
Abstract
The molecular mass of the purified killer toxin from the marine killer yeast YF07b was estimated to be 47.0 kDa. The optimal pH and temperature of the purified killer toxin were 4.5 and 40°C, respectively. The toxin was activated by Ca2+, K+, Na+, Mg2+, Na+, and Co2+. However, Fe2+, Fe3+, Hg2+, Cu2+, Mn2+, Zn2+, and Ag+ acted as inhibitors in decreasing activity of the toxin. The toxin was strongly inhibited by phenylmethanesulphonyl fluoride (PMSF), iodoacetic acid, ethylenediaminetetraacetic acid, and 1,10-phenanthroline. The Km of the toxin for laminarin was 1.17 g L−1. The toxin also actively hydrolyzed laminarin and killed the whole cells of the pathogenic yeast in crab.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Up to now, toxin-producing killer yeasts have been identified in many genera of yeasts [17], and killer toxins from many species of terrestrial yeasts have been purified and characterized [5, 7, 12].
In recent years, much evidence have shown that some marine yeasts are pathogenic to some marine animals and have caused large economic losses in maricultural industry in some regions of China [17]. A pathogenic yeast strain, WCY, which could cause milky disease in Portunus trituberculatus, was identified to be Metschnikowia bicuspidate [17]. It was found that nystatin and other chemicals are active against the pathogenic yeast. However, the compounds with minimum inhibitory concentration are toxic to the crab and it is impossible to apply the expensive antibiotics to the open sea. It was found that the yeast Torulopsis mogii is the pathogen to some shrimp in China [15]. The yeast Metschnikowia bicuspidate var. bicuspidate, a pathogenic yeast of aquatic invertebrates, was capable of infecting aquaculture-reared, disease-free Artemia [10]. A new species of marine yeast, Kluyveromyces penaeid, was isolated from the heart tissue of subadult shrimp Penaeus chinensis during tissue culture [16]. In our previous study, we found that marine yeast strain YF07b, which was identified to be Pichia anomala, had the highest ability to produce killer toxin against the pathogenic yeast [17]. In this study, our main purposes are to purify and characterize killer toxin from Pichia anomala YF07b and use the purified killer toxin for hydrolysis of laminarin (β-1, 3-glucan) and the pathogenic yeast cells.
Materials and Methods
Yeast Strains
P. anomala YF07b was the killer toxin producer. Metschnikowia bicuspidate WCY, which could cause the milky disease in Portunus trituberculatus, was used as the sensitive yeast strain used in this study [17]. The two yeast strains were maintained in YPD medium (prepared with seawater) containing 20.0 g L−1 glucose, 10.0 g L−1 yeast extract, and 20.0 g L−1 polypeptone at 4°C.
Media
The assay medium (pH 4.5) was used according to [12]. Killer toxin production medium (pH 4.5) was used according to [17].
Killer Toxin Production
The marine yeast strain YF07b was grown in 50.0 ml of the production medium by shaking at 130 rpm and 23.0°C for 3 days [17]. The fermented broth was centrifuged at 2823×g and 4°C for 5 min, and the supernatant obtained was taken as the crude killer toxin.
Measurement of Killer Toxin Activity
Two hundreds microliters of the crude killer toxin or the eluted killer toxin were added to each sterile Oxford-cup, which was put on the assay medium seeded with the pathogenic yeast strain WCY and incubated at 15°C for 72 h, and the diameter of the inhibition zone was used as a measure of the yeast killer activity [12, 17].
Determination of β-1,3-d-Glucanase Activity of the Killer Toxin
It has been reported that killer toxin from P. anomala has β-1,3-d-glucanase activity [5, 6]. β-1,3-d-glucanase activity of the killer toxin was determined according to the methods [6]. The amount of reducing sugar in the reaction mixture was assayed by the method of Nelson-Somogyi [14]. One β-1,3-d-glucanase unit (U) was defined as the amount of enzyme that produces 1 μg of reducing sugar per min under the assay conditions used in this study.
Killer Toxin Purification
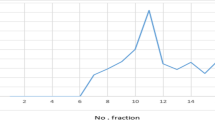
Killer toxin purification was carried out at 4°C according to the methods [9]. Six hundred milliliters of the culture of the marine killer yeast strain YF07b grown for 72 h was used as the starting material for the killer toxin purification. Protein concentration in the elute was measured by the method of Bradford, and bovine serum albumin served as standard [2]. The purity and molecular mass of the purified killer toxin in the concentrated fractions showing both the killer toxin activity and β-1,3-d-glucanase activity were analyzed in noncontinuous denaturing sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) [8] with a Two-Dimensional Electrophoresis System (Amersham Biosciences, Sweden) and stained by Coomassie Brilliant Blue R-250 [3]. The molecular mass standards for SDS-PAGE are indicated in Figure 1.

Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (12%) of the fractions showing β-1,3-d-glucanase activity obtained during the purification. The lanes are as follows: M, marker proteins with relative molecular masses indicated on the left; 1, the elute from DEAE Sepharose Fast Flow anion exchange chromatography; 2, the elute from gel filtration chromatography (SephadexTM G-75); 3, the concentrated supernatant of the culture
Effects of pH and Temperature on β-1,3-d-Glucanase Activity and Stability of the Killer Toxin
The effect of pH on β-1,3-d-glucanase activity of the purified killer toxin was determined by incubating the purified killer toxin between pH 3.0 and 8.0 using the standard assay conditions. The buffer used was 0.1 m citric disodium phosphate buffer (pH 3.0–8.0). The pH stability was tested by 24 h pre-incubation of the purified killer toxin in appropriate buffers that had the same ionic concentrations at different pH values ranging from 3.0 to 8.0 at temperature of 4°C. The remaining activities of β-1,3-d-glucanase were measured immediately after this treatment with the standard method as mentioned above. The optimal temperature for β-1,3-d-glucanase activity of the purified killer toxin was determined at temperatures from 20 to 90°C in the same buffer as described above. Temperature stability of the purified killer toxin was tested by pre-incubating the killer toxin at different temperatures ranging from 20 to 90°C during 60 min; residual activity was measured as described above immediately. Here, the relative β-1,3-d-glucanase activity of pre-incubated sample at 4°C was regarded as 100%.
Effects of Different Metal Ions and Protein Inhibitors on β-1,3-d-Glucanase Activity of the Killer Toxin
The assay of β-1,3-d-glucanase activity of the killer toxin was performed for 1 h in the reaction mixture as described above with various metal ions at a final concentration of 5.0 mm. The activity assayed in the absence of metal ions was defined as control. The effects of protein inhibitors at a final concentration of 5.0 mm on β-1,3-d-glucanase activity of the purified killer toxin were measured in the reaction mixture as described above. The purified killer toxin was pre-incubated with the respective compound for 1 h at 0°C, followed by the standard enzyme assay as described above. The activity assayed in the absence of the protein inhibitors was defined as control.
Determination of Kinetics Parameters
Two hundred fifty microliters of laminarin solution in 0.05 m citric acid–disodium phosphate buffer (pH 4.5) were mixed with 20 μL of the purified killer toxin (the final laminarin concentrations were 0.16, 0.31, 0.63, 1.25, and 2.5 g L−1, respectively) and the mixture was incubated at 30°C for 60 min. Km values were obtained from Lineweaver-Burk plot and expressed as the mean of the three different experiments.
Laminarin Hydrolysis
The reaction mixture containing 20 μL of 26.8 U mL−1 of the purified killer toxin and 0.25% laminarin in 0.1 mL of the buffer (0. 05 m, pH 4.5) was incubated at 30°C for 1 h. After that, the toxin in the mixture was inactivated by heating at 100°C for 10 min immediately [4]. The end products of laminarin hydrolysis were determined by thin layer chromatography [4].
Results and Discussion
Purification of the Killer Toxin
The results in this study show that the killer toxin was purified to homogeneity with a 180-fold increase in specific β-1,3-d-glucanase activity with a yield of about 14% as compared to that in the supernatant from the cell culture by concentration, gel filtration chromatography (SephadexTM G-75, Sigma-Aldrich), and DEAE Sepharose Fast Flow anion exchange chromatography (Sigma-Aldrich) (data not shown). SDS-PAGE was used to determine protein purity and estimate molecular mass of the finally concentrated elute as described by Laemmli [8]. The results in Figure 1 indicate that there was one single protein band from the finally concentrated elute, and the relative molecular mass of the purified killer toxin was estimated to be 47.0 kDa by SDS-PAGE. Therefore, it may be concluded that the killer toxin from the marine killer yeast strain was a monomer of 47.0 kDa protein. It has also been reported that the extracellular killer toxin from the terrestrial yeasts, P. anomala NCYC432 and P. anomala NCYC434 K5 type, had 47 kDa and 49 kDa of molecular mass, respectively, whereas the killer toxin from P. anomala WC 65 had 83.3 kDa of molecular mass [5, 6]. Usually, most of the killer toxins from Pichia spp are monomer. However, a novel type of killer toxin (salt-mediated killer toxin) produced by the halotolerant yeast P. farinosa, is a heterodimer (14.214 kDa) [7]. Our results show that N-terminal amino acid sequence of the purified killer toxin had 100% match of that of killer toxin from P. anomala NCYC 432 [6] (data not shown).
Optimum Temperature and Thermal Stability
The β-1,3-d-glucanase activity of the purified killer toxin measured as a function of temperature from 20°C to 90°C shows that the activity was the highest at 40°C (Figure 2). The thermostability was investigated by pre-incubating the enzyme in the same buffer as described in Materials and Methods for 60 min and the remaining activity was determined. As shown in Figure 2, the residual β-1,3-d-glucanase activity still kept 93.0% of the control after treatment at 60°C for 1 h, indicating that the enzyme was very stable up to 60°C. Figure 2 also reveals that the killer toxin was inactivated rapidly at temperature higher than 60°C. From these results, the toxin seemed to have considerable thermostability. The toxins from P. anomala showed high stability up to 37°C [5, 6]. At pH 4, optimal killer activity of killer toxin produced by Pichia membranifaciens CYC 1106 was observed at temperatures up to 20.6°C [12]. This means that the killer toxin from the marine killer yeast strain had higher thermostability than that from other Pichia spp.

Effects of different temperatures on β-1,3-d-glucanase activity (▲) and stability (■) of the purified killer toxin. Data are given as means ± SD, n = 3
Optimum pH and pH Stability
Our results (Figure 3) show that the maximum activity was observed at pH 4.5. It can be seen from the results in Figure 3 that the activity profile of the killer toxin was stable in the range of pH 3.0–5.0. For example, the residual activity still remained more than 91.5% after the treatment at pH 5.0 and 4°C for 24 h. Naturally, the toxin shows its maximum killer activity at pH 2.5–4.0, its toxicity steeply decreases with increasing pH, and is finally lost at pH 6.0. Killer toxin from Pichia kluyveri was active at pH 2.5–4.7 and the killer toxin from Pichia inositovora was active at pH 3.4–4.2 [6]. The toxin from P. anomala showed high stability at pH values between 3 and 5.5 and up to 37°C [5, 6]. The killer toxin produced by P. membranifaciens CYC 1106 was stable only within a narrow pH range (3.0–4.8) [12]. This results show that the killer toxin from the marine killer yeast strain had similar pH stability to that from most terrestrial yeasts. However, the most stable killer toxins are those of Hansenula mrakii (stable at pH 2–11) and H. saturnus (stable at pH 3–11) [7].

Effects of different pH on β-1,3-d-glucanase activity (■) and stability (▲) of the purified killer toxin. Data are given as means ± SD, n = 3
Effects of Different Cations and Protein Inhibitors on Activity of the Purified β-1,3-d-Glucanase
It was found that Ca2+, Co2+, K+, Na+, and Mg2+ (at the concentrations of 5.0 mm) activated the activity of β-1,3-d-glucanase of the purified killer toxin (data not shown). However, Fe2+, Fe3+, Hg2+, Cu2+, Mn2+, Zn2+, and Ag+ (at the concentrations of 5.0 mm) acted as inhibitors in decreasing β-1,3-d-glucanase activity of the purified killer toxin, with Fe2+ and Cu2+ (at the concentration of 5.0 mm) showing the lowest rank (11.9%) (data not shown), suggesting that they were able to alter the enzyme conformation [13]. The inhibition by mercuric ions may indicate the importance of thiol-containing amino acid residues in the killer toxin function [1]. The killing effect of killer toxin from P. membranifaciens CYC 1106 is enhanced with increased NaCl or KCl concentrations [12]. However, our results were different from those reported by Izgu et al. [5].
The presence of the chelating agents, ethylenediaminetetraacetic acid (EDTA) and 1,10-phenanthroline, inhibited the toxin activity, demonstrating that the purified killer toxin was metalloenzyme [11]. The toxin activity was strongly inhibited by PMSF, indicating that Ser residues were essential for the killer toxin active sites (data not shown). The results in this study also show that iodoacetic acid had a negative effect on the killer toxin activity, suggesting that Cys residues were important for active sites of the toxin (data not shown). However, EDTA had no effects on activity of the killer toxin from P. anomala 434 K5 type [5].
Kinetics Parameters
Lineweaver-Burk plots in Figure 4 show that apparent Km value of the killer toxin for laminarin was 1.17 g L−1. So far, several killer toxins have been found to have exo-β-1, 3-glucanase activity. The apparent Km of the toxin from P. anomala NCYC 432 and P. anomala NCYC 434 K5 type was 0.3 g L−1 and 0.25 g L−1, respectively [5, 6]. This suggests that the killer toxin from the marine yeast displayed lower affinity for laminarin than that from P. anomala.

Lineweaver-Burk plot for Km values of the purified killer toxin in the presence of different concentrations of laminarin. Data are given as means ± SD, n = 3
Laminarin Hydrolysis
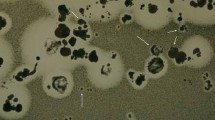
The results in Figure 5 demonstrate that only monosaccharides and disaccharides were released from laminarin after hydrolysis for 1 h with the purified killer toxin. The results imply that the killer toxin had very high exo-β-1, 3-glucanase activity. The results in Figure 6 show that the purified killer toxin gave a clear inhibition zone on the plate seeded with the pathogenic yeast, indicating that it could actively kill the intact cells of the pathogenic yeast WCY. This may be related to very high exo-β-1,3-glucanase activity of the killer toxin.

Thin-layer chromatogram of the hydrolysis products of laminarin with the purified killer toxin. Lane 1: Control (laminarin + inactivated killer toxin by heating at 100°C for 10 min); lane 2: the hydrolysis products for 1 h; lane 3: glucose; lane 4: maltose; lane 5: kestose (trisaccharides); lane 6: nystose (tetrasaccharides)

Killer activity of the purified killer toxin. Two hundred microliters of the purified killer toxin was added to the Oxford cup, which was put on the assay medium seeded with the pathogenic yeast strain WCY and incubated at 15°C for 72 h, and the diameter of the inhibition zone was measured
References
Barth G, Gaillardin C (1997) Physiology and genetics of the dimorphic fungus Yarrowia lipolytica. FEMS Microbiol Rev 19:219–237
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
George V, Diwan AM (1983) Simultaneous staining of proteins during polyacrylamide gel electrophoresis in acidic gels by countermigration of Coomassie brilliant blue R-250. Anal Biochem 132:481–483
Gong F, Sheng J, Chi ZM, Li J (2007) Inulinase production by a marine yeast Pichia guilliermondii and inulin hydrolysis by the crude inulinase. J Ind Microbiol Biotechnol 34:179–185
Izgu F, Altınbay D, Sertkaya A (2005) Enzymic activity of the K5-type yeast killer toxin and its characterization. Biosci Biotechnol Biochem 69:2200–2206
Izgu F, Altınbay D, Acun T (2006) Killer toxin of Pichia anomala NCYC 432; purification, characterization and its exo-β-1,3-glucanase activity. Enzyme Microb Tech 39:669–676
Kashiwagi T, Kunishima N, Suzuki C, Tsuchiya F, Nikkuni S, Arata Y, Morikawa K (1997) The novel acidophilic structure of the killer toxin from halotolerant yeast demonstrates remarkable folding similarity with a fungal killer toxin. Structure 5:81–93
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Ma CL, Ni XM, Chi ZM, Ma LY, Gao LM (2007) Purification and characterization of an alkaline protease from the marine yeast Aureobasidium pullulans for bioactive peptide production from different sources. Mar Biotechnol 9:343–351
Moore MM, Strom MB (2003) Infection and mortality by the yeast Metschnikowia bicuspidata var, bicuspidate in Chinook salmon fed live adult brine shrimp (Artemia franciscana). Aquaculture 220:43–57
Ramirez-Zavala B, Mercado-Flores Y, Hernadez-Rodriguez C, Villa-Tanaca L (2004) Purification and characterization of lysine aminopeptidase from Kluyveromyces marxiamus. FEMS Microbiol Lett 235:369–375
Santos A, Marquina D, Leal JA, Peinado JM (2000) (1,6)-β-D-glucan as cell wall receptor for Pichia membranifaciens killer toxin. Appl Environ Microbiol 66:1809–1813
Sharon C, Furugoh S, Yamakido T, Ogawa H, Kato Y (1998) Purification and characterization of a lipase from Pseudomonas aeruginosa KKA-5 and its role in castor oil hydrolysis. J Ind Microbiol Biotechnol 20:304–307
Spiro RG (1966) Analysis of sugars found in glycoproteins. Method Enzymol 8:3–26
Sun YH, Sun QH (1998) Studies on pathogenic organism and prevention and cure for explosive epidemic disease of parent prawn of Macrobrachium rosenbergh. J Fish China 22:56–60
Tong SL, Miao HZ (1999) A new species of marine yeast Kluyveromyces penaeid isolated from the heart of penaeid shrimp Pennaeus chinensis. J Mar Biol Ass U K 79:559–561
Wang XH, Chi ZM, Yue LX, Li J, Li MJ, Wu LF (2007) A marine killer yeast against the pathogenic yeast strain in crab (Portunus trituberculatus) and an optimization of the toxin production. Microbiol Res 162:77–85
Acknowledgment
This research was supported by Grant 30670058 from National Natural Science Foundation of China.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, X., Chi, Z., Yue, L. et al. Purification and Characterization of Killer Toxin from a Marine Yeast Pichia anomala YF07b Against the Pathogenic Yeast in Crab. Curr Microbiol 55, 396–401 (2007). https://doi.org/10.1007/s00284-007-9010-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-007-9010-y