapisispora phaffii is active under such conditions against apiculate yeast species, including Hanseniaspora uvarum, which dominate on grapes and grape juice (Ciani and Fatichenti 2001). During experimental wine fermentation, KpKt was found to display inhibitory activity against H. uvarum comparable to the routinely applied SO2. It was suggested that KpKt could substitute for SO2, thereby eliminating undesired or harmful residual traces of SO2 in the final product (Ciani and Fatichenti 2001; Comitini et al. 2004b). As an alternative of using the killer strain, a production strain for production of recombinant KpKt (rKpKt) was recently developed (Chessa et al. 2017). As a further benefit, rKpKt was found to exhibit a broadened spectrum of target yeasts, killing not only Kloeckera/Hanseniaspora and Zygosaccharomyces but also D. bruxellensis (Chessa et al. 2017).
Other killer toxins such as PiKt (Wickerhamomyces anomalus), KwKt (Kluyveromyces wickerhamii), and CpKT1/CpKT2 (Candida pyralidae) and a KP6-related toxin (Ustilago maydis) were also shown to be active and stable in wine environment and are capable of inhibiting Dekkera/Brettanomyces spoilage yeasts, indicating an application potential in wine industry for these toxins as well (Comitini et al. 2004a; Santos et al. 2011; Mehlomakulu et al. 2014, 2017).
In addition to the mentioned non-Saccharomyces killer toxins, also the virus-encoded K1/K2 toxins of S. cerevisiae have application potential in wine industry. Wine fermentation is typically started using defined S. cerevisiae strains optimized for fermentation performance and able to dominate native yeasts in the grape must (Pretorius 2000). Specific starter yeast strains were engineered by cytoduction to possess the L-A and M viruses and the corresponding killer phenotype (Ouchi and Akiyama 1976; Hara et al. 1980; Seki et al. 1985; Boone et al. 1990; Sulo et al. 1992; Sulo and Michalcáková 1992; Michalcáková et al. 1994). Since K2 displays a higher activity at wine pH (~3.5) compared to K1 (Pfeiffer and Radler 1984), it is considered to be most suitable for biocontrol in the wine environment. As an alternative to strain engineering, fermentation starters which naturally express the K2-type killer phenotype as well as desired fermentation characteristics can also be directly selected from the population of indigenous yeasts (Lopes et al. 2007). Such selected or engineered S. cerevisiae killer strains typically retain desired flavor and fermentation characteristics and are able to suppress indigenous S. cerevisiae strains due to toxin production. Since K2 killer strains are frequent among the natural population on grape surfaces, the use of defined K2 killer-positive fermentation starters, which also display K2 immunity, additionally prevents overgrowth of the starter strain by the indigenous killer (Jacobs and Van Vuuren 1991). A limitation of the S. cerevisiae killer toxins in wine and fermentation industry, however, is the relatively narrow spectrum of sensitive target yeast species for these toxins. In particular, non-Saccharomyces yeasts present at grape surfaces are routinely insensitive to the S. cerevisiae killer toxins (Young and Yagiu 1978) and thus are largely restricting the biocontrol potential to Saccharomyces contaminants.
Prevention of spoilage in other fermentation products by killer toxins was also investigated. K. lactis zymocin and Cyb. mrakiiHM-1 can potentially be used in controlling silage spoilage (Kitamoto et al. 1993, 1999; Lowes et al. 2000).
Besides application in fermentation industries, killer yeasts are also attractive agents for biocontrol purposes in agriculture. Several of the the glucanase toxins from different strains of Wickerhamomyces anomalus (formerly Pichia anomala) are characterized by a broad antimicrobial activity which is directed not only against other yeasts but also inhibits pathogenic bacteria or mycelial fungi and even protozoans (Sawant et al. 1989; Walker et al. 1995; Jijakli and Lepoivre 1998; Izgü et al. 2007a, b; Wang et al. 2007a; Muccilli et al. 2013; Valzano et al. 2016). The activity against mycelial fungi has been exploited for biocontrol of postharvest diseases caused by plant pathogenic fungi on commercially important fruits (Walker et al. 1995; Santos et al. 2004; Santos and Marquina 2004b; Platania et al. 2012; Aloui et al. 2015; Perez et al. 2016). In particular, green mold disease caused by Penicillium digitatum developing on citrus fruit during postharvest storage could be controlled by W. anomalus toxin (Platania et al. 2012; Perez et al. 2016). Currently, efforts are undertaken to embed killer yeasts in edible coatings made of sodium alginate and locust bean gum, which results in high retention of the killer strain on the fruit surface and was shown to strongly reduce green mold development (Aloui et al. 2015). In a related application, P. membranifaciens toxin was shown to be applicable against Botrytis cinerea, the causal agent of gray mold disease on grapes. Treatment of Vitis vinifera plants with either purified toxin or the P. membranifaciens killer strain protected against B. cinerea (Santos and Marquina 2004b). The strains of this killer species were also active in suppressing B. cinerea growth on apples or pears following harvest, identifying a general application potential for P. membranifaciens killer toxins or strains in biocontrol agent of gray mold disease (Santos et al. 2004; Lutz et al. 2013). It was suggested that biocontrol efficiency is not only determined by the production of killer toxin but influenced by the ability to colonize wounds, production of other hyrolytic enzymes such as chitinase or protease, and the inhibition of spore germination (Lutz et al. 2013).
A W. anomalus strain isolated from marine environment was shown to be of potential use in the biocontrol of a crab pathogenic yeast, Metschnikowia bicuspidata. Infection of the commercially important crab species Portunus trituberculatus by the pathogenic yeast has caused severe economic losses in aquacultures of this species in China. The identification and preliminary characterization of the toxin suggested that it could be used for inhibiting growth of M. bicuspidata in aquaculture (Wang et al. 2007a, b).
More recently, W. anomalus glucanase killer strains were isolated from Anopheles mosquitoes. Since the toxin has demonstrated activity against the rodent malaria parasite Plasmodium berghei, novel strategies to utilize such strains to control the spread of plasmodium infection in malaria mosquitoes were proposed (Valzano et al. 2016).
Lastly, killer toxins were expressed in transgenic plants, leading to disease resistant crops. For example, transgenic maize plants were constructed expressing KP4 toxin from Ustilago maydis, leading to robust resistance against infection by U. maydis (Allen et al. 2011). Similar approaches were also followed in other plants or using differents toxins (U. maydis KP6, W. anomala KP) (Kinal et al. 1995; Park et al. 1996b; Donini et al. 2005).
V. Concluding Remarks
Yeast killer toxins are thought to serve the purpose of competitor killing and thereby provide a selective advantage to the producing species. However, since a number of such toxins are encoded on selfish genetic elements of viral origin, they may also serve the purpose of genetic stabilization of the viruslike element in the cell. Even though known killer toxins are most heterogenous with respect to protein primary and tertiary structures, some common features can be recognized. This includes common toxin maturation principles involving processing in the ER of the producer cell and the utilization of similar mechanisms to first interact with the target cell and subsequently target an essential biological process either inside or outside of it. Several strategies are currently followed to exploit such natural antimicrobials acting on eukaryotic target cells for application in medicine or agriculture and food industries.
References
Allen A, Islamovic E, Kaur J, Gold S, Shah D, Smith TJ (2011) Transgenic maize plants expressing the Totivirus antifungal protein, KP4, are highly resistant to corn smut. Plant Biotechnol J 9:857–864
Allen A, Chatt E, Smith TJ (2013) The atomic structure of the virally encoded antifungal protein, KP6. J Mol Biol 425:609–621
Aloui H, Licciardello F, Khwaldia K, Hamdi M, Restuccia C (2015) Physical properties and antifungal activity of bioactive films containing Wickerhamomyces anomalus killer yeast and their application for preservation of oranges and control of postharvest green mold caused by Penicillium digitatum. Int J Food Microbiol 200:22–30
Antuch W, Güntert P, Wüthrich K (1996) Ancestral beta gamma-crystallin precursor structure in a yeast killer toxin. Nat Struct Biol 3:662–665
Ashida S, Shimazaki T, Kitano K, Hara S (1983) New killer toxin of Hansenula mrakii. Agric Biol Chem 47:2953–2955
Bär C, Zabel R, Liu S, Stark MJ, Schaffrath R (2008) A versatile partner of eukaryotic protein complexes that is involved in multiple biological processes: Kti11/Dph3. Mol Microbiol 69:1221–1123
Becker B, Blum A, Gießelmann E, Dausend J, Rammo D, Müller NC, Tschacksch E, Steimer M, Spindler J, Becherer U, Rettig J, Breinig F, Schmitt MJ (2016) H/KDEL receptors mediate host cell intoxication by a viral A/B toxin in yeast. Sci Rep 6:31105
Belda I, Ruiz J, Alonso A, Marquina D, Santos A (2017) The biology of Pichia membranifaciens killer toxins. Toxins (Basel) 9:pii: E112
Bevan EA, Makower M (1963) The physiological basis of the killer character in yeast. Proc XIth Int Congr Genet 1:202–203
Bevan EA, Herring AJ, Mitchell DJ (1973) Preliminary characterization of two species of dsRNA in yeast and their relationship to the “killer” character. Nature 245:81–86
Bolen PL, Kurtzman CP, Ligon JM, Mannarelli BM, Bothast RJ (1992) Physical and genetic characterization of linear DNA plasmids from the heterothallic yeast Saccharomycopsis crataegensis. Antonie Van Leuwenhoek 61:195–295
Boone C, Sdicu AM, Wagner J, Degré R, Sanchez C, Bussey H (1990) Integration of the yeast K1 killer toxin gene into the genome of marked wine yeasts and its effect on vinifcation. Am J Enol Vitic 41:37–42
Bostian KA, Elliott Q, Bussey H, Burn V, Smith A, Tipper DJ (1984) Sequence of the preprotoxin dsRNA gene of type I killer yeast: multiple processing events produce a two-component toxin. Cell 36:741–751
Breinig F, Tipper DJ, Schmitt MJ (2002) Kre1p, the plasma membrane receptor for the yeast K1 viral toxin. Cell 108:395–405
Breinig F, Schleinkofer K, Schmitt MJ (2004) Yeast Kre1p is GPI-anchored and involved in both cell wall assembly and architecture. Microbiology 150:3209–3218
Breinig F, Sendzik T, Eisfeld K, Schmitt MJ (2006) Dissecting toxin immunity in virus-infected killer yeast uncovers an intrinsic strategy of self-protection. Proc Natl Acad Sci U S A 103:3810–3815
Bussey H (1991) K1 killer toxin, a pore-forming protein from yeast. Mol Microbiol 5:2339–2343
Butler AR, O’Donnell RW, Martin VJ, Gooday GW, Stark MJ (1991a) Kluyveromyces lactis toxin has an essential chitinase activity. Eur J Biochem 199:483–488
Butler AR, Porter M, Stark MJR (1991b) Intracellular expression of Kluyveromyces lactis toxin γ subunit mimics treatment with exogenous toxin and distinguishes two classes oftoxin-resistant mutant. Yeast 7:617–625
Butler AR, White JH, Folawiyo Y, Edlin A, Gardiner D, Stark MJ (1994) Two Saccharomyces cerevisiae genes which control sensitivity to G1 arrest induced by Kluyveromyces lactis toxin. Mol Cell Biol 14:6306–6316
Buzzini P, Corazzi L, Turchetti B, Buratta M, Martini A (2004) Characterization of the in vitro antimycotic activity of a novel killer protein from Williopsis saturnus DBVPG 4561 against emerging pathogenic yeasts. FEMS Microbiol Lett 238:359–365
Carroll SY, Stirling PC, Stimpson HE, Giesselmann E, Schmitt MJ, Drubin DG (2009) A yeast killer toxin screen provides insights into a/b toxin entry, trafficking, and killing mechanisms. Dev Cell 17(4):552–560
Castón JR, Trus BL, Booy FP, Wickner RB, Wall JS, Steven AC (1997) Structure of L-A virus: a specialized compartment for the transcription and replication of double-stranded RNA. J Cell Biol 138:975–985
Cenci E, Bistoni F, Mencacci A, Perito S, Magliani W, Conti S, Polonelli L, Vecchiarelli A (2004) A synthetic peptide as a novel anticryptococcal agent. Cell Microbiol 6:953–961
Chakravarty AK, Smith P, Jalan R, Shuman S (2014) Structure, mechanism, and specificity of a eukaryal tRNA restriction enzyme involved in self-nonself discrimination. Cell Rep 7:339–347
Chen WB, Han JF, Jong SC, Chang SC (2000) Isolation, purification, and characterization of a killer protein from Schwanniomyces occidentalis. Appl Environ Microbiol 66:5348–5352
Chessa R, Landolfo S, Ciani M, Budroni M, Zara S, Ustun M, Cakar ZP, Mannazzu I (2017) Biotechnological exploitation of Tetrapisispora phaffii killer toxin: heterologous production in Komagataella phaffii (Pichia pastoris). Appl Microbiol Biotechnol 101:2931–2942
Ciani M, Fatichenti F (2001) Killer toxin of Kluyveromyces phaffii DBVPG 6076 as a biopreservative agent to control apiculate wine Yeasts. Appl Environ Microbiol 67:3058–3063
Comitini F, Ciani M (2011) Kluyveromyces wickerhamii killer toxin: purification and activity towards Brettanomyces/Dekkera yeasts in grape must. FEMS Microbiol Lett 316:77–82
Comitini F, De Ingeniis J, Pepe L, Mannazzu I, Ciani M (2004a) Pichia anomala and Kluyveromyces wickerhamii killer toxins as new tools against Dekkera/Brettanomyces spoilage yeasts. FEMS Microbiol Lett 238:235–240
Comitini F, Di Pietro N, Zacchi L, Mannazzu I, Ciani M (2004b) Kluyveromyces phaffii killer toxin active against wine spoilage yeasts: purification and characterization. Microbiology 150:2535–2541
Comitini F, Mannazzu I, Ciani M (2009) Tetrapisispora phaffii killer toxin is a highly specific beta-glucanase that disrupts the integrity of the yeast cell wall. Microb Cell Factories 8:55
Cong YS, Yarrow D, Li YY, Fukuhara H (1994) Linear DNA plasmids from Pichia etchellsii, Debaryomyces hansenii and Wingea robertsiae. Microbiology 140:1327–1335
da Silva S, Calado S, Lucas C, Aguiar C (2008) Unusual properties of the halotolerant yeast Candida nodaensis Killer toxin, CnKT. Microbiol Res 163:243–251
de la Peña P, Barros F, Gascón S, Lazo PS, Ramos S (1981) Effect of yeast killer toxin on sensitive cells of Saccharomyces cerevisiae. J Biol Chem 256:10420–10425
Dignard D, Whiteway M, Germain D, Tessier D, Thomas DY (1991) Expression in yeast of a cDNA copy of the K2 killer toxin gene. Mol Gen Genet 227:127–136
Dinman JD, Icho T, Wickner RB (1991) A -1 ribosomal frameshift in a double-stranded RNA virus of yeast forms a gag-pol fusion protein. Proc Natl Acad Sci U S A 88:174–178
Donini M, Lico C, Baschieri S, Conti S, Magliani W, Polonelli L, Benvenuto E (2005) Production of an engineered killer peptide in Nicotiana benthamiana by using a potato virus X expression system. Appl Environ Microbiol 71:6360–6367
Eisfeld K, Riffer F, Mentges J, Schmitt MJ (2000) Endocytotic uptake and retrograde transport of a virally encoded killer toxin in yeast. Mol Microbiol 37:926–940
Fichtner L, Schaffrath R (2002) KTI11 and KTI13, Saccharomyces cerevisiae genes controlling sensitivity to G1 arrest induced by Kluyveromyces lactis zymocin. Mol Microbiol 44:865–875
Fichtner L, Frohloff F, Burkner K, Larsen M, Breunig KD, Schaffrath R (2002) Molecular analysis of KTI12/TOT4, a Saccharomyces cerevisiae gene required for Kluyveromyces lactis zymocin action. Mol Microbiol 43:783–791
Fichtner L, Jablonowski D, Schierhorn A, Kitamoto HK, Stark MJR, Schaffrath R (2003) Elongator’s toxin-target (TOT) function is nuclear localization sequence dependent and suppressed by post-translational modification. Mol Microbiol 49:1297–1307
Frohloff F, Fichtner L, Jablonowski D, Breuning KD, Schaffrath R (2001) Saccharomyces cerevisiae elongator mutations confer resistance to the Kluyveromyces lactis zymocin. EMBO J 20:1993–2003
Fuentefria AM, Suh SO, Landell MF, Faganello J, Schrank A, Vainstein MH, Blackwell M, Valente P (2008) Trichosporon insectorum sp. nov., a new anamorphic basidiomycetous killer yeast. Mycol Res 112:93–99
Fukuhara H (1995) Linear DNA plasmids of yeasts. FEMS Microbiol Lett 131:1–9
Golubev WI (2006) Antagonistic interactions among yeasts. In: Rosa CA, Péter G (eds) Biodiversity and ecophysiology of yeasts, The yeast handbook. Springer, Berlin, pp 197–219
Golubev WI (2015) Intraspecific and intrageneric antagonistic activity of Wickerhamomyces anomalus. Microbiol (Moscow) 84:193–193
Golubev W, Shabalin Y (1994) Microcin production by the yeast Cryptococcus humicola. FEMS Microbiol Lett 119:105–110
Golubev WI, Pfeifer I, Golubeva E (2002) Mycocin production in Trichosporon pullulans populations colonizing tree exudates in the spring. FEMS Microbiol Ecol 40:151–157
Golubev WI, Pfeiffer I, Churkina LG, Golubeva EW (2003) Double-stranded RNA viruses in a mycocinogenic strain of Cystofilobasidium infirmominiatum. FEMS Yeast Res 3:63–68
Goto K, Iwatuki Y, Kitano K, Obata T, Hara S (1990) Cloning and nucleotide sequence of the KHR killer gene of Saccharomyces cerevisiae. Agric Biol Chem 54:979–984
Goto K, Fukuda H, Kichise K, Kitano K, Hara S (1991) Cloning and nucleotide sequence of the KHS killer gene of Saccharomyces cerevisiae. Agric Biol Chem 55:1953–1958
Gu F, Khimani A, Rane SG, Flurkey WH, Bozarth RF, Smith TJ (1995) Structure and function of a virally encoded fungal toxin from Ustilago maydis: a fungal and mammalian Ca2+ channel inhibitor. Structure 3:805–814
Gunge N, Tamaru A, Ozawa F, Sakaguchi K (1981) Isolation and characterization of linear deoxyribonucleic acid plasmids from Kluyveromyces lactis and the plasmid-associated killer character. J Bacteriol 145:382–390
Guyard C, Séguy N, Lange M, Ricard I, Polonelli L, Cailliez JC (1999) First steps in the purification and characterization of a Pichia anomala killer toxin. J Eukaryot Microbiol 46:144S
Guyard C, Séguy N, Cailliez JC, Drobecq H, Polonelli L, Dei-Cas E, Mercenier A, Menozzi FD (2002a) Characterization of a Williopsis saturnus var. mrakii high molecular weight secreted killer toxin with broad-spectrum antimicrobial activity. J Antimicrob Chemother 49:961–971
Guyard C, Dehecq E, Tissier JP, Polonelli L, Dei-Cas E, Cailliez JC, Menozzi FD (2002b) Involvement of β-glucans in the wide-spectrum antimicrobial activity of Williopsis saturnus var. mrakii MUCL 41968 killer toxin. Mol Med 8:686–694
Hara S, Iimura Y, Otsuka K (1980) Breeding of useful killer wine yeasts. Am J Enol Vitic 31(1):28–33
Hayman GT, Bolen BL (1991) Linear DNA plasmids of Pichia inositovora are associated with a novel killer toxin activity. Curr Genet 19:389–393
Heiligenstein S, Eisfeld K, Sendzik T, Jimenéz-Becker N, Breinig F, Schmitt MJ (2006) Retrotranslocation of a viral A/B toxin from the yeast endoplasmic reticulum is independent of ubiquitination and ERAD. EMBO J 25:4717–4727
Hishinuma F, Hirai K (1991) Genome organization of the linear plasmid, pSKL, isolated from Saccharomyces kluyveri. Mol Gen Genet 226:97–106
Hishinuma F, Nakamura K, Hirai K, Nishizawa R, Gunge N, Maeda T (1984) Cloning and nucleotide sequence of the DNA killer plasmids from yeast. Nucleic Acids Res 12:l7581–l7597
Hodgson VJ, Button D, Walker GM (1995) Anti-Candida activity of a novel killer toxin from the yeast Williopsis mrakii. Microbiology 141:2003–2012
Huang B, Johansson MJ, Bystrom AS (2005) An early step in wobble uridine tRNA modification requires the Elongator complex. RNA 11:424–436
Huang B, Lu J, Byström AS (2008) A genome-wide screen identifies genes required for formation of the wobble nucleoside 5-methoxycarbonylmethyl-2-thiouridine in Saccharomyces cerevisiae. RNA 14:2183–2194
Hutchins K, Bussey H (1983) Cell wall receptor for yeast killer toxin: involvement of (1-6)-β-D-glucan. J Bacteriol 154:161–169
Icho T, Wickner RB (1989) The double-stranded RNA genome of yeast virus L-A encodes its own putative RNA polymerase by fusing two open reading frames. J Biol Chem 264:6716–6723
Izgü F, Altinbay D (2004) Isolation and characterization of the K5-type yeast killer protein and its homology with an exo-β-1,3-glucanase. Biosci Biotechnol Biochem 68:685–693
Izgü F, Altinbay D, Sağiroğlu AK (1999) Isolation and characterization of the K6 type yeast killer protein. Microbios 99:161–172
Izgü F, Altinbay D, Türeli AE (2007a) In vitro susceptibilities of Candida spp. to Panomycocin, a novel exo-β-1,3-glucanase isolated from Pichia anomala NCYC 434. Microbiol Immunol 51:797–803
Izgü F, Altinbay D, Türeli AE (2007b) In vitro activity of panomycocin, a novel exo-β-1,3-glucanase isolated from Pichia anomala NCYC 434, against dermatophytes. Mycoses 50:31–34
Jablonowski D, Schaffrath R (2007) Zymocin, a composite chitinase and tRNase killer toxin from yeast. Biochem Soc Trans 35:1533–1537
Jablonowski D, Fichtner L, Martin VJ, Klassen R, Meinhardt F, Stark MJR, Schaffrath R (2001) Saccharomyces cerevisiae cell wall chitin, the potential Kluyveromyces lactis zymocin receptor. Yeast 18:1285–1299
Jablonowski D, Zink S, Mehlgarten C, Daum G, Schaffrath R (2006) tRNAGlu wobble uridine methylation by Trm9 identifies Elongator’s key role for zymocin-induced cell death in yeast. Mol Microbiol 59:677–688
Jacobs CJ, Van Vuuren HJJ (1991) Effects of different killer yeasts on wine fermentations. Am J Enol Vitic 42(4):295–300
Jain R, Poulos MG, Gros J, Chakravarty AK, Shuman S (2011) Substrate specificity and mutational analysis of Kluyveromyces lactisgamma-toxin, a eukaryal tRNA anticodon nuclease. RNA 17:1336–1343
Jeske S, Meinhardt F (2006) Autonomous cytoplasmic linear plasmid pPac1-1 of Pichia acaciae: molecular structure and expression studies. Yeast 23:479–486
Jeske S, Meinhardt F, Klassen R (2006) Extranuclear inheritance: virus-like DNA-elements in yeast. In: Esser K, Lüttge U, Kadereit J, Beyschlag W (eds) Progress in botany, vol 68. Springer, Berlin, pp 98–129
Jijakli MH, Lepoivre P (1998) Characterization of an exo-β-1,3-glucanase produced by Pichia anomala strain K, antagonist of Botrytis cinerea on apples. Phytopathology 88:335–343
Kabir ME, Karim N, Krishnaswamy S, Selvakumar D, Miyamoto M, Furuichi Y, Komiyama T (2011) Peptide derived from anti-idiotypic single-chain antibody is a potent antifungal agent compared to its parent fungicide HM-1 killer toxin peptide. Appl Microbiol Biotechnol 92:1151–1160
Kagan B (1983) Mode of action of yeast killer toxins: channel formation in lipid bilayer membranes. Nature 302:709–711
Kalhor HR, Clarke S (2003) Novel methyltransferase for modified uridine residues at the wobble position of tRNA. Mol Cell Biol 23:9283–9292
Kämper J, Meinhardt F, Gunge N, Esser K (1989a) New recombinant linear DNA-elements derived from Kluyveromyces lactis killer plasmids. Nucleic Acids Res 17:1781
Kämper J, Meinhardt F, Gunge N, Esser K (1989b) In vivo construction of linear vectors based on killer plasmids from Kluyveromyces lactis: selection of a nuclear gene results in attachment of telomeres. Mol Cell Biol 9:3931–3937
Kämper J, Esser K, Gunge N, Meinhardt F (1991) Heterologous gene expression on the linear DNA killer plasmid from Kluyveromyces lactis. Curr Genet 19:109–118
Kasahara S, Ben Inoue S, Mio T, Yamada T, Nakajima T, Ichishima E, Furuichi Y, Yamada H (1994) Involvement of cell wall β-glucan in the action of HM-1 killer toxin. FEBS Lett 348:27–32
Kashiwagi T, Kunishima N, Suzuki C, Tsuchiya F, Nikkuni S, Arata Y, Morikawa K (1997) The novel acidophilic structure of the killer toxin from halotolerant yeast demonstrates remarkable folding similarity with a fungal killer toxin. Structure 5:81–94
Kast A, Klassen R, Meinhardt F (2014) rRNA fragmentation induced by a yeast killer toxin. Mol Microbiol 91:606–617
Kast A, Voges R, Schroth M, Schaffrath R, Klassen R, Meinhardt F (2015) Autoselection of cytoplasmic yeast virus like elements encoding toxin/antitoxin systems involves a nuclear barrier for immunity gene expression. PLoS Genet 11:e1005005
Keppetipola N, Jain R, Meineke B, Diver M, Shuman S (2009) Structure-activity relationships in Kluyveromyces lactis γ-toxin, a eukaryal tRNA anticodon nuclease. RNA 15:1036–1044
Keszthelyi A, Ohkusu M, Takeo K, Pfeiffer I, Litter J, Kucsera J (2006) Characterisation of the anticryptococcal effect of the FC-1 toxin produced by Filobasidium capsuligenum. Mycoses 49:176–183
Kimura T, Kitamoto N, Matsuoka K, Nakamura K, Iimura Y, Kito Y (1993) Isolation and nucleotide sequences of the genes encoding killer toxins from Hansenula mrakii and H. saturnus. Gene 137:265–270
Kimura T, Komiyama T, Furuichi Y, Iimura Y, Karita S, Sakka K, Ohmiya K (1999) N-Glycosylation is involved in the sensitivity of Saccharomyces cerevisiae to HM-1 killer toxin secreted from Hansenula mrakii IFO 0895. Appl Microb Biotechnol 51:176–184
Kinal H, Park CM, Berry JO, Koltin Y, Bruenn JA (1995) Processing and secretion of a virally encoded antifungal toxin in transgenic tobacco plants: evidence for a Kex2p pathway in plants. Plant Cell 7:677–688
Kitada K, Hishinuma H (1987) A new linear plasmid isolated from the yeast Saccharomyces kluyveri. Mol Gen Genet 206:377–381
Kitamoto HK, Ohmomo S, Nakahara T (1993) Selection of killer yeasts (Kluyveromyces lactis) to prevent aerobic deterioration in silage making. J Dairy Sci 76:803–811
Kitamoto HK, Hasebe A, Ohmomo S, Suto EG, Muraki M, Iimura Y (1999) Prevention of aerobic spoilage of maize silage by a genetically modified killer yeast, Kluyveromyces lactis, defective in the ability to grow on lactic acid. Appl Environ Microbiol 65:4697–4700
Klassen R, Meinhardt F (2002) Linear plasmids pWR1A and pWR1B of the yeast Wingea robertsiae are associated with a killer phenotype. Plasmid 48:142–148
Klassen R, Meinhardt F (2003) Structural and functional analysis of the killer element pPin1-3 from Pichia inositovora. Mol Gen Genomics 270:190–199
Klassen R, Meinhardt F (2007) Linear protein-primed replicating plasmids in eukaryotic microbes. In: Meinhardt F, Klassen R (eds) Microbial linear plasmids, microbiology monographs, vol 7. Springer, Berlin, pp 187–226
Klassen R, Tontsidou L, Larsen M, Meinhardt F (2001) Genome organization of the linear cytoplasmic element pPE1B from Pichia etchellsii. Yeast 18:953–961
Klassen R, Jablonowski D, Schaffrath R, Meinhardt F (2002) Genome organization of the linear Pichia etchellsii plasmid pPE1A: evidence for expression of an extracellular chitin-binding protein homologous to the alpha-subunit of the Kluyveromyces lactis killer toxin. Plasmid 47:224–233
Klassen R, Teichert S, Meinhardt F (2004) Novel yeast killer toxins provoke S-phase arrest and DNA damage checkpoint activation. Mol Microbiol 53:263–273
Klassen R, Paluszynski J, Wemhoff S, Pfeiffer A, Fricke J, Meinhardt F (2008) The primary target of the killer toxin from Pichia acaciae is tRNAGln. Mol Microbiol 69:681–697
Klassen R, Wemhoff S, Krause J, Meinhardt F (2011) DNA repair defects sensitize cells to anticodon nuclease yeast killer toxins. Mol Gen Genomics 285:185–195
Klassen R, Kast A, Wünsche G, Paluszynski J, Meinhardt F (2014) Immunity factors for two related tRNAGln targeting killer toxins distinguish cognate and non-cognate toxic subunits. Curr Genet 60:213–222
Klassen R, Schaffrath R, Buzzini P, Philip Ganter PF (2017) Antagonistic interactions and killer yeasts. In: Buzzini P, Lachance MA, Yurkov A (eds) Yeasts in natural ecosystems: ecology. Springer, Cham, pp 229–275
Koltin Y, Day PR (1976) Inheritance of killer phenotypes and double-stranded RNA in Ustilago maydis. Proc Natl Acad Sci U S A 73:594–598
Komiyama T, Ohta T, Furuichi Y, Ohta Y, Tsukada Y (1995) Structure and activity of HYI killer toxin from Hansenula saturnus. Biol Pharm Bull 18:1057–1059
Komiyama T, Ohta T, Urakami H, Shiratori Y, Takasuka T, Satoh M, Watanabe T, Furuichi Y (1996) Pore formation on proliferating yeast Saccharomyces cerevisiae cell buds by HM-1 killer toxin. J Biochem 119:731–736
Komiyama T, Shirai T, Ohta T, Urakami H, Furuichi Y, Ohta Y, Tsukada Y (1998) Action properties of HYI killer toxin from Williopsis saturnus var. saturnus, and antibiotics, aculeacin A and papulacandin B. Biol Pharm Bull 21:1013–1019
Komiyama T, Kimura T, Furuichi Y (2002) Round shape enlargement of the yeast spheroplast of Saccharomyces cerevisiae by HM-1 toxin. Biol Pharm Bull 25:959–965
Kono I, Himeno K (1997) A novel killer yeast effective on Schizosaccharomyces pombe. Biosci Biotechnol Biochem 61:563–564
Larsen M, Gunge N, Meinhardt F (1998) Kluyveromyces lactis killer plasmid pGKL2: evidence for a viral-like capping enzyme encoded by OFR3. Plasmid 40:243–246
Leidel S, Pedrioli PG, Bucher T, Brost R, Costanzo M, Schmidt A, Aebersold R, Boone C, Hofmann K, Peter M (2009) Ubiquitin-related modifier Urm1 acts as a sulphur carrier in thiolation of eukaryotic transfer RNA. Nature 458:228–232
Li N, Erman M, Pangborn W, Duax WL, Park CM, Bruenn J, Ghosh D (1999) Structure of Ustilago maydis killer toxin KP6 alpha-subunit. A multimeric assembly with a central pore. J Biol Chem 4:20425–20431
Ligon JM, Bolen PL, Hill DS, Bothast RJ, Kurtzman CP (1989) Physical and biological characterization of linear DNA plasmids of the yeast Pichia inositovora. Plasmid 2:185–194
Lopes CA, Rodríguez ME, Sangorrín M, Querol A, Caballero AC (2007) Patagonian wines: the selection of an indigenous yeast starter. J Ind Microbiol Biotechnol 34:539–546
Lowes KF, Shearman CA, Payne J, MacKenzie D, Archer DB, Merry RJ, Gasson MJ (2000) Prevention of yeast spoilage in feed and food by the yeast mycocin HMK. Appl Environ Microbiol 66:1066–1076
Lu J, Huang B, Esberg A, Johansson MJ, Bystrom AS (2005) The Kluyveromyces lactis γ-toxin targets tRNA anticodons. RNA 11:1648–1654
Lu J, Esberg A, Huang B, Byström AS (2008) Kluyveromyces lactisgamma-toxin, a ribonuclease that recognizes the anticodon stem loop of tRNA. Nucleic Acids Res 36:1072–1080
Lukša J, Podoliankaité M, Vepštaite I, Strazdaité-Žieliene Z, Urbonavičius J, Serviené E (2015) Yeast-1,6-glucan is a primary target for the Saccharomyces cerevisiae K2 toxin. Eukaryot Cell 14:406–414
Lutz MC, Lopes CA, Rodriguez ME, Sosa MC, Sangorrín MP (2013) Efficacy and putative mode of action of native and commercial antagonistic yeasts against postharvest pathogens of pear. Int J Food Microbiol 164:166–172
Magliani W, Conti S, Gerloni M, Bertolotti D, Polonelli L (1997) Yeast killer systems. Clin Microbiol Rev 10:369–400
Magliani W, Conti S, Salati A, Vaccari S, Ravanetti L, Maffei DL, Polonelli L (2004) Therapeutic potential of yeast killer toxin-like antibodies and mimotopes. FEMS Yeast Res 5:11–18
Magliani W, Conti S, Giovati L, Zanello PP, Sperindè M, Ciociola T, Polonelli L (2012) Antibody peptide based antifungal immunotherapy. Front Microbiol 3:190
Marquina D, Peres C, Caldas FV, Marques JF, Peinado JM, Spencer-Martins I (1992) Characterization of the yeast populations in olive brines. Lett Appl Microbiol 14:279–283
Martinac B, Zhu H, Kubalski A, Zhou XL, Culbertson M, Bussey H, Kung C (1990) Yeast K1 killer toxin forms ion channels in sensitive yeast spheroplasts and in artificial liposomes. Proc Natl Acad Sci U S A 87:6228–6232
Mehlgarten C, Schaffrath R (2003) Mutant casein kinase I (Hrr25p/Kti14p) abrogates the G1 cell cycle arrest induced by Kluyveromyces lactis zymocin in budding yeast. Mol Gen Genomics 269:188–196
Mehlgarten C, Schaffrath R (2004) After chitin docking, toxicity of Kluyveromyces lactis zymocin requires Saccharomyces cerevisiae plasma membrane H+-ATPase. Cell Microbiol 6:569–580
Mehlomakulu NN, Setati ME, Divol B (2014) Characterization of novel killer toxins secreted by wine-related non-Saccharomyces yeasts and their action on Brettanomyces spp. Int J Food Microbiol 188:83–91
Mehlomakulu NN, Prior KJ, Setati ME, Divol B (2017) Candida pyralidae killer toxin disrupts the cell wall of Brettanomyces bruxellensis in red grape juice. J Appl Microbiol 122:747–758
Meineke B, Kast A, Schwer B, Meinhardt F, Shuman S, Klassen R (2012) A fungal anticodon nuclease ribotoxin exploits a secondary cleavage site to evade tRNA repair. RNA 18:1716–1724
Meinhardt F, Schaffrath R (2001) Extranuclear inheritance: cytoplasmic linear double-stranded DNA killer elements of the dairy yeast Kluyveromyces lactis. In: Esser K, Lüttge U, Kadereit JW, Beyschlag W (eds) Progress in botany, vol 62. Springer, Berlin, pp 51–70
Meinhardt F, Larsen M, Wodara C, Schickel J (1994) A novel approach to express a heterologous gene on linear killer plasmids: expression of the bacterial aph gene from a cytoplasmic promoter fragment without in-phase fusion to the plasmid open reading frame. Plasmid 32:318–327
Meskauskas A, Citavicius D (1992) The K2-type killer toxin- and immunity-encoding region from Saccharomyces cerevisiae: structure and expression in yeast. Gene 111:135–139
Michalcáková S, Sturdík E, Sulo P (1994) Construction and properties of K2 and K3 type killer Saccharomyces wine yeasts. Wein-Wissenschaft 49:130–132
Middelbeek EJ, Hermans JM, Stumm C (1979) Production, purification and properties of a Pichia kluyveri killer toxin. Antonie Van Leeuwenhoek 45:437–450
Middelbeek EJ, van de Laar HH, Hermans JM, Stumm C, Vogels GD (1980a) Physiological conditions affecting the sensitivity of Saccharomyces cerevisiae to a Pichia kluyveri killer toxin and energy requirement for toxin action. Antonie Van Leeuwenhoek 46:483–497
Middelbeek EJ, Crützen QH, Vogels GD (1980b) Effects of potassium and sodium ions on the killing action of a Pichia kluyveri toxin in cells of Saccharomyces cerevisiae. Antimicrob Agents Chemother 18:519–524
Miyamoto M, Onozato N, Selvakumar D, Kimura T, Furuichi Y, Komiyama T (2006) The role of the histidine-35 residue in the cytocidal action of HM-1 killer toxin. Microbiology 152:2951–2958
Miyamoto M, Furuichi Y, Komiyama T (2011) Genome-wide screen of Saccharomyces cerevisiae for killer toxin HM-1 resistance. Yeast 28:27–41
Miyamoto M, Furuichi Y, Komiyama T (2012) The high-osmolarity glycerol- and cell wall integrity-MAP kinase pathways of Saccharomyces cerevisiae are involved in adaptation to the action of killer toxin HM-1. Yeast 29:475–485
Muccilli S, Wemhoff S, Restuccia C, Meinhardt F (2013) Exoglucanase-encoding genes from three Wickerhamomyces anomalus killer strains isolated from olive brine. Yeast 30:33–43
Nakatsukasa K, Brodsky JL (2008) The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic 9:861–870
Nandakumar J, Schwer B, Schaffrath R, Shuman S (2008) RNA repair: an antidote to cytotoxic eukaryal RNA damage. Mol Cell 31:278–286
Noma A, Sakaguchi Y, Suzuki T (2009) Mechanistic characterization of the sulfur-relay system for eukaryotic 2-thiouridine biogenesis at tRNA wobble positions. Nucleic Acids Res 37:1335–1352
Novotná D, Flegelová H, Janderová B (2004) Different action of killer toxins K1 and K2 on the plasma membrane and the cell wall of Saccharomyces cerevisiae. FEMS Yeast Res 4:803–813
Orentaite I, Poranen MM, Oksanen HM, Daugelavicius R, Bamford DH (2016) K2 killer toxin-induced physiological changes in the yeast Saccharomyces cerevisiae. FEMS Yeast Res 16:fow003
Oro L, Zara S, Fancellu F, Mannazzu I, Budroni M, Ciani M, Comitini F (2014) TpBGL2 codes for a Tetrapisispora phaffii killer toxin active against wine spoilage yeasts. FEMS Yeast Res 14:464–471
Ouchi K, Akiyama H (1976) Breeding of useful killer sake yeasts by repeated back-crossing. J Ferment Technol 54:615
Pagé N, Gérard-Vincent M, Ménard P, Beaulieu M, Azuma M, Dijkgraaf GJ, Li H, Marcoux J, Nguyen T, Dowse T, Sdicu AM, Bussey H (2003) A Saccharomyces cerevisiaegenome-wide mutant screen for altered sensitivity to K1 killer toxin. Genetics 163:875–894
Paluszynski JP, Klassen R, Meinhardt F (2007) Pichia acaciae killer system: genetic analysis of toxin immunity. Appl Environ Microbiol 73:4373–4378
Park CM, Bruenn JA, Ganesa C, Flurkey WF, Bozarth RF, Koltin Y (1994) Structure and heterologous expression of the Ustilago maydis viral toxin KP4. Mol Microbiol 11:155–164
Park CM, Banerjee N, Koltin Y, Bruenn JA (1996a) The Ustilago maydis virally encoded KP1 killer toxin. Mol Microbiol 20:957–963
Park CM, Berry JO, Bruenn JA (1996b) High-level secretion of a virally encoded anti-fungal toxin in transgenic tobacco plants. Plant Mol Biol 30:359–366
Peery T, Shabat-Brand T, Steinlauf R, Koltin Y, Bruenn J (1987) Virus-encoded toxin of Ustilago maydis: two polypeptides are essential for activity. Mol Cell Biol 7:470–477
Perez MF, Contreras L, Garnica NM, Fernández-Zenoff MV, Farías ME, Sepulveda M, Ramallo J, Dib JR (2016) Native killer yeasts as biocontrol agents of postharvest fungal diseases in lemons. PLoS One 11(10):e0165590
Pfeiffer P, Radler F (1984) Comparison of the killer toxin of several yeasts and the purification of a toxin of type K2. Arch Microbiol 137:357–361
Pfeiffer I, Golubev WI, Farkas Z, Kucsera J, Golubev N (2004) Mycocin production in Cryptococcus aquaticus. Antonie Van Leeuwenhoek 86:369–375
Platania C, Restuccia C, Muccilli S, Cirvilleri G (2012) Efficacy of killer yeasts in the biological control of Penicillium digitatum on Tarocco orange fruits (Citrus sinensis). Food Microbiol 30:219–225
Polonelli L, Morace G (1986) Reevaluation of the yeast killer phenomenon. J Clin Microbiol 24:866–869
Polonelli L, Morace G (1987) Production and characterization of yeast killer toxin monoclonal antibodies. J Clin Microbiol 25:460–462
Polonelli L, Morace G (1988) Yeast killer toxin-like anti-idiotypic antibodies. J Clin Microbiol 26:602–604
Polonelli L, Fanti F, Conti S, Campani L, Gerloni M, Castagnola M, Morace G, Chezzi C (1990) Detection by immunofluorescent anti-idiotypic antibodies of yeast killer toxin cell wall receptors of Candida albicans. J Immunol Methods 132:205–209
Polonelli L, Lorenzini R, De Bernardis F, Gerloni M, Conti S, Morace G, Magliani W, Chezzi C (1993) Idiotypic vaccination: immunoprotection mediated by anti-idiotypic antibodies with antibiotic activity. Scand J Immunol 37:105–110
Polonelli L, De Bernardis F, Conti S, Boccanera M, Gerloni M, Morace G, Magliani W, Chezzi C, Cassone A (1994) Idiotypic intravaginal vaccination to protect against candidal vaginitis by secretory, yeast killer toxin-like anti-idiotypic antibodies. J Immunol 152:3175–3182
Polonelli L, Séguy N, Conti S, Gerloni M, Bertolotti D, Cantelli C, Magliani W, Cailliez JC (1997) Monoclonal yeast killer toxin-like candidacidal anti-idiotypic antibodies. Clin Diagn Lab Immunol 4:142–146
Polonelli L, Magliani W, Conti S, Bracci L, Lozzi L, Neri P, Adriani D, De Bernardis F, Cassone A (2003) Therapeutic activity of an engineered synthetic killer antiidiotypic antibody fragment against experimental mucosal and systemic candidiasis. Infect Immun 71:6205–6212
Pretorius IS (2000) Tailoring wine yeast for the new millennium: novel approaches to the ancient art of winemaking. Yeast 16:675–729
Puchkov EO, Wiese A, Seydel U, Kulakovskaya TV (2001) Cytoplasmic membrane of a sensitive yeast is a primary target for Cryptococcus humicola mycocidal compound (microcin). Biochim Biophys Acta 1512:239–250
Puchkov EO, Zähringer U, Lindner B, Kulakovskaya TV, Seydel U, Wiese A (2002) The mycocidal, membrane-active complex of Cryptococcus humicola is a new type of cellobiose lipid with detergent features. Biochim Biophys Acta 1558:161–170
Puhalla JE (1968) Compatibility reactions on solid medium and interstrain inhibition in Ustilago maydis. Genetics 60:461–474
Radler F, Schmitt MJ, Meyer B (1990) Killer toxin of Hanseniaspora uvarum. Arch Microbiol 154:175–178
Rep M, Krantz M, Thevelein JM, Hohmann S (2000) The transcriptional response of Saccharomyces cerevisiae to osmotic shock. Hot1p and Msn2p/Msn4p are required for the induction of subsets of high osmolarity glycerol pathway-dependent genes. J Biol Chem 275:8290–8300
Riffer F, Eisfeld K, Breinig F, Schmitt MJ (2002) Mutational analysis of K28 preprotoxin processing in the yeast Saccharomyces cerevisiae. Microbiology 148:1317–1328
Rodríguez-Cousiño N, Esteban R (2017) Relationships and evolution of double-stranded RNA totiviruses of yeasts inferred from analysis of L-A-2 and L-BC variants in wine yeast strain populations. Appl Environ Microbiol 83:pii: e02991-16
Rodríguez-Cousiño N, Maqueda M, Ambrona J, Zamora E, Esteban R, Ramírez M (2011) A new wine Saccharomyces cerevisiae killer toxin (Klus), encoded by a double-stranded rna virus, with broad antifungal activity is evolutionarily related to a chromosomal host gene. Appl Environ Microbiol 77:1822–1832
Rodríguez-Cousiño N, Gómez P, Esteban R (2013) L-A-lus, a new variant of the L-A totivirus found in wine yeasts with Klus killer toxin-encoding Mlus double-stranded RNA: possible role of killer toxin-encoding satellite RNAs in the evolution of their helper viruses. Appl Environ Microbiol 79:4661–4674
Romanos M, Boyd A (1988) A transcriptional barrier to expression of cloned toxin genes of the linear plasmid k1 of Kluyveromyces lactis: evidence that native k1 has novel promoters. Nucleic Acids Res 16:7333–7350
Santos A, Marquina D (2004a) Ion channel activity by Pichia membranifaciens killer toxin. Yeast 21:151–162
Santos A, Marquina D (2004b) Killer toxin of Pichia membranifaciens and its possible use as a biocontrol agent against grey mould disease of grapevine. Microbiology 150:2527–2534
Santos A, Marquina D, Leal JA, Peinado JM (2000) (1-6)-β-D-glucan as cell wall receptor for Pichia membranifaciens killer toxin. Appl Environ Microbiol 66:1809–1813
Santos A, Marquina D, Barroso J, Peinado JM (2002) (1-6)-β-D-glucan as the cell wall binding site for Debaryomyces hansenii killer toxin. Lett Appl Microbiol 34:95–99
Santos A, Sánchez A, Marquina D (2004) Yeasts as biological agents to control Botrytis cinerea. Microbiol Res 159:331–338
Santos A, Del Mar Alvarez M, Mauro MS, Abrusci C, Marquina D (2005) The transcriptional response of Saccharomyces cerevisiae to Pichia membranifaciens killer toxin. J Biol Chem 280:41881–41892
Santos A, San Mauro M, Abrusci C, Marquina D (2007) Cwp2p, the plasma membrane receptor for Pichia membranifaciens killer toxin. Mol Microbiol 64:831–843
Santos A, San Mauro M, Bravo E, Marquina D (2009) PMKT2, a new killer toxin from Pichia membranifaciens, and its promising biotechnological properties for control of the spoilage yeast Brettanomyces bruxellensis. Microbiology 155:624–634
Santos A, Navascués E, Bravo E, Marquina D (2011) Ustilago maydis killer toxin as a new tool for the biocontrol of the wine spoilage yeast Brettanomyces bruxellensis. Int J Food Microbiol 145:147–154
Santos A, Alonso A, Belda I, Marquina D (2013) Cell cycle arrest and apoptosis, two alternative mechanisms for PMKT2 killer activity. Fungal Genet Biol 50:44–54
Satwika D, Klassen R, Meinhardt F (2012a) Anticodon nuclease encoding virus like elements in yeast. Appl Microbiol Biotechnol 96:345–356
Satwika D, Klassen R, Meinhardt F (2012b) Repeated capture of a cytoplasmic linear plasmid by the host nucleus in Debaryomyces hansenii. Yeast 29:145–154
Sawant AD, Abdelal AT, Ahearn DG (1989) Purification and characterization of the anti-Candida toxin of Pichia anomala WC 65. Antimicrob Agents Chemother 33:48–52
Schaffrath R, Leidel SA (2017) Wobble uridine modifications – a reason to live, a reason to die?! RNA Biol 14:1209–1222
Schaffrath R, Meacock PA (1996) A cytoplasmic gene shuffle system in Kluyveromyces lactis: use of epitope-tagging to detect a killer plasmid-encoded gene product. Mol Microbiol 19:545–554
Schaffrath R, Meinhardt F (2005) Kluyveromyces lactis zymocin and other plasmid-encoded yeast killer toxins. In: Schmitt MJ, Schaffrath R (eds) Microbial protein toxins, vol 11. Springer, Berlin, pp 133–155
Schaffrath R, Soond SM, Meacock PA (1995) Cytoplasmic gene expression in yeast: a plasmid-encoded transcription system in Kluyveromyces lactis. Biochem Soc Trans 23:128
Schaffrath R, Meinhardt F, Meacock PA (1996) Yeast killer plasmid pGKL2: molecular analysis of UCS5, a cytoplasmic promoter element essential for ORF5 gene function. Mol Gen Genet 250:286–294
Schaffrath R, Sasnauskas K, Meacock PA (2000) Use of gene shuffles to study the cytoplasmic transcription system operating on Kluyveromyces lactis linear DNA plasmids. Enzym Microb Technol 26:664–670
Schickel J, Helmig C, Meinhardt F (1996) Kluyveromyces lactis killer system. Analysis of cytoplasmic promoters of linear plasmids. Nucleic Acids Res 24:1879–1886
Schmitt MJ, Breinig F (2002) The viral killer system in yeast: from molecular biology to application. FEMS Microbiol Rev 26:257–276
Schmitt MJ, Breinig F (2006) Yeast viral killer toxins: lethality and self-protection. Nat Rev Microbiol 4:212–221
Schmitt MJ, Neuhausen F (1994) Killer toxin-secreting double-stranded RNA mycoviruses in the yeasts Hanseniaspora uvarum and Zygosaccharomyces bailii. J Virol 68:1765–1772
Schmitt M, Radler F (1987) Mannoprotein of the yeast cell wall as primary receptor for the killer toxin of Saccharomyces cerevisiae strain 28. J Gen Microbiol 133:3347–3354
Schmitt MJ, Tipper DJ (1990) K28, a unique double-stranded RNA killer virus of Saccharomyces cerevisiae. Mol Cell Biol 10:4807–4815
Schmitt MJ, Tipper DJ (1992) Genetic analysis of maintenance and expression of L and M double-stranded RNAs from yeast killer virus K28. Yeast 8:373–384
Schmitt MJ, Tipper DJ (1995) Sequence of the M28 dsRNA: preprotoxin is processed to an α/β heterodimeric protein toxin. Virology 213:341–351
Schmitt MJ, Klavehn P, Wang J, Schönig I, Tipper DJ (1996) Cell cycle studies on the mode of action of yeast K28 killer toxin. Microbiology 142:2655–2662
Schmitt MJ, Poravou O, Trenz K, Rehfeldt K (1997) Unique double-stranded RNAs responsible for the anti-Candida activity of the yeast Hanseniaspora uvarum. J Virol 71:8852–8855
Schründer J, Meinhardt F (1995) An extranuclear expression system for analysis of cytoplasmic promoters of yeast linear killer plasmids. Plasmid 33:139–151
Seki T, Choi EH, Ryu D (1985) Construction of killer wine yeast strain. Appl Environ Microbiol 49:1211–1215
Selvakumar D, Zhang QZ, Miyamoto M, Furuichi Y, Komiyama T (2006a) Identification and characterization of a neutralizing monoclonal antibody for the epitope on HM-1 killer toxin. J Biochem 139:399–406
Selvakumar D, Miyamoto M, Furuichi Y, Komiyama T (2006b) Inhibition of fungal β-1,3-glucan synthase and cell growth by HM-1 killer toxin single-chain anti-idiotypic antibodies. Antimicrob Agents Chemother 50:3090–3097
Selvakumar D, Miyamoto M, Furuichi Y, Komiyama T (2006c) Inhibition of β-1,3-glucan synthase and cell growth of Cryptococcus species by recombinant single-chain anti-idiotypic antibodies. J Antibiot (Tokyo) 59:73–79
Semenza JC, Hardwick KG, Dean N, Pelham HR (1990) ERD2, a yeast gene required for the receptor-mediated retrieval of luminal ER proteins from the secretory pathway. Cell 61:1349–1357
Servienė E, Lukša J, Orentaitė I, Lafontaine DL, Urbonavičius J (2012) Screening the budding yeast genome reveals unique factors affecting K2 toxin susceptibility. PLoS One 7:e50779
Sor F, Fukuhara H (1985) Structure of a linear plasmid of the yeast Kluyveromyces lactis: compact organization of the killer genome. Curr Genet 9:147–155
Stark MJR, Boyd A (1986) The killer toxin of Kluyveromyces lactis: characterization of the toxin subunits and identification of the genes which encode them. EMBO J 5:1995–2002
Stark MJ, Mileham AJ, Romanos MA, Boyd A (1984) Nucleotide sequence and transcription analysis of a linear DNA plasmid associated with the killer character of the yeast Kluyveromyces lactis. Nucleic Acids Res 12:6011–6030
Stark MJR, Boyd A, Mileham AJ, Romanos MA (1990) The plasmid encoded killer system of Kluyveromyces lactis: a review. Yeast 6:1–29
Steinberg G, Perez-Martin J (2008) Ustilago maydis, a new fungal model system for cell biology. Trends Cell Biol 18:61–67
Steinlauf R, Peery T, Koltin Y, Bruenn J (1988) The Ustilago maydis virus encoded toxin—effect of KP6 on cells and spheroplasts. Exp Mycol 12:264–274
Studte P, Zink S, Jablonowski D, Bär C, von der Haar T, Tuite MF, Schaffrath R (2008) tRNA and protein methylase complexes mediate zymocin toxicity in yeast. Mol Microbiol 69:1266–1277
Sulo P, Michalcáková S (1992) The K3 type killer strains of genus Saccharomyces for wine production. Folia Microbiol (Praha) 37:289–294
Sulo P, Michalcakova S, Reiser V (1992) Construction and properties of K1 type killer wine yeasts. Biotechnol Lett 14:55–60
Suzuki C, Nikkuni S (1994) The primary and subunit structure of a novel type killer toxin produced by a halotolerant yeast, Pichia farinosa. J Biol Chem 269:3041–3046
Suzuki C, Kashiwagi T, Tsuchiya F, Kunishima N, Morikawa K, Nikkuni S, Arata Y (1997) Circular dichroism analysis of the interaction between the α and β subunits in a killer toxin produced by a halotolerant yeast, Pichia farinosa. Protein Eng 10:99–101
Suzuki C, Ando Y, Machida S (2001) Interaction of SMKT, a killer toxin produced by Pichia farinosa, with the yeast cell membranes. Yeast 18:1471–1478
Suzuki Y, Schwartz SL, Mueller NC, Schmitt MJ (2017) Cysteine residues in a yeast viral A/B toxin crucially control host cell killing via pH-triggered disulfide rearrangements. Mol Biol Cell 28:1123–1131
Takasuka T, Komiyama T, Furuichi Y, Watanabe T (1995) Cell wall synthesis specific cytocidal effect of Hansenula mrakiitoxin-1 on Saccharomyces cerevisiae. Cell Mol Biol Res 41:575–581
Tamás MJ, Luyten K, Sutherland FC, Hernandez A, Albertyn J, Valadi H, Li H, Prior BA, Kilian SG, Ramos J, Gustafsson L, Thevelein JM, Hohmann S (1999) Fps1p controls the accumulation and release of the compatible solute glycerol in yeast osmoregulation. Mol Microbiol 31:1087–1104
Tao J, Ginsberg I, Banerjee N, Held W, Koltin Y, Bruenn JA (1990) Ustilago maydis KP6 killer toxin: structure, expression in Saccharomyces cerevisiae, and relationship to other cellular toxins. Mol Cell Biol 10:1373–1381
Theisen S, Molkenau E, Schmitt MJ (2000) Wicaltin, a new protein toxin secreted by the yeast Williopsis californica and its broad-spectrum antimycotic potential. J Microbiol Biotechnol 10:547–550
Tiggemann M, Jeske S, Larsen M, Meinhardt F (2001) Kluyveromyces lactis cytoplasmic plasmid pGKL2: heterologous expression of Orf3p and prove of guanylyltransferase and mRNA-triphosphatase activities. Yeast 18:815–825
Tipper DJ, Bostian KA (1984) Double-stranded ribonucleic acid killer systems in yeasts. Microbiol Rev 48:125–156
Tokunaga M, Wada N, Hishinuma F (1987) Expression and identification of immunity determinants on linear DNA killer plasmids pGKL1 and pGKL2 in Kluyveromyces lactis. Nucleic Acids Res 15:1031–1046
Tokunaga M, Kawamura A, Hishinuma F (1989) Expression of pGKL killer 28K subunit in Saccharomyces cerevisiae: identification of 28K subunit as a killer protein. Nucleic Acids Res 17:3435–3446
Tommasino M, Ricci S, Galeotti C (1988) Genome organization of the killer plasmid pGKL2 from Kluyveromyces lactis. Nucleic Acids Res 16:5863–5978
Travassos LR, Silva LS, Rodrigues EG, Conti S, Salati A, Magliani W, Polonelli L (2004) Therapeutic activity of a killer peptide against experimental paracoccidioidomycosis. J Antimicrob Chemother 54:956–958
Valzano M, Cecarini V, Cappelli A, Capone A, Bozic J, Cuccioloni M, Epis S, Petrelli D, Angeletti M, Eleuteri AM, Favia G, Ricci I (2016) A yeast strain associated to Anopheles mosquitoes produces a toxin able to kill malaria parasites. Malar J 15:21
Walker GM, McLeod AH, Hodgson VJ (1995) Interactions between killer yeasts and pathogenic fungi. FEMS Microbiol Lett 127:213–222
Wang X, Chi Z, Yue L, Li J, Li M, Wu L (2007a) A marine killer yeast against the pathogenic yeast strain in crab (Portunus trituberculatus) and an optimization of the toxin production. Microbiol Res 162:77–85
Wang X, Chi Z, Yue L, Li J (2007b) Purification and characterization of killer toxin from a marine yeast Pichia anomala YF07b against the pathogenic yeast in crab. Curr Microbiol 55:396–401
Wedral D, Shewfelt R, Frank J (2010) The challenge of Brettanomyces in wine. LWT-Food Sci Technol 43:1474–1479
Weiler F, Schmitt MJ (2003) Zygocin, a secreted antifungal toxin of the yeast Zygosaccharomyces bailii, and its effect on sensitive fungal cells. FEMS Yeast Res 3:69–76
Weiler F, Rehfeldt K, Bautz F, Schmitt MJ (2002) The Zygosaccharomyces bailii antifungal virus toxin zygocin: cloning and expression in a heterologous fungal host. Mol Microbiol 46:1095–1105
Wemhoff S, Klassen R, Meinhardt F (2014) Site-directed mutagenesis of the heterotrimeric killer toxin zymocin identifies residues required for early steps in toxin action. Appl Environ Microbiol 80:6549–6559
Wemhoff S, Klassen R, Meinhardt F (2016a) DNA damage induced by the anticodon nuclease from a Pichia acaciae killer strain is linked to ribonucleotide reductase depletion. Cell Microbiol 18:211–222
Wemhoff S, Klassen R, Beetz A, Meinhardt F (2016b) DNA damage responses are induced by tRNA anticodon nucleases and hygromycin B. PLoS One 11:e0157611
Wickner RB (1992) Double-stranded and single-stranded RNA viruses of Saccharomyces cerevisiae. Annu Rev Microbiol 46:347–375
Wickner RB (1996) Double-stranded RNA viruses of Saccharomyces cerevisiae. Microbiol Rev 60:250–265
Wilson DW, Meacock PA (1988) Extranuclear gene expression in yeast: evidence for a plasmid encoded RNA-polymerase of unique structure. Nucleic Acids Res 16:8097–8112
Wistow GJ, Piatigorsky J (1988) Lens crystallins: the evolution and expression of proteins for a highly specialized tissue. Annu Rev Biochem 57:479–504
Woods DR, Bevan EA (1968) Studies on the nature of the killer factor produced by Saccharomyces cerevisiae. J Gen Microbiol 51:115–126
Worsham PL, Bolen PL (1990) Killer toxin production in Pichia acaciae is associated with linear DNA plasmids. Curr Genet 18:77–80
Yamamoto T, Iratani T, Hirata H, Imai M, Yamaguchi H (1986a) Killer toxin from Hansenula mrakii selectively inhibits cell wall synthesis in a sensitive yeast. FEBS Lett 197:50–54
Yamamoto T, Imai M, Tachibana K, Mayumi M (1986b) Application of monoclonal antibodies to the isolation and characterization of a killer toxin secreted by Hansenula mrakii. FEBS Lett 195:253–257
Yamamoto T, Uchida K, Hiratani T, Miyazaki T, Yagiu J, Yamaguchi H (1988) In vitro activity of the killer toxin from yeast Hansenula mrakii against yeasts and molds. Antibiot (Tokyo) 41:398–403
Young TW, Yagiu M (1978) A comparison of the killer character in different yeasts and its classification. Antonie Van Leeuwenhoek 44(1):59–77
Zabel R, Bär C, Mehlgarten C, Schaffrath R (2008) Yeast alpha-tubulin suppressor Ats1/Kti13 relates to the Elongator complex and interacts with Elongator partner protein Kti11. Mol Microbiol 69:175–187
Zhu H, Bussey H (1991) Mutational analysis of the functional domains of yeast K1 killer toxin. Mol Cell Biol 11:175–181
Zhu YS, Zhang XY, Cartwright CP, Tipper DJ (1992) Kex2-dependent processing of yeast K1 killer preprotoxin includes cleavage at ProArg-44. Mol Microbiol 6:511–520
Zhu YS, Kane J, Zhang XY, Zhang M, Tipper DJ (1993) Role of the gamma component of preprotoxin in expression of the yeast K1 killer phenotype. Yeast 9:251–266
Zink S, Mehlgarten C, Kitamoto HK, Nagase J, Jablonowski D, Dickson RC, Stark MJR, Schaffrath R (2005) Mannosyl-diinositolphospho-ceramide, the major yeast plasma membrane sphingolipid, governs toxicity of Kluyveromyces lactis zymocin. Eukaryot Cell 4:879–889
Zorg J, Kilian S, Radler F (1988) Killer toxin producing strains of the yeasts Hanseniaspora uvarum and Pichia kluyveri. Arch Microbiol 149:261–267
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Schaffrath, R., Meinhardt, F., Klassen, R. (2018). Yeast Killer Toxins: Fundamentals and Applications. In: Anke, T., Schüffler, A. (eds) Physiology and Genetics. The Mycota, vol 15. Springer, Cham. https://doi.org/10.1007/978-3-319-71740-1_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-71740-1_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-71739-5
Online ISBN: 978-3-319-71740-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)