
Abstract
Osteoclasts are monocyte/macrophage arising cells with the classical function of bone resorption, thus fulfilling the bone remodelling process in concert with osteoblasts. The correct balance between osteogenic functions and osteoclast activity is mandatory to prevent skeletal diseases. While an exacerbated bone resorption is associated with bone loss, eventually leading to osteoporosis, the lack of osteoclast activity is responsible for osteopetrosis, a rare genetic disorder characterised by increased bone density and a wide heterogeneity in terms of severity, ranging from asymptomatic to fatal in infancy. Besides this well-established role in bone resorption, new functions have been recently attributed to the osteoclast. Indeed there is a reciprocal crosstalk between osteoclasts and osteoblasts which influence each other, in case of osteoclasts by releasing factors from the resorbing matrix and by secreting the so-called clastokines. Another recently discovered function of osteoclasts is haematopoiesis regulation. This draws to the obvious consequence that any osteoclast dysfunction would not cause exclusively a bone phenotype. As for other cell types, the knowledge of osteoclast biology has benefited from the study of skeletal diseases in which their formation and function are compromised. Furthermore, well-established methods are available to perform osteoclast primary cultures, and the identification of macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor ĸB ligand (RANKL) as pro-osteoclastogenic factors fostered their employment. Therefore, nowadays the preferential way to obtain purified differentiated osteoclasts is to isolate osteoclast precursors from the bone marrow or peripheral blood mononuclear cells and treat with the above-mentioned pro-osteoclastogenic cytokines.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Osteoclasts are monocyte/macrophage arising cells with the classical function of bone resorption, thus fulfilling the bone remodelling process in concert with osteoblasts. The correct balance between osteogenic functions and osteoclast activity is mandatory to prevent skeletal diseases. While an exacerbated bone resorption is associated with bone loss, eventually leading to osteoporosis, the lack of osteoclast activity is responsible for osteopetrosis, a rare genetic disorder characterised by increased bone density and a wide heterogeneity in terms of severity, ranging from asymptomatic to fatal in infancy. Besides this well-established role in bone resorption, new functions have been recently attributed to the osteoclast. Indeed there is a reciprocal crosstalk between osteoclasts and osteoblasts which influence each other, in case of osteoclasts by releasing factors from the resorbing matrix and by secreting the so-called clastokines. Another recently discovered function of osteoclasts is haematopoiesis regulation. This draws to the obvious consequence that any osteoclast dysfunction would not cause exclusively a bone phenotype. As for other cell types, the knowledge of osteoclast biology has benefited from the study of skeletal diseases in which their formation and function are compromised. Furthermore, well-established methods are available to perform osteoclast primary cultures, and the identification of macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor ĸB ligand (RANKL) as pro-osteoclastogenic factors fostered their employment. Therefore, nowadays the preferential way to obtain purified differentiated osteoclasts is to isolate osteoclast precursors from the bone marrow or peripheral blood mononuclear cells and treat with the above-mentioned pro-osteoclastogenic cytokines.
3.1 Osteoclast Biology
Osteoclasts are classically described as the cells of the bone tissue devoted to destroy the mineralised matrix, thus accomplishing an apparently damaging function that actually is crucial for the correct homeostasis of this hard tissue [1]. In fact, bone resorption is a necessary step that, when perfectly balanced by the osteogenic function of osteoblasts, fulfils the bone remodelling process, which ensures the maintenance of a correct bone mass throughout the life of each individual, in terms of both quality and quantity. When this delicate osteoblast-osteoclast coupling is unbalanced, it causes bone diseases.
As for many other cell types of the body, the biology of osteoclasts has benefited from the study of skeletal diseases in which their formation and function are compromised. The classical example is osteopetrosis (also known as marble bone disease), a rare genetic disorder characterised by increased bone density accompanied by a wide range of complications, such as bone marrow failure, compressive neuropathies, hypocalcaemia and fractures, most of them resulting from the lack of bone resorption [2].
3.1.1 Osteoclastogenesis
One of the first experiments suggesting the actual origin of osteoclasts dates back to the late 1970s, when it was shown that bone resorption could be restored in osteopetrotic mice by bone marrow transplantation or by parabiosis, thus suggesting a haematopoietic origin and a circulating ability of osteoclast precursors [3]. These results came after other studies that, in contrast, had hypothesised a common origin for osteoblasts and osteoclasts [4]. Therefore, as stated by Chambers in his recent review [5], «the osteoclasts are not really bone cells, but blood-borne immigrants into bone».
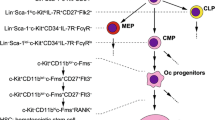
By now, it is known that osteoclasts arise from the monocyte/macrophage cell line through a step-by-step process requiring the sequential activation of specific pathways (◘ Fig. 3.1). First, there is the switch-on of the transcription factor PU.1 [6], which drives the positive regulation of the receptor of the macrophage colony-stimulating factor (M-CSF), c-fms, expressed by the haematopoietic stem cells (HSCs). This event eventually leads to cell commitment towards a common progenitor for macrophage and osteoclasts, belonging to the granulocyte macrophage colony-forming unit (CFU-GM) lineage [7, 8]. M-CSF is necessary for proliferation and survival of these macrophage/osteoclast progenitors. Moreover, it promotes the expression of the receptor activator of nuclear factor κB (RANK) [9]. This is a very crucial step due to the fundamental role of the RANK/RANKL/osteoprotegerin (OPG) triad in osteoclastogenesis [10,11,12,13]. Moreover, the appearance of RANK also marks the transition from the CFU-GM cells to a committed osteoclast precursor [14]. The pro-osteoclastogenic ligand of RANK (RANKL) is produced by lymphocytes, stromal cells, osteoblasts and osteocytes, preferentially as a transmembrane cytokine, requiring a cell-cell contact, and, in lower quantities, as a soluble factor released through the proteolytic cleavage of the active ectodomain. In both circumstances, RANKL primes intracellular pathways in the osteoclast precursors that definitely promote their full differentiation (◘ Fig. 3.1).

Osteoclast differentiation . Schematic representation of the multistep process of osteoclast differentiation. For each step, the genes crucial for the process are reported (boxes). HSC haematopoietic stem cells, GM-CFU granulocyte/monocyte colony-forming unit, OCL osteoclast, M-CSF macrophage colony-stimulating factor, OPG osteoprotegerin, RANK receptor activator of nuclear factor κB, RANKL RANK ligand
As mentioned above, another player in the regulation of osteoclastogenesis is OPG, a secreted glycoprotein belonging to the TNF receptor superfamily [10]. It shares the same extracellular domain of RANK, which allows binding of RANKL, thus preventing its interaction with RANK (◘ Fig. 3.1). It is therefore described as a decoy receptor that negatively affects osteoclastogenesis [10].
Going deeply inside this pathway, RANKL binding to RANK induces the subsequent interaction of the cytoplasmic tail of RANK with the TNF receptor-associated factor (TRAF) 6, which in turn activates the transcription factor nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB). This is a dimeric transcription factor pivotal for osteoclastogenesis, since the double knockout of its subunits prevents osteoclast formation [15]. NF-κB in turn upregulates the nuclear factor of activated T cells and cytoplasmic, calcineurin-dependent (NFATc) 1, which undergoes auto-amplification. The cooperation among NF-kB, NFATc1, activator protein 1 (AP1), PU.1 and microphthalmia-associated transcription factor (MITF) finally promotes the transcription of specific downstream genes necessary for osteoclast differentiation and function [14]. These include tartrate-resistant acid phosphatase (TRAcP), cathepsin K, matrix metalloprotease 9 (MMP-9), calcitonin receptor (CTR) and dendritic cell-specific transmembrane protein (DC-STAMP), the latter pivotal for fusion of preosteoclasts into multinucleated cells [16]. Although the discovery of the RANKL/RANK pathway represents a milestone in osteoclastogenesis, this also requires the involvement of two immune co-receptors displaying the classical immunoreceptor tyrosine-based activation motif (ITAM): Fc receptor common γ signalling chain (FcRγ) and DNAX-activating protein of 12 kDa (DAP12). These co-receptors interact with osteoclast-associated receptor (OSCAR) and trigger receptor expressed on myeloid cells 2 (TREM2), with a resulting activation of the phospholipase Cγ (PLCγ) eventually leading to intracellular Ca2+ oscillations that mediate calcineurin-dependent activation of NFATc1 [17].
3.1.2 Osteoclast Functions
Apart from the well-known activity of bone resorption, the picture of osteoclast duties has changed over the years, thus delineating a new profile including unexpected functions for this very versatile cell, as we will describe in the next paragraphs.
3.1.2.1 Osteoclast Bone Resorption
The machinery of bone resorption is now well known and requires mature and polarised multinucleated cells, firmly adhering to the bone surface in order to isolate the underlying matrix that will be digested. A mature osteoclast is a polarised cell with plasma membrane domains associated with specific functions. The specialised domain facing the bone matrix, characterised by extensive folding of the plasma membrane, is named «ruffled border». The «sealing membrane», a circular outer domain-containing adhesion structures, is crucial for the tight sealing of the bone area to be resorbed [1]. These adhesions are called podosomes [18] and are constituted by dynamic actin microfilaments, actin-binding proteins and signalling molecules, which move to the periphery of the osteoclast forming a podosomal belt [19]. A further step of cytoskeletal rearrangement before starting resorption is the gathering of podosomes in hooplike structures named actin rings [19]. Finally, the tight sealing is guaranteed by the integrin receptors, mainly αVβ3 and, to a lesser extent, α2β1 and αVβ5, which ensure the tight anchorage of microfilaments with the extracellular matrix.
The portion of the osteoclast membrane facing the vascular compartment represents the basolateral domain [20], which again participates to the bone resorption function since it is rich of molecules involved in ion transport, and in the response to extracellular stimuli. Just opposite to the ruffled border, the basolateral membrane displays the functional secretory domain [21] that contributes to the release of the bone degradation products into the circulation, through intense vesicular trafficking and transcytosis processes [22].
Bone resorption is a step-by-step process (◘ Fig. 3.2). After adhesion, osteoclasts dissolve first the inorganic components of the bone matrix. To this aim, the carbonic anhydrase II (CAII) accelerates the hydration of carbonic anhydride (CO2) into carbonic acid (H2CO3), which spontaneously dissociates in bicarbonate (HCO3 −) and proton (H+) ions. The latter are actively transported in the extracellular microenvironment underneath the cells, called resorption lacuna, by means of a specialised vacuolar-type proton (H+)-ATPase located in the ruffled border, while the HCO3 − is exchanged with chloride (Cl−) through the HCO3−/Cl− anion exchanger 2 (AE2) [23, 24]. The Cl− ions are then moved in the resorption lacuna by a 2Cl−/1H+ antiporter, and the result is the presence of hydrochloric acid (HCl) in the lacuna. This acidic microenvironment dissolves the hydroxyapatite, exposing the organic bone matrix, which can now be digested by proteolytic enzymes, including cathepsin K [25], released by lysosomal exocytosis. Finally, debris deriving from the digested matrix is removed by the osteoclast through the functional secretory domain by transcytosis [26].

Osteoclast bone resorption . Schematic diagram showing the molecular machinery of bone resorption
The mechanism of bone resorption has recently been further enriched by other molecules (◘ Fig. 3.2). Pleckstrin homology domain-containing family M (with RUN domain) member 1 (PLEKHM1) [27] is likely involved in vesicular trafficking, while osteopetrosis-associated transmembrane protein 1 (OSTM1) [28] represents the β subunit of the 2Cl−/1H+ antiporter, ensuring its correct placing in the lysosomal and ruffled border membranes.
3.1.2.2 Osteoclast Regulation of Osteoblasts
It is well known that a close crosstalk between osteoclasts and osteoblasts is crucial to maintain a correct balance between resorption and formation in the bone remodelling process. Although the paracrine regulation of osteoclasts by osteogenic cells is well described, the reciprocal regulation has become apparent only recently.
Definitely, osteoclasts concur to regulate osteoblast formation and recruitment at the sites of bone remodelling through the release of factors stored in the bone matrix, such as transforming growth factor (TGF) β, insulin-like growth factor 1 (IGF1) and bone morphogenetic proteins (BMPs) [29, 30], which recruit and activate osteoblasts in the resorbed area. In addition, what emerged recently is that osteoclasts directly regulate osteoblast differentiation by secreting coupling factors, collectively called clastokines [31,32,33]. This was previously suggested by the observation that transgenic mice in which osteoclast formation is severely affected present with impaired osteoblast function and decreased bone mineralisation [34, 35]. Conversely, in the osteoclast-rich osteopetrosis models, bone formation rate is not affected or is even increased [36, 37].
One of the first clastokines identified so far is sphingosine 1-phosphate (S1P), which was found to induce in vitro osteoblast differentiation [38, 39]. Consistently, in vivo studies performed with myeloid-specific cathepsin K knockout mice showed that their osteoclasts had increased levels of S1P with a consequent increase of osteoblast number [31].
TRAcP is another evoked clastokine, which likely promotes osteoblast differentiation. Indeed, TRAcP-overexpressing mice have an increased bone formation rate [40]. Similar effects were observed with the collagen triple repeat containing 1 (CTHRC1) [41, 42] and the complement factor 3a (C3a), the latter recently identified in osteoclast-conditioned medium [43]. Interestingly, it has been observed that while CTHRC1 deletion in osteoblasts does not induce a bone phenotype, its conditional knockout in osteoclasts resulted in reduced bone mass and bone formation rate [44].
3.1.2.3 The Osteoclast Niche: Regulation of Haematopoietic Stem Cells
Another recently discovered function of osteoclasts is the regulation of haematopoiesis, which highlights the high versatility of this cell. An indirect role of HSC regulation for the osteoclast has been ascertained by the means of the MMP-9 and cathepsin K enzymes released during resorption, which regulate the activation of some cytokines crucial for HSC homeostasis. In particular, cathepsin K cleaves the CXC chemokine ligand 12 (CXCL12), responsible for the anchorage of the HSC to the niche, causing the mobilisation of immature haematopoietic progenitor cells [45]. Likewise, MMP-9 allows the release of soluble Kit-ligand (sKit-L), thus promoting the transfer of HSC from the quiescent to proliferative niche. Consistently, in MMP-9 knockout mice, both sKit-L release and HSC motility are impaired, resulting in the failure of haematopoietic recovery [46]. Interestingly, it has been observed that increased levels of local calcium, which could derive from the osteoclast bone-resorbing activity, promote HSC engraftment at the endosteal niche [47].
Other clues suggesting that osteoclasts could regulate HSC rely on the evidence that some treatments affecting osteoclasts also influence HSC homeostasis and vice versa. As an example, bisphosphonates augmented peripheral HSC numbers after their mobilisation with granulocyte colony-stimulating factor (G-CSF) [48], while Sim et al. showed that alendronate increased the long-term engraftment potential and stress resistance of HSCs [49]. Strontium ranelate, which also inhibits osteoclast function, delayed the recovery of HSC after bone marrow transplantation [50]. Likewise, bone marrow HSCs are increased after prostaglandin E2 (PGE2) administration [51]. Finally, mobilisation of HSC after G-CSF treatment is further increased in mice carrying osteopetrosis due to mutation of genes involved in osteoclast differentiation, including M-CSF, c-Fos and RANKL, while it is reduced in osteoprotegerin (OPG) knockout mice [52], characterised by high numbers of osteoclasts.
3.2 Osteoclast Deregulation and Related Pathologies
The picture of osteoclast physiology has undoubtedly benefited from the study of diseases in which their formation and function are deregulated. In fact, the correct balance between osteogenic functions and osteoclast activity is mandatory to preserve the bone and prevent skeletal diseases. Herein we will describe three main pathologies due to the failure of osteoblast-osteoclast coupling.
3.2.1 Osteoporosis
This is a systemic and progressive bone disease characterised by a decrease in bone mass and density, eventually leading to a higher risk of fracture [53, 54]. It has been estimated that approximately 30% of postmenopausal women develop osteoporosis in the United States and in Europe, and at least 40% of these women and 15–30% of men will experience one or more fractures in their remaining lifetime [55, 56]. Furthermore, increase of life expectancy worldwide will be responsible for a steadily increase in the incidence of this disease in the years to come.
The term «primary» osteoporosis refers to a condition related to elderly people and is further classified as type I (i.e. postmenopausal) and type II (age related) [57]. This condition is generally due to an exacerbated osteoclast activity that cannot be compensated by a suitable deposition of new bone by osteoblasts. As a matter of fact, age-related oestrogen and androgen withdrawal is the main guilty of bone mass defaillance, since these hormones physiologically act on two fronts: promoting osteoblast survival and function to one side and restraining bone resorption by favouring osteoclast apoptosis to the other [58, 59]. Oestrogens also reduce osteoblast production of pro-osteoclastogenic cytokines, such as tumour necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6 and RANKL, and increase the secretion of OPG [60, 61].
Secondary osteoporosis includes a broad range of osteoporotic features arising from a number of chronic diseases with the onset at any ages [62]. Indeed, bone mass loss could be secondary to the four following disease conditions:
-
Endocrine diseases, such as hypogonadism and, to a lesser extent, hyperthyroidism. Glucocorticoid treatment, which is often indicated in the anti-inflammatory therapies, is also responsible for bone loss [63].
-
Environmental and lifestyle factors, which include sedentary life, alcohol and use of drugs [64].
-
Chronic inflammatory diseases, such as the rheumatoid arthritis [65].
-
Reduced mobility as consequence of cerebrovascular accident, spinal cord injury and weightlessness, the latter condition experienced by astronauts living for months at 0 gravity [66, 67].
According to the knowledge of the onset of osteoporosis, animal models have been developed in order to experimentally mimic this disease. With regard to the oestrogen withdrawal-induced osteoporosis, the best model employed is the ovariectomy of adult (age 4–8 weeks old) female mice and rats [68]. Ovariectomized (OVX) animals show a dramatic decrease of trabecular bone mass along with an increase of the osteoclast numbers and of the serum levels of the bone resorption marker carboxy-terminal collagen cross-links (CTX) [69].
A successful in vivo model of secondary osteoporosis is the hindlimb suspension [70]. This model mimics the bone loss induced by mechanical unloading. Mice or rats are subjected to hindlimb suspension by means of their tail, which is hanged to a swivel apparatus (approximately 30° angle), thus allowing animals to move freely into the cage using their forelimbs and to readily access food and water. After 21 days of suspension, animals present a decrease in their bone volume, due to an increase of osteoclast numbers [71].
Finally, another model useful to mimic disuse osteoporosis is the botulin toxin A (Botox) treatment, which consists in the injection of Botox (2.0 unit/100 g) into the right quadriceps and the posterior compartment of the right calf (targeting gastrocnemius, plantaris and soleus) [72]. Therefore, this treatment induces a transient and local paralysis, eventually leading to hindlimb bone loss, which becomes overt after 21 from treatment [71, 72].
3.2.2 Bone Metastases
Bone metastases represent the fatal destiny of several oncologic patients, especially those affected by breast and prostate carcinomas, in which the incidence of relapse in bone can reach 70 and 90%, respectively [73]. Once bone metastases come up, the chance of survival dramatically drops, and the quality of life deteriorates, eventually leading to a severe morbidity characterised by pain, fractures, nerve compression and, not least, hypercalcemia, due to exacerbated bone resorption [74]. From the clinical and radiographic points of view, bone metastases can be classified into (i) osteosclerotic, in which there is an abnormal deposition of a woven bone, very poor in quality; (ii) osteolytic, due to prominent bone resorption; and (iii) mixed, in which both features coexist in the same metastatic site [75]. Osteolytic lesions are most frequently observed in breast cancer patients, and as suggested by the name, bone erosion is extensive, allowing the tumour cells to create a physical space into a hard tissue, where they can survive and proliferate [75]. Therefore, this pathological condition typically evokes an exacerbated osteoclast activity. As a matter of fact, there is a general consensus about the fact that tumour cells are not able per se to resorb the bone matrix, while they can produce factors that directly and/or indirectly stimulate osteoclast formation and activity, thus incepting the so-called vicious cycle [75]. Several studies tried to picture this complex and tight crosstalk between osteoclasts and cancer cells, paving the way for the identification of new therapeutic strategies able to affect both cell types [76, 77]. Most of these studies relied on the possibility to reproduce the bone metastatic disease in mouse models, through the intracardiac injection of osteotropic tumour cells. This technique, developed by Arguello [78] and implemented by Yoneda [79], implies the injection of tumour cells that have a specific propensity to colonise the bone, into the left ventricle of 4-week-old female mice, the latter being immunocompromised if the tumour cells are of human origin. This allows cells to spread in the systemic circulation and colonise the bone. Generally, after 3–4 weeks from cell inoculation, it is possible to appreciate the presence of osteolytic lesions in the hindlimbs of mice by X-ray analysis [80]. Further processing of the hindlimbs for histochemical and histomorphometric analysis allows to determine other features of the bone metastases, which include the increase of osteoclast number and surface over the bone surface by histomorphometric analysis, and the worsening of the tumour burden, by histological staining with haematoxylin and eosin or by immunohistochemistry of specific tumour markers [80].
Among the factors involved in the fuelling of the vicious circle, the parathyroid hormone-related protein (PTHrP) was one of the first spotted protagonists, proven to exert a key role in the development of bone metastases [81]. Moreover, its production by tumour cells is further stimulated by TGFβ [82]. In turn, PTHrP induces osteoblasts and stromal cells to produce RANKL, thus promoting osteoclastogenesis [83].
Other pro-osteolytic factors produced by tumour cells in the bone microenvironment are IL-6, IL-8 and IL-11 [84, 85], cyclooxygenase-2 (COX-2) [86], hypoxia inducible factor (HIF)-1α [87] and TNF-α, all promoting osteoclast formation and resorption. Moreover, a recent study from Sethi et al. [88] demonstrated that tumour-derived Jagged promotes osteolytic bone metastases by the activation of the NOTCH pathway in osteoclasts [88].
What further consolidates the vicious circle is that osteoclasts, while destroying the bone matrix, release and activate several growth factors therein stored, such as TGFβ, BMPs, IGF-1, VEGF and PDGF, which support tumour cell survival and growth. Therefore, it is conceivable that, in order to fight the vicious circle, it is necessary to act on two fronts: (i) inhibit osteoclast activity by antiresorptive therapy and (ii) block local tumour growth, thus counteracting this dangerous synergy.
3.2.3 Osteopetrosis
This pathology features the other side of the coin, that is, when osteoclasts do not work at all. In fact, osteopetrosis (i.e. marble bone disease) is an onomatopoeic term to describe a rare genetic disorder characterised by increased bone density at radiography, now known to be due to the impairment of osteoclast function [89]. The first description of the clinical features of osteopetrosis came from Albers-Schönberg in 1904, who also gave the name to this pathology [90].
Osteopetrosis is a very heterogeneous disease in terms of severity, ranging from asymptomatic to fatal in infancy. Four forms of osteopetrosis are currently classified: autosomal dominant, autosomal recessive, intermediate autosomal recessive and X-linked osteopetrosis [2]. The hallmarks for all these variants are the increase of bone mass, eventually leading to frequent fractures due to a poor quality of the bone, and reduced skull cavities, nerve foramina and bone marrow space. Extra skeletal symptoms are also present, such as anaemia, pancytopenia and hepatosplenomegaly, all due to bone marrow failure [89]. The autosomal recessive osteopetrosis is generally the most severe form, with an incidence of up to 1 in 250,000 births. It is also known as malignant infantile osteopetrosis , due to the lethal outcome and the early onset in the first year of life. It is characterised by dense and fragile bones, deformities, short stature, deafness and blindness, bone marrow failure and impaired immune function, with sepsis and secondary infections and, in some cases, mental retardation due to primary neurodegeneration [91].
Osteopetrosis is a typical osteoclast failure disease, and depending on whether it is due to a defect in osteoclast formation or in osteoclast function, it is classified as osteoclast-rich or osteoclast-poor subtypes. The former is the most common form. In this case, osteoclasts are generated normally, but they do not degrade bone due to loss-of-function mutations in genes encoding for key factors responsible for bone resorption, such as the TCIRG1, which encodes the a3 subunit of the H+-ATPase pump, accounting for the 50% of autosomal recessive cases [92, 93]. Mutations in the CLCN7 and the CA II genes, coding for the 2Cl−/1H+ antiporter and the carbonic anhydrase type II, respectively, are also implicated in this disease [94, 95]. Osteopetrosis due to CAII mutations is characterised also by renal tubular acidosis and has an intermediate severity [95]. Loss-of-function mutations of the CLCN7 gene cause a severe autosomal recessive form characterised by lysosomal storage diseases often leading to primary neuropathy [94], while heterozygous missense mutations cause autosomal dominant osteopetrosis, characterised by a milder course.
Other genes recently found to be involved in osteopetrosis are OSTM1, which has the likely role to act as a β-subunit to stabilise the ClC-7 protein [96], and sorting nexin 10 (SNX10), whose product has been suggested to interact with the proton pump [97]. PLEKHM1 is another protein whose deficiency induces an intermediate form of osteopetrosis, due to a defective vesicular trafficking in osteoclasts, eventually leading to impairment of their activity [27].
With regard to the osteoclast-poor type, Sobacchi et al. [98] found in 2007 that mutations in the TNFSF11 gene, coding for RANKL, caused autosomal recessive osteopetrosis characterised by a complete absence of osteoclasts. Unfortunately, patients carrying this mutation cannot be treated with HSC transplantation, the only therapeutic option for the management of infantile osteopetrosis, because the molecular defect is not intrinsic in the osteoclast but affects RANKL-producing cells, including osteoblasts and stromal cells. Consistently, another study identified mutations in the TNFRSF11A gene coding for RANK, giving rise to an autosomal recessive form with a phenotype similar to that caused by RANKL mutations [99] but treatable with HSC transplantation given that this defect is osteoclast autonomous.
3.3 Osteoclast Pharmacology
When osteoclast bone resorption exceeds osteoblast bone formation, a net loss of bone mass occurs. Therefore, these cells are targeted by various drugs in order to rescue a balanced bone turnover. The most prevalent treatments are herein described.
3.3.1 Bisphosphonates
Bisphosphonates include a group of compounds that strongly inhibit osteoclast function, and over the past two decades, they have been largely employed to treat osteoporosis and bone metastases [100]. What made bisphosphonates a good therapeutic strategy is their selective affinity for the bone, due to their P-C-P backbone structure and ability to chelate calcium ions, thus binding to exposed bone mineral and being internalised by bone-resorbing osteoclasts that then undergo apoptosis [101]. Indeed, the simplest bisphosphonates, clodronate and etidronate, employed since 1989, induce massive osteoclast apoptosis because they are incorporated into non-hydrolysable analogues of adenosine triphosphate. The more powerful next-generation nitrogen-containing bisphosphonates (pamidronate, alendronate, ibandronate, risedronate, zoledronate) instead inhibit a key enzyme of the mevalonate pathway, the farnesyl pyrophosphate (FPP) synthase, thus resulting in the accumulation of the isopentenyl diphosphate, which is incorporated into the toxic nucleotide metabolite ApppI, eventually leading to osteoclast apoptosis as well [101]. Moreover, bisphosphonates prevent the prenylation of small GTPases, hereby disrupting the production of isoprenoid lipids in the mevalonate pathway and affecting osteoclast function [102].
With regard to the therapeutic application in the treatment of postmenopausal osteoporosis, while the first generation of bisphosphonates showed a moderate effect on bone resorption, the second- and third-generation compounds were much more potent, as shown by several clinical trials performed during the last 15 years, with a reduction of fracture rates up to 50% [103]. However, recently, an association between long-term bisphosphonate use and atypical femoral fracture risk has emerged [55, 104]. Moreover, due to the high affinity of bisphosphonates for the bone mineral, they accumulate in the bone matrix in long-term treatments, «freezing» the bone and causing it to become much more static [105].
The use of bisphosphonates in the management of bone metastases has beneficial effects not only for osteolytic lesions but also for osteosclerotic metastases, efficiently counteracting skeletal morbidity and improving the quality of life [106]. However, benefits in the increase overall survival of patients need to be more deeply ascertained, and likely as a consequence of the massive dose administered in these patients, some of them develop, as an adverse effect, the osteonecrosis of the jaw [107].
3.3.2 Denosumab
Given the pivotal role of the RANKL/RANK/OPG axis in the biology of the osteoclast, many therapeutic strategies were diverted to block this pathway. A recent drug developed for this purpose is denosumab, a human monoclonal IgG2 antibody specifically raised against the soluble and cell surface RANKL, thus inhibiting its binding to RANK with a resulting block of osteoclastogenesis [108]. Overall, the effectiveness of this drug in postmenopausal osteopenic women seems to be not inferior, or even greater, than that of the reference drug alendronate, with a better patient compliance, since administration is performed subcutaneously once every 6 months [109,110,111]. Denosumab treatment also improves bone mechanical properties [112], and its effect on bone remodelling seems to be reversible [113] after stopping the treatment, thus avoiding a permanent loss of dynamicity of the bone. Finally, it has been reported that denosumab has the same adverse effect of alendronate [114], while other studies found hypocalcaemia in few treated patients [109].
3.3.3 New Antiresorptive Agents
Due to the possible side effects observed in the currently available antiresorptive therapies, the need to identify new targets for alternative treatments is still present [115]. In this respect, the most recently identified strategies are described below.
3.3.3.1 Cathepsin K Inhibitors
Cathepsin K belongs to a large family of at least 11 cysteine proteases in humans. As already mentioned, it is highly expressed and released by osteoclasts inside the resorption lacuna where it breaks down type I collagen. Indeed, the pivotal role of this enzyme in osteoclasts has been clearly demonstrated by the fact that mutations of its gene lead to pycnodysostosis (i.e. Toulouse-Lautrec syndrome), a rare genetic disease characterised by an impairment of bone organic matrix resorption, while osteoclasts form and demineralise the bone matrix normally [116, 117]. From a therapeutic point of view, the fact that cathepsin K inhibitors block osteoclast activity and do not prevent osteoclast formation is a remarkable advantage, given that it allows the osteoclast to perform other physiologic functions, one for all the regulations of osteoblasts. Odanacatib (MK-0822) is a cathepsin K inhibitor that has been employed in phase I and II clinical trials in postmenopausal women, proving to be effective in preventing bone resorption [118]. This compound has also been found to reduce serum bone resorption biomarkers in breast cancer patients with bone metastases [119]. Another cathepsin K inhibitor under phase I and II clinical trial is ONO-5334, which shows an antiresorptive activity similar to that of bisphosphonates [120].
3.3.3.2 αVβ3 Integrin Inhibitors
αVβ3 integrin is also a potential antiresorptive and antitumoural target, since this receptor participates in osteoclast adhesion, triggering a complex intracellular signalling involving tyrosine kinases c-Src and Syk [121], a crucial step for the assessment of bone resorption. αVβ3 integrin inhibitors (i.e. S247, ATN-161, PSK1404) have been mainly employed in preclinical trials to treat bone metastases, providing the double advantage to target osteoclasts and tumour cells, since in the latter this integrin can be highly expressed and provide a pro-invasive phenotype [122]. L-000845704 is another αVβ3 antagonist already employed in clinical trials, which proved to inhibit bone resorption in women with postmenopausal osteoporosis [123].
3.4 Methods for Osteoclast Primary Cultures
First attempts to obtain osteoclast primary cultures, dating back more than 30 years ago, include mechanical disaggregation of the long bones of newborn animals (i.e. rabbits and rats), which provided short living mature osteoclasts [124, 125]. Successful mature osteoclasts were also isolated from the long bones of laying hens kept in low-calcium diet, a condition that enhances osteoclast formation and activity [126].
The identification of M-CSF and RANKL as powerful pro-osteoclastogenic factors has greatly contributed to the generation of osteoclast primary cultures from different sources. In fact, nowadays, the preferential way to obtain primary osteoclasts is by isolating osteoclast precursors from the bone marrow or peripheral blood mononuclear cells and differentiating them using these two pro-osteoclastogenic cytokines. This method results in a large number of purified differentiated osteoclasts.
3.4.1 Osteoclast Isolation from Mouse Bone Marrow Cells
The fulfilment of this strategy has been supported by the ascertainment that osteoblasts and stromal cells inside the bone marrow produce a plethora of factors that positively regulate osteoclast differentiation from the monocyte/macrophage lineage. As described below, two main protocols can be used to obtain osteoclasts from bone marrow cells, one mimicking an osteoblast-preosteoclast coculture and the other requiring the purification of osteoclast precursors from the bone marrow.
3.4.1.1 Osteoclast Primary Culture from Unfractionated Bone Marrow Cells
This method starts from a bone marrow culture, which contains osteoclast precursors and mesenchymal stromal cells supporting osteoclastogenesis by producing RANKL and M-CSF under the stimulation of 1α,25-dihydroxy-vitamin D3 [1,25(OH)2D3] added to the culture [127, 128]. Seven-day-old mice are euthanized by CO2 inhalation, and then the fore- and hindlimbs are excised and put in HANK’s balanced salt solution (HBSS). A gross cleaning with a blade is performed to remove the surrounding soft tissues; hence, the long bones are finely minced into small pieces, and bone marrow is mechanically flushed out by repeated pipetting with a Pasteur pipette. Collected bone marrow cells are cultured in Dulbecco’s modified eagle medium (DMEM) plus 10% foetal bovine serum (FBS); then, the day after, medium is replaced by fresh DMEM supplemented with 10% FBS and 10−8M 1,25(OH)2D3. After 8–10 days of culture, multinucleated osteoclasts appear and can be subjected to characterisation [69].
3.4.1.2 Osteoclast Primary Culture from Purified Bone Marrow Macrophages
This method requires the isolation of osteoclast precursors from the bone marrow and their subsequent differentiation by treatment with M-CSF and RANKL. Briefly, the bone marrow is flushed out from the limbs of 7-day-old mice as described above; then it is diluted 1:1 in HBSS and stratified over Ficoll/Histopaque 1077. After centrifugation at 400 g for 30 min without brake, the solution appears stratified in three layers. Osteoclast precursors are recovered from the white ringlike intermediate layer, resuspended in DMEM containing 10% FBS and plated. After 3 h, cultures are washed to remove non-adherent cells, and then DMEM supplemented with 10% FBS, 50 ng/ml mouse recombinant (mr) M-CSF and 50 ng/ml mrRANKL are added. After 5–7 days of culture, it is possible to observe an enriched multinucleated osteoclast population [69].
3.4.2 Osteoclast Isolation from Human Peripheral Blood
The setup of this primary culture has greatly contributed to the study of the biology of osteoclasts in normal and pathological conditions, allowing to recover osteoclasts from an available human source, the peripheral blood, instead of bone marrow biopsies [93, 129].
Human peripheral blood mononuclear cells are obtained by density gradient of fresh peripheral blood [93]. This sample is first diluted 1:1 with PBS or HBSS and then stratified over Ficoll/Histopaque 1077 and centrifuged at 800 g for 30 min with the brake off. This procedure will again result in three layers, of which the middle white contains the osteoclast precursors. Isolated buffy coat cells are then washed with HBSS and centrifuged and resuspended in DMEM plus 10% FBS and plated. After 3 h, plates are rinsed to remove non-adherent cells and cultured in the aforementioned medium in the presence of 50 ng/ml human recombinant (hr) M-CSF and 30 ng/ml hrRANKL. The culture requires at least 2 weeks in order to provide multinucleated osteoclasts and 3 weeks to assess bone resorption, during which the medium should be replaced every 3–4 days.
3.4.3 Evaluation of Osteoclast Differentiation
Three main determinants demonstrate that bona fide osteoclasts have been formed in the primary culture: (1) the presence of giant multinucleated cells (more than three nuclei/cell) which can be easily observed by phase contrast microscopy, (2) the positivity of these cells to the osteoclast marker TRAcP and (3) their ability to resorb the bone. This latter will be discussed in the next paragraph.
3.4.3.1 Histochemical TRAcP Assay
As suggested by the name, TRAcP belongs to a class of metalloenzymes that catalyse the hydrolysis of various phosphate esters and anhydrides under acidic conditions. Although it has always been considered a classical marker of osteoclasts, TRAcP is also expressed by inflammatory macrophages and dendritic cells. TRAcP activity can be easily evaluated in osteoclast cultures fixed in 4% paraformaldehyde, by a histochemical assay using a commercially available kit (Sigma-Aldrich #387A) according to the manufacturer’s instruction. TRAcP-positive osteoclasts appear as purple stained cells with three or more nuclei (◘ Fig. 3.3a, b).

a Picture showing an osteoclast primary culture obtained from purified bone marrow macrophages and subjected to cytochemical assay for the tartrate-resistant acid phosphatase (TRAcP) activity (magnification, 25×). b Magnification of a (100×). c Picture of a bone slice stained with toluidine blue showing several resorption pits evidenced as blue spot (magnification = 100×)
3.4.3.2 Transcriptional Evaluation of Osteoclast-Specific Genes
As described above, during osteoclast differentiation, the activated NF-kB transcription factor promotes the transcriptional expression of downstream osteoclast-specific genes, which can be evaluated by RT-PCR. Typical genes whose expression increases during osteoclastogenesis are cathepsin K, CTR, MMP-9, RANK and TRAcP [69]. Genes whose expression is associated with preosteoclast fusion can also be evaluated, such as DC-STAMP, CD44 and macrophage fusion receptor (MFR).
3.4.4 Evaluation of Osteoclast Function
In vitro osteoclast activity can be easily assessed by the resorption pit assay. Osteoclast precursors are plated on dentine/bone slices (commercially available) and differentiated into mature osteoclasts as described above. Alternatively, after their differentiation in plastic dishes, mature osteoclasts are detached and replated onto dentine/bone slices for at least 48 h [66]. Osteologic dishes covered with a layer of hydroxyapatite can also be used. In both cases, at the end of the resorption period (48 h for rodent osteoclasts and 1 week for human osteoclasts), cells are mechanically removed from the bone surface, and sections are stained with toluidine blue or Coomassie blue or observed by scanning electron microscopy. Toluidine blue and Coomassie blue staining are very easy procedures, which exploit the higher affinity of these dyes for the organic bone matrix components that are exposed after removal of the mineral by the action of osteoclasts. These areas appear more intensely stained, and in the case of toluidine blue, they are metachromatic for the content of glycosaminoglycans (◘ Fig. 3.3c). Scanning electron microscopy is more time-consuming but can provide more detailed information on pit shape, size and depth. Total pit area and volume are then quantified by light and by scanning electron microscopy, respectively, using a morphometric software. Alternatively, pits are classified in small (<10 μm diameter), medium (10–30 μm diameter) and large (>30 μm diameter), and their number is multiplied for a size score of 0.3 for small pits, 1 for medium pits and 3 for large pits. The sum of the three scores provides the final pit index that is proportional of the pit number and size [130]. Other methods make use of dentin/bone sections pre-stained with calcium-binding fluorofores (calcein, alizarin red or inactive fluorescent bisphosphonates). Fluorofores are removed during bone resorption, and in this case, the resorption pits appear as dark areas in a fluorescent background. They are then evaluated by fluorescence or confocal microscopy using the same parameters described above.
3.5 Conclusions
Osteoclasts are bone-resorbing cells that largely contribute to bone remodelling. Their deregulated activity impacts the bone health, and for this reason, they are considered important targets for therapy. Osteoclast biotechnology has largely furthered the knowledge on their biology and added to the development of therapies to block their exacerbated activity (i.e. in osteoporosis, bone metastases) or, conversely, re-establish their impaired formation or resorption (i.e. osteopetrosis). More is expected in the years to come to broaden the impact of their therapeutic management in other pathological conditions that currently have no cure.
Take-Home Messages
-
The osteoclast is a monocyte/macrophage arising cell whose differentiation is a fine-tuned process regulated by systemic and local factors, among which the RANKL/RANK pathway plays a crucial role.
-
The osteoclast is more than a hungry cell that eats bone, showing a versatile profile in terms of functions, which include the regulation of osteoblastogenesis and haematopoiesis.
-
Several skeletal diseases are the result of a dysfunction of osteoclast formation and activity, which makes this cell a useful target for therapy.
-
The investigation of osteoclast pathophysiology can rely on well-established in vitro methods of precursor isolation from mouse bone marrow and human peripheral blood.
References
Cappariello A, Maurizi A, Veeriah V, Teti A. The Great Beauty of the osteoclast. Archiv Biochem Biophys. 2014;558:70–8.
Del Fattore A, Cappariello A, Teti A. Genetics, pathogenesis and complications of osteopetrosis. Bone. 2008;42(1):19–29.
Walker DG. Bone resorption restored in osteopetrotic mice by transplant of normal bone marrow and spleen cells. Science. 1975;190(4216):784–5.
Rasmussen H, Bordier P. The cellular basis of metabolic bone disease. New Engl J Med. 1973;289(1):25–32.
Chambers TJ. The birth of the osteoclast. Ann N Y Acad Sci. 2010;1192:19–26.
Tondravi MM, McKercher SR, Anderson K, Erdmann JM, Quiroz M, Maki R, et al. Osteopetrosis in mice lacking hematopoietic transcription factor PU.1. Nature. 1997;386(6620):81–4.
Van de Wijngaert FP, Tas MC, Burger EH. Conditioned medium of fetal mouse long bone rudiments stimulates the formation of osteoclast precursor-like cells from mouse bone marrow. Bone. 1989;10(1):61–8.
Menaa C, Kurihara N, Roodman GD. CFU-GM-derived cells from osteoclasts at a very high efficiency. Biochem Biophys Res Commun. 2000;267(3):943–6.
Biskobing DM, Fan X, Rubin J. Characterization of MCSF-induced proliferation and subsequent osteoclast formation in murine marrow culture. J Bone Miner Res. 1995;10(7):1025–32.
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89(2):309–19.
Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93(2):165–76.
Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95(7):3597–602.
Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13(18):2412–24.
Teitelbaum SL. Osteoclasts: what do they do and how they do it? Am J Pathol. 2007;170(2):427–35.
Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, et al. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 1997;11(24):3482–96.
Kukita T, Wada N, Kukita A, Kakimoto T, Sandra F, Toh K, et al. RANKL-induced DC-STAMP is essential for osteoclastogenesis. J Exp Med. 2004;200(7):941–6.
Nakashima T, Hayashi M, Takayanagi H. New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol Metab. 2012;23(11):582–90.
Marchisio PC, Cirillo D, Naldini L, Primavera MV, Teti A, Zambonin Zallone A. Cell-substratum interaction of cultured avian osteoclasts is mediated by specific adhesion structures. J Cell Biol. 1984;99(5):1696–705.
Saltel F, Chabadel A, Bonnelye E, Jurdic P. Actin cytoskeletal organisation in osteoclasts: a model to decipher transmigration and matrix degradation. Eur J Cell Biol. 2008;87(8–9):459–68.
Supanchart C, Kornak U. Ion channels and transporters in osteoclasts. Arch Biochem Biophys. 2008;47(2):161–5.
Vääräniemi J, Halleen JM, Kaarlonen K, Ylipahkala H, Alatalo SL, Andersson G, et al. Intracellular machinery for matrix degradation in bone-resorbing osteoclasts. J Bone Miner Res. 2004;19(9):1432–40.
Hirvonen MJ, Fagerlund K, Lakkakorpi P, Väänänen HK, Mulari MT. Novel perspectives on transcytotic route in osteoclasts. Bonekey Rep. 2013;2:306.
Teti A, Blair HC, Teitelbaum SL, Kahn AJ, Koziol C, Konsek J, et al. Cytoplasmic pH regulation and chloride/bicarbonate exchange in avian osteoclasts. J Clin Invest. 1989;83(1):227–33.
Lindsey AE, Schneider K, Simmons DM, Baron R, Lee BS, Kopito RR. Functional expression and subcellular localization of an anion exchanger cloned from choroid plexus. Proc Natl Acad Sci U S A. 1990;87(14):5278–82.
Henriksen K, Sorensen MG, Nielsen RH, Gram J, Schaller S, Dziegel MH, et al. Degradation of the organic phase of bone by osteoclasts: a secondary role for lysosomial acidification. J Bone Miner Res. 2006;21(1):58–88.
Nesbitt SA, Horton MA. Trafficking of matrix collagens through bone-resorbing osteoclasts. Science. 1997;276(5310):266–9.
Van Wesenbeck L, Odgren PR, Coxon FP, Frattini A, Moens P, Perdu B, et al. Involvement of PLEKHM1 in osteoclastic vesicular transport and osteopetrosis in incisors absent rats and humans. J Clin Invest. 2007;117(4):919–30.
Lange PF, Wartosch L, Jentsch TJ, Fuhrmann JC. ClC7 requires Ostm1 as a beta-subunit to support bone resorption and lysosomal function. Nature. 2006;440(7081):220–3.
Xian L, Wu X, Pang L, Lou M, Rosen CJ, Qiu T, et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat Med. 2012;18(7):1095–101.
Pfeilschifter J, D’Souza SM, Mundy GR. Effect of transforming growth factor-beta on osteoblastic osteosarcoma cells. Endocrinology. 1987;121(1):212–8.
Lotinun S, Kiviranta R, Matsubara T, Alzare JA, Neff L, Luth A, et al. Osteoclast-specific cathepsin K deletion stimulates S1P-dependent bone formation. J Clin Invest. 2013;123(2):666–81.
Teti A. Mechanisms of osteoclast-dependent bone formation. Bonekey Rep. 2013;2:449.
Drissi H, Sanjay A. The multifaceted osteoclast; far and beyond bone resorption. J Cell Biochem. 2016;117(8):1753–6.
Dai XM, Zong XH, Akhter MP, Stanley ER. Osteoclast deficiency results in disorganized matrix, reduced mineralization, and abnormal osteoblast behavior in developing bone. J Bone Miner Res. 2004;19(9):1441–51.
Sakagami N, Amizuka N, Li M, Takeuchi K, Hoshino M, Nakamura M, et al. Reduced osteoblastic population and defective mineralization in osteopetrotic (op/op) mice. Micron. 2005;36(7–8):688–95.
Del Fattore A, Peruzzi B, Rucci N, Recchia I, Cappariello A, Longo M, et al. Clinical, genetic, and cellular analysis of 49 osteopetrotic patients: implication for diagnosis and treatment. J Med Genet. 2006;43(4):315–25.
Pennypacker B, Shea M, Liu Q, Masarachia P, Saftig P, Rodan S, et al. Bone density, strength, and formation in adult cathepsin K−/− mice. Bone. 2009;44(2):199–207.
Ruy J, Kim HJ, Chang EJ, Huang H, Banno Y, Kim HH, et al. Sphingosine 1-phosphatase as a regulator of osteoclast differentiation and osteoclast-osteoblast coupling. EMBO J. 2006;25(24):5840–51.
Pederson L, Ruan M, Westendorf JJ, Khosla S, Oursler MJ. Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphatase. Proc Natl Acad Sci U S A. 2008;105(52):29764–9.
Hayman AR, Cox TM. Tartrate-resistant acid phosphatase knockout mice. J Bone Miner Res. 2003;18(10):1905–7.
Kimura H, Kwan KM, Zhang Z, Deng JM, Darnay BG, Behringer RR, et al. Cthrc1 is a positive regulator of osteoblastic bone formation. PLoS One. 2008;3(9):e3174.
Takeshita S, Fumoto T, Matsuoka K, Park KA, Aburatani H, Kato S, et al. Osteoclast-secreted CTHRC1 in the coupling of bone resorption and bone formation. J Clin Invest. 2013;123(9):3914–24.
Matsuoka K, Park KA, Ito M, Ikeda K, Takeshita S. Osteoclast-derived complement component 3a stimulates osteoblast differentiation. J Bone Miner Res. 2014;29(7):1522–30.
Charles JF, Aliprantis AO. Osteoclasts, more than «bone eaters». Trends in Mol Med. 2014;20(8):449–59.
Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y, et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med. 2006;12(6):657–64.
Heissig B, Hattori K, Dias S, Friedrich M, Ferris B, Hackett NR, et al. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell. 2002;109(5):625–37.
Adams GB, Chabner KT, Alley IR, Olson DP, Szczepiorkowski ZM, Poznansky MC, et al. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature. 2006;439(7076):599–603.
Takamatsu Y, Simmons PJ, Moore RJ, Morris HA, To IB, Lè Vesque JP. Osteoclast-mediated bone resorption is stimulated during short-term administration of granulocyte colony-stimulating factor but is not responsible for hematopoietic progenitor cell mobilization. Blood. 1998;92(9):3465–73.
Sim HJ, Kook SH, Yun CY, Bhattarai G, Cho ES, Lee JC. Brief report: consecutive alendronate administration-mediated inhibition of osteoclasts improves long-term engraftment potential and stress resistance of HSCs. Stem Cells. 2016;34(10):2601–7.
Lymperi S, Horwood N, Marley S, Gordon MY, Cope AP, Dazzi F. Strontium can increase some osteoblasts without increasing hematopoietic stem cells. Blood. 2008;111(3):1173–8.
Frisch BJ, Porter RL, Gigliotti BJ, Olm-Shipman AJ, Weber JM, O’Keefe RJ, et al. In vivo prostaglandin E2 treatment alters the bone marrow microenvironment and preferentially expands short-term hematopoietic stem cells. Blood. 2009;114(19):4054–63.
Miyamoto T. Role of osteoclasts in regulating hematopoietic stem and progenitor cells. World J Orthop. 2013;4(4):198–206.
O’Connor KM. Evolution and treatment of osteoporosis. Med Clin North Am. 2016;100(4):807–26.
Kruger MC, Wolber FM. Osteoporosis: modern paradigms for last century’s bones. Forum Nutr. 2016;8(6):E376.
Compston J. Pathophysiology of atypical femoral fractures and osteonecrosis of the jaw. Osteoporos Int. 2011;22(12):2951–61.
Sattui SE, Saag KG. Fracture mortality: associations with epidemiology and osteoporosis treatment. Nat Rev Endocrinol. 2014;10(10):592–602.
Hendrickx G, Boudin E, Van Hul W. A look behind the scenes: the risk and pathogenesis of primary osteoporosis. Nat Rev Rheumatol. 2015;11(8):462–74.
Hughes DE, Dai A, Tiffee JC, Li HH, Mundy GR, Boyce BF. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-beta. Nat Med. 1996;2(10):1132–6.
Xing L, Boyce BF. Regulation of apoptosis in osteoclasts and osteoblastic cells. Biochem Biophys Res Commun. 2005;328(3):709–20.
Hofbauer LC, Khosla S, Dunstain CR, Lacey DL, Spelsberg TC, Riggs BL. Estrogen stimulates gene expression and protein production of osteoprotegerin in human osteoblastic cells. Endocrinology. 1999;140(9):4367–70.
Shevde NK, Bendixen AC, Dienger KM, Pike JW. Estrogens suppress rankl-induced osteoclast differentiation via a stromal cell independent mechanism involving c-jun repression. Proc Natl Acad Sci U S A. 2000;97(14):7829–34.
Diab DL, Watts NB. Secondary osteoporosis: differential diagnosis and workup. Clin Obstet Gynecol. 2013;56(4):686–93.
Seibel MJ, Cooper MS, Zhou H. Glucocorticoid-induced osteoporosis: mechanisms, management and future perspectives. Lancet Diabetes Endocrinol. 2013;1(1):59–70.
Sapre S, Thakur R. Lifestyle and dietary factors determine age at natural menopause. J Midlife Health. 2014;5(1):3–5.
Węgierska M, Dura M, Blumfield E, Żuchowski P, Waszczak M, Jeka S. Osteoporosis diagnostics in patients with rheumatoid arthritis. Rheumatologia. 2016;54(1):29–34.
Edwards WB, Schnitzer TJ, Troy KL. The mechanical consequence of actual bone loss and simulated bone recovery in acute spinal cord injury. Bone. 2014;60:141–7.
Rucci N, Rufo A, Alamanou M, Teti A. Modeled microgravity stimulates osteoclastogenesis and bone resorption by increasing osteoblast RANKL/OPG ratio. J Cell Biochem. 2007;100(2):464–73.
Lelovas PP, Xanthos TT, Thoma SE, Lyritis GP, Dontas IA. The laboratory rat as an animal model for osteoporosis research. Comp Med. 2008;58(5):424–30.
Rucci N, Rufo A, Alamanou M, Capulli M, Del Fattore A, Åhrman E, et al. The glycosaminoglycan-binding domain of PRELP acts as a cell type-specific NF-κB inhibitor that impairs osteoclastogenesis. J Cell Biol. 2009;187(5):669–83.
Sakata T, Sakai A, Tsurukami H, Okimoto N, Okazaki Y, Ikeda S, et al. Trabecular bone turnover and bone marrow cell development in tail-suspended mice. J Bone Miner Res. 1999;14(9):1596–604.
Rucci N, Capulli M, Piperni SG, Cappariello A, Lau P, Frings-Meuthen P, et al. Lipocalin 2: a new mechanoresponding gene regulating bone homeostasis. J Bone Miner Res. 2015;30(2):357–68.
Warner SE, Sanford DA, Becker BA, Bain SD, Srinivasan S, Gross TS. Botox induced muscle paralysis rapidly degrades bone. Bone. 2006;38(2):257–64.
Kan C, Vargas G, Pape FL, Clézardin P. Cancer cell colonisation in the bone microenvironment. Int J Mol Sci. 2016;17(10):E1674.
Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res. 2006;12(20 Pt 2):6243s–9s.
Roodman GD. Mechanisms of bone metastasis. New Engl J Med. 2004;350(16):1655–64.
Clézardin P, Teti A. Bone metastasis: pathogenesis and therapeutic implications. Clin Exp Metastasis. 2007;24(8):599–608.
Jehn CF, Diel IJ, Overkamp F, Kurth A, Schaefer R, Miller K, et al. Management of metastatic bone disease algorithms for diagnostics and treatment. Anticancer Res. 2016;36(6):2631–7.
Arguello F, Baggs RB, Frantz CN. A murine model of experimental metastasis to bone and bone marrow. Cancer Res. 1988;48(3):6876–81.
Yoneda T, Sasaki A, Dunstan C, Williams PJ, Bauss F, De Clerck YA, et al. Inhibition of osteolytic bone metastasis of breast cancer by combined treatment with the bisphosphonate ibandronate and tissue inhibitor of the matrix metalloproteinase-2. J Clin Invest. 1997;99(10):2509–17.
Rucci N, Recchia I, Angelucci A, Alamanou M, Del Fattore A, Fortunati D, et al. Inhibition of protein kinase c-Src reduces the incidence of breast cancer metastases and increases survival in mice: implications for therapy. J Pharmacol Exp Ther. 2006;318(1):161–72.
Guise TA, Yin JJ, Taylor SD, Kumagai Y, Dallas M, Boyce BF, et al. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J Clin Invest. 1996;98(7):1544–9.
Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, et al. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest. 1999;103(2):197–206.
Weilbaecher KN, Guise TA, McCauley LK. Cancer and bone: a fatal attraction. Nat Rev Cancer. 2011;11(7):411–25.
Zhang Y, Fujita N, Oh-hara T, Morinaga Y, Nakagawa T, Yamada M, et al. Production of interleukin-11 in bone-derived endothelial cells and its role in the formation of osteolytic bone metastases. Oncogene. 1998;16(3):693–703.
Bendre MS, Montague DC, Peer T, Akel NS, Gaddy D, Suva LJ. Interleukin-8 stimulation of osteoclastogenesis and bone resorption is a mechanism for the increased osteolysis of metastastic bone disease. Bone. 2003;33(1):28–37.
Singh B, Berry JA, Shoher A, Ayers GD, Wei C, Lucci A. COX-2 involvement in breast cancer metastasis to bone. Oncogene. 2007;26(26):3789–96.
Hiraga T, Kizaka-Kondoh S, Hirota K, Hiraoka M, Yoneda T. Hypoxia and hypoxia- inducible factor-1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 2007;67(9):4157–63.
Sethi N, Dai X, Winter CG, Kang Y. Tumor-derived JAGGED1 promotes osteolytic bone metastases of breast cancer by engaging notch signaling in bone cells. Cancer Cell. 2011;19(2):192–205.
Tolar J, Teitelbaum SL, Orchad PJ. Osteopetrosis. N Engl J Med. 2004;351(27):2839–49.
Albers-Schönberg HE. Röntgenbilder einer seltenen Knock-enerkrankung. Munch Med Wochenscher. 1904;5:365–8.
Villa A, Guerrini MM, Cassani B, Pangrazio A, Sobacchi C. Infantile malignant, autosomal recessive osteopetrosis: the rich and the poor. Calcif Tissue Int. 2009;84(1):1–12.
Sobacchi C, Frattini A, Orchard P, Porras O, Tezcan I, Andolina M, et al. The mutational spectrum of human malignant autosomal recessive osteopetrosis. Human Mol Genet. 2001;10(17):1767–73.
Taranta A, Migliaccio S, Recchia I, Caniglia M, Luciani M, De Rossi G, et al. Genotype-phenotype relationship in human ATP6i-dependent autosomal recessive osteopetrosis. Am J Pathol. 2003;162(1):57–68.
Kornak U, Kasper D, Bosl MR, Kaiser E, Schweizer M, Schulz A, et al. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 2001;104(2):205–15.
Sly WS, Hewett-Emmett D, Whyte MP, Yu YS, Tashian RE. Carbonic anhydrase II deficiency identified as the primary defect in the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. Proc Natl Acad Sci U S A. 1983;80(9):2752–6.
Pangrazio A, Poliani PL, Megarbane A, Lefranc G, Lanino E, Di Rocco M, et al. Mutations in OSTM1 (grey lethal) define a particularly severe form of autosomal recessive osteopetrosis with neural involvement. J Bone Miner Res. 2006;21(7):1098–105.
Pangrazio A, Fasth A, Sbardellati A, Orchard PJ, Kasow KA, Raza J, et al. SNX10 mutations define a subgroup of human autosomal recessive osteopetrosis with variable clinical severity. J Bone Miner Res. 2013;28(5):1041–9.
Sobacchi C, Frattini A, Guerrini MM, Abinun M, Pangrazio A, Susani L, et al. Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nat Genet. 2007;39(8):960–2.
Guerrini MM, Sobacchi C, Cassani B, Abinun M, Kilic SS, Pangrazio A, et al. Human osteoclast-poor osteopetrosis with hypogamma-globulinemia due to TNFRSF11A (RANK) mutations. Am J Human Genet. 2008;83(1):64–76.
Zhou J, Ma X, Wang T, Zhai S. Comparative efficacy of bisphosphonates in short-term fracture prevention for primary osteoporosis: a systemic review with network meta-analyses. Osteoporosis Int. 2016;27(11):3289–300.
Rogers MJ. New insights into the molecular mechanisms of action of bisphosphonates. Curr Pharm Des. 2003;9(32):2643–58.
Rogers MJ, Crockett JC, Coxon FP, Mönkkönen J. Biochemical and molecular mechanisms of action of bisphosphonates. Bone. 2011;49(16):34–41.
Eriksen EF, Diez-Perez A, Boonen S. Update of long-term treatment with bisphosphonates for postmenopausal osteoporosis: a systematic review. Bone. 2014;58:126–35.
Odvina CV, Zerwekh JE, Rao DS, Maalouf N, Gottschalk FA, Pak CY, et al. Severely suppressed bone turnover: a potential complication of alendronate therapy. J Clin Endocrinol Metab. 2005;90(3):1294–301.
Pazianas M, van der Geest S, Miller P. Bisphosphonates and bone quality. Bonekey Rep. 2014;3:529.
Young RJ, Coleman RE. Zoledronic acid to prevent and treat cancer metastasis: new perspects for an old drug. Future Oncol. 2013;9(5):633–43.
Filleul O, Crompot E, Saussez S. Bisphosphonate-induced osteonecrosis of the jaw: a review of 2400 patient cases. J Cancer Res Clin Oncol. 2010;136(8):1117–24.
Kostenuik PJ, Nguyen HQ, McCabe J, Warmington KS, Kurahara C, Sun N, et al. Denosumab, a fully human monoclonal antibody to RANKL, inhibits bone resorption and increases BMD in knock-out mice that express chimeric (murine/human) RANKL. J Bone Miner Res. 2009;24(2):182–95.
McClung MR, Lewiecki EM, Cohen SB, Bolognese MA, Woodson GC, Moffett AH, et al. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med. 2006;354(8):821–31.
Lewiecki EM, Miller PD, McClung MR, Cohen SB, Bolognese MA, Liu Y, et al. Two-year treatment with denosumab (AMG162) in a randomized phase 2 study of postmenopausal women with low BMD. J Bone Miner Res. 2007;22(12):1832–41.
Cummings SR, San Martin J, McClung MR, Siris ES, Eastell R, Reid IR, et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361(8):756–65.
Beck TJ, Lewiecki EM, Miller PD, Felsenberg D, Liu Y, Ding B, et al. Effects of denosumab on the geometry of the proximal femur in postmenopausal women in comparison with alendronate. J Clin Densitom. 2008;11(3):351–9.
Farrier AJ, Sanchez Franco LC, Shoaib A, Gulati V, Johnson N, Uzoigwe CE, et al. New anti-resorptives and antibody mediated anti-resorptive therapy. Bone Joint J. 2016;98-B(2):160–5.
Trouvin AP, Goëb V. Receptor activator of nuclear factor-kB ligand and osteoprotegerin: maintaining the balance to prevent bone loss. Clin Interv Aging. 2010;5:345–54.
Harslof T, Langdahl BL. New horizons in osteoporosis therapies. Curr Opin Pharmacol. 2016;28:38–42.
Hodder A, Huntley C, Aronson JK, Ramachandran M. Pycnodysostosis and the making of an artist. Gene. 2014;119(14):1109–13.
Gelb BD, Shi GP, Chapman HA, Desnick RJ. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science. 1996;273(5279):1236–8.
Mukherjee K, Chattopadhyay N. Pharmacological inhibition of cathepsin K: a promising novel approach for postmenopausal osteoporosis therapy. Biochem Pharmacol. 2016;117:10–9.
Jensen AB, Wynne C, Ramirez G, He W, Song Y, Berd Y, et al. The cathepsin K inhibitor odanacatib suppresses bone resorption in women with breast cancer and established bone metastases: results of a 4 week, double-bind, randomized, controlled trial. Clin Breast Cancer. 2010;10(6):452–8.
Eastell R, Nagase S, Ohyama M, Small M, Sawyer J, Boonen S, et al. Safety and efficacy of the cathepsin K inhibitor ONO-5334 in postmenopausal osteoporosis: the OCEAN study. J Bone Miner Res. 2012;26(6):1303–12.
Zou W, Kitaura H, Reeve J, Long F, Tybulewicz VLJ, Shattil SJ, et al. Syk, c-Src, the αvβ3 integrin, and ITAM immunoreceptors, in concert, regulate osteoclastic bone resorption. J Cell Biol. 2007;176(6):877–88.
Clézardin P. Integrins in bone metastasis formation and potential therapeutic implications. Curr Cancer Drug Targets. 2009;9(7):801–6.
Murphy MG, Cerchio K, Stoch SA, Gottesdiener K, Wu M, Recker R. Effect of L-0845704, an aVb3 integrin antagonist, on markers of bone turnover and bone mineral density in postmenopausal osteoporotic women. J Clin Endocrinol Metab. 2005;90(4):2022–8.
Chambers TJ. Phagocytosis and trypsin-resistant glass adhesion by osteoclasts in culture. J Pathol. 1979;127(2):55–60.
Chambers TJ. Resorption of bone by mouse peritoneal macrophages. J Pathol. 1981;135(4):295–9.
Zambonin ZA, Teti A, Primavera MV. Isolated osteoclasts in primary culture: first observations on structure and survival in culture media. Anat Embryol (Berl). 1982;165(3):405–13.
David JP, Neff L, Chen Y, Rincon M, Horne W, Baron R. A new method to isolate large numbers of rabbit osteoclasts and osteoclast like cells: application to the characterization of serum response element binding proteins during osteoclast differentiation. J Bone Miner Res. 1998;13(11):1730–8.
Teti A, Taranta A, Villanova I, Recchia I, Migliaccio S. Osteoclast isolation: new developments and methods. J Bone Miner Res. 1999;14(7):1251–2.
Matayoshi A, Brown C, Di Persio JF, Haug J, Abu-Amer Y, Liapis H, et al. Human blood-mobilized hematopoietic precursors differentiate into osteoclasts in the absence of stromal cells. Proc Natl Acad Sci U S A. 1996;93(20):10785–90.
Caselli G, Mantovanini M, Gandolfi CA, Allegretti M, Fiorentino S, Pellegrini L, et al. Tartronates: a new generation of drugs affecting bone metabolism. J Bone Miner Res. 1997;12(6):972–81.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Rucci, N., Teti, A. (2017). Osteoclasts: Essentials and Methods. In: Pietschmann, P. (eds) Principles of Bone and Joint Research. Learning Materials in Biosciences. Springer, Cham. https://doi.org/10.1007/978-3-319-58955-8_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-58955-8_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-58954-1
Online ISBN: 978-3-319-58955-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)