
Abstract
HER2 is a member of the ErbB/HER family of receptor tyrosine kinases that promotes the proliferation of a subset of human breast cancers. HER2 is over-expressed in 20–25% of breast cancers and high expression is associated with poor clinical outcomes if not treated with an HER2-targeted drug. Although HER2 does not have a known extracellular ligand, HER2 is the preferred heterodimerization partner for the other HER-family receptors and HER2-containing heterodimers have increased affinity for ligands that bind to those heterodimer partners. The downstream effect is strong signal transduction and tumor cell proliferation. Even in the absence of a ligand, HER2 over-expression by itself can drive tumorigenesis and tumor growth. Given its elevated expression and oncogenic activity, its preferred status as a HER-family signaling partner, and the enhanced activity of HER2-containing heterodimers, HER2 is a valuable pharmacological target for the treatment of HER2+ breast cancer. HER2-targeted agents fall into three general categories: therapeutic antibodies, antibody-drug conjugates, and tyrosine kinase inhibitors. The resistance mechanisms associated with each of these classes of drugs will be reviewed here.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- HER2
- Resistance
- Trastuzumab
- Lapatinib
- Small molecule inhibitors
- Antibody inhibitors
- Antibody-drug conjugates
- PI3K/Akt
- PTEN
- Breast cancer
HER2 as a Therapeutic Target
The human HER (ErbB) gene family codes for four receptor tyrosine kinases (RTKs): HER1 (EGFR, ErbB-1), HER2 (Neu, ErbB-2), HER3 (ErbB-3), and HER4 (ErbB-4). As typical RTKs, the receptors consist of an extracellular ligand binding domain (except for HER2, which has no known ligand), a single transmembrane domain, and an intracellular tyrosine kinase domain (except for HER3, which does not have intrinsic kinase activity) (see Fig. 1). Growth factor ligand binding leads to a series of conformational changes that result in homodimer and heterodimer activation and ensuing phosphorylation at specific tyrosine residues that reside in the intracellular tail region of each receptor. This, in turn, leads to binding of SH2-containing docking proteins, the links between activated receptors and their downstream signaling pathways, eventually producing enhanced proliferation, migration, angiogenesis, and survival [1].

Diagrammatic representation of the HER2 receptor and its interaction with trastuzumab (red antibody), pertuzumab (orange antibody), and tyrosine kinase inhibitors (blue hexagon). The diagram illustrates trastuzumab’s inability (due to its binding location) to prevent HER2 dimerization with HER3 (and HER1) compared to pertuzumab’s inhibition of ligand-mediated heterodimerization. The natural HER3 ligand, heregulin, is represented by the green triangle. The oblong segments within each receptor represent folded domains and the yellow balls connected by lines represent the plasma membrane of the cell
HER2 was first described as an oncoprotein when the rat homolog (encoded by neu) was identified as a 185 kDa receptor in a group of chemically-induced neuroblastomas and glioblastomas and shown to confer loss of contact inhibition when expressed in mouse fibroblasts [2]. In humans, HER2 is over-expressed in 20–25% of breast cancers and high expression is associated with poor clinical outcomes if not treated with an HER2-targeted drug [3]. Cell-surface HER2 exists in a constitutively active configuration that allows it to homodimerize and stimulate downstream signaling even without a known ligand [4]. At the same time, HER2 is the preferred heterodimerization partner for the other HER family receptors and HER2-containing heterodimers have increased affinity for the respective partner’s ligands, resulting in strong downstream signaling activity by those heterodimers. HER2/HER3 heterodimers are the most active signaling complex in the HER family and the most effective activator of phosphatidylinositol 3-kinase (PI3K)/AKT, the crucial signal transduction pathway in HER2-positive breast cancer [5, 6]. Given its elevated expression and oncogenic activity, its preferred status as an HER family signaling partner, and the enhanced activity of HER2-containing heterodimers, HER2 represents an attractive pharmacological target for the treatment of HER2-positive (HER2+) breast cancer. HER2-targeted agents fall into three general categories: therapeutic antibodies, antibody-drug conjugates, and tyrosine kinase inhibitors (TKIs). The resistance mechanisms associated with each of these classes of drugs will be reviewed here. Figure 2 shows a diagrammatic representation of the most significant molecular mechanisms described herein, using trastuzumab (antibody inhibitor) and lapatinib (TKI) as the reference drugs. Table 1 provides a summary of molecular mechanisms with literature citations.

Molecular model of the HER2 kinase domain based on the crystal structure of the domain in complex with SYR127063 (pdb ID 3PPO) [137]. SY127063 is a pyrrolo[3,2-d]pyrimidine-based selective TKI of HER2, which binds HER2 in an active conformation. SY127063 is shown in CPK coloring format (left panel); the panel on the right has the TKI ligand hidden. The green in both panels shows the C-helix; the yellow shows the A-loop; the blue shows the side chains of amino acid residues, L755 and T798, which are known to affect lapatinib resistance when they are mutated (see text)
Antibody Inhibitors
Trastuzumab (Herceptin)
Early studies showed that monoclonal antibodies directed at the extracellular domain of HER2 can dramatically inhibit proliferation of HER2+ breast cancer cells in culture and reduce their growth as xenografts in animal models [7,8,9,10]. Using HER2+ breast cancer cell lines, later studies showed that a humanized form of an anti-HER2 monoclonal antibody enhances the anti-tumor effect of cytotoxic chemotherapy drugs cisplatin, paclitaxel, and doxorubicin [9, 11].
From these observations emerged the development of trastuzumab (Genentech, Inc.), a humanized, recombinant IgG1 monoclonal antibody with high affinity to the extracellular domain of HER2. Following a successful clinical trial that showed an increase in both time to disease progression and response rate [12], trastuzumab was approved by the U.S. Food and Drug Adminisration (FDA) for use, in combination with the microtubule inhibitor paclitaxel, for first-line treatment of HER2+ metastatic breast cancer. Trastuzumab is also recommended for use in the adjuvant setting for early breast cancer, either as monotherapy or in combination with chemotherapy, and a number of trials point to benefit when trastuzumab is added to neoadjuvant chemotherapy as well [13,14,15,16].
The mechanism by which trastuzumab acts in patients is still not entirely known, but it most likely has a combination of effects subsequent to HER2 binding. The most well-documented mechanisms are antibody-dependent cell-mediated cytotoxicity (ADCC); inhibition of cleavage of the extracellular domain of HER2, which prevents the formation of a p95 truncated form of HER2 that is constitutively active (and resistant to antibody binding); and inhibition of downstream signal transduction (predominantly through blockage of the PI3K/AKT pathway), which in turn disrupts normal cell cycle control, survival/apoptosis responses, and DNA repair [5]. These effects on apoptosis and DNA repair may be particularly important in patients who are treated with conventional cytotoxic chemotherapy or radiation along with trastuzumab, possibly accounting for synergistic effects of the combination therapies. Trastuzumab also appears to inhibit angiogenesis as a downstream effect of signal transduction inhibition, by lowering levels of pro-angiogenic factors such as VEGF, and upregulating the anti-angiogenic factor thrombospondin-1 (see [17] for a recent review of trastuzumab mechanisms).
Despite significant impact on outcomes for HER2+ breast cancers, inherent and acquired resistance to trastuzumab remain a considerable clinical barrier. Approximately 75–85% of patients fail to respond to trastuzumab monotherapy due to inherent resistance [18,19,20]. Response rates increase to 50–75% when trastuzumab is combined with conventional chemotherapy [12, 18, 21], but most patients who respond to monotherapy or combination therapies will nevertheless experience disease progression and resistance within 1 year [12, 15, 22]. The mechanisms of resistance are varied, but the bulk of findings from in vitro and pre-clinical model systems indicate that sustained activation of the primary downstream signaling pathway, PI3K/AKT, is crucial. The molecular mechanisms for achieving such sustained signaling can be divided into three categories: (1) Steric effects that prevent HER2 inhibition – i.e., modification to the trastuzumab binding site due to changes in the HER2 structure or masking of its extracellular binding site (2) Activation of alternative receptors – i.e., compensation for blocking of HER2 signaling by activation of other receptor tyrosine kinases and (3) Modification to downstream effectors – i.e., alteration to effectors downstream of HER2, allowing for constitutive activation of PI3K/AKT and other pro-survival pathways. Each of these will be discussed next.
Steric Effects
Elimination or masking of the extracellular region of HER2 can prevent trastuzumab from binding to the receptor and, thus, promotes resistance. An amino-terminally truncated form of HER2, called p95HER2, results from the alternative translation of HER2 mRNA and/or proteolytic shedding of the extracellular domain of full-length HER2 [23, 24]. This truncated variant has constitutive tyrosine kinase activity and it appears to confer resistance to non-trastuzumab treatment. A retrospective study of specimens from breast cancer patients treated with non-trastuzumab regimens found a reduced time for disease free survival (DFS) for patients with high p95HER2 expression levels (median of 32 months) vs. patients with low p95HER2 expression levels (median of 139 months) (p < 0.0001) [25].
p95HER2 can heterodimerize with HER3, but not HER1, and trastuzumab is ineffective against this receptor complex [26] and p95HER2 generally [27]. A retrospective examination of metastatic tumors from patients treated with trastuzumab found a significant inverse relationship between p95HER2 expression and responsiveness to trastuzumab [27]. Of nine patients with p95HER2 expression, only one (11%) showed a partial response to trastuzumab therapy, whereas 19/37 (51%) of patients with tumors expressing full-length HER2 (and not p95HER2) exhibited complete or partial response (p = 0.029). In a separate study, a specific antibody targeting p95HER2 was used to quantify its expression in 93 specimens from patients with trastuzumab-treated metastatic breast cancer. A correlation was found between high p95HER2 levels and shorter progression-free survival (PFS) (hazard ratio (HR) = 1.9; p < 0.05) and lower overall survival (OS) (HR = 2.2; p ≤ 0.01) [28].
A second variant, Δ16HER-2, is created by a splice variant of HER2 mRNA that encodes a receptor with an altered conformation in the extracellular domain. Δ16HER-2 forms stable homodimers that are highly oncogenic and resistant to trastuzumab binding and growth inhibition, with possible coupling to Src kinase as an important mediator of Δ16HER-2 efficacy [29, 30]. Δ16HER-2 has been detected in 52% of HER2+ primary breast cancers and 89% of Δ16HER-2+ patients present with lymph node-positive disease [30]. The association between Δ16HER-2 and patient response to trastuzumab has not been reported.
A steric effect on trastuzumab binding can also be achieved by over-expression of non-receptor proteins such as Mucin-4 (MUC4), an O-glycosylated membrane protein that binds to HER2 [31]. MUC4 over-expression in breast cancer cell lines has been shown to decrease the accessibility of trastuzumab to HER2 through partial masking of its extracellular domain [31, 32]. A recent retrospective analysis of 78 HER2+ patients who had received adjuvant trastuzumab therapy found that MUC4+ tumors are associated with significantly shorter DFS in univariate analysis (HR = 4.40; p = 0.018) and MUC4 is an independent predictor of poor DFS in multivariate analysis after adjusting for stage and nodal status (HR = 5.43; p = 0.008) [33].
Activation of Alternative Receptors
Another mechanism for sustained PI3K/AKT signaling in the face of trastuzumab is through compensatory activation of alternative RTKs, either other members of the HER family or non-HER receptors that signal through the same or parallel pathways (see [34] for a recent review).
HER1 and HER3
Perhaps the most likely compensatory receptors for HER2 inhibition are family members HER1 and HER3, both of which are seen to be up-regulated in different models of trastuzumab resistance [35,36,37,38]. These receptors can function either as HER1/HER1 homodimers or as HER1/HER3 heterodimers but there is also evidence to suggest that HER1/HER2 and HER2/HER3 heterodimer levels are an important determinant of both primary (de novo) and acquired trastuzumab resistance [39]. Increased HER1 and HER3 levels in resistant cells are often accompanied by increased levels of their specific ligands, resulting in increased signaling through these receptors and increased sensitivity to their respective HER inhibitors (tyrosine kinase inhibitors or antibodies). This is true in cultured cell lines (in vitro selection) and in xenografts (in vivo selection) [35, 37, 40]. Several studies have shown a synergistic or enhanced effect of HER1 inhibition in combination with trastuzumab on trastuzumab-resistant breast cancer cell lines [35, 36, 41], further suggesting that compensatory signaling through HER1 is part of the resistance mechanism. Additional evidence comes from the efficacy of HER2-targeted drugs that also inhibit HER1 or prevent ligand-dependent HER2/HER3 signaling, which will both be discussed later.
The implication from the observations about HER1 and HER3 compensatory signaling is that relative HER family expression levels (not just HER2), and possibly their dimerization patterns, could be important in determining the primary response to trastuzumab and HER-targeted combinations, as well as the response to these agents in the relapse/resistance setting. Clinical data on this point are scarce. In the primary setting, only HER2 is used for therapeutic decision-making, and clinical trials testing newer HER2-targeted antibodies or combinations of trastuzumab with small molecule HER inhibitors (both of which should prevent HER-family compensatory activity) generally do not consider different HER expression levels or dimerization patterns as part of the trial design. In an analysis of biomarkers in the TRYPHAENA trial – which looked at trastuzumab plus pertuzumab (see below), sequential or simultaneous with chemotherapy, in the neoadjuvant setting – high levels of membrane-bound HER1 correlated with lower pathological complete response (pCR) rate (p = 0.0157 by chi-squared test) only when patients from all arms of the study were pooled [42]. The conclusion from this small study was that HER1 is not a suitable biomarker for patient selection at this time, but larger studies should be conducted before ruling out the importance of HER1 and HER3 as predictors of trastuzumab response.
Insulin-Like Growth Factor-1 Receptor
The insulin-like growth factor-1 receptor (IGF-1R) is another alternative RTK that can be activated to overcome HER2 inhibition. IGF-1R is not a native dimerization partner of HER2, but its signaling cascade shares several key features with HER2, most notably the PI3K/AKT pathway. IGF-1R over-expression commonly occurs in breast tumors [43] and in trastuzumab-resistant cancer cells [38, 44]. Early indication of a connection to trastuzumab resistance came from a study of MCF7 cells (which naturally express high levels of IGF-1R) transfected with HER2 cDNA. These cells show trastuzumab resistance in the presence but not the absence of the IGF-1R ligand, IGF-1 [45]. This same study showed that exogenous over-expression of IGF-1R in SK-Br-3 cells (which naturally express low levels of IGF-1R and are HER2+) is sufficient to confer trastuzumab resistance and that resistance can be abrogated by adding IGFBP3, a protein that binds IGF-1 and sequesters it from IGF-1R [45]. SK-Br-3 cells selected for trastuzumab resistance do not have elevated IGF-1R, but they do display a IGF-1R/HER2 dimer that is not seen in parental SK-Br-3 cells [46]. In these cells, IGF-1R activation with IGF-1 results in HER2 phosphorylation/activation and specific IGF-1R tyrosine kinase inhibitors reduce the HER2 activation state only in the resistant cells. Taken together, these studies suggest that up-regulation or dysregulation of the IGF-1R signaling could be a mechanism for conferring trastuzumab resistance.
The cross-talk between IGF-1R and HER2 in trastuzumab-resistant cells in culture prompted investigation of the predictive value of IGF-1R expression for therapeutic response. However, at least four different studies have found no correlation between IGF-1R or phospho-IGF-1R and survival or trastuzumab response/resistance in treatment-naïve patients (see [47]). It is possible that IGF-1R is more important as a mechanism of acquired resistance, rather than being a determinant of initial response, but there have been no reports to date on possible changes in IGF-1R levels or activity after trastuzumab therapy and the possible contribution of IGF-1R to the acquired resistance phenotype.
MET
The mesenchymal-epithelial transition (MET) RTK and its ligand, hepatocyte growth factor (HGF), are often over-expressed in breast cancer and MET over-expression is an independent predictor of poor prognosis in breast cancer [48,49,50,51,52]. A subset of HER2+ breast cancers is positive for MET over-expression and there is evidence that co-expression might contribute to a more invasive phenotype [53, 54]. Similar to the IGF-1R story, ligand-mediated activation of MET confers resistance to trastuzumab in HER2+ cell lines and MET inhibition sensitizes them to the drug. MET activation results in increased signaling through AKT in the presence of trastuzumab, suggesting that MET signaling is able to compensate for HER2 inhibition as a mechanism for MET’s involvement in drug resistance [52]. In a retrospective analysis of 130 HER2+ metastatic breast cancers, increased c-MET and HGF gene copy number, as determined by fluorescence in situ hybridization (FISH), were observed in about 25% of cases. c-MET FISH-positivity (n = 36) was associated with higher trastuzumab failure rate (p = 0.0001) and shorter time to progression (HR = 1.74; p = 0.001), compared to c-MET FISH-negative cases. HGF FISH-positivity (n = 33) was associated with higher trastuzumab failure rate (p = 0.007), compared with HGF FISH-negative cases [55].
VEGFR
The angiogenesis pathway stimulated by vascular endothelial growth factor (VEGF) and its receptor (VEGFR) is strongly implicated in HER2+ breast cancer growth and trastuzumab’s mechanism of action. HER2 expression and signaling are associated with high VEGF expression in cell lines [56]. Clinically, the majority of HER2+ breast cancers exhibit VEGF over-expression and co-expression of HER2 and VEGF predicts worse clinical outcomes in patients with primary breast cancer [57, 58]. As mentioned earlier, one mechanism of trastuzumab action appears to be an inhibition of angiogenesis mediated by reduced levels of VEGF and VEGFR signaling [17, 59].
Further evidence of VEGF-mediated trastuzumab resistance comes from inhibitor studies. Cell line models of inherent and acquired trastuzumab resistance have high VEGF expression. Bevacizumab, a monoclonal antibody targeted to VEGF, or sorafinib, a relatively non-specific inhibitor of VEGFR kinase activity, can reverse the resistance phenotype in these cell line models, both in vitro and as xenografts [60,61,62]. Sunitinib, another non-specific inhibitor of VEGFR kinase, has shown activity in patients with HER2+ breast cancer who had previously received trastuzumab [63]. In a phase I dose-escalation study of bevacizumab in combination with trastuzumab and the dual HER1/HER2 inhibitor lapatinib (see below for a further discussion of lapatinib), 50% of the 26 breast cancer patients had partial response, complete response or stable disease for ≥6 months. This was true even for the patients who had received therapy with trastuzumab and/or lapatinib within the previous year [64].
Modification to Downstream Effectors
The same effect achieved through activation of parallel RTKs to compensate for HER2 inhibition can be achieved by modifying downstream effectors of their common signaling pathways in a way that allows for signal transduction even when HER2 and other receptors are inhibited. There are several key regulators of HER2 signaling that are associated with trastuzumab resistance.
PI3K and PTEN
PI3K activates the AKT signaling pathway by phosphorylating the signaling lipid phosphatidylinositol-4,5-bisphosphate (PIP2) and converting it to phosphatidylinositol-3,4,5-trisphosphate (PIP3), which in turn phosphorylates and activates AKT (for a review, see [65]). The PI3K family consists of multiple members divided into three main classes. HER2 exerts its oncogenic function primarily through the p110α catalytic isoform encoded by the PIK3CA gene [66,67,68]. Activating mutations in PIK3CA, including “hotspot” mutations in exons 9 and 20, are found in approximately 20% of HER2+ tumors, and there is preclinical evidence linking such mutations to trastuzumab resistance [66].
Phosphatase and tensin homolog (PTEN) is a lipid phosphatase that dephosphorylates PIP3 and is thus an inhibitor of the PI3K/AKT pathway [65]. Activating mutations in PI3K or loss of PTEN function result in hyperactive PI3K/AKT signaling that should counteract HER2 inhibition by trastuzumab. PTEN activity is crucial to AKT dephosphorylation following treatment with trastuzumab. Trastuzumab resistance can be achieved in cell culture and in mouse xenografts by antisense-mediated inhibition of PTEN and the resistance phenotype can be rescued by inhibition of PI3K [69]. Moreover, a large-scale RNA interference screen performed on the HER2+ BT474 cell line found that expression of a shRNA targeted to PTEN leads to a decreased growth inhibitory effect of trastuzumab, which can be reversed by the expression of constitutively active PI3K [66]. PTEN protein is lost or low in approximately 40% of HER2+ breast cancers [70, 71].
The contribution of PI3K/PTEN alterations to clinical trastuzumab resistance has been difficult to tease out. Activating mutations in PIK3CA or loss of PTEN function would each result in increased PI3K/AKT signaling, so they are largely mutually exclusive occurrences in cancer patients [72]. Attempts to correlate PIK3CA mutation or PTEN loss with patient response or outcomes have produced varying results, most likely due to small sample sizes, different treatment backgrounds and tumor status of the patients, and different study designs. Several retrospective analyses of specimens from HER2+ patients treated with trastuzumab-based regimens have shown correlations between PTEN loss and poor trastuzumab response or worse patient outcomes [66, 69, 73]. PIK3CA mutations by themselves are not associated with patient outcomes, but the combination of PTEN loss and PIK3CA mutation (i.e., PI3K pathway activation) is even more significantly associated with shorter PFS than PTEN loss alone [66]. Studies analyzing PTEN and PIK3CA status in patients presenting with metastatic breast cancer and treated with first-line trastuzumab-based regimens have generally reported associations between PIK3CA mutation and/or PTEN loss and shorter time to progression or survival, to varying degrees of statistical significance [74,75,76].
In the adjuvant setting, there might be a trend towards an association between PTEN positivity and metastasis-free survival [77], but PIK3CA mutation does not appear to be predictive of response or outcome [77,78,79]. Perhaps the most pertinent results come from a set of neoadjuvant trials in which trastuzumab is compared with lapatinib or the combination of trastuzumab + lapatinib, a small molecule inhibitor of HER2 and HER1 tyrosine kinase activity (see section “Small-Molecule Inhibitors” below). Data from the trastuzumab arms of those trials showed that low PTEN or PIK3CA mutations are associated with lower pCR rate [80,81,82], but statistical significance is achieved only when data for low PTEN and PIK3CA mutation are combined [80]. PIK3CA mutations alone are significantly associated with pCR rate when data from multiple treatment arms (trastuzumab, lapatinib, trastuzumab + lapatinib) and trials are combined [81, 83]. A similar finding emerged from the analysis of metastatic breast cancer patients treated with first-line trastuzumab or trastuzumab + pertuzumab, an antibody that prevents HER2 dimerization with other HER-family receptors and IGF-1R (see below). PIK3CA mutations are associated with lower PFS with either treatment, but statistical significance is achieved only when data from both treatments are combined [74].
Taken together, there is growing evidence to suggest a mechanistic role for the PI3K pathway in trastuzumab response/resistance, with perhaps the greatest effect when multiple HER receptors are inhibited by trastuzumab in combination with other HER-targeted agents. The clinical value of PIK3CA or PTEN as predictive biomarkers has yet to be definitively established, however.
Src
The Src tyrosine kinase lies downstream of multiple RTKs, including HER-family receptors, and it is a common node in the PI3K/AKT, ERK, STAT3, and FAK pathways, thereby playing a role in major cellular functions such as proliferation, survival, angiogenesis, motility and adhesion [84, 85]. Its role in breast cancer has recently been reviewed [86]. Src is activated in a number of cell line models of acquired trastuzumab resistance, most likely downstream of RTK compensatory signaling as described earlier [38, 87], and when resistance is artificially created by PTEN knockdown [38]. In one recent model, trastuzumab resistance conferred by physical contact between breast cancer cells and mesenchymal stem cells appears to occur by Src activation [88]. Over-expression of either wild type Src or a constitutively active Src mutant is sufficient to confer trastuzumab resistance, whereas expression of a dominant-negative form of Src leads to enhanced trastuzumab sensitivity. Saracatinib, a small molecule inhibitor of Src, reverses trastuzumab resistance associated with RTK activation, PTEN knockdown, and constitutive Src activation. In retrospective analyses of primary breast tumors from patients treated with trastuzumab, high levels of Y416-phosphorylated (active) Src are found to be associated with low clinical response rate and lower OS compared to tumors with low levels of phosphorylated Src [38, 87].
p27kip1
As noted earlier, one of the downstream effects of trastuzumab is G1 cell-cycle arrest. This observation has been correlated to a dose-dependent effect of trastuzumab on accumulation of the CDK2 inhibitor p27kip1 by mechanisms that involve multiple signaling pathways known to be impacted by trastuzumab binding (including PI3K/AKT) [89, 90]. Moreover, HER2+ breast cancer cell lines selected for trastuzumab resistance express lower levels of p27kip1 and exogenous expression of p27kip1 in resistant cells renders them once again susceptible to growth inhibition by trastuzumab [91]. This indicates an important role for p27kip1 in trastuzumab-mediated cell cycle arrest and resistance and raises interesting questions about whether p27kip1 might be used as a predictive or prognostic biomarker and whether one or more mechanism of trastuzumab-mediated p27kip1 accumulation can be exploited therapeutically (see section “Implications for New Targets and Drug Combinations”).
Protein Kinase a (PKA) and t-Darpp
Microarray analysis of trastuzumab-resistant cell lines has revealed significant differences, relative to trastuzumab-sensitive parental cells, in the expression of several genes involved in the regulation of PKA signaling [92]. These same trastuzumab-resistant cells have increased PKA activity (as measured by CREB DNA binding activity) and enforced down-regulation of a PKA regulatory subunit that normally keeps PKA catalytic activity in check is sufficient to confer increased PKA activity and partial trastuzumab resistance [92]. PKA activation was further implicated in trastuzumab resistance in a screen that examined the effect of exogenously expressing a panel of kinases on breast cancer cell proliferation in the presence of a HER2-targeted shRNA [93]. This study suggested that PKA activation does not result in increased signaling through the PI3K or ERK pathways, but rather confers survival through inactivation of the proapoptotic protein BAD.
Another gene involved in PKA regulation is PPP1R1B, which codes for two transcriptional variants, Darpp-32 and its truncated isoform known as t-Darpp. Several studies have identified t-Darpp as being up-regulated in trastuzumab-resistant breast cancer cell lines and over-expression of exogenous t-Darpp is sufficient to confer trastuzumab resistance [92, 94,95,96]. Cells that over-express exogenous t-Darpp have elevated PKA activity and trastuzumab-resistant PI3K/AKT signaling [95]. The mechanism by which t-Darpp activates PKA and/or PI3K/AKT signaling and confers trastuzumab resistance is not entirely known, but models involving stabilization of HER1 signaling, inhibition of apoptosis, and a direct effect on the PKA holoenzyme have all been proposed [36,94; D. Theile, manuscript in preparation]. Regardless of the mechanism, t-Darpp may ultimately represent a resistance-specific factor and new target for overcoming or preventing trastuzumab resistance.
Pertuzumab
The breakthrough in targeted HER2 therapy achieved by trastuzumab and the ensuing resistance to its growth inhibitory effects encouraged the development of a second generation of HER2-targeted drugs. Pertuzumab (Genentech, Inc.) is one of many second-generation antibody inhibitors of HER2. Unlike trastuzumab, which only disrupts the ligand-independent homodimerization of HER2, pertuzumab binds to the extracellular dimerization domain of the receptor and eliminates dimerization with other HER-family receptors, as well as IGF-1R [46, 97,98,99].
As discussed above, a key driver of trastuzumab resistance is increased signaling by heterodimerization with HER3, HER1 and IGF-1R. Pertuzumab’s ability to prevent such heterodimerization has made it a candidate for combination therapy along with trastuzumab. Preclinical studies showed a synergistic effect of combining trastuzumab with pertuzumab in BT474 breast cancer cells [99] and in xenografts of HER2+ breast cancer cells in mice [100]. The same xenograft study also demonstrated that a combination of the two drugs can inhibit the growth of tumors following acquired resistance to trastuzumab [100].
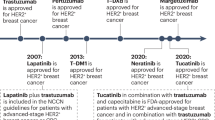
In early clinical trials, pertuzumab showed great potential as a second-line therapy in patients who progressed on trastuzumab regimens, and results are even more impressive when trastuzumab and pertuzumab are used in combination as second-line therapy [101, 102]. In 2013, pertuzumab was approved by the FDA for first-line treatment of metastatic, HER2+ breast cancer in combination with trastuzumab + docetaxel. This approval was granted following the CLEOPATRA phase III clinical trial in which patients received either standard first-line treatment with trastuzumab + docetaxel (control arm) or a combination of pertuzumab + trastuzumab + docetaxel. The pertuzumab arm of the study resulted in a PFS of 18.5 months and 80.2% objective response (OR) rate, compared to 12.4 months PFS and 69.3% OR rate in the control arm (p < 0.001) [103]. A follow-up a year later also demonstrated significant improvement in OS in patients treated with pertuzumab + trastuzumab + docetaxel [104]. Pertuzumab + trastuzumab + docetaxel also showed significantly improved pCR rate (p = 0.0141) and trends towards better 5-year PFS and DFS rates over trastuzumab + docetaxel in neoadjuvant treatment of HER2+ breast cancer patients in the NeoSphere trial [105, 106].
Taken together, the data on pertuzumab suggest that this antibody holds great promise for treating trastuzumab-resistant disease or perhaps preventing a major mechanism of resistance. The most common resistance mechanisms acquired by tumors following treatment with trastuzumab involve activation of alternative signaling pathways in order to maintain activity in the PI3K/AKT pathway. Pertuzumab has the ability to overcome or prevent many of these mechanisms by denying HER2 heterodimerization with other receptors. This idea is supported by biomarker studies alluded to earlier, in which PIK3CA mutations, which act downstream of receptor dimerization, were found to be associated with poorer response to pertuzumab/trastuzumab regimens in the CLEOPATRA trial [74], consistent with earlier trends seen in smaller trials, NeoSphere and TRYPHAENA [42, 107].
Other Antibodies
There are two new HER2-targeted antibodies currently in clinical trials for treatment of HER2+ cancers, with limited information on their efficacy and mechanisms of resistance.
Margetuximab
Margetuximab (MGAH22, MacroGenics, Inc.) is a HER2-targeted antibody with a Fc-domain (non-targeting end of the antibody) engineered to bind to and activate immune effector cells and induce ADCC. Preclinical trials have shown it can induce ADCC better than HER2-targeted antibodies with wild type Fc domains [108]. Phase I clinical trials concluded that margetuximab treatment is tolerable at all doses examined and shows promising activity in HER2+ tumors, including breast cancer [109]. Margetuximab efficacy is being examined in SOPHIA (clinicaltrials.gov identifier NCT02492711), a phase III trial testing margetuximab in combination with chemotherapy compared to trastuzumab + chemotherapy for third-line metastatic breast cancer, with no outcome data yet available [110].
FS102
FS102 (F-Star Biotechnology, Ltd. in partnership with Bristol-Myers Squibb) is a HER2-targeted Fc fragment with an antigen binding domain that recognizes a unique site on the HER2 receptor and induces programmed cell death and marked HER2 internalization and degradation. Preclinical studies have shown that SK-Br-3 cells exposed to FS102 go through apoptosis [111], as opposed to the G1 phase cell cycle arrest that trastuzumab causes in these cells. Such studies have also demonstrated complete tumor regression, apoptosis induction and reduction of HER2 levels in patient-derived xenograft models of breast and gastric cancer. The effect of FS102 is superior to the effects of trastuzumab or trastuzumab + pertuzumab studied in parallel and FS102 is able to inhibit the growth of xenografts that progress on trastuzumab or trastuzumab + pertuzumab, suggesting non-overlapping mechanisms of resistance to these agents [111]. FS102 is being tested in a dose-escalating phase I clinical trial for treatment of HER2+ solid tumors (NCT02286219).
Induction of apoptosis overcomes a serious flaw in the G1 arrest mechanism mediated by trastuzumab. Cells arrested in G1 remain viable and can escape cell cycle arrest by activating alternative signaling pathways or the HER2 signaling pathway further downstream. Although not well studied, resistance to FS102, in contrast, would likely arise through mechanisms of avoidance -- either through modifications to the HER2 receptor or masking of the binding site -- or through mechanisms of apoptosis inhibition. This, along with the preclinical studies with patient-derived xenografts [111], raises the possibility that trastuzumab resistance might not result in cross-resistance to FS102, thus allowing for FS102 as second-line therapy or even in combination with standard first-line therapies.
Antibody-Drug Conjugates
T-DM1
T-DM1 (Genentech, Inc.) is the prototypical antibody-drug conjugate that targets HER2. The antibody component is trastuzumab and the drug component is emtansine (DM1), a small molecule that binds tubulin and prevents microtubule assembly, thus inducing mitotic arrest and cell death in dividing cells. DM1 is derived from maytansine, which was originally found to be highly potent but with unacceptable systemic toxicity due to lack of specificity to tumor cells [112,113,114,115]. DM1 was specifically designed to be used as a conjugate with trastuzumab, which allows for more focused drug activity in the cytoplasm of target cells following HER2-mediated endocytosis [116,117,118].
In the original pre-clinical studies of T-DM1, it was found to be a potent inhibitor of growth in an array of HER2+ cells, regardless of whether those cells respond to trastuzumab alone and by a mechanism that results in cell death. In HER2+ xenograft models, T-DM1 was shown to inhibit tumor growth and promote tumor regression [119]. Following these initial studies, a multitude of phase I/II clinical trials in patients previously treated with trastuzumab reported significantly reduced toxicity, compared to DM1, with levels tolerable beyond the known clinical dose. These trials also found an overall response rate of at least 40%, indicating the potential of T-DM1 as second-line therapy of HER2+ breast cancer [120,121,122,123,124,125].
The success of these trials led to EMILIA, a phase III trial testing the efficacy and safety of T-DM1 in patients with locally-advanced or metastatic HER2+ breast cancer previously treated with trastuzumab + taxanes. The trial included 991 patients divided into two arms, one in which patients received a common second-line therapy of lapatinib + capecitabine and the other in which patients received T-DM1 as second-line therapy. The study demonstrated improvements in both OS (30.9 months with T-DM1 vs. 25.1 months in the control arm) and PFS (9.6 months with T-DM1 vs. 6.4 months in the control arm) while also exhibiting reduced toxicity and adverse effects for patients [126, 127].
The EMILIA trial led to FDA approval of T-DM1 in February, 2013, for the treatment of metastatic HER2+ breast cancer. A more recent phase III trial named TH3RESA compared the efficacy of T-DM1 to treatment of physician choice in similar second-line therapy and reconfirmed the improved PFS, OS and tolerability of T-DM1 compared to current treatment options, thus solidifying its position as the standard second-line therapy for HER2+ breast cancer patients [128].
The MARIANNA trial examined the efficacy of T-DM1 alone and T-DM1 + pertuzumab in first-line therapy of HER2+ breast cancer, compared with the current standard of care of trastuzumab + taxane. Although the results did show reduced toxicity of T-DM1, it failed to show superiority in either PFS or OS to the current standard regimen [129]. Taking in consideration the results of the CLEOPATRA trials discussed above, which show superiority of pertuzumab to trastuzumab + taxane therapy, T-DM1 is unlikely to be used as first-line standard of care for HER2+ breast cancer [130].
The overall response rate to T-DM1 is 80% in patients with HER2+ breast cancer who progress on prior HER2-directed therapy [121]. This suggests that primary resistance to T-DM1 is relatively infrequent, but all of the clinical trials discussed above indicate that patients eventually cease to respond to T-DM1 therapy due to acquired resistance. Since T-DM1 is a relatively new drug, little is known about the mechanisms involved in acquired resistance. However, we can speculate on some likely resistance factors, based on what is known about the molecular mechanism of T-DM1 action:
a) Reduced binding and internalization. Since T-DM1 is dependent on binding to HER2 and receptor-mediated endocytosis to enter the cytoplasm, any changes to HER2 that prevent trastuzumab from binding to the extracellular region would result in reduced sensitivity to T-DM1 as well. As discussed earlier, mechanisms that affect binding to HER2 include expression of the p95HER2 or Δ16HER-2 isoforms and over-expression of MUC4 that result in masking of the trastuzumab binding site. In addition, the internalization of HER2 following trastuzumab binding is a regulated process that depends on clathrin activity (see [131] for a review on HER2 trafficking), suggesting that inhibition or down-regulation of clathrin-dependent endocytosis would also lower intracellular levels of DM1.
b) Reduced lysosomal processing. Following internalization of the T-DM1/HER2 complex into endosomes, DM1 needs to be released from trastuzumab by lysosomal degradation and accumulate in the cytoplasm at a concentration that meets the threshold needed to promote cell death. Changes to endosomal trafficking, which are known to occur following endocytosis-mediated therapy [132, 133], could result in increased re-shuttling of the complex to the plasma membrane and decreased lysosomal trafficking. At the same time, modification to the lysosomal degradation machinery could result in decreased degradation of T-DM1. Both mechanisms would result in reduced cytoplasmic concentrations of DM1.
c) Reduced intracellular activity. Once in the cytoplasm, DM1 binds to tubulin and inhibits microtubule polymerization. Any modifications to tubulin or to enzymes involved in the microtubule dynamics could impact the efficacy of the drug [112, 134]. DM1 is also a substrate for the efflux pump P-glycoprotein encoded by the MDR1 gene [135]. MDR1 over-expression may result in reduced sensitivity to DM1 as is the case for several other drugs that are exported from the cell via efflux pumps [136].
Small-Molecule Inhibitors
Pharmacological and clinical activity of antibody-based drugs mainly relies on their extracellular interaction with HER2. An alternative approach is to inhibit the kinase moiety located intracellularly, especially under certain conditions of trastuzumab resistance. The kinase domains of the HER family are structurally quite similar to other kinases [137, 138]. They contain two large lobes (called the N-lobe and the C-lobe), with the actual kinase activity site located in the cleft (called the hinge region) between the two lobes. The kinase activity site harbors an ATP-binding pocket, a flexible substrate binding site, and two regulatory regions called the activation loop (A-loop) and the C-helix (see Fig. 2). In the inactive conformation of the kinase domain, the activation loop hinders the binding of substrates. Upon activation, the loop is structurally altered leading to an open substrate binding site.
In the early 1990s, natural compounds such as erbstatin and synthetic biosimilars like the ‘tyrphostins’ were shown to inhibit HER-family function, but they were found to interact with the substrate binding pocket [139] and had rather poor HER kinase selectivity [140]. Subsequent enzymological studies of HER1 determined that an intermediate ternary complex forms during catalysis, in which the kinase domain interacts with the γ-phosphate of ATP and a tyrosyl hydroxyl and tyrosyl aromatic ring of the peptide substrate (the target of the RTK phosphorylating activity) [141]. This information was used to search a database of predicted three-dimensional structures for compounds that mimic these interactions. The goal was to identify compounds that act similarly to and compete with ATP but do not lead to receptor phosphorylation. From this, the 4-anilino-quinazolines emerged as low nanomolar ATP-competitive inhibitors of HER1 [142]. Because the kinase domains of the four HER receptors show a high degree of identity (59%–81%), inhibitors that selectively inhibit only one of the four potential kinase activities are hard to design [143]. Structure-activity relationship studies determined that substitutions on the 4-anilino ring of the 4-anilino-quinazolines play a role in selectivity, with large substitutions being correlated with increased affinity for HER2 [142]. At least four other classes of bicyclic compounds – pyridopyrimidines, pyrrolopyrimidines, pyrrolotriazines, and cyanoquinolines – have been developed as HER kinase inhibitors. Although less is known about their structure-activity relationships, they appear to follow the same principles for target binding as the quinazolines. In the following sections we will review the dominant small molecule drugs targeting HER2 and other HER family members.
Lapatinib (See Fig. 3)
Lapatinib targets HER2 and HER1 and was the first TKI approved by the FDA for the treatment of patients with metastatic HER2+ breast cancer. Numerous clinical trials evaluated its efficacy in various patient cohorts (HER2+ or HER2−; early or advanced breast cancer; pretreated or therapy-naïve). Taken together, these studies (recently reviewed in [144]) showed that (1) monotherapy with lapatinib is effective in heavily pre-treated HER2+ but not HER2− patients; (2) lapatinib shows some, but rather minor, clinical efficacy in trastuzumab-resistant advanced or metastatic breast cancer; (3) lapatinib enhances the efficacy of other concurrently administered cytotoxic drugs that target different sites within the cell (e.g. taxanes, capecitabine etc); (4) when directly compared to mono-ty of molecular mechanisms with literature citationst cancer without prior therapy, lapatinib is equally effective as trastuzumab; but (5) trials evaluating lapatinib + chemotherapy vs. trastuzumab + chemotherapy in HER2+ advanced breast cancer revealed that the latter is superior. Based on these clinical trials, lapatinib is currently mostly used in combination with capecitabine for palliation of patients who were previously treated first-line with a combination of trastuzumab, an anthracycline (doxorubicin or epirubicin) and a taxane (docetaxel or paclitaxel).

Schematic representation of the most significant molecular resistance mechanisms to trastuzumab (left) and lapatinib (right). Additional information and corresponding reference citations can be found in the text. Arrowheads (→) and bars (--) attached to solid lines indicate known activation or inhibition effects, respectively, on the indicated pathway or protein. The dotted-line arrow from t-Darpp indicates an indirect mechanism of activation when t-Darpp is over-expressed
Thus, although lapatinib is a potent inhibitor of HER2 kinase activity, its clinical impact, over and above that of trastuzumab, is limited. This is in part due to the compensatory nature of HER3 upregulation and its heterodimerization with HER2, leading to activation of PI3K/AKT signaling despite HER2 inhibition (see below). As a consequence, supra-therapeutic concentrations would be needed for a complete inhibition of HER2/HER3 signaling by lapatinib. Such high concentrations are not tolerable given the severe dermatologic and gastro-intestinal toxicities that result from HER1 and HER2 inhibition by lapatinib. Dividing doses into two applications per day, in contrast to once daily dosing, has been suggested to increase total drug exposure while minimizing toxicity. Even 7000 mg per day can be safely administered in this manner without evidence of dose-limiting toxicity and there is preliminary indication of therapeutic efficacy [145].
Given these findings and the considerable amount of pre-clinical work done with lapatinib, it is worth exploring the mechanisms of lapatinib resistance that probably represent mechanisms that will be shared by other less-studied TKIs as well. We describe molecular mechanisms of resistance, as we did in Sections “Antibody Inhibitors” and “Antibody-Drug Conjugates”, and also discuss pharmacological factors that affect drug efficacy in the clinical setting.
Steric Effects
Since lapatinib targets the kinase domains of HER1 and HER2, mutations that render the kinases constitutively active or prevent TKIs from binding are likely mechanisms of resistance. Experimental approaches screening for HER2 mutations associated with lapatinib resistance have revealed amino acid substitutions at 16 different HER2 residues, with 12 mutated amino acids mapping to the kinase domain [146]. Mutations with the highest impact on lapatinib efficacy cluster in the N-lobe and hinge region. L755S and T798I mutations are associated with considerable lapatinib resistance. Notably, a T790 M mutation in HER1 also confers resistance to lapatinib at a level that is comparable to the T798I mutation (the analogous site) in HER2. The L755S and T798I amino acid substitutions most likely lead to steric hindrance of the lapatinib-receptor interaction and diminish the structural flexibility of the drug binding site. As a result, the inactive conformation of the kinase domain required for lapatinib binding becomes energetically unfavorable and, thus, is less likely to form. Interestingly, EXEL-7647, an experimental TKI that targets both inactive and active receptor confirmations, potently inhibits receptor function and downstream signaling by wild type but also mutant forms of HER2 [146].
Activation of Alternative Receptors
There is a large body of evidence indicating that alternative receptor pathways are switched on to compensate for lapatinib’s inhibitory effect on HER2 signaling. The most salient of these are discussed here.
HER1 and HER3
As with trastuzumab, the most effective means of compensating for HER2 inhibition by lapatinib most likely comes from the HER family of receptors themselves and several lines of evidence support this idea. Apart from the receptors themselves, over-expression of their respective ligands can mediate lapatinib resistance. Using a comprehensive set of protein- and DNA-based methodologies, one study showed that lapatinib-resistant cells have up-regulated expression of the HER3 ligand heregulin (HRG) and that this functions in an autocrine fashion to stimulate signal transduction [147]. The observation in this system is that HER2 is inhibited by lapatinib, but HER1 phosphorylation is only partially inhibited in the lapatinib-resistant cells. This slightly sustained HER1 signaling is apparently sufficient to detour signaling to a HRG-driven HER1/HER3 pathway. Gefitinib and erlotinib, two HER1-specific TKIs, cannot overcome HRG-HER3-mediated activation of HER1, nor reverse lapatinib resistance in this model system, whereas neratinib, an irreversible pan-HER TKI (see below), can overcome lapatinib resistance [147]. This same report suggests the clinical relevance of HRG expression. In a study of 204 HER2+ breast cancers, HRG mRNA levels were found to correlate significantly with risk of recurrence (p = 0.0036) and there is a statistically significant association between high HRG expression and decreased DFS. High HRG expressers have a median DFS of 2.84 years and intermediate + low expressers have a median DFS of 10.04 years (p = 0.0034) [147].
Other HER ligands have been implicated in lapatinib resistance. For example, exposing SK-Br-3 cells to transforming growth factor-α (TGF-α) increases phosphorylation of HER1 and its downstream targets and decreases the sensitivity of these cells to lapatinib [148]. Notably, high serum levels of TGF-α and amphiregulin, another HER1 ligand, predict poor response to gefitinib in patients with advanced non-small cell lung cancer [149]. Thus, it is possible that high TGF-α levels could also associate with poorer outcome in breast cancer patients treated with lapatinib. Indeed, in a study of 64 patients treated with lapatinib + capecitabine, the response rate was found to be higher in individuals with low serum TGF-α compared to those with high levels (p = 0.001). There was a trend towards shorter time-to-progression in patients with high serum TGF-α compared to low TGF-α (p = 0.067) [148].
Other means of activating HER1 have also been implicated in resistance. For example, heparanase has been suggested to modulate lapatinib efficacy by promoting signaling through HER1. Heparanase is an endoglycosidase that cleaves heparin sulfate to biologically active fragments that promote proliferation or angiogenesis, and this activity has been implicated in metastatic potential, mostly to the brain [150, 151]. Heparanase can also affect HER1 phosphorylation by a mechanism that appears to be independent from its enzymatic activity [152], suggesting that heparanase could play a role in lapatinib resistance. Such a role was demonstrated using a potent inhibitor of heparanase activity, Roneparstat, and a cell line that over-expresses HER1 and HER2 and was selected for lapatinib resistance. These cells have elevated heparanase levels, activity and secretion, compared with lapatinib-sensitive parental cells, and elevated signaling through HER1, FAK and ERK1/2 pathways, even in the presence of lapatinib. Roneparstat inhibits HER1 phosphorylation and downstream signaling through FAK and ERK1/2, thereby reversing the lapatinib resistance phenotype, both in vitro and in mice [153]. This study again underlines the importance of alternative signaling/survival pathways that cancer cells use to withstand HER2 or HER1 inhibition by lapatinib. The clinical significance of heparanase has been determined and a variety of heparanase inhibitors are being studied as therapeutic agents [150, 151], but a link between heparanase and clinical lapatinib resistance has not yet been established.
MET and AXL
MET, described earlier in the context of trastuzumab resistance, also appears to affect response to lapatinib. MET’s ability to mediate lapatinib resistance was demonstrated in gastric cancer cells. Exposure of these cells to HGF induces MET phosphorylation and leads to ERK and AKT signaling and lapatinib resistance, whereas down-regulation or selective inhibition of MET re-sensitizes resistant cells to lapatinib [154]. Although these data pertain to HER2+ gastric cancer cells, it seems probable that the same mechanism of lapatinib resistance also occurs in breast cancer, where MET and HER2 are frequently co-expressed and appear to compensate for each other in activating downstream signaling [155].
AXL is an RTK that exhibits tumorigenic potential, most likely related to its kinase domain that can be activated independent of ligand binding by simple over-expression [156,157,158]. AXL over-expression is associated with poor prognosis of numerous human cancers including tumors of the breast, colon, esophagus, thyroid, ovaries, stomach, kidney, brain, or lung [159]. Increased AXL expression might potentially play a role in resistance to a c-KIT TKI, imatinib, in gastrointestinal stromal tumors that express c-KIT [160] and in chemotherapy resistance in acute myeloid leukemia [161], lung cancer [162] and ovarian cancer [163]. In addition, when HER2+ breast cancer cells (BT474) are exposed to lapatinib for long periods of time, surviving lapatinib-resistant subclones significantly over-express AXL. Subsequent inhibition of AXL expression by RNAi or function by treatment with foretinib (a multi-kinase inhibitor of AXL, MET, and VEGFR) restores lapatinib sensitivity in this model, whereas more specific MET and VEGFR inhibitors do not [159]. Interestingly, AXL expression can also be diminished and lapatinib sensitivity restored when cells are deprived of estrogen or treated with the estrogen receptor (ER) antagonist fulvestrant, indicating that the ER pathway stimulates AXL expression and in turn promotes lapatinib resistance. Indeed, up-regulation or enhancement of the ER pathway is considered a redundant survival mechanism contributing to lapatinib resistance [165] (see below).
Modification to Downstream Effectors
PI3K and PTEN
Mutations in downstream mediators of HER2 signaling can play a significant role in lapatinib resistance. For instance, PTEN loss and/or PIK3CA mutations not only mediate trastuzumab resistance, as described earlier, but they also cause lapatinib resistance. The E545K and H1047R amino acid substitutions in PI3K have repeatedly been associated with lapatinib resistance, both in vitro and in animal models [166, 167]. Notably, expression of the H1047R mutant markedly up-regulates HRG, discussed earlier as a HER3 ligand that contributes to lapatinib resistance, and corresponding cells have elevated phospho-HER3 levels [168]. However, such cells actually maintain cell growth and AKT activation through a pathway that does not completely depend on HER3 [168]. Only combined inhibition of PI3K and HER2 with BEZ235 (see Section “Implications for New Targets and Drug Combinations”) and lapatinib, respectively, completely inhibits growth of cells with PTEN deficiency or PIK3CA mutation [166, 168].
The role of increased PI3K activity in lapatinib’s clinical efficacy is not entirely clear. On the one hand, activation of the PI3K pathway (through activating PIK3CA mutations or PTEN loss) was found to be associated with lower lapatinib efficacy in patients with metastatic breast cancer treated with lapatinib + capecitabine. Clinical benefit rate was 36% and OR rate was 9% in patients with PI3K activation (n = 22), compared to 61% and 31%, respectively, in PI3K non-activated tumors (n = 35) [164]. On the other hand, PTEN status was not associated with response in a phase II trial of lapatinib monotherapy in patients with recurrent HER2+ inflammatory breast cancer [169, 170]. The potential involvement of PI3K activation in lapatinib response has nevertheless prompted the idea that combined inhibition of PI3K and HER2 could be a preferred approach against cancers that contain both HER2 gene amplification and PIK3CA activating mutations (see Section 5).
mTOR, the primary downstream mediator of PI3K/AKT signaling, can be a marker of enhanced signaling through this pathway due to alternative RTK activation or PI3K/PTEN mutation, and mTOR thereby becomes a potential target for overcoming HER2-targeted resistance (see Section 5). But there also appears to be a role for mTOR in lapatinib resistance that is independent of upstream modulators of mTOR activity. This was shown originally in a SK-Br-3 cell line selected for lapatinib resistance [171] and recently confirmed in an AU565 cell lines selected for lapatinib resistance [172], and it may also be the case in a MCF7-based model of acquired lapatinib resistance [173]. The resistant SK-Br-3 and AU565 cells show sustained activation of mTOR that does not depend on signaling through upstream RTKs or PI3K/AKT, or modulation of other known PI3K/AKT-independent regulators of mTOR activity (Erk, IKKβ, AMPK, RHEB, GSK-3β, and PRAS40, among others). Importantly, lapatinib resistance in these systems is reversible with pharmacological inhibitors of mTOR [171, 172]. The mechanism by which mTOR is constitutively activated in these lapatinib-resistant cells is not known but it does not appear to involve mutation of mTOR or its known regulators [172].
Src
In several examples of breast cancer cell lines selected for lapatinib resistance, HER2 auto-phosphorylation is inhibited by lapatinib but considerable signaling via the PI3K/AKT axis is nevertheless sustained through a mechanism that involves up-regulated Src and Src family kinases [174, 175]. In such cells, Src inhibition by saracatinib abolishes PI3K/AKT signaling and re-sensitizes cells and their respective tumor xenografts to lapatinib. The importance of Src for lapatinib resistance has been confirmed in breast cancer and esophageal cancer cell lines and xenografts [176, 177]. On a molecular level, certain activating mutations in Src can mediate lapatinib resistance. In one model system, breast cancer cells selected for lapatinib resistance carry an E527K mutation in Src and ectopic expression of this mutant is sufficient to confer lapatinib resistance in previously lapatinib-sensitive cells [178].
Besides constitutive activation through mutation, Src can be activated via signals from integrin proteins such as β1-integrin [179], a mechanoreceptor that promotes breast cancer initiation and progression and may be coupled to the HER1/HER2 pathway when breast cancer cells are grown in 3-dimensional systems [180]. A role for β1-integrin in lapatinib resistance was demonstrated by diminishing β1-integrin activity (anti-β1 antibody or RNAi), with subsequent restoration of lapatinib efficacy, although a mechanistic connection to Src was not made in this study [181].
Ras
The Ras family of small GTP binding proteins represents another well-known mechanism of malignant transformation that might also mediate lapatinib resistance. Mutations in Ras can affect its GTP binding property and thus lead to constitutively active Ras proteins, eventually leading to tumorigenesis through activation of MEK and ERK (see [182]). Although Ras mutations are rarely seen in breast cancer, elevated Ras signaling is frequently observed in HER2+ breast cancers [183, 184]. Ras might be a mediator of lapatinib resistance given the high likelihood that the MEK/ERK axis is an alternative signaling pathway when HER2/PI3K/AKT signaling is disrupted [86]. Over-expression of either a wild type H-Ras or an oncogenic H-Ras allele (Ras G12 V) is sufficient to reduce lapatinib efficacy in two different HER2+ cell lines and resistance can be reversed by a MEK inhibitor (U0126) [185].
Estrogen Receptor
Signaling through the ER pathway as a mechanism of acquired lapatinib resistance has been observed both in tissue culture and in mice. Unlike trastuzumab, lapatinib causes up-regulation of ER and progesterone receptor (PR) expression and activity. In breast cancer cell lines (UACC-812 and BT474), ER mRNA levels are increased 6-fold and PR levels are increased 15-fold by lapatinib. This up-regulation is also reflected at the protein level. Fulvestrant, an ER-targeted agent that blocks estrogen binding and causes ER degradation, accordingly suppresses elevated ER levels induced by lapatinib and re-sensitizes lapatinib-resistant cells and xenografts to lapatinib. Alternative approaches to deplete cells of estrogen (estrogen-free cell culture media) confirm ER signaling as a mediator of de novo or acquired lapatinib resistance [159, 186].
Molecularly, the mechanism by which ER signaling can lead to lapatinib resistance has not been directly demonstrated, but it seems reasonable to suggest that this occurs through ER’s effect on genes that promote the cell cycle, proliferation and anti-apoptosis [187]. In BT474 cells, 24-hour exposure to lapatinib leads to increased ER signaling (increased levels of ER-target genes such as PR and bcl-2) that appears to be mediated by induction of FOXO3a (a transcription factor that promotes ER expression and is inactivated by PI3K/AKT signaling) in response to the acute inhibitory effect of lapatinib on PI3K/AKT activity [165]. These same molecular changes (increased FOXO3a and enhanced ER signaling activity) are seen in BT474 cells selected for lapatinib resistance and in early-stage breast tumors after 14 days of neoadjuvant lapatinib therapy [165].
Hormone receptor status (ER and PR) seems to be of general clinical relevance in the context of lapatinib response. In a retrospective analysis of HER2+ metastatic breast cancer patients treated with lapatinib + paclitaxel, event-free survival in women with HER2+ and concurrently ER+/PR+ tumors was only 5.7 months, compared to a previous study showing 8.3 months for women with HER2+ but ER−/PR− tumors [188]. Moreover, the pCR rate during neoadjuvant treatment with lapatinib is clearly lower in patients with ER+/PR+ cancers (16.1%) than in women with ER−/PR− tumors (33.7%). This does not seem to be restricted to lapatinib, as the same negative influence of ER/PR expression is seen with trastuzumab alone and with the combination of trastuzumab + lapatinib [189].
Pharmacological Factors of Resistance
Several clinical pharmacological factors can also contribute to ineffectiveness of HER2-targeting small molecules such as lapatinib. Although the exposure-response relationship for lapatinib is not entirely clear, preliminary data from clinical trials suggest that there is a threshold level for therapeutic efficacy. In a phase I dose-escalation study evaluating women with advanced HER2+ breast cancer, a relationship between lapatinib plasma concentration and clinical response or biological activity (transient tumor reduction during lapatinib therapy) was reported [145]. The most striking responses were seen in patients who achieve a maximum lapatinib plasma concentration (Cmax) approaching 10,000 ng/ml, whereas patients with lapatinib Cmax levels of 3500 ng/ml all had progressive disease at 2 months after the start of treatment [145]. This suggests a dose-response relationship, although definite conclusions cannot be drawn because Cmax does not reliably indicate drug exposure. A trial evaluating heavily pretreated patients with HER1+ and/or HER2+ metastatic cancers (breast, colorectal, head and neck, lung, and ovarian cancers) demonstrated that steady-state trough levels (Cmin) of lapatinib below 1000 ng/ml are associated with non-response [190], again suggesting a threshold level of lapatinib exposure that needs to be met for minimum clinical efficacy and consistent with the idea that ineffectiveness can arise from under-exposure (plasma concentrations below efficient levels). The factors that might limit plasma concentrations are discussed next.
Non-adherence
Non-adherence to orally administered anti-cancer drugs is frequently observed and well documented. Non-adherence occurs when drug doses are skipped, additional doses are taken, or doses are taken in the wrong quantity or at the wrong time [191]. Incidence of non-adherence among patients taking orally administered anti-cancer drugs range from 0% to 84% [191, 192], depending on the definition of non-adherence, the tool used to measure non-adherence, and the type of therapy (therapy complexity; patterns and kind of adverse drug effects).
Although studies evaluating non-adherence and associated outcomes among breast cancer patients on lapatinib have not been reported, some indication of the importance of non-adherence for efficacy of the general class of TKIs comes from trials with chronic myeloid leukemia (CML) patients given imatinib [193]. For instance, in a study of 87 patients with chronic-phase CML treated with imatinib for 6 years, adherence rates monitored during a three-month period significantly (p < 0.001) correlated with the 6-year probability of a 1000-fold reduction in BCR-ABL (imatinib target) transcripts. Such a reduction is considered to be a major molecular response and an important predictor of overall survival. Multivariate analysis additionally identified adherence as an independent predictor for major molecular response (relative risk, 11.7; p = 0.001). Moreover, no molecular responses were observed when adherence was below 80% (p < 0.001). It was concluded that in patients with CML treated with imatinib, poor adherence might be the predominant reason for inability to obtain adequate molecular responses [194]. Educational or behavioral approaches are recommended to ensure high therapy adherence of patients treated with oral TKIs [191].
Pharmacokinetics
Pharmacokinetic disturbances can also lead to underexposure and thus potentially mediate clinical resistance to TKIs. TKIs show high inter-individual variability in pharmacokinetics and are expected to exhibit considerable pre-systemic clearance (first-pass effect) and, thus, poor oral bioavailability [195, 196]. Data on absolute bioavailability is scarce, however, given the lack of drug formulations suitable for intravenous delivery for most TKIs. All TKIs (including lapatinib) are extensively metabolized by the cytochrome P-450 isoenzyme 3A4 (CYP3A4) [197]. Given the high propensity for CYP3A4 to be induced or inhibited by co-medications, drug-drug interactions with concomitantly administered therapeutics or over-the-counter drugs are very likely. Since under-exposure is most relevant for clinical non-responsiveness, drug-drug interactions with inducers of lapatinib disposition are highlighted here. Anti-epileptic drugs substantially lower exposure to lapatinib. For example, when patients with recurrent glioblastoma multiforme are treated with lapatinib, its apparent oral clearance increases by about ten-fold when patients are also treated with CYP3A4-inducing anti-epileptics such as phenytoin or carbamazepine [198]. The latter has been confirmed to lower lapatinib exposure in a study examining the effect of carbamazepine titrated up to 200 mg (BID) over 20 days on a single 250 mg dose of lapatinib. Carbamazepine decreases lapatinib area under the time-concentration curve (AUC) by 72%, Cmax by 59% and absorption rate by 28%, but has no effect on drug half-life [199]. This suggests that the major site of interaction is the intestine, with only minor effects on hepatic drug metabolism. A likely explanation is that carbamazepine also induces P-glycoprotein, the product of the MDR1 gene that is a drug efflux pump and a known transporter of lapatinib [200]. Assuming dose-linear pharmacokinetics, the magnitude of carbamazepine-lapatinib interaction (72% AUC decrease) might also be critical when clinical doses of lapatinib (>1250 mg per day) are administered.
The aqueous solubility of lapatinib declines when pH is >4 [201, 202], suggesting that acid-reducing drugs might also lower absorption, exposure and subsequent efficacy of lapatinib. When women with metastatic HER2+ breast cancer are treated with 1250 mg lapatinib once daily in the morning with or without esomeprazole (a proton pump inhibitor) before bed time, AUC of lapatinib is significantly decreased by 26% (ranging from 6% to 49%) in the patients receiving esomeprazole [202]. Although the magnitude of this effect might have only minor clinical relevance, the time point of dosing should be considered when co-administration of acid-reducing drugs is needed. For example, concurrent dosing might avoid this interaction because the stomach (where the drugs are most likely to interact) has a pH <4, where lapatinib is most soluble.
Herbal drugs are commonly consumed by cancer patients to increase quality of life or to manage adverse effects of therapy. A survey of 2000 cancer patients reported that 49% of breast cancer patients consume herbal drugs [203]. The most relevant herbal drug for lapatinib is St. John’s Wort, which has substantial impact on CYP3A4- and CYP2C9-mediated metabolism and P-glycoprotein-mediated drug transport. St. John’s Wort can lower exposure of co-administered drugs by up to 70%. A considerable interaction between St. John’s Wort and imatinib has been demonstrated, and interactions with other TKIs are likely, but current evidence is limited [204].
Given the high inter-individual variability of TKI pharmacokinetics and the suggested concentration threshold for efficacy, adjustment of drug dosing according to therapeutic drug monitoring (TDM) leading to optimization of lapatinib pharmacotherapy seems appropriate in some circumstances. Indeed, TDM has been suggested for a subset of TKIs, albeit with different levels of evidence supporting its routine clinical application. Although there is good evidence for the meaningful role of imatinib TDM, for other TKIs, including lapatinib, data is insufficient to incorporate TDM into clinical routine. The use of TDM during targeted therapy regimens might best be reserved for situations pertaining to lack of therapeutic response, unexpected toxicity, anticipated drug-drug interactions or concerns over treatment adherence. In the future, concentration–effect relationships should be evaluated in more detail. For example, performing randomized trials comparing classic dosing with pharmacokinetics-guided adaptive dosing will help to establish target plasma concentrations and eventually individualize pharmacotherapy to maximize efficacy and prevent toxicity [205].
Other Small Molecule Inhibitors
Because significant mechanisms of TKI resistance arise from pathway switching and over-expression of targeted receptors and their ligands (leading to auto-activation), a long-lasting inhibition of the RTK kinase activity is desired. Irreversible TKIs are considered advantageous compared to reversible inhibitors in this regard [206]. Several irreversible inhibitors of HER2 are currently under investigation for their clinical efficacy, although little information is so far available on mechanisms of resistance to these newer agents. Following is a summary of findings with three irreversible inhibitors that have particular relevance to HER2+ breast cancer.
Neratinib
Neratinib (HKI-272, Puma Biotechnology) is an irreversible, pan-HER (HER1, −2, −4) TKI [207,208,209]. In pre-clinical studies, neratinib was shown to overcome trastuzumab resistance, both in cell culture and in xenograft models [210]. In the clinic, neratinib has shown substantial activity in patients with advanced HER2+ breast cancer (78% 16-week PFS rate, median PFS of 39.6 weeks, OR rate of 56% in a cohort of 64 patients who had not received prior trastuzumab therapy). Response rates are lower for patients who previously received trastuzumab (16-week PFS of 59%, median PFS of 22.3 months, 24% OR rate among a cohort of 63 patients) [211]. This perhaps suggests a trastuzumab resistance mechanism in these patients that is downstream of HER2 and thus shared by neratinib. This low response was also evident in a phase II randomized trial of HER2+ breast cancer patients with locally advanced or metastatic disease and prior trastuzumab therapy that compared lapatinib + capecitabine (n = 116) with neratinib (n = 117) as second-line therapy. Median OS for the neratinib arm was 19.7 months vs. 23.6 months for the lapatinib + capecitabine arm and clinical benefit rate was lower with neratinib (44% versus 64%; p = 0.003) [212]. In a phase III trial comparing neratinib (n = 1420) with placebo (n = 1420) in patients with HER2+ breast cancer who had previously received neoadjuvant or adjuvant trastuzumab, neratinib was associated with improved two-year invasive DFS (HR = 0.67, p = 0.0091). There was a greater benefit to patients with ER+/PR+ tumors (HR = 0.51, p = 0.0013 relative to placebo) than patients with ER−/PR− tumors (HR = 0.93, p = 0.74) (pinteraction = 0.054) [213]. OS data from this trial have not yet been reported.
Canertinib
Canertinib (CI-1033, Pfizer) is a 4-anilinoquinazoline that irreversibly inhibits HER1, HER2 and HER4 through interaction with the ATP binding site of the respective receptor kinase domains, leading to inhibition of receptor auto-phosphorylation when cell lines are stimulated with EGF (HER1) or HRG (HER2, HER3, HER4) [214,215,216]. Canertinib inhibits tumor growth in xenograft models and it was the first pan-HER inhibitor to undergo clinical trial. Canertinib resistance appears to occur by sustained PI3K/AKT/mTOR signaling, with the dual PI3K/mTOR inhibitor BEZ235 able to overcome the resistance phenotype [217]. In a randomized, phase II, dose-finding study of patients with measurable, progressive or recurrent metastatic breast cancer, HER2+ patients who received 450 mg canertinib once daily for 14 days every 21 days had a response rate of 18.8% and one-year OS rate of 86.7%. The drug was well tolerated only at the 50 mg dose, however, thus potentially limiting the clinical utility of this drug [218].
Afatinib
Afatinib (BIBW-2992, Boehringer Ingelheim) also binds covalently to HER1, HER2 and HER4 and inhibits signaling through all HER-family homodimers and heterodimers [86, 216, 219]. Afatinib is currently approved for first-line treatment of metastatic non-small cell lung carcinomas that contain HER1 mutations known to sensitize the receptor to TKI inhibition [219]. In the HER2+ breast cancer setting, afatinib has shown modest clinical benefit in phase II studies of women with metastatic disease who had progressed during or after trastuzumab and/or lapatinib therapy [220, 221]. The DAFNE phase II trial looked at neoadjuvant afatinib + trastuzumab + conventional chemotherapy in previously untreated HER2+ patients (n = 65) [222]. The overall pCR rate was 49.2%, below the target rate of 55% that would have advanced the regimen to phase III trials. ER−/PR− patients (n = 19; pCR = 63.2%) performed slightly better than ER+/PR+ patients (n = 46; pCR = 43.5%), a difference that did not achieve statistical significance (p = 0.153), and there was no statistical difference between PIK3CA-wild type (n = 48; pCR = 54.2%) and PIK3CA-mutant (n = 13; pCR = 38.5%) patients (p = 0.363). Statistical significance in these cases might have been compromised by the small number of patients in the study. In a smaller neoadjuvant trial comparing afatinib (n = 10) to trastuzumab (n = 11) and lapatinib (n = 8), the objective response was seen in 8 of the afatinib-treated patients, comparable to lapatinib (6 objective responses) and better than trastuzumab (4 objective responses), but the study was terminated early due to slow enrollment [223]. Finally, in a phase III study of patients with HER2+ metastatic breast cancer who had progressed on trastuzumab (n = 508), afatinib + vinorelbine (a tubulin-targeted drug) did not improve PFS or OS and was less tolerable than trastuzumab + vinorelbine [224]. Hormone receptor status did not affect outcomes in either treatment arm.
Implications for New Targets and Drug Combinations
Understanding mechanisms of resistance raises the possibility of using corresponding mechanism-based inhibitors either to prevent the initial development of resistance or to restore HER2-targeted drug efficacy once resistance has emerged. Only a relatively small number of such mechanism-based inhibitors have made it into clinical phases of development and most of these newer agents are being tested in combination with trastuzumab, either as front-line combination therapy or in patients who have progressed from prior trastuzumab therapy. This is because of pre-clinical and clinical evidence that trastuzumab-resistant cell lines and tumors continue to be dependent on HER2 signaling and that combining trastuzumab with other targeted agents is clinically beneficial in patients who have progressed on trastuzumab [35, 225,226,227,228]. The possibility of combining three or more targeted agents is also being explored. The following sections describe the mechanism-based targets and corresponding drugs that are the most promising in the setting of HER2+ breast cancer. Several other proteins discussed earlier as resistance mechanisms are not included in this section either because corresponding drugs have not yet advanced clinically or because such drugs have not shown enough clinical benefit to warrant further development for treating HER2+ breast cancer.
Targeting Alternative Receptors
HER1 and HER3
Pan-specific agents that target HER2 plus HER1 and/or HER3 have been discussed earlier in the context of possible resistance mechanisms pertaining to these multi-receptor agents. The trials testing combinations of such drugs are too numerous to summarize in the current chapter, but the predominant drugs to date have been pertuzumab and lapatinib. Pertuzumab showed clear clinical benefit when added to trastuzumab, both in patients who had progressed on prior trastuzumab and in trastuzumab-naïve patients [101, 103, 104], leading to FDA approval for use in combination with trastuzumab to treat HER2+ breast cancer in the neoadjuvant and metastatic settings. Likewise, lapatinib + trastuzumab was shown to be beneficial in patients who had progressed on trastuzumab [189, 225, 226] and it is notably effective in patients with p95HER2, a key resistance factor for trastuzumab [229, 230]. Lapatinib, in combination with capecitabine, was approved in 2007 for the treatment of advanced HER2+ breast cancer.
IGF-1R
IGF-1R can confer trastuzumab resistance but it does not appear to be involved in lapatinib resistance. Recent reviews of development and clinical trials of IGF-1R inhibitors can be found in [231,232,233,234,235]. These inhibitors include monoclonal antibodies and TKIs, the most notable of which in the context of breast cancer and trastuzumab resistance are cixutumumab (IMC-A12, ImClone) and BMS-754807 (Bristol-Myers Squibb). Cixutumumab is a monoclonal antibody with high selectivity and affinity for IGF-1R. It has undergone phase II clinical trial (NCT00684983) in combination with capecitabine + lapatinib in HER2+ metastatic breast cancer patients who had progressed on trastuzumab-based regimens [235]. BMS-754807 is a selective, non ATP-competitive IGF-1R TKI that shows strong synergy with trastuzumab in inhibiting proliferation of a HER2+ breast cancer cell line and with lapatinib in inhibiting a lung cancer cell line [236]. A phase I/II trial (NCT00788333) evaluated BMS-754807 in combination with trastuzumab in patients with HER2+ metastatic breast cancer. Results from NCT00684983 and NCT00788333 are still pending.
A newer TKI, KW-2450 (Kyowa Kakko Kirin Co. Ltd.), inhibits IGF-1R and insulin receptor kinases with similar efficacy in vitro [237]. It showed promise in an initial phase I trial (NCT00921336), with four of 10 evaluable patients having stable disease as best response [238]. However, a follow-on phase I/II trial (NCT01199367) to evaluate KW-2450 in combination with lapatinib + letrozole in patients with advanced or metastatic HER2+ breast cancer was terminated in phase I because a well-tolerated dose permitting further phase II study was not identified.
MET, AXL, VEGFR
MET, AXL and VEGFR are RTKs that have all been implicated in trastuzumab and/or lapatinib resistance, as described earlier. Foretinib (GSK1363089, GlaxoSmithKlein), which inhibits all three receptors, has proven effective in vitro (see above) [159]. In phase I and II trials, efficacy was highly variable, ranging from stable disease as best response [239, 240] to 43% of head and neck cancer patients experiencing tumor shrinkage [241]. Foretinib was considered to be of potential clinical value, especially when combined with other (HER-targeting) drugs [241]. Pazopanib, another multi-kinase inhibitor that targets VEGFR, PDGFR and FGFR, has shown clinical activity in combination with lapatinib [242]. In this study, patients with HER2+ breast cancer who had not previously received trastuzumab or chemotherapy were treated with pazopanib + lapatinib (n = 69) or lapatinib alone (n = 72). The 12-week response rate was 36.2% for the combination treatment arm vs. 22.2% for the lapatinib treatment arm but the week-12 progressive disease rates, PFS and OS rates were not statistically different between the two arms [242].
In an attempt to target the VEGFR pathway more specifically, several clinical trials have evaluated the efficacy of bevacizumab, a VEGF-targeted monoclonal antibody, in combination with lapatinib or trastuzumab. A phase II study of lapatinib + bevacizumab in HER2+ metastatic breast cancer patients (n = 52, 90% of whom had received prior trastuzumab therapy) found a 12-week PFS rate of 69.2% and median PFS of 24.7 weeks. The conclusion from this trial was that lapatinib + bevacizumab is active in patients with HER2+ breast tumors [243]. Efficacy of lapatinib + bevacizumab was even higher when combined with trastuzumab: half of the 23 patients in the study showed complete response, partial response or stable disease for more than 6 months. This relatively high rate of stable disease was not negatively influenced by prior treatment with any of the study drugs [64].
AVEREL (NCT00391092) is a large (n = 424) phase III trial testing the addition of bevacizumab to first-line trastuzumab + docetaxel therapy in patients with locally recurrent or metastatic HER2+ breast cancer who had not received prior therapy for their metastatic disease. At a median follow-up of 26 months, there was a trend towards longer PFS (16.5 months in the bevacizumab arm vs. 13.7 months in the non-bevacizumab arm; HR = 0.82) that did not reach statistical significance (p = 0.0775) [244]. Further analysis of this trial’s outcomes is pending.
Targeting Downstream Effectors
PI3K/AKT/ mTOR
As described earlier, the PI3K/AKT pathway is an attractive target to inhibit cancer cell growth or to restore efficacy in HER2+ breast cancers that have become resistant to HER2-targeted drugs. mTOR is the primary downstream effector of PI3K/AKT signaling and is thought to be a key target for short-circuiting multiple mechanisms of HER2-related drug resistance [245,246,247,248]. mTOR is the catalytic subunit of mTOR complex-1 (mTORC1) and mTOR complex-2 (mTORC2), both of which act downstream of PI3K/AKT to regulate cell growth. mTORC1, the target of the original mTOR inhibitor known as rapamycin, phosphorylates S6 K1 ribosomal protein and 4EBP1 translation initiation factor to promote mRNA translation. mTORC2 phosphorylates AKT and controls the cytoskeletal network, and it is critical for cancer survival and progression [249,250,251,252]. mTORC1 and mTORC2 are both valid targets in the context of breast cancer and HER2-based therapies [253]. Inhibitors of PI3K, AKT, mTOR and dual PI3K/mTOR inhibitors have all been explored clinically and are discussed here in the context of HER2+ breast cancer.
mTOR Inhibitors
The most advanced mTOR-targeted drug is everolimus (RAD001, Novartis Pharmaceuticals), which functions as an allosteric inhibitor of mTOR in the context of mTORC1. Everolimus inhibits the growth of HER2+ breast cancer cell lines and enhances the effectiveness of trastuzumab and lapatinib in cell lines and human xenografts in mice [246, 247]. Two phase III clinical trials, Bolero-1 and Bolero-3, have demonstrated clear clinical benefit of combining everolimus with trastuzumab, most particularly for patients with ER−/PR− tumors. In Bolero-3 (n = 569) the addition of everolimus to trastuzumab + vinorelbine prolongs PFS in patients with trastuzumab-resistant advanced HER2+ breast cancer [254]. Median PFS in the everolimus group was 7.00 months compared with 5.78 months in the placebo group (HR = 0.78; p = 0.0067). The benefit is seen in ER+/PR+ and ER−/PR− patients but was more pronounced in the ER−/PR− group (HR = 0.65 in the ER−/PR− group; HR = 0.93 in the ER+/PR+ group). Bolero-1 (n = 719) studied the effect of adding everolimus (vs. placebo) to trastuzumab + paclitaxel as first-line treatment for advanced HER2+ breast cancer [255]. The addition of everolimus had only a modest effect on PFS in the entire population of patients (median PFS of 14.95 months in the everolimus group vs. 14.49 months in the placebo group) that was not statistically significant (HR = 0.89; p = 0.1166). The benefit of everolimus was much more profound in the ER−/PR− sub-population -- median PFS of 20.27 months for the everolimus group versus 13.08 months for the placebo group (HR = 0.66; p = 0.0049). Taken together, the findings from these two studies suggest that mTOR is most important as a mechanism of trastuzumab resistance when the ER/PR pathway is not available as an alternative signaling pathway, and that the use of mTOR-targeted drugs should be confined to this group of patients.
There are several newer mTOR inhibitors that compete with ATP binding to mTOR and thereby inhibit both mTORC1 and mTORC2. These include AZD8055 (AstraZeneca), AZD2014 (AstraZeneca), TAK-228 (developed by Millenium, also known as MLN01 and INK-228), CC-223 (Celgene), and the recently described MTI-31 (Chinese Academy of Sciences), some of which have been shown to restore sensitivity to trastuzumab or lapatinib in cell line models of resistance [172, 245]. A phase I clinical trial of CC-223 identified PR or stable disease in two of three patients with advanced breast cancer (unknown HER2 status) [256]. Nevertheless, mTOR inhibitors by themselves might be of relatively minor clinical value in HER2+ breast cancer.
PI3K/mTOR Inhibitors
Most PI3K inhibitors are actually dual inhibitors of PI3K and mTOR. The dual inhibition is both for the purpose of short-circuiting multiple mechanisms of PI3K/AKT/mTOR activation and for the apparent need to inhibit feedback up-regulation of PI3K signaling when mTOR is blocked [247, 257,258,259]. Most of these dual PI3K/mTOR inhibitors are still early in their clinical testing, but we describe several here because of the importance of the PI3K/mTOR pathway and the promise shown for this class of drugs overall. We also include brief discussion of a pan-PI3K inhibitor that is not in the dual PI3K/mTOR class because of its potential relevance to HER2+ breast cancer.
BEZ235 (Novartis Pharmaceuticals) is an early example of a dual PI3K/mTOR inhibitor. Pre-clinical studies with this agent indicated that BEZ235 is a potent inhibitor of breast cancer cell lines across a spectrum of molecular sub-types, including lines with inherent and acquired trastuzumab resistance. Growth inhibition is via growth arrest and apoptosis. The drug also inhibits tumor progression in xenograft models, both as a single agent and even more effectively in combination with trastuzumab [247]. Although several early-phase trials of BEZ235 have been conducted, no results from trials with HER2+ patients have been reported and clinical efficacy with BEZ235 appears to be limited to disease stabilization. Although BEZ235 is considered to be of minor clinical value and is no longer in clinical development, it nevertheless represents an important advance in targeted efforts to manage trastuzumab (and other drug) resistance resulting from PI3K/AKT activation.
SAR245409 (Sanofi) is another PI3K/mTOR inhibitor. When it was combined with erlotinib, an HER1-targeted TKI, in a phase I trial with 46 patients (mostly lung cancer), stable disease (in 37% of patients) was the best response. Analysis of pharmacodynamic markers in serial skin samples showed that the maximum inhibition of PI3K, HER1 and MAPK pathways was only between 37% and 75%. Based on these results, it was speculated that other dosing regimens leading to higher systemic drug exposure might be needed to achieve better clinical efficacy [260].
PKI-587 (PF-05212384, Pfizer) is a dual PI3K/mTOR inhibitor that was evaluated in a phase I trial with 77 solid tumor patients, including four with breast cancer [261]. Treatment with PKI-587 lowered pAKT levels in post-treatment biopsies compared with paired pre-treatment biopsies, as well as other PI3K/mTOR pathway markers to varying degrees. Two patients experienced objective PR and 27 exhibited stable disease, with eight patients having stable disease for >6 months. PKI-587 is currently under investigation in phase II trials in endometrial and non-small cell lung cancer patients and should perhaps be tested in breast cancer settings as well.
Buparlisib (BKM120, Novartis Pharmaceuticals) is a pan-class 1A PI3K inhibitor that was combined with carboplatin + paclitaxel in a phase I trial that included patients with solid tumors [262]. Of 25 patients with measurable disease, five exhibited objective responses (one CR, four PR) and the three patients with known loss of PTEN expression all benefited from the treatment. Dose-limiting toxicities included paclitaxel-typical uncomplicated neutropenia. It was concluded from this trial that addition of buparlisib to carboplatin + paclitaxel is safe and might increase the efficacy of this cytotoxic combination therapy, especially in tumors with loss of PTEN expression. NeoPHOEBE is a phase II trial (NCT01816594) investigating the effect of adding buparlisib to trastuzumab + paclitaxel for the neoadjuvant treatment of HER2+ primary breast cancer. Results are still pending.
AKT Inhibitors
AKT is a serine-threonine kinase that lies downstream of PI3K and upstream of mTORC1. AKT phosphorylation results in a cascade of downstream molecular events that ultimately result in cell proliferation and survival, and the turning off of AKT phosphorylation is an essential element of effective HER2 inhibition mediated by the HER2-targeted drugs described in this chapter. Initial enthusiasm for development of AKT-targeted drugs was perhaps dampened by the discovery that AKT inhibition causes release of a negative feedback loop on expression and phosphorylation of multiple upstream RTKs, including IGF-1R and the HER family of receptors [263, 264]. The end result in the context of HER2+ breast cancer is increased levels of ligand-activated HER1, HER3 and HER4, and increased dimerization with and signaling through HER2. Importantly, trastuzumab and lapatinib are both capable of reversing the feedback effect and the combination of AKT inhibition with HER2 inhibition is effective in reducing the growth of HER2+ cells in culture and as xenografts in mice [263, 264]. Based on this, a few AKT-targeted drugs are in development for use in combination with HER2 targeting.
The most advanced AKT inhibitor is MK-2206 (Merck), which shows potent and selective inhibition of AKT and anti-proliferation activity both in vitro and in vivo against HER2+ cells and xenografts [265, 266]. Its growth inhibitory effect is enhanced in cells with PTEN loss or PIK3CA mutation [266], which perhaps are more dependent on PI3K/AKT signaling for proliferation. Phase I clinical trials tested MK-2206 in combination with trastuzumab [267] or trastuzumab + lapatinib [268] in patients who had progressed on HER2-targeted therapy. Results showed that the combinations are safe and demonstrate some clinical activity in a pre-treated patient population, thus warranting further investigation. Ongoing clinical trials include NCT01042379 (a phase II trial testing MK-2206 with or without trastuzumab, along with several other targeted drugs, using a tumor’s molecular profile to assign each patient to a selected protocol) and NCT01277757 (a phase II trial testing MK-2206 in patients with stage III or stage IV breast cancer who have failed prior therapy and have tumors with a PIK3CA mutation, an AKT mutation, and/or PTEN loss/PTEN mutation).
ER Pathway
Given the role of ER in lapatinib resistance described earlier, combining anti-HER2 drugs with therapeutics targeting the ER pathway (ER modulators such as tamoxifen or fulvestrant; aromatase inhibitors such as anastrozole, letrozole or exemestane) might be a promising approach in HER2+/ER+ cancers. In one such trial, adding lapatinib to fulvestrant did not improve PFS or OS, but patients with ER+/PR+/HER2+ and ER+/PR+/HER2− tumors were all included in the trial, meaning that outcomes might have been affected by the non-expression of one of the main targets of lapatinib (HER2) [269]. In an earlier trial, letrozole showed a beneficial effect in combination with lapatinib (enhanced PFS and clinical benefit rate) in patients with metastatic breast cancer that is both ER+/PR+ and HER2+ [270]. Thus, the combination of lapatinib and letrozole seems to be a rational and clinically effective strategy to improve outcomes in patients with HER2+, ER+/PR+ breast cancer and potentially to combat ER-mediated resistance against HER2-directed therapies [271]. Additional discussion of ER pathway inhibitors and resistance can be found in the chapter on “Resistance to Endocrine Therapy”.
Concluding Remarks
This review chapter has focused on molecular mechanisms of resistance, but clinical resistance is ultimately a manifestation of a selection process that happens at the cellular level based on the phenotype(s) conferred by an accumulation of molecular changes in a population of malignant cells. In our case, the molecular changes include mutations and gene expression or activity alterations that compensate for the therapy-mediated inactivation of HER2. In a pre-treatment cancer, intratumor heterogeneity -- the varied molecular landscape that results from genomic instability in all cancers -- hinders the efficacy of therapeutics that are targeted to a single oncogenic driver (such as HER2). Even when a targeted therapy is initially effective, a subset of cells in the population with the greatest growth advantage – due to their particular combination of pre-existing and adaptive molecular changes – will emerge as therapy-resistant disease. This, too, is heterogeneous in nature, due to the multiple molecular mechanisms that can promote survival [272, 273].
Proper analysis of inherent and adaptive heterogeneity will likely be required to develop effective patient-specific therapeutics. To be most effective, such analysis should occur at multiple time points to account for ongoing selection/evolution as patients undergo treatment [273]. This is becoming increasingly feasible with the development of fast and inexpensive sequencing techniques, even at the single cell level. Thus we are poised to make further advances in cancer therapy based on molecular characterizations, a concept now known as precision medicine. Indeed, knowledge about particular molecular mechanisms of resistance described in this chapter – and our ability to detect such mechanisms – has led to the identification of new targets and corresponding new drugs to prevent or counteract resistance in patients with HER2+ breast cancer. Agents like pertuzumab and lapatinib arose from an understanding that HER1 and HER3 are significant compensatory signaling mechanisms that contribute to trastuzumab resistance. Given the central role of common downstream nodes in multiple signaling pathways, proteins such as PI3K/AKT and mTOR represent the next generation of targets for combating resistance.
We still have much to learn about the best combinations to use in the clinic and whether to deploy the combinations concurrently as a means of preventing the emergence of one or more resistance mechanism, or sequentially as a way to overcome acquired resistance. It can be argued that both approaches will be necessary, depending on the molecular details of an individual cancer at the time of diagnosis and the likely mechanisms of resistance that will emerge as an adaptation to first-line therapy. The presence of PI3K/AKT aberrations in a primary tumor, for example, might argue for concurrent treatment with a HER2-targeted drug and a PI3K/AKT/mTOR inhibitor. On the other hand, this first-line approach might simply lead to the emergence of other, unknown resistance mechanisms against which we have no therapeutic options. Given the evolutionary nature of resistance, it has been suggested that it might be more effective to take a sequential approach, in an attempt to anticipate the evolution of a cancer and maintain it in a state that is treatable with available drugs to known adaptive mechanisms of resistance [273]. In this latter case, it will be important to customize second- and third-line therapies to the molecular features of resistance in each individual and to optimize timing of the sequential deliveries. The solutions to these therapeutic problems will only come from further understanding of mechanisms of drug action and resistance that we discern from laboratory studies and of drugs’ clinical activity, tolerability and pharmacokinetic interactions that we discern from clinical trials. The future of precision medicine will thus depend on a better knowledge of the evolution of resistance mechanisms and knowing how to apply that information to the selection and administration of targeted drugs.
Abbreviations
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- AUC:
-
Area under the curve
- CDK:
-
Cyclin dependent kinase
- CML:
-
Chronic myelogenous leukemia
- CYP:
-
Cytochrome P450
- DFS:
-
Disease-free survival
- EGF:
-
Epidermal growth factor
- ER:
-
Estrogen receptor
- ERK:
-
Extracellular signal regulated kinase
- FAK:
-
Focal adhesion kinase
- FDA:
-
United States Food and Drug Administration
- HER1:
-
Human ErbB receptor 1 also known as EGF receptor
- HER2:
-
Human ErbB receptor 2
- HER3:
-
Human ErbB receptor 3
- HGF:
-
Hepatocyte growth factor
- HR:
-
Hazard ratio
- HRG:
-
Heregulin
- IGF:
-
Insulin-like growth factor
- IGF-1R:
-
IGF-1 receptor
- MEK:
-
ERK kinase
- MET:
-
Receptor tyrosine kinase encoded by c-MET gene, also known as hepatocyte growth factor receptor
- mTOR:
-
Mammalian target of rapamycin
- mTORC:
-
mTOR complex
- MUC4:
-
mucin-4
- OS:
-
Overall survival
- pCR:
-
Pathological complete response
- PFS:
-
Progression-free survival
- PI3K:
-
Phosphatidylinositol 3-kinase
- PIK3CA :
-
Gene encoding the p110α catalytic subunit of PI3K
- PIP2 :
-
Phosphatidylinositol-4,5-bisphosphate
- PIP3 :
-
Phosphatidylinositol-3,4,5-trisphosphate
- PKA:
-
Protein kinase A
- PR:
-
Progesterone receptor
- PTEN:
-
Phosphatase and tensin homolog
- RTK:
-
Receptor tyrosine kinase
- STAT3:
-
Signal transducer and activator of transcription 3
- TDM:
-
Therapeutic drug monitoring
- T-DM1:
-
Trastuzumab-emtansine antibody drug conjugate
- TGF-α:
-
Transforming growth factor-α
- TKI:
-
Tyrosine kinase inhibitor
- VEGF:
-
Vascular endothelial growth factor
- VEGFR:
-
VEGF receptor
References
Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–34.
Schechter AL, Stern DF, Vaidyanathan L, Decker SJ, Drebin JA, Greene MI, Weinberg RA. The neu oncogene: an erb-B-related gene encoding a 185,000-Mr tumour antigen. Nature. 1984;312:513–6.
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82.
Lonardo F, Di Marco E, King CR, Pierce JH, Segatto O, Aaronson SA, Di Fiore PP. The normal erbB-2 product is an atypical receptor-like tyrosine kinase with constitutive activity in the absence of ligand. New Biol. 1990;2:992–1003.
Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21:177–84.
Wallasch C, Weiss FU, Niederfellner G, Jallal B, Issing W, Ullrich A. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J. 1995;14:4267–75.
Hancock MC, Langton BC, Chan T, Toy P, Monahan JJ, Mischak RP, Shawver LKA. Monoclonal antibody against the c-erbB-2 protein enhances the cytotoxicity of cis-diamminedichloroplatinum against human breast and ovarian tumor cell lines. Cancer Res. 1991;51:4575–80.
Hudziak RM, Lewis GD, Winget M, Fendly BM, Shepard HM, Ullrich A. p185HER2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor. Mol Cell Biol. 1989;9:1165–72.
Pietras RJ, Fendly BM, Chazin VR, Pegram MD, Howell SB, Slamon DJ. Antibody to HER-2/neu receptor blocks DNA repair after cisplatin in human breast and ovarian cancer cells. Oncogene. 1994;9:1829–38.
Stancovski I, Hurwitz E, Leitner O, Ullrich A, Yarden Y, Sela M. Mechanistic aspects of the opposing effects of monoclonal antibodies to the ERBB2 receptor on tumor growth. Proc Natl Acad Sci U S A. 1991;88:8691–5.
Baselga J, Norton L, Albanell J, Kim YM, Mendelsohn J. Recombinant humanized anti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res. 1998;58:2825–31.
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92.
Gianni L, Eiermann W, Semiglazov V, Lluch A, Tjulandin S, Zambetti M, Moliterni A, Vazquez F, Byakhov MJ, Lichinitser M, Climent MA, et al. Neoadjuvant and adjuvant trastuzumab in patients with HER2-positive locally advanced breast cancer (NOAH): follow-up of a randomised controlled superiority trial with a parallel HER2-negative cohort. Lancet Oncol. 2014;15:640–7.
Kim MM, Allen P, Gonzalez-Angulo AM, Woodward WA, Meric-Bernstam F, Buzdar AU, Hunt KK, Kuerer HM, Litton JK, Hortobagyi GN, Buchholz TA, et al. Pathologic complete response to neoadjuvant chemotherapy with trastuzumab predicts for improved survival in women with HER2-overexpressing breast cancer. Ann Oncol. 2013;24:1999–2004.
Nahta R, Esteva FJ. Herceptin: mechanisms of action and resistance. Cancer Lett. 2006;232:123–38.
Piccart-Gebhart MJ, Procter M, Leyland-Jones B, Goldhirsch A, Untch M, Smith I, Gianni L, Baselga J, Bell R, Jackisch C, Cameron D, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353:1659–72.
Spector NL, Blackwell KL. Understanding the mechanisms behind trastuzumab therapy for human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol. 2009;27:5838–47.
Baselga J. Herceptin alone or in combination with chemotherapy in the treatment of HER2-positive metastatic breast cancer: pivotal trials. Oncology. 2001;61(Suppl 2):14–21.
Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, Wolter JM, Paton V, Shak S, Lieberman G, Slamon DJ. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17:2639–48.
Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore M, Shak S, et al. First-line Herceptin monotherapy in metastatic breast cancer. Oncology. 2001;61(Suppl 2):37–42.
Eiermann W. Trastuzumab combined with chemotherapy for the treatment of HER2-positive metastatic breast cancer: pivotal trial data. Ann Oncol. 2001;12(Suppl 1):S57–62.
Esteva FJ, Valero V, Booser D, Guerra LT, Murray JL, Pusztai L, Cristofanilli M, Arun B, Esmaeli B, Fritsche HA, Sneige N, et al. Phase II study of weekly docetaxel and trastuzumab for patients with HER-2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:1800–8.
Anido J, Scaltriti M, Bech Serra JJ, Santiago Josefat B, Todo FR, Baselga J, Arribas J. Biosynthesis of tumorigenic HER2 C-terminal fragments by alternative initiation of translation. EMBO J. 2006;25:3234–44.
Christianson TA, Doherty JK, Lin YJ, Ramsey EE, Holmes R, Keenan EJ. Clinton GM. NH2-terminally truncated HER-2/neu protein: relationship with shedding of the extracellular domain and with prognostic factors in breast cancer. Cancer Res. 1998;58:5123–9.
Saez R, Molina MA, Ramsey EE, Rojo F, Keenan EJ, Albanell J, Lluch A, Garcia-Conde J, Baselga J, Clinton GM. p95HER-2 predicts worse outcome in patients with HER-2-positive breast cancer. Clin Cancer Res. 2006;12:424–31.
Xia W, Liu LH, Ho P, Spector NL. Truncated ErbB2 receptor (p95ErbB2) is regulated by heregulin through heterodimer formation with ErbB3 yet remains sensitive to the dual EGFR/ErbB2 kinase inhibitor GW572016. Oncogene. 2004;23:646–53.
Scaltriti M, Rojo F, Ocana A, Anido J, Guzman M, Cortes J, Di Cosimo S, Matias-Guiu X. Ramon y Cajal S, Arribas J, Baselga J. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst. 2007;99:628–38.
Sperinde J, Jin X, Banerjee J, Penuel E, Saha A, Diedrich G, Huang W, Leitzel K, Weidler J, Ali SM, Fuchs EM, et al. Quantitation of p95HER2 in paraffin sections by using a p95-specific antibody and correlation with outcome in a cohort of trastuzumab-treated breast cancer patients. Clin Cancer Res. 2010;16:4226–35.
Castiglioni F, Tagliabue E, Campiglio M, Pupa SM, Balsari A, Menard S. Role of exon-16-deleted HER2 in breast carcinomas. Endocr Relat Cancer. 2006;13:221–32.
Mitra D, Brumlik MJ, Okamgba SU, Zhu Y, Duplessis TT, Parvani JG, Lesko SM, Brogi E, Jones FE. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol Cancer Ther. 2009;8:2152–62.
Price-Schiavi SA, Jepson S, Li P, Arango M, Rudland PS, Yee L, Carraway KL. Rat Muc4 (sialomucin complex) reduces binding of anti-ErbB2 antibodies to tumor cell surfaces, a potential mechanism for herceptin resistance. Int J Cancer. 2002;99:783–91.
Nagy P, Friedlander E, Tanner M, Kapanen AI, Carraway KL, Isola J, Jovin TM. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. 2005;65:473–82.
Mercogliano MF, De Martino M, Venturutti L, Rivas MA, Proietti CJ, Inurrigarro G, Frahm I, Allemand DH, Gil Deza E, Ares S, Gercovich FG et al. TNFalpha-induced mucin 4 expression elicits trastuzumab resistance in HER2-positive breast cancer. Clin Cancer Res. 2017;23:636–48.
Nahta R. Pharmacological strategies to overcome HER2 cross-talk and Trastuzumab resistance. Curr Med Chem. 2012;19:1065–75.
Chan CT, Metz MZ, Kane SE. Differential sensitivities of trastuzumab (Herceptin)-resistant human breast cancer cells to phosphoinositide-3 kinase (PI-3K) and epidermal growth factor receptor (EGFR) kinase inhibitors. Breast Cancer Res Treat. 2005;91:187–201.
Denny EC, Kane SE. T-Darpp promotes enhanced EGFR activation and new drug synergies in Her2-positive breast cancer cells. PLoS One. 2015;10:e0132267.
Ritter CA, Perez-Torres M, Rinehart C, Guix M, Dugger T, Engelman JA, Arteaga CL. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res. 2007;13:4909–19.
Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, Xiong Y, Tseng LM, Li SH, Ding Z, Sahin AA, et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med. 2011;17:461–9.
Baselga J. A new anti-ErbB2 strategy in the treatment of cancer: prevention of ligand-dependent ErbB2 receptor heterodimerization. Cancer Cell. 2002;2:93–5.
Narayan M, Wilken JA, Harris LN, Baron AT, Kimbler KD, Maihle NJ. Trastuzumab-induced HER reprogramming in “resistant” breast carcinoma cells. Cancer Res. 2009;69:2191–4.
O'Donovan N, Byrne AT, O'Connor AE, McGee S, Gallagher WM, Crown J. Synergistic interaction between trastuzumab and EGFR/HER-2 tyrosine kinase inhibitors in HER-2 positive breast cancer cells. Investig New Drugs. 2011;29:752–9.
Schneeweiss A, Chia S, Hegg R, Tausch C, Deb R, Ratnayake J, McNally V, Ross G, Kiermaier A, Cortes J. Evaluating the predictive value of biomarkers for efficacy outcomes in response to pertuzumab- and trastuzumab-based therapy: an exploratory analysis of the TRYPHAENA study. Breast Cancer Res. 2014;16:R73.
Papa V, Gliozzo B, Clark GM, McGuire WL, Moore D, Fujita-Yamaguchi Y, Vigneri R, Goldfine ID, Pezzino V. Insulin-like growth factor-I receptors are overexpressed and predict a low risk in human breast cancer. Cancer Res. 1993;53:3736–40.
Huang X, Gao L, Wang S, McManaman JL, Thor AD, Yang X, Esteva FJ, Liu B. Heterotrimerization of the growth factor receptors erbB2, erbB3, and insulin-like growth factor-i receptor in breast cancer cells resistant to herceptin. Cancer Res. 2010;70:1204–14.
Lu Y, Zi X, Zhao Y, Mascarenhas D, Pollak M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J Natl Cancer Inst. 2001;93:1852–7.
Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005;65:11118–28.
Browne BC, Eustace AJ, Kennedy S, O'Brien NA, Pedersen K, McDermott MS, Larkin A, Ballot J, Mahgoub T, Sclafani F, Madden S, et al. Evaluation of IGF1R and phosphorylated IGF1R as targets in HER2-positive breast cancer cell lines and tumours. Breast Cancer Res Treat. 2012;136:717–27.
Camp RL, Rimm EB, Rimm DL. Met expression is associated with poor outcome in patients with axillary lymph node negative breast carcinoma. Cancer. 1999;86:2259–65.
Ghoussoub RA, Dillon DA, D'Aquila T, Rimm EB, Fearon ER, Rimm DL. Expression of c-met is a strong independent prognostic factor in breast carcinoma. Cancer. 1998;82:1513–20.
Jin L, Fuchs A, Schnitt SJ, Yao Y, Joseph A, Lamszus K, Park M, Goldberg ID, Rosen EM. Expression of scatter factor and c-met receptor in benign and malignant breast tissue. Cancer. 1997;79:749–60.
Nagy J, Curry GW, Hillan KJ, McKay IC, Mallon E, Purushotham AD, George WD. Hepatocyte growth factor/scatter factor expression and c-met in primary breast cancer. Surg Oncol. 1996;5:15–21.
Shattuck DL, Miller JK, Carraway KL III, Sweeney C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 2008;68:1471–7.
Khoury H, Naujokas MA, Zuo D, Sangwan V, Frigault MM, Petkiewicz S, Dankort DL, Muller WJ, Park MHGF. Converts ErbB2/Neu epithelial morphogenesis to cell invasion. Mol Biol Cell. 2005;16:550–61.
Lindemann K, Resau J, Nahrig J, Kort E, Leeser B, Annecke K, Welk A, Schafer J, Vande Woude GF, Lengyel E, Harbeck N. Differential expression of c-met, its ligand HGF/SF and HER2/neu in DCIS and adjacent normal breast tissue. Histopathology. 2007;51:54–62.
Minuti G, Cappuzzo F, Duchnowska R, Jassem J, Fabi A, O'Brien T, Mendoza AD, Landi L, Biernat W, Czartoryska-Arlukowicz B, Jankowski T, et al. Increased MET and HGF gene copy numbers are associated with trastuzumab failure in HER2-positive metastatic breast cancer. Br J Cancer. 2012;107:793–9.
Yen L, You XL, Al Moustafa AE, Batist G, Hynes NE, Mader S, Meloche S, Alaoui-Jamali MA. Heregulin selectively upregulates vascular endothelial growth factor secretion in cancer cells and stimulates angiogenesis. Oncogene. 2000;19:3460–9.
Konecny GE, Meng YG, Untch M, Wang HJ, Bauerfeind I, Epstein M, Stieber P, Vernes JM, Gutierrez J, Hong K, Beryt M, et al. Association between HER-2/neu and vascular endothelial growth factor expression predicts clinical outcome in primary breast cancer patients. Clin Cancer Res. 2004;10:1706–16.
Tai W, Qin B, Cheng K. Inhibition of breast cancer cell growth and invasiveness by dual silencing of HER-2 and VEGF. Mol Pharm. 2010;7:543–56.
Izumi Y, Xu L, di Tomaso E, Fukumura D, Jain RK. Tumour biology: herceptin acts as an anti-angiogenic cocktail. Nature. 2002;416:279–80.
du Manoir JM, Francia G, Man S, Mossoba M, Medin JA, Viloria-Petit A, Hicklin DJ, Emmenegger U, Kerbel RS. Strategies for delaying or treating in vivo acquired resistance to trastuzumab in human breast cancer xenografts. Clin Cancer Res. 2006;12:904–16.
Oliveras-Ferraros C, Vazquez-Martin A, Martin-Castillo B, Perez-Martinez MC, Cufi S, Del Barco S, Bernado L, Brunet J, Lopez-Bonet E, Menendez JA. Pathway-focused proteomic signatures in HER2-overexpressing breast cancer with a basal-like phenotype: new insights into de novo resistance to trastuzumab (Herceptin). Int J Oncol. 2010;37:669–78.
Valabrega G, Capellero S, Cavalloni G, Zaccarello G, Petrelli A, Migliardi G, Milani A, Peraldo-Neia C, Gammaitoni L, Sapino A, Pecchioni C, et al. HER2-positive breast cancer cells resistant to trastuzumab and lapatinib lose reliance upon HER2 and are sensitive to the multitargeted kinase inhibitor sorafenib. Breast Cancer Res Treat. 2011;130:29–40.
Burstein HJ, Elias AD, Rugo HS, Cobleigh MA, Wolff AC, Eisenberg PD, Lehman M, Adams BJ, Bello CL, DePrimo SE, Baum CM, et al. Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol. 2008;26:1810–6.
Falchook GS, Moulder SL, Wheler JJ, Jiang Y, Bastida CC, Kurzrock R. Dual HER2 inhibition in combination with anti-VEGF treatment is active in heavily pretreated HER2-positive breast cancer. Ann Oncol. 2013;24:3004–11.
Leslie NR, Downes CP. PTEN: the down side of PI 3-kinase signalling. Cell Signal. 2002;14:285–95.
Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, Beijersbergen RL, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402.
Hanker AB, Pfefferle AD, Balko JM, Kuba MG, Young CD, Sanchez V, Sutton CR, Cheng H, Perou CM, Zhao JJ, Cook RS, et al. Mutant PIK3CA accelerates HER2-driven transgenic mammary tumors and induces resistance to combinations of anti-HER2 therapies. Proc Natl Acad Sci U S A. 2013;110:14372–7.
Wang Q, Liu P, Spangle JM, Von T, Roberts TM, Lin NU, Krop IE, Winer EP, Zhao JJ. PI3K-p110alpha mediates resistance to HER2-targeted therapy in HER2+, PTEN-deficient breast cancers. Oncogene. 2016;35:3607–12.
Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–27.
Perren A, Weng LP, Boag AH, Ziebold U, Thakore K, Dahia PL, Komminoth P, Lees JA, Mulligan LM, Mutter GL, Eng C. Immunohistochemical evidence of loss of PTEN expression in primary ductal adenocarcinomas of the breast. Am J Pathol. 1999;155:1253–60.
Singh B, Ittmann MM, Krolewski JJ. Sporadic breast cancers exhibit loss of heterozygosity on chromosome segment 10q23 close to the Cowden disease locus. Genes Chromosomes Cancer. 1998;21:166–71.
Saal LH, Holm K, Maurer M, Memeo L, Su T, Wang X, JS Y, Malmstrom PO, Mansukhani M, Enoksson J, Hibshoosh H, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005;65:2554–9.
Esteva FJ, Guo H, Zhang S, Santa-Maria C, Stone S, Lanchbury JS, Sahin AA, Hortobagyi GN, PTEN YD. PIK3CA, p-AKT, and p-p70S6K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am J Pathol. 2010;177:1647–56.
Baselga J, Cortes J, Im SA, Clark E, Ross G, Kiermaier A, Swain SM. Biomarker analyses in CLEOPATRA: a phase III, placebo-controlled study of pertuzumab in human epidermal growth factor receptor 2-positive, first-line metastatic breast cancer. J Clin Oncol. 2014;32:3753–61.
Park YH, Jung HA, Choi MK, Chang W, Choi YL, Do IG, Ahn JS, Im YH. Role of HER3 expression and PTEN loss in patients with HER2-overexpressing metastatic breast cancer (MBC) who received taxane plus trastuzumab treatment. Br J Cancer. 2014;110:384–91.
Razis E, Bobos M, Kotoula V, Eleftheraki AG, Kalofonos HP, Pavlakis K, Papakostas P, Aravantinos G, Rigakos G, Efstratiou I, Petraki K, et al. Evaluation of the association of PIK3CA mutations and PTEN loss with efficacy of trastuzumab therapy in metastatic breast cancer. Breast Cancer Res Treat. 2011;128:447–56.
Adamczyk A, Niemiec J, Janecka A, Harazin-Lechowska A, Ambicka A, Grela-Wojewoda A, Domagala-Haduch M, Cedrych I, Majchrzyk K, Kruczak A, Rys J, et al. Prognostic value of PIK3CA mutation status, PTEN and androgen receptor expression for metastasis-free survival in HER2-positive breast cancer patients treated with trastuzumab in adjuvant setting. Pol J Pathol. 2015;66:133–41.
Loi S, Haibe-Kains B, Majjaj S, Lallemand F, Durbecq V, Larsimont D, Gonzalez-Angulo AM, Pusztai L, Symmans WF, Bardelli A, Ellis P, et al. PIK3CA mutations associated with gene signature of low mTORC1 signaling and better outcomes in estrogen receptor-positive breast cancer. Proc Natl Acad Sci U S A. 2010;107:10208–13.
Pogue-Geile KL, Song N, Jeong JH, Gavin PG, Kim SR, Blackmon NL, Finnigan M, Rastogi P, Fehrenbacher L, Mamounas EP, Swain SM, et al. Intrinsic subtypes, PIK3CA mutation, and the degree of benefit from adjuvant trastuzumab in the NSABP B-31 trial. J Clin Oncol. 2015;33:1340–7.
Dave B, Migliaccio I, Gutierrez MC, MF W, Chamness GC, Wong H, Narasanna A, Chakrabarty A, Hilsenbeck SG, Huang J, Rimawi M, et al. Loss of phosphatase and tensin homolog or phosphoinositol-3 kinase activation and response to trastuzumab or lapatinib in human epidermal growth factor receptor 2-overexpressing locally advanced breast cancers. J Clin Oncol. 2011;29:166–73.
Loibl S, von Minckwitz G, Schneeweiss A, Paepke S, Lehmann A, Rezai M, Zahm DM, Sinn P, Khandan F, Eidtmann H, Dohnal K, et al. PIK3CA mutations are associated with lower rates of pathologic complete response to anti-human epidermal growth factor receptor 2 (her2) therapy in primary HER2-overexpressing breast cancer. J Clin Oncol. 2014;32:3212–20.
Majewski IJ, Nuciforo P, Mittempergher L, Bosma AJ, Eidtmann H, Holmes E, Sotiriou C, Fumagalli D, Jimenez J, Aura C, Prudkin L, et al. PIK3CA mutations are associated with decreased benefit to neoadjuvant human epidermal growth factor receptor 2-targeted therapies in breast cancer. J Clin Oncol. 2015;33:1334–9.
Loibl S, Majewski I, Guarneri V, Nekljudova V, Holmes E, Bria E, Denkert C, Schem C, Sotiriou C, Loi S, Untch M, et al. PIK3CA mutations are associated with reduced pathological complete response rates in primary HER2-positive breast cancer: pooled analysis of 967 patients from five prospective trials investigating lapatinib and trastuzumab. Ann Oncol. 2016;27:1519–25.
Elsberger B. Translational evidence on the role of Src kinase and activated Src kinase in invasive breast cancer. Crit Rev Oncol Hematol. 2014;89:343–51.
Roskoski R Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol Res. 2015;94:9–25.
Roskoski R Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014;79:34–74.
Peiro G, Ortiz-Martinez F, Gallardo A, Perez-Balaguer A, Sanchez-Paya J, Ponce JJ, Tibau A, Lopez-Vilaro L, Escuin D, Adrover E, Barnadas A, et al. Src, a potential target for overcoming trastuzumab resistance in HER2-positive breast carcinoma. Br J Cancer. 2014;111:689–95.
Daverey A, Drain AP, Kidambi S. Physical intimacy of breast cancer cells with mesenchymal stem cells elicits Trastuzumab resistance through Src activation. Sci Rep. 2015;5:13744.
Le XF, Claret FX, Lammayot A, Tian L, Deshpande D, LaPushin R, Tari AM, Bast RC Jr. The role of cyclin-dependent kinase inhibitor p27Kip1 in anti-HER2 antibody-induced G1 cell cycle arrest and tumor growth inhibition. J Biol Chem. 2003;278:23441–50.
Le XF, Pruefer F, Bast RC Jr. HER2-targeting antibodies modulate the cyclin-dependent kinase inhibitor p27Kip1 via multiple signaling pathways. Cell Cycle. 2005;4:87–95.
Nahta R, Takahashi T, Ueno NT, Hung MC, Esteva FJ. P27(kip1) down-regulation is associated with trastuzumab resistance in breast cancer cells. Cancer Res. 2004;64:3981–6.
Gu L, Lau SK, Loera S, Somlo G, Kane SE. Protein kinase a activation confers resistance to trastuzumab in human breast cancer cell lines. Clin Cancer Res. 2009;15:7196–206.
Moody SE, Schinzel AC, Singh S, Izzo F, Strickland MR, Luo L, Thomas SR, Boehm JS, Kim SY, Wang ZC, Hahn WC. PRKACA mediates resistance to HER2-targeted therapy in breast cancer cells and restores anti-apoptotic signaling. Oncogene. 2015;34:2061–71.
Belkhiri A, Dar AA, Peng DF, Razvi MH, Rinehart C, Arteaga CL, El-Rifai W. Expression of t-DARPP mediates trastuzumab resistance in breast cancer cells. Clin Cancer Res. 2008;14:4564–71.
Gu L, Waliany S, Kane SE. Darpp-32 and its truncated variant t-Darpp have antagonistic effects on breast cancer cell growth and herceptin resistance. PLoS One. 2009;4:e6220.
Hamel S, Bouchard A, Ferrario C, Hassan S, Aguilar-Mahecha A, Buchanan M, Quenneville L, Miller W, Basik M. Both t-Darpp and DARPP-32 can cause resistance to trastuzumab in breast cancer cells and are frequently expressed in primary breast cancers. Breast Cancer Res Treat. 2010;120:47–57.
Agus DB, Akita RW, Fox WD, Lewis GD, Higgins B, Pisacane PI, Lofgren JA, Tindell C, Evans DP, Maiese K, Scher HI, et al. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell. 2002;2:127–37.
Metzger-Filho O, Winer EP, Krop I. Pertuzumab: optimizing HER2 blockade. Clin Cancer Res. 2013;19:5552–6.
Nahta R, Hung MC, Esteva FJ. The HER-2-targeting antibodies trastuzumab and pertuzumab synergistically inhibit the survival of breast cancer cells. Cancer Res. 2004;64:2343–6.
Scheuer W, Friess T, Burtscher H, Bossenmaier B, Endl J, Hasmann M. Strongly enhanced antitumor activity of trastuzumab and pertuzumab combination treatment on HER2-positive human xenograft tumor models. Cancer Res. 2009;69:9330–6.
Baselga J, Gelmon KA, Verma S, Wardley A, Conte P, Miles D, Bianchi G, Cortes J, McNally VA, Ross GA, Fumoleau P, et al. Phase II trial of pertuzumab and trastuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer that progressed during prior trastuzumab therapy. J Clin Oncol. 2010;28:1138–44.
Cortes J, Fumoleau P, Bianchi GV, Petrella TM, Gelmon K, Pivot X, Verma S, Albanell J, Conte P, Lluch A, Salvagni S, et al. Pertuzumab monotherapy after trastuzumab-based treatment and subsequent reintroduction of trastuzumab: activity and tolerability in patients with advanced human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol. 2012;30:1594–600.
Baselga J, Cortes J, Kim SB, Im SA, Hegg R, Im YH, Roman L, Pedrini JL, Pienkowski T, Knott A, Clark E, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366:109–19.
Swain SM, Kim SB, Cortes J, Ro J, Semiglazov V, Campone M, Ciruelos E, Ferrero JM, Schneeweiss A, Knott A, Clark E, et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA study): overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2013;14:461–71.
Gianni L, Pienkowski T, Im YH, Roman L, Tseng LM, Liu MC, Lluch A, Staroslawska E. De la Haba-Rodriguez J, Im SA, Pedrini JL et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13:25–32.
Gianni L, Pienkowski T, Im YH, Tseng LM, Liu MC, Lluch A, Staroslawska E. De la Haba-Rodriguez J, Im SA, Pedrini JL, Poirier B et al. 5-year analysis of neoadjuvant pertuzumab and trastuzumab in patients with locally advanced, inflammatory, or early-stage HER2-positive breast cancer (NeoSphere): a multicentre, open-label, phase 2 randomised trial. Lancet Oncol. 2016;17:791–800.
Gollamudi J, Parvani JG, Schiemann WP, Vinayak S. Neoadjuvant therapy for early-stage breast cancer: the clinical utility of pertuzumab. Cancer Manag Res. 2016;8:21–31.
Nordstrom JL, Gorlatov S, Zhang W, Yang Y, Huang L, Burke S, Li H, Ciccarone V, Zhang T, Stavenhagen J, Koenig S, et al. Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcgamma receptor binding properties. Breast Cancer Res. 2011;13:R123.
Burris HA, Im SA, Bauer TM, DY O, Jones SF, Nordstrom JL, Li H, Carlin DA, Baughman JE, Lechleider RJ, Bang YJ. Updated findings of a first-in-human, phase I study of margetuximab (M), an fc-optimized chimeric monoclonal antibody (MAb), in patients (pts) with HER2-positive advanced solid tumors. J Clin Oncol. 2015;33:523.
Jiang H, Rugo HS. Human epidermal growth factor receptor 2 positive (HER2+) metastatic breast cancer: how the latest results are improving therapeutic options. Ther Adv Med Oncol. 2015;7:321–39.
Leung KM, Batey S, Rowlands R, Isaac SJ, Jones P, Drewett V, Carvalho J, Gaspar M, Weller S, Medcalf M, Wydro MM, et al. A HER2-specific modified fc fragment (Fcab) induces antitumor effects through degradation of HER2 and apoptosis. Mol Ther. 2015;23:1722–33.
Cabral F, Sobel ME, Gottesman MM. CHO mutants resistant to colchicine, colcemid or griseofulvin have an altered beta-tubulin. Cell. 1980;20:29–36.
Lopus M, Oroudjev E, Wilson L, Wilhelm S, Widdison W, Chari R, Jordan MA. Maytansine and cellular metabolites of antibody-maytansinoid conjugates strongly suppress microtubule dynamics by binding to microtubules. Mol Cancer Ther. 2010;9:2689–99.
Neidhart JA, Laufman LR, Vaughn C, McCracken JD. Minimal single-agent activity of maytansine in refractory breast cancer: a southwest oncology group study. Cancer Treat Rep. 1980;64:675–7.
Ravry MJ, Omura GA, Birch R. Phase II evaluation of maytansine (NSC 153858) in advanced cancer. A Southeastern Cancer Study Group trial. Am J Clin Oncol. 1985;8:148–50.
Barok M, Joensuu H, Isola J. Trastuzumab emtansine: mechanisms of action and drug resistance. Breast Cancer Res. 2014;16:209.
Chari RV. Targeted cancer therapy: conferring specificity to cytotoxic drugs. Acc Chem Res. 2008;41:98–107.
Erickson HK, Park PU, Widdison WC, Kovtun YV, Garrett LM, Hoffman K, Lutz RJ, Goldmacher VS, Blattler WA. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006;66:4426–33.
Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, Blattler WA, Lambert JM, Chari RV, Lutz RJ, Wong WL, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68:9280–90.
Beeram M, Krop IE, Burris HA, Girish SR, Yu W, Lu MW, Holden SN, Modi S. A phase 1 study of weekly dosing of trastuzumab emtansine (T-DM1) in patients with advanced human epidermal growth factor 2-positive breast cancer. Cancer. 2012;118:5733–40.
Burris HA III, Rugo HS, Vukelja SJ, Vogel CL, Borson RA, Limentani S, Tan-Chiu E, Krop IE, Michaelson RA, Girish S, Amler L, et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J Clin Oncol. 2011;29:398–405.
Gupta M, Wang B, Carrothers TJ, LoRusso PM, Chu YW, Shih T, Loecke D, Joshi A, Saad O, Yi JH, Girish S. Effects of Trastuzumab Emtansine (T-DM1) on QT interval and safety of Pertuzumab plus T-DM1 in patients with previously treated human epidermal growth factor receptor 2-positive metastatic breast cancer. Clin Pharmacol Drug Dev. 2013;2:11–24.
Hurvitz SA, Dirix L, Kocsis J, Bianchi GV, Lu J, Vinholes J, Guardino E, Song C, Tong B, Ng V, Chu YW, et al. Phase II randomized study of trastuzumab emtansine versus trastuzumab plus docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol. 2013;31:1157–63.
Krop IE, Beeram M, Modi S, Jones SF, Holden SN, Yu W, Girish S, Tibbitts J, Yi JH, Sliwkowski MX, Jacobson F, et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clin Oncol. 2010;28:2698–704.
Krop IE, LoRusso P, Miller KD, Modi S, Yardley D, Rodriguez G, Guardino E, Lu M, Zheng M, Girish S, Amler L, et al. A phase II study of trastuzumab emtansine in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer who were previously treated with trastuzumab, lapatinib, an anthracycline, a taxane, and capecitabine. J Clin Oncol. 2012;30:3234–41.
Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, DY O, Dieras V, Guardino E, Fang L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783–91.
Welslau M, Dieras V, Sohn JH, Hurvitz SA, Lalla D, Fang L, Althaus B, Guardino E, Miles D. Patient-reported outcomes from EMILIA, a randomized phase 3 study of trastuzumab emtansine (T-DM1) versus capecitabine and lapatinib in human epidermal growth factor receptor 2-positive locally advanced or metastatic breast cancer. Cancer. 2014;120:642–51.
Krop IE, Kim SB, Gonzalez-Martin A, LoRusso PM, Ferrero JM, Smitt M, Yu R, Leung AC, Wildiers H. Trastuzumab emtansine versus treatment of physician's choice for pretreated HER2-positive advanced breast cancer (TH3RESA): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:689–99.
Ellis PA, Barrios CH, Eiermann W, Toi M, Im SA, Conte PF, Martin M, Pienkowski T, Pivot XB, Burris HA, Strasak A, et al. Phase III, randomized study of trastuzumab emtansine (T-DM1) ± pertuzumab (P) vs trastuzumab + taxane (HT) for first-line treatment of HER2-positive MBC: primary results from the MARIANNE study. J Clin Oncol. 2015;33:507.
Swain SM, Baselga J, Kim SB, Ro J, Semiglazov V, Campone M, Ciruelos E, Ferrero JM, Schneeweiss A, Heeson S, Clark E, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med. 2015;372:724–34.
Bertelsen V, Stang E. The mysterious ways of ErbB2/HER2 trafficking. Membranes (Basel). 2014;4:424–46.
Barok M, Tanner M, Koninki K, Isola J. Trastuzumab-DM1 causes tumour growth inhibition by mitotic catastrophe in trastuzumab-resistant breast cancer cells in vivo. Breast Cancer Res. 2011;13:R46.
Ritchie M, Tchistiakova L, Scott N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. MAbs. 2013;5:13–21.
Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer. 2010;10:194–204.
Kovtun YV, Audette CA, Mayo MF, Jones GE, Doherty H, Maloney EK, Erickson HK, Sun X, Wilhelm S, Ab O, Lai KC, et al. Antibody-maytansinoid conjugates designed to bypass multidrug resistance. Cancer Res. 2010;70:2528–37.
Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist. 2003;8:411–24.
Aertgeerts K, Skene R, Yano J, Sang BC, Zou H, Snell G, Jennings A, Iwamoto K, Habuka N, Hirokawa A, Ishikawa T, et al. Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J Biol Chem. 2011;286:18756–65.
Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277:46265–72.
Posner I, Engel M, Gazit A, Levitzki A. Kinetics of inhibition by tyrphostins of the tyrosine kinase activity of the epidermal growth factor receptor and analysis by a new computer program. Mol Pharmacol. 1994;45:673–83.
Osherov N, Gazit A, Gilon C, Levitzki A. Selective inhibition of the epidermal growth factor and HER2/neu receptors by tyrphostins. J Biol Chem. 1993;268:11134–42.
Ward WH, Cook PN, Slater AM, Davies DH, Holdgate GA, Green LR. Epidermal growth factor receptor tyrosine kinase. Investigation of catalytic mechanism, structure-based searching and discovery of a potent inhibitor. Biochem Pharmacol. 1994;48:659–66.
Arkin M, Moasser MM. HER-2-directed, small-molecule antagonists. Curr Opin Investig Drugs. 2008;9:1264–76.
Schroeder RL, Stevens CL, Sridhar J. Small molecule tyrosine kinase inhibitors of ErbB2/HER2/Neu in the treatment of aggressive breast cancer. Molecules. 2014;19:15196–212.
Segovia-Mendoza M, Gonzalez-Gonzalez ME, Barrera D, Diaz L, Garcia-Becerra R. Efficacy and mechanism of action of the tyrosine kinase inhibitors gefitinib, lapatinib and neratinib in the treatment of HER2-positive breast cancer: preclinical and clinical evidence. Am J Cancer Res. 2015;5:2531–61.
Chien AJ, Munster PN, Melisko ME, Rugo HS, Park JW, Goga A, Auerback G, Khanafshar E, Ordovas K, Koch KM, Moasser MM. Phase I dose-escalation study of 5-day intermittent oral lapatinib therapy in patients with human epidermal growth factor receptor 2-overexpressing breast cancer. J Clin Oncol. 2014;32:1472–9.
Trowe T, Boukouvala S, Calkins K, Cutler RE Jr, Fong R, Funke R, Gendreau SB, Kim YD, Miller N, Woolfrey JR, Vysotskaia V, et al. EXEL-7647 inhibits mutant forms of ErbB2 associated with lapatinib resistance and neoplastic transformation. Clin Cancer Res. 2008;14:2465–75.
Xia W, Petricoin EF III, Zhao S, Liu L, Osada T, Cheng Q, Wulfkuhle JD, Gwin WR, Yang X, Gallagher RI, Bacus S, et al. An heregulin-EGFR-HER3 autocrine signaling axis can mediate acquired lapatinib resistance in HER2+ breast cancer models. Breast Cancer Res. 2013;15:R85.
Rhee J, Han SW, Cha Y, Ham HS, Kim HP, DY O, Im SA, Park JW, Ro J, Lee KS, Park IH, et al. High serum TGF-alpha predicts poor response to lapatinib and capecitabine in HER2-positive breast cancer. Breast Cancer Res Treat. 2011;125:107–14.
Ishikawa N, Daigo Y, Takano A, Taniwaki M, Kato T, Hayama S, Murakami H, Takeshima Y, Inai K, Nishimura H, Tsuchiya E, et al. Increases of amphiregulin and transforming growth factor-alpha in serum as predictors of poor response to gefitinib among patients with advanced non-small cell lung cancers. Cancer Res. 2005;65:9176–84.
Arvatz G, Weissmann M, Ilan N, Vlodavsky I. Heparanase and cancer progression: new directions, new promises. Hum Vaccin Immunother. 2016;12:2253–6.
Fux L, Ilan N, Sanderson RD, Vlodavsky I. Heparanase: busy at the cell surface. Trends Biochem Sci. 2009;34:511–9.
Cohen-Kaplan V, Doweck I, Naroditsky I, Vlodavsky I, Ilan N. Heparanase augments epidermal growth factor receptor phosphorylation: correlation with head and neck tumor progression. Cancer Res. 2008;68:10077–85.
Zhang L, Ngo JA, Wetzel MD, Marchetti D. Heparanase mediates a novel mechanism in lapatinib-resistant brain metastatic breast cancer. Neoplasia. 2015;17:101–13.
Chen CT, Kim H, Liska D, Gao S, Christensen JG, Weiser MR. MET activation mediates resistance to lapatinib inhibition of HER2-amplified gastric cancer cells. Mol Cancer Ther. 2012;11:660–9.
Paulson AK, Linklater ES, Berghuis BD, App CA, Oostendorp LD, Paulson JE, Pettinga JE, Melnik MK, Vande Woude GF, Graveel CR. MET and ERBB2 are coexpressed in ERBB2+ breast cancer and contribute to innate resistance. Mol Cancer Res. 2013;11:1112–21.
Burchert A, Attar EC, McCloskey P, Fridell YW, Liu ET. Determinants for transformation induced by the Axl receptor tyrosine kinase. Oncogene. 1998;16:3177–87..
Janssen JW, Schulz AS, Steenvoorden AC, Schmidberger M, Strehl S, Ambros PF, Bartram CRA. Novel putative tyrosine kinase receptor with oncogenic potential. Oncogene. 1991;6:2113–20.
O'Bryan JP, Frye RA, Cogswell PC, Neubauer A, Kitch B, Prokop C, Espinosa R III, Le Beau MM, Earp HS, Liu ET. Axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biol. 1991;11:5016–31.
Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, Halsey W, Sathe GM, Martin AM, Gilmer TM. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res. 2009;69:6871–8.
Mahadevan D, Cooke L, Riley C, Swart R, Simons B, Della Croce K, Wisner L, Iorio M, Shakalya K, Garewal H, Nagle R, et al. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene. 2007;26:3909–19.
Hong CC, Lay JD, Huang JS, Cheng AL, Tang JL, Lin MT, Lai GM, Chuang SE. Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett. 2008;268:314–24.
Lay JD, Hong CC, Huang JS, Yang YY, Pao CY, Liu CH, Lai YP, Lai GM, Cheng AL, IJ S, Chuang SE. Sulfasalazine suppresses drug resistance and invasiveness of lung adenocarcinoma cells expressing AXL. Cancer Res. 2007;67:3878–87.
Macleod K, Mullen P, Sewell J, Rabiasz G, Lawrie S, Miller E, Smyth JF, Langdon SP. Altered ErbB receptor signaling and gene expression in cisplatin-resistant ovarian cancer. Cancer Res. 2005;65:6789–800.
Wang L, Zhang Q, Zhang J, Sun S, Guo H, Jia Z, Wang B, Shao Z, Wang Z, Hu X. PI3K pathway activation results in low efficacy of both trastuzumab and lapatinib. BMC Cancer. 2011;11:248.
Xia W, Bacus S, Hegde P, Husain I, Strum J, Liu L, Paulazzo G, Lyass L, Trusk P, Hill J, Harris J, et al. A model of acquired autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proc Natl Acad Sci U S A. 2006;103:7795–800.
Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, Beijersbergen RL, Valero V, Seoane J, Bernards R, Baselga J. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008;68:9221–30.
Koninki K, Barok M, Tanner M, Staff S, Pitkanen J, Hemmila P, Ilvesaro J, Isola J. Multiple molecular mechanisms underlying trastuzumab and lapatinib resistance in JIMT-1 breast cancer cells. Cancer Lett. 2010;294:211–9.
Chakrabarty A, Rexer BN, Wang SE, Cook RS, Engelman JA, Arteaga CL. H1047R phosphatidylinositol 3-kinase mutant enhances HER2-mediated transformation by heregulin production and activation of HER3. Oncogene. 2010;29:5193–203.
Johnston S, Trudeau M, Kaufman B, Boussen H, Blackwell K, LoRusso P, Lombardi DP, Ben Ahmed S, Citrin DL, DeSilvio ML, Harris J, et al. Phase II study of predictive biomarker profiles for response targeting human epidermal growth factor receptor 2 (HER-2) in advanced inflammatory breast cancer with lapatinib monotherapy. J Clin Oncol. 2008;26:1066–72.
Xia W, Husain I, Liu L, Bacus S, Saini S, Spohn J, Pry K, Westlund R, Stein SH, Spector NL. Lapatinib antitumor activity is not dependent upon phosphatase and tensin homologue deleted on chromosome 10 in ErbB2-overexpressing breast cancers. Cancer Res. 2007;67:1170–5.
Jegg AM, Ward TM, Iorns E, Hoe N, Zhou J, Liu X, Singh S, Landgraf R, Pegram MD. PI3K independent activation of mTORC1 as a target in lapatinib-resistant ERBB2+ breast cancer cells. Breast Cancer Res Treat. 2012;136:683–92.
Brady SW, Zhang J, Tsai MH, Yu D. PI3K-independent mTOR activation promotes lapatinib resistance and IAP expression that can be effectively reversed by mTOR and Hsp90 inhibition. Cancer Biol Ther. 2015;16:402–11.
Vazquez-Martin A, Oliveras-Ferraros C, Colomer R, Brunet J, Menendez JA. Low-scale phosphoproteome analyses identify the mTOR effector p70 S6 kinase 1 as a specific biomarker of the dual-HER1/HER2 tyrosine kinase inhibitor lapatinib (Tykerb) in human breast carcinoma cells. Ann Oncol. 2008;19:1097–109.
Formisano L, Nappi L, Rosa R, Marciano R, D'Amato C, D'Amato V, Damiano V, Raimondo L, Iommelli F, Scorziello A, Troncone G, et al. Epidermal growth factor-receptor activation modulates Src-dependent resistance to lapatinib in breast cancer models. Breast Cancer Res. 2014;16:R45.
Rexer BN, Ham AJ, Rinehart C, Hill S, Granja-Ingram Nde M, Gonzalez-Angulo AM, Mills GB, Dave B, Chang JC, Liebler DC, Arteaga CL. Phosphoproteomic mass spectrometry profiling links Src family kinases to escape from HER2 tyrosine kinase inhibition. Oncogene. 2011;30:4163–74.
De Luca A, D'Alessio A, Gallo M, Maiello MR, Bode AM, Normanno N. Src and CXCR4 are involved in the invasiveness of breast cancer cells with acquired resistance to lapatinib. Cell Cycle. 2014;13:148–56.
Nam HJ, Im SA, DY O, Elvin P, Kim HP, Yoon YK, Min A, Song SH, Han SW, Kim TY, Bang YJ. Antitumor activity of saracatinib (AZD0530), a c-Src/Abl kinase inhibitor, alone or in combination with chemotherapeutic agents in gastric cancer. Mol Cancer Ther. 2013;12:16–26.
Hong YS, Kim J, Pectasides E, Fox C, Hong SW, Ma Q, Wong GS, Peng S, Stachler MD, Thorner AR, Van Hummelen P, et al. Src mutation induces acquired lapatinib resistance in ERBB2-amplified human gastroesophageal adenocarcinoma models. PLoS One. 2014;9:e109440.
Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–23.
Wang F, Weaver VM, Petersen OW, Larabell CA, Dedhar S, Briand P, Lupu R, Bissell MJ. Reciprocal interactions between beta1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: a different perspective in epithelial biology. Proc Natl Acad Sci U S A. 1998;95:14821–6.
Huang C, Park CC, Hilsenbeck SG, Ward R, Rimawi MF, Wang YC, Shou J, Bissell MJ, Osborne CK, Schiff R. beta1 integrin mediates an alternative survival pathway in breast cancer cells resistant to lapatinib. Breast Cancer Res. 2011;13:R84.
Li T, Sparano JA. Inhibiting Ras signaling in the therapy of breast cancer. Clin Breast Cancer. 2003;3:405–16. discussion 417-420
Janes PW, Daly RJ, deFazio A, Sutherland RL. Activation of the Ras signalling pathway in human breast cancer cells overexpressing erbB-2. Oncogene. 1994;9:3601–8.
von Lintig FC, Dreilinger AD, Varki NM, Wallace AM, Casteel DE, Boss GR. Ras activation in human breast cancer. Breast Cancer Res Treat. 2000;62:51–62.
Zoppoli G, Moran E, Soncini D, Cea M, Garuti A, Rocco I, Cirmena G, Grillo V, Bagnasco L, Icardi G, Ansaldi F, et al. Ras-induced resistance to lapatinib is overcome by MEK inhibition. Curr Cancer Drug Targets. 2010;10:168–75.
Wang YC, Morrison G, Gillihan R, Guo J, Ward RM, Fu X, Botero MF, Healy NA, Hilsenbeck SG, Phillips GL, Chamness GC, et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2-positive breast cancers--role of estrogen receptor and HER2 reactivation. Breast Cancer Res. 2011;13:R121.
Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 2003;144:4562–74.
Finn RS, Press MF, Dering J, Arbushites M, Koehler M, Oliva C, Williams LS, Di Leo A. Estrogen receptor, progesterone receptor, human epidermal growth factor receptor 2 (HER2), and epidermal growth factor receptor expression and benefit from lapatinib in a randomized trial of paclitaxel with lapatinib or placebo as first-line treatment in HER2-negative or unknown metastatic breast cancer. J Clin Oncol. 2009;27:3908–15.
Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, de Azambuja E, Aura C, Gomez H, Dinh P, Fauria K, Van Dooren V, Aktan G, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet. 2012;379:633–40.
Burris HA 3rd, Hurwitz HI, Dees EC, Dowlati A, Blackwell KL, O'Neil B, Marcom PK, Ellis MJ, Overmoyer B, Jones SF, Harris JL, et al. Phase I safety, pharmacokinetics, and clinical activity study of lapatinib (GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J Clin Oncol. 2005;23:5305–13.
Ruddy K, Mayer E, Partridge A. Patient adherence and persistence with oral anticancer treatment. CA Cancer J Clin. 2009;59:56–66.
Foulon V, Schoffski P, Wolter P. Patient adherence to oral anticancer drugs: an emerging issue in modern oncology. Acta Clin Belg. 2011;66:85–96.
Verbrugghe M, Duprez V, Beeckman D, Grypdonck M, Quaghebeur M, Verschueren C, Verhaeghe S, Van Hecke A. Factors influencing adherence in cancer patients taking oral tyrosine kinase inhibitors: a qualitative study. Cancer Nurs. 2016;39:153–62.
Marin D, Bazeos A, Mahon FX, Eliasson L, Milojkovic D, Bua M, Apperley JF, Szydlo R, Desai R, Kozlowski K, Paliompeis C, et al. Adherence is the critical factor for achieving molecular responses in patients with chronic myeloid leukemia who achieve complete cytogenetic responses on imatinib. J Clin Oncol. 2010;28:2381–8.
Duckett DR, Cameron MD. Metabolism considerations for kinase inhibitors in cancer treatment. Expert Opin Drug Metab Toxicol. 2010;6:1175–93.
van Erp NP, Gelderblom H, Guchelaar HJ. Clinical pharmacokinetics of tyrosine kinase inhibitors. Cancer Treat Rev. 2009;35:692–706.
Scheffler M, Di Gion P, Doroshyenko O, Wolf J, Fuhr U. Clinical pharmacokinetics of tyrosine kinase inhibitors: focus on 4-anilinoquinazolines. Clin Pharmacokinet. 2011;50:371–403.
Thiessen B, Stewart C, Tsao M, Kamel-Reid S, Schaiquevich P, Mason W, Easaw J, Belanger K, Forsyth P, McIntosh L, Eisenhauer E. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother Pharmacol. 2010;65:353–61.
Smith DA, Koch KM, Arya N, Bowen CJ, Herendeen JM, Beelen A. Effects of ketoconazole and carbamazepine on lapatinib pharmacokinetics in healthy subjects. Br J Clin Pharmacol. 2009;67:421–6.
Dai CL, Tiwari AK, Wu CP, Su XD, Wang SR, Liu DG, Ashby CR Jr, Huang Y, Robey RW, Liang YJ, Chen LM, et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68:7905–14.
Bence AK, Anderson EB, Halepota MA, Doukas MA, DeSimone PA, Davis GA, Smith DA, Koch KM, Stead AG, Mangum S, Bowen CJ, et al. Phase I pharmacokinetic studies evaluating single and multiple doses of oral GW572016, a dual EGFR-ErbB2 inhibitor, in healthy subjects. Investig New Drugs. 2005;23:39–49.
Koch KM, Im YH, Kim SB, Urruticoechea Ribate A, Stephenson J, Botbyl J, Cartee L, Holshouser J, Ridgway D. Effects of esomeprazole on the pharmacokinetics of Lapatinib in breast cancer patients. Clin Pharmacol Drug Dev. 2013;2:336–41.
Morris KT, Johnson N, Homer L, Walts DA. Comparison of complementary therapy use between breast cancer patients and patients with other primary tumor sites. Am J Surg. 2000;179:407–11.
Haefeli WE, Carls A. Drug interactions with phytotherapeutics in oncology. Expert Opin Drug Metab Toxicol. 2014;10:359–77.
Widmer N, Bardin C, Chatelut E, Paci A, Beijnen J, Leveque D, Veal G, Astier A. Review of therapeutic drug monitoring of anticancer drugs part two--targeted therapies. Eur J Cancer. 2014;50:2020–36.
Ocana A, Amir E. Irreversible pan-ErbB tyrosine kinase inhibitors and breast cancer: current status and future directions. Cancer Treat Rev. 2009;35:685–91.
Rabindran SK. Antitumor activity of HER-2 inhibitors. Cancer Lett. 2005;227:9–23.
Rabindran SK, Discafani CM, Rosfjord EC, Baxter M, Floyd MB, Golas J, Hallett WA, Johnson BD, Nilakantan R, Overbeek E, Reich MF, et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004;64:3958–65.
Tsou HR, Overbeek-Klumpers EG, Hallett WA, Reich MF, Floyd MB, Johnson BD, Michalak RS, Nilakantan R, Discafani C, Golas J, Rabindran SK, et al. Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity. J Med Chem. 2005;48:1107–31.
Canonici A, Gijsen M, Mullooly M, Bennett R, Bouguern N, Pedersen K, O'Brien NA, Roxanis I, Li JL, Bridge E, Finn R, et al. Neratinib overcomes trastuzumab resistance in HER2 amplified breast cancer. Oncotarget. 2013;4:1592–605.
Burstein HJ, Sun Y, Dirix LY, Jiang Z, Paridaens R, Tan AR, Awada A, Ranade A, Jiao S, Schwartz G, Abbas R, et al. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J Clin Oncol. 2010;28:1301–7.
Martin M, Bonneterre J, Geyer CE Jr, Ito Y, Ro J, Lang I, Kim SB, Germa C, Vermette J, Wang K, Wang K, et al. A phase two randomised trial of neratinib monotherapy versus lapatinib plus capecitabine combination therapy in patients with HER2+ advanced breast cancer. Eur J Cancer. 2013;49:3763–72.
Chan A, Delaloge S, Holmes FA, Moy B, Iwata H, Harvey VJ, Robert NJ, Silovski T, Gokmen E, von Minckwitz G, Ejlertsen B, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17:367–77.
Allen LF, Eiseman IA, Fry DW, Lenehan PF. CI-1033, an irreversible pan-erbB receptor inhibitor and its potential application for the treatment of breast cancer. Semin Oncol. 2003;30:65–78.
Slichenmyer WJ, Elliott WL, Fry DW. CI-1033, a pan-erbB tyrosine kinase inhibitor. Semin Oncol. 2001;28:80–5.
Smaill JB, Gonzales AJ, Spicer JA, Lee H, Reed JE, Sexton K, Althaus IW, Zhu T, Black SL, Blaser A, Denny WA, et al. Tyrosine kinase inhibitors. 20. Optimization of substituted Quinazoline and Pyrido[3,4-d]pyrimidine derivatives as orally active, irreversible inhibitors of the epidermal growth factor receptor family. J Med Chem. 2016;59:8103–24.
Brunner-Kubath C, Shabbir W, Saferding V, Wagner R, Singer CF, Valent P, Berger W, Marian B, Zielinski CC, Grusch M, Grunt TW. The PI3 kinase/mTOR blocker NVP-BEZ235 overrides resistance against irreversible ErbB inhibitors in breast cancer cells. Breast Cancer Res Treat. 2011;129:387–400.
Rixe O, Franco SX, Yardley DA, Johnston SR, Martin M, Arun BK, Letrent SP, Rugo HS. A randomized, phase II, dose-finding study of the pan-ErbB receptor tyrosine-kinase inhibitor CI-1033 in patients with pretreated metastatic breast cancer. Cancer Chemother Pharmacol. 2009;64:1139–48.
Hirsh V. Next-generation covalent irreversible kinase inhibitors in NSCLC: focus on Afatinib. BioDrugs. 2015;29:167–83.
Cortes J, Dieras V, Ro J, Barriere J, Bachelot T, Hurvitz S, Le Rhun E, Espie M, Kim SB, Schneeweiss A, Sohn JH, et al. Afatinib alone or afatinib plus vinorelbine versus investigator's choice of treatment for HER2-positive breast cancer with progressive brain metastases after trastuzumab, lapatinib, or both (LUX-breast 3): a randomised, open-label, multicentre, phase 2 trial. Lancet Oncol. 2015;16:1700–10.
Lin NU, Winer EP, Wheatley D, Carey LA, Houston S, Mendelson D, Munster P, Frakes L, Kelly S, Garcia AA, Cleator S, et al. A phase II study of afatinib (BIBW 2992), an irreversible ErbB family blocker, in patients with HER2-positive metastatic breast cancer progressing after trastuzumab. Breast Cancer Res Treat. 2012;133:1057–65.
Hanusch C, Schneeweiss A, Loibl S, Untch M, Paepke S, Kummel S, Jackisch C, Huober J, Hilfrich J, Gerber B, Eidtmann H, et al. Dual blockade with AFatinib and Trastuzumab as NEoadjuvant treatment for patients with locally advanced or operable breast cancer receiving Taxane-Anthracycline containing chemotherapy-DAFNE (GBG-70). Clin Cancer Res. 2015;21:2924–31.
Rimawi MF, Aleixo SB, Rozas AA, Nunes de Matos Neto J, Caleffi M, Figueira AC, Souza SC, Reiriz AB, Gutierrez C, Arantes H, Uttenreuther-Fischer MM, et al. A neoadjuvant, randomized, open-label phase II trial of afatinib versus trastuzumab versus lapatinib in patients with locally advanced HER2-positive breast cancer. Clin Breast Cancer. 2015;15:101–9.
Harbeck N, Huang CS, Hurvitz S, Yeh DC, Shao Z, Im SA, Jung KH, Shen K, Ro J, Jassem J, Zhang Q, et al. Afatinib plus vinorelbine versus trastuzumab plus vinorelbine in patients with HER2-overexpressing metastatic breast cancer who had progressed on one previous trastuzumab treatment (LUX-breast 1): an open-label, randomised, phase 3 trial. Lancet Oncol. 2016;17:357–66.
Blackwell KL, Burstein HJ, Storniolo AM, Rugo H, Sledge G, Koehler M, Ellis C, Casey M, Vukelja S, Bischoff J, Baselga J, et al. Randomized study of Lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J Clin Oncol. 2010;28:1124–30.
Blackwell KL, Burstein HJ, Storniolo AM, Rugo HS, Sledge G, Aktan G, Ellis C, Florance A, Vukelja S, Bischoff J, Baselga J, et al. Overall survival benefit with lapatinib in combination with trastuzumab for patients with human epidermal growth factor receptor 2-positive metastatic breast cancer: final results from the EGF104900 study. J Clin Oncol. 2012;30:2585–92.
von Minckwitz G, du Bois A, Schmidt M, Maass N, Cufer T, de Jongh FE, Maartense E, Zielinski C, Kaufmann M, Bauer W, Baumann KH, et al. Trastuzumab beyond progression in human epidermal growth factor receptor 2-positive advanced breast cancer: a german breast group 26/breast international group 03-05 study. J Clin Oncol. 2009;27:1999–2006.
von Minckwitz G, Schwedler K, Schmidt M, Barinoff J, Mundhenke C, Cufer T, Maartense E, de Jongh FE, Baumann KH, Bischoff J, Harbeck N, et al. Trastuzumab beyond progression: overall survival analysis of the GBG 26/BIG 3-05 phase III study in HER2-positive breast cancer. Eur J Cancer. 2011;47:2273–81.
Hutchinson L. Targeted therapies: Lapatinib is effective in patients with p95HER2-positive tumors. Nat Rev Clin Oncol. 2010;7:358.
Scaltriti M, Chandarlapaty S, Prudkin L, Aura C, Jimenez J, Angelini PD, Sanchez G, Guzman M, Parra JL, Ellis C, Gagnon R, et al. Clinical benefit of lapatinib-based therapy in patients with human epidermal growth factor receptor 2-positive breast tumors coexpressing the truncated p95HER2 receptor. Clin Cancer Res. 2010;16:2688–95.
Janssen JA, Varewijck AJ. IGF-IR targeted therapy: past, present and future. Front Endocrinol (Lausanne). 2014;5:224.
King ER, Wong KK. Insulin-like growth factor: current concepts and new developments in cancer therapy. Recent Pat Anticancer Drug Discov. 2012;7:14–30.
McKian KP, Haluska P. Cixutumumab. Expert Opin Investig Drugs. 2009;18:1025–33.
Rodon J, DeSantos V, Ferry RJ Jr, Kurzrock R. Early drug development of inhibitors of the insulin-like growth factor-I receptor pathway: lessons from the first clinical trials. Mol Cancer Ther. 2008;7:2575–88.
Vu T, Sliwkowski MX, Claret FX. Personalized drug combinations to overcome trastuzumab resistance in HER2-positive breast cancer. Biochim Biophys Acta. 1846;2014:353–65.
Carboni JM, Wittman M, Yang Z, Lee F, Greer A, Hurlburt W, Hillerman S, Cao C, Cantor GH, Dell-John J, Chen C, et al. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol Cancer Ther. 2009;8:3341–9.
Buck E, Mulvihill M. Small molecule inhibitors of the IGF-1R/IR axis for the treatment of cancer. Expert Opin Investig Drugs. 2011;20:605–21.
Schwartz GK, Dickson MA, LoRusso PM, Sausville EA, Maekawa Y, Watanabe Y, Kashima N, Nakashima D, Akinaga S. Preclinical and first-in-human phase I studies of KW-2450, an oral tyrosine kinase inhibitor with insulin-like growth factor receptor-1/insulin receptor selectivity. Cancer Sci. 2016;107:499–506.
Shah MA, Wainberg ZA, Catenacci DV, Hochster HS, Ford J, Kunz P, Lee FC, Kallender H, Cecchi F, Rabe DC, Keer H, et al. Phase II study evaluating 2 dosing schedules of oral foretinib (GSK1363089), cMET/VEGFR2 inhibitor, in patients with metastatic gastric cancer. PLoS One. 2013;8:e54014.
Shapiro GI, McCallum S, Adams LM, Sherman L, Weller S, Swann S, Keer H, Miles D, Muller T, Lorusso P. A phase 1 dose-escalation study of the safety and pharmacokinetics of once-daily oral foretinib, a multi-kinase inhibitor, in patients with solid tumors. Investig New Drugs. 2013;31:742–50.
Seiwert T, Sarantopoulos J, Kallender H, McCallum S, Keer HN, Blumenschein G Jr, Phase II. Trial of single-agent foretinib (GSK1363089) in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. Investig New Drugs. 2013;31:417–24.
Johnston SR, Gomez H, Stemmer SM, Richie M, Durante M, Pandite L, Goodman V, Slamon D. A randomized and open-label trial evaluating the addition of pazopanib to lapatinib as first-line therapy in patients with HER2-positive advanced breast cancer. Breast Cancer Res Treat. 2013;137:755–66.
Rugo HS, Chien AJ, Franco SX, Stopeck AT, Glencer A, Lahiri S, Arbushites MC, Scott J, Park JW, Hudis C, Nulsen B, et al. A phase II study of lapatinib and bevacizumab as treatment for HER2-overexpressing metastatic breast cancer. Breast Cancer Res Treat. 2012;134:13–20.
Gianni L, Romieu GH, Lichinitser M, Serrano SV, Mansutti M, Pivot X, Mariani P, Andre F, Chan A, Lipatov O, Chan S, et al. AVEREL: a randomized phase III trial evaluating bevacizumab in combination with docetaxel and trastuzumab as first-line therapy for HER2-positive locally recurrent/metastatic breast cancer. J Clin Oncol. 2013;31:1719–25.
Garcia-Garcia C, Ibrahim YH, Serra V, Calvo MT, Guzman M, Grueso J, Aura C, Perez J, Jessen K, Liu Y, Rommel C, et al. Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to anti-HER2 therapy. Clin Cancer Res. 2012;18:2603–12.
Miller TW, Forbes JT, Shah C, Wyatt SK, Manning HC, Olivares MG, Sanchez V, Dugger TC, de Matos Granja N, Narasanna A, Cook RS, et al. Inhibition of mammalian target of rapamycin is required for optimal antitumor effect of HER2 inhibitors against HER2-overexpressing cancer cells. Clin Cancer Res. 2009;15:7266–76.
O'Brien NA, McDonald K, Tong L, von Euw E, Kalous O, Conklin D, Hurvitz SA, di Tomaso E, Schnell C, Linnartz R, Finn RS, et al. Targeting PI3K/mTOR overcomes resistance to HER2-targeted therapy independent of feedback activation of AKT. Clin Cancer Res. 2014;20:3507–20.
Rosen N, She QBAKT. Cancer--is it all mTOR? Cancer Cell. 2006;10:254–6.
Gingras AC, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001;15:807–26.
Huang K, Fingar DC. Growing knowledge of the mTOR signaling network. Semin Cell Dev Biol. 2014;36:79–90.
Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–8.
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101.
Qian J, Chen Y, Meng T, Ma L, Meng L, Wang X, Yu T, Zask A, Shen J, Yu K. Molecular regulation of apoptotic machinery and lipid metabolism by mTORC1/mTORC2 dual inhibitors in preclinical models of HER2+/PIK3CAmut breast cancer. Oncotarget. 2016;7:67071–86.
Andre F, O'Regan R, Ozguroglu M, Toi M, Xu B, Jerusalem G, Masuda N, Wilks S, Arena F, Isaacs C, Yap YS, et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014;15:580–91.
Hurvitz SA, Andre F, Jiang Z, Shao Z, Mano MS, Neciosup SP, Tseng LM, Zhang Q, Shen K, Liu D, Dreosti LM, et al. Combination of everolimus with trastuzumab plus paclitaxel as first-line treatment for patients with HER2-positive advanced breast cancer (BOLERO-1): a phase 3, randomised, double-blind, multicentre trial. Lancet Oncol. 2015;16:816–29.
Bendell JC, Kelley RK, Shih KC, Grabowsky JA, Bergsland E, Jones S, Martin T, Infante JR, Mischel PS, Matsutani T, Xu S, et al. A phase I dose-escalation study to assess safety, tolerability, pharmacokinetics, and preliminary efficacy of the dual mTORC1/mTORC2 kinase inhibitor CC-223 in patients with advanced solid tumors or multiple myeloma. Cancer. 2015;121:3481–90.
Chen SM, Guo CL, Shi JJ, Xu YC, Chen Y, Shen YY, Su Y, Ding J, Meng LH. HSP90 inhibitor AUY922 abrogates up-regulation of RTKs by mTOR inhibitor AZD8055 and potentiates its antiproliferative activity in human breast cancer. Int J Cancer. 2014;135:2462–74.
O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8.
Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, Poulikakos PI, Scaltriti M, Moskatel E, Baselga J, Guichard S, Rosen N. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011;1:248–59.
Janne PA, Cohen RB, Laird AD, Mace S, Engelman JA, Ruiz-Soto R, Rockich K, Xu J, Shapiro GI, Martinez P, Felip E. Phase I safety and pharmacokinetic study of the PI3K/mTOR inhibitor SAR245409 (XL765) in combination with erlotinib in patients with advanced solid tumors. J Thorac Oncol. 2014;9:316–23.
Shapiro GI, Bell-McGuinn KM, Molina JR, Bendell J, Spicer J, Kwak EL, Pandya SS, Millham R, Borzillo G, Pierce KJ, Han L, et al. First-in-human study of PF-05212384 (PKI-587), a small-molecule, intravenous, dual inhibitor of PI3K and mTOR in patients with advanced cancer. Clin Cancer Res. 2015;21:1888–95.
Hyman DM, Snyder AE, Carvajal RD, Gerecitano JF, Voss MH, Ho AL, Konner J, Winkelmann JL, Stasi MA, Monson KR, Iasonos A, et al. Parallel phase Ib studies of two schedules of buparlisib (BKM120) plus carboplatin and paclitaxel (q21 days or q28 days) for patients with advanced solid tumors. Cancer Chemother Pharmacol. 2015;75:747–55.
Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71.
Gijsen M, King P, Perera T, Parker PJ, Harris AL, Larijani B, Kong A. HER2 phosphorylation is maintained by a PKB negative feedback loop in response to anti-HER2 herceptin in breast cancer. PLoS Biol. 2010;8:e1000563.
Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, Kotani H. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9:1956–67.
Sangai T, Akcakanat A, Chen H, Tarco E, Wu Y, Do KA, Miller TW, Arteaga CL, Mills GB, Gonzalez-Angulo AM, Meric-Bernstam F. Biomarkers of response to Akt inhibitor MK-2206 in breast cancer. Clin Cancer Res. 2012;18:5816–28.
Hudis C, Swanton C, Janjigian YY, Lee R, Sutherland S, Lehman R, Chandarlapaty S, Hamilton N, Gajria D, Knowles J, Shah J, et al. A phase 1 study evaluating the combination of an allosteric AKT inhibitor (MK-2206) and trastuzumab in patients with HER2-positive solid tumors. Breast Cancer Res. 2013;15:R110.
Han HS, Swanton C, Janjigian YY, Sutherland SC, Chandarlapaty S, Lehman R, Hamilton N, Knowles J, Lee R, Yan L, Sullivan D, et al. A phase I study of the AKT inhibitor (MK-2206) with concurrent trastuzumab and lapatinib in patients with HER2-positive solid tumors. J Clin Oncol. 2011;29:3028.
Burstein HJ, Cirrincione CT, Barry WT, Chew HK, Tolaney SM, Lake DE, Ma C, Blackwell KL, Winer EP, Hudis CA. Endocrine therapy with or without inhibition of epidermal growth factor receptor and human epidermal growth factor receptor 2: a randomized, double-blind, placebo-controlled phase III trial of fulvestrant with or without lapatinib for postmenopausal women with hormone receptor-positive advanced breast cancer-CALGB 40302 (alliance). J Clin Oncol. 2014;32:3959–66.
Johnston S, Pippen J Jr, Pivot X, Lichinitser M, Sadeghi S, Dieras V, Gomez HL, Romieu G, Manikhas A, Kennedy MJ, Press MF, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol. 2009;27:5538–46.
Riemsma R, Forbes CA, Amonkar MM, Lykopoulos K, Diaz JR, Kleijnen J, Rea DW. Systematic review of lapatinib in combination with letrozole compared with other first-line treatments for hormone receptor positive(HR+) and HER2+ advanced or metastatic breast cancer(MBC). Curr Med Res Opin. 2012;28:1263–79.
Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–45.
Burrell RA, Swanton C. Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol Oncol. 2014;8:1095–111.
Scott GK, Robles R, Park JW, Montgomery PA, Daniel J, Holmes WE, Lee J, Keller GA, Li WL, Fendly BM, et al. A truncated intracellular HER2/neu receptor produced by alternative RNA processing affects growth of human carcinoma cells. Mol Cell Biol. 1993;13:2247–57.
Bendell JC, Kurkjian C, Infante JR, Bauer TM, Burris HA III, Greco FA, Shih KC, Thompson DS, Lane CM, Finney LH, Jones SF. A phase 1 study of the sachet formulation of the oral dual PI3K/mTOR inhibitor BEZ235 given twice daily (BID) in patients with advanced solid tumors. Investig New Drugs. 2015;33:463–71.
Christenson JL, Denny EC, Kane SE. T-Darpp overexpression in HER2-positive breast cancer confers a survival advantage in lapatinib. Oncotarget. 2015;6:33134–45.
Acknowledgements
J.A.M. and S.E.K. are supported by a grant from the National Institutes of Health (GM105898).
No Conflict Statement
“No potential conflicts of interest were disclosed.”
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter

Cite this chapter
Theile, D., Lenz, G., Momand, J.A., Kane, S.E. (2017). Resistance to HER2-Targeted Therapy. In: Prosperi, J. (eds) Resistance to Targeted Therapies in Breast Cancer. Resistance to Targeted Anti-Cancer Therapeutics, vol 16. Springer, Cham. https://doi.org/10.1007/978-3-319-70142-4_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-70142-4_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-70141-7
Online ISBN: 978-3-319-70142-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)