
Abstract
Hyperactivation of the PI3K pathway is frequent in human cancer. Whether it occurs via overexpression/phosphorylation of upstream receptors that promote the binding and activation of PI3K, or as a consequence of activating alterations of the nodes of the signaling cascade, deregulated PI3K signaling can promote tumor growth and survival. This provided the rationale to develop inhibitors targeting virtually all the components of this pathway. Despite these efforts, however, the responses in the clinic have been anecdotal and short lived for most of these agents.
In the last few years, clinical studies have demonstrated that specific compounds can elicit strong antitumor activity if administered to selected patients. For example, AKT catalytic inhibitors and specific PI3Kα inhibitors have shown promising clinical responses in patients with tumors bearing activating mutations of AKT and PIK3CA, respectively. Nevertheless, the intrinsic or acquired resistance to PI3K/AKT/mTOR inhibitors limits the activity of these agents. The mechanisms that tumor cells adopt to by-pass pharmacological inhibition of PI3K/AKT/mTOR are tissue-dependent and can be the results of either pre-existing conditions that rapidly compensate for the therapeutic pressure or the acquisition of genomic and/or epigenomic changes that confer fitness over time even upon PI3K full blockade. In both cases, combinatorial strategies seem to be necessary to prevent or delay the emergence of drug resistance, and many of these therapeutic options are currently being tested in the clinic.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
6.1 The PI3K/AKT Pathway
6.1.1 Overview
Receptor tyrosine kinases (RTKs) are large proteins that exist as monomers, dimers, or multimers and can be found embedded in the plasma membrane through a relatively short transmembrane-spanning domain. While the amino-terminal portion of these proteins is mainly involved in the recognition of extracellular ligands, the carboxy terminus is intracellular and has been shown to serve as a docking platform to many signaling molecules, especially upon phosphorylation [1]. Therefore, RTKs are generally associated with proteins that are responsible for triggering the downstream signal transduction when the receptor has been stimulated/activated. There are several signaling pathways that are activated by RTKs, including the mitogen-activated protein kinase (MAPK) , phosphatidylinositol 3-kinase (PI3K) , Src, phospholipase C (PLC), and Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathways , among others [2]. In this chapter, we will describe the basic knowledge regarding the biochemistry and signal transduction of the PI3K pathway, the current pharmacological strategies aimed to target this pathway, and we will discuss in detail the current mechanisms of resistance to this family of inhibitors.
The PI3K family is composed by eight members with catalytic lipid kinase activity classified in three groups according to their substrate specificity and structure. Class I PI3K use phosphatidylinositol (PI)-(4,5)-bisphosphate (PIP2) as a substrate in order to generate phosphatidylinositol-(3,4,5)-trisphosphate (PIP3). Both class II and III give rise to phosphatidylinositol 3-phosphate from the unphosphorylated substrate PI. While the Class I of PI3K is mainly involved in the signal transduction downstream of receptors such as RTK and G-protein coupled receptors (GPCR), class II and III appear to be related with vesicular trafficking. Intensive work in the field of class II and III PI3K is currently undergoing to better understand the physiological and pathophysiological roles of these lipid kinases [3,4,5]. For the purpose of this chapter, we will focus on the class I PI3K and use the term “PI3K” to refer specifically to this class of kinases (Table 6.1).
6.1.2 Biochemistry and Genetics
PI3K enzymes are defined by their lipid kinase activity, required to phosphorylate the 3-OH’ residue of the inositol ring of PIP2. This enzymatic activity is carried out by the catalytic subunit (p110), which is normally associated with a regulatory subunit. Each subunit contains different protein domains [6, 7]. These domains are important for the biochemical and structural functions of the protein and, in the case of p110, include:
-
Kinase domain : Required for the enzymatic activity of the protein.
-
Helical domain : It is used as an interacting interphase not only with other proteins but also within the structure of p110.
-
C2 domain : It is thought to participate in the phospholipid binding required for plasma membrane targeting.
-
Ras-binding domain (RBD) : This region exhibits high affinity towards the GTP-loaded small GTPase Ras.
-
p85-binding domain : Also known as the N-terminal adaptor-binding domain (ABD) , this region is responsible for the binding to the regulatory subunit.
The regulatory subunit of PI3K (p85) is characterized by the presence of two different SH2 domains (nSH2 and cSH2), an SH3 domain, a BH domain, and an inter-SH2 domain (iSH2). While the SH2 domains are required for binding to the activated phospho-Tyr residues of RTK, the inter-SH2 domain seems to be responsible for the interaction with the ABD domain of p110. The BH domain has been shown to interact to small GTP-ases Rac1 and Cdc42. The regulatory subunits of PI3K have multiple functions. They stabilize the catalytic subunit, inhibit the basal kinase activity, and they also engage the activation of the catalytic subunit downstream of the phosphorylated tyrosine motifs as a result of the interaction with their SH2 domains [8].
The crystal structure of PI3Kα in complex with the regulatory subunit p85α was initially solved in 2007 [9], and provided deep insight into the domain distribution, catalytic mechanism, and the template for rational drug design. X-ray crystallography and hydrogen-deuterium exchange mass spectrometry have shown that there are several inhibitory interfaces between p85 and p110. For instance, the p85 nSH2 domain creates inhibitory interfaces with the C2, helical, and C-lobe kinase domains of p110, the p85 iSH2 domain with the p110 C2 domain, and the p85 cSH2 domain with the p110 C-lobe kinase domain. This last interaction is, in fact, an isoform-specific regulatory mechanism, since only the p110β and p110δ exhibit this contact [10].
There are four different genes that encode the Class I PI3K family (PIK3CA, PIK3CB, PIK3CD, and PIK3CG). A lot of the knowledge regarding the functions of each catalytic and regulatory isoform of PI3K has been achieved through the generation of transgenic mice that either lack a gene (knock-out) or express a kinase inactive version of these genes (knock-in). These models are not only important to elucidate the contribution of each isoform into the normal biology, but also to gain insights into the possible secondary effects that prolonged inhibition of these kinases could lead to [5, 11]. For instance, using these genetic mouse models it has been elucidated that Pik3ca inhibition results in major defects in the generation of the vascular system [12]. Pik3ca knock-out mice die at the embryonic stage (E9.5) from severe vascular defects and impaired proliferation [13]. Similar results have been observed with Pik3ca knock-in mice, where the D933A mutation renders the kinase inactive. Although heterozygous mice are viable, vascular defects are still observed, together with a metabolic impairment [14]. In the case of Pik3cb, the difference between the two strategies to generate mouse models is more accentuated. While the knock-out mice die at E3.5, the knock-in version only remains partially lethal, suggesting a non-catalytic function of this isoform during the development [3, 14]. Additionally, these mice exhibit impaired insulin and GPCR-dependent signaling resulting in a metabolic phenotype. Pik3cd and Pik3cg mouse models evidence an important contribution of both kinases in the immunological response. None of the strains reported are embryonically lethal, but adults either carrying the kinase-inactive mutation or lacking the gene have impaired signaling and functions in the B and T cells, neutrophils, and macrophages [15,16,17,18]. Importantly, Pik3cd mice also display allergy and have been recently linked to decreased T-cell-dependent cancer immunotolerance, encouraging the use of PI3Kδ inhibitors as immunotherapy [19].
6.1.3 Signal Transduction by the PI3K Pathway
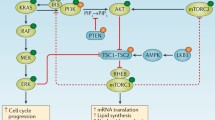
A schematic of the PI3K pathway is depicted in Fig. 6.1. Upon catalytic activity of PI3K, the levels of PIP3 in the membrane raise drastically. The presence of PIP3 at the plasma membrane triggers the rapid activation of downstream effectors that are involved in cell survival, proliferation, motility, control of metabolism, and gene expression among others [3]. The key molecular features involved in the recognition of PIP3 are the phosphoinositide-binding domains, which are present in several proteins that are part of the PI3K pathway and involved in the subcellular localization or the activation of these proteins. Specifically, the pleckstrin homology domain (PH) has been shown to preferentially bind PIP3 over other phosphoinositides. There are over 250 proteins that contain identifiable PH domains, underscoring the complexity of the lipid signal cascade [20]. Among all these proteins, the better characterized PH domain-containing effectors of the PI3K pathway are the RHO and ARF Guanine Nucleotide Exchange Factors (GEFs), PLC, and the kinases AKT and 3-phosphoinositide dependent protein kinase-1 (PDK1), among others. In terms of the cellular signaling in cancer, PDK1 appears to play a major role. Experiments using Pdpk1 (the gene coding PDK1) knock-out mice have shown to reduce tumor burden when crossed with Pten (the gene coding for Phosphatase and tensin homolog, PTEN) heterozygous mice, which are prone to malignancies such as lymphoma and prostate cancer in a PI3K-dependent manner [21]. PDK1 contains a high affinity PH domain that has the ability to recognize PIP3 upon PI3K activity. Although PDK1 is constitutively active, the PH domain provides substrate specificity upon translocation into the plasma membrane, where some of the PDK1 targets are recruited. This is the case of AKT, which contains a PH domain that requires the interaction with PIP3 in order to first unfold the kinase domain due to a conformational change and second interact with PDK1 in the plasma membrane [22]. The presence of the lipid phosphatase PTEN is sufficient to revert this event, by decreasing the levels of PIP3 in the plasma membrane [23].

The PI3K/AKT pathway. Activation of the PI3K/AKT pathway is the result of several stimuli. However, it is commonly associated to RTKs activation through the binding of the regulatory subunit p85 to RTK phosphorylated residues. The lipid kinase activity of the catalytic subunit of PI3K (p110) generates the second messenger PIP3, which recruits different effectors to propagate the downstream signaling cascade. PDK1 and mTORC2 activate AKT by two phosphorylations, allowing this kinase to phosphorylate an array of molecules involved in cell death, cell cycle control, metabolism, and other cellular effects. Here, we have represented some downstream effectors that include, but are not limited to, PRAS40 and TSC2 (negative regulators of mTORC1), BAD (pro-apoptotic BH3-containing protein), p21CIP1 and p27KIP1 (CDK inhibitors and negative regulators of the cell cycle), and FOXO transcription factor (apoptosis and cell cycle transcriptional regulator)
At the plasma membrane, AKT is phosphorylated at the activation loop (T308 in AKT1) by PDK1 [24], and this appears to be sufficient to partially activate the kinase. Additionally, the mTOR complex 2 (mTORC2) phosphorylates AKT at the hydrophobic motif (S473 in AKT1), providing increased activity and/or substrate specificity [25]. mTORC2 is a large protein complex formed by the kinase mTOR and several proteins required for the proper assembly and substrate recognition, such as RICTOR, SIN1, MLST8, DEPTOR, and PROTOR among others [26]. mTORC2 is required for the hydrophobic motif phosphorylation of several protein kinases, including AKT, and some evidences also suggest that mTORC2 is able to phosphorylate AKT and PKC at the turn motif (T450) during protein translation [27, 28]. The mechanism of activation of mTORC2 remains elusive, although it has been proposed that the complex would be activated in the presence of PIP3 due to the presence of a PH domain in the subunit SIN1 [29]. It has been suggested that other kinases might be responsible for the phosphorylation of the hydrophobic motif of AKT; however the cumulative evidence using genetic and pharmacologic tools suggest that mTORC2 is the main upstream kinase, if not the only one.
The AKT family of serine/threonine protein kinases contains three isoforms that are encoded by the genes AKT1, 2, and 3. Activation of AKT is considered a key output of the PI3K pathway due to the large number of substrates interacting with this kinase, including mediators of apoptosis, cell cycle, metabolism, and others that contain the consensus motif RXRXX(S/T) [30, 31]. For instance, AKT is able to phosphorylate and inhibit BAD, a proapoptotic member of the BCL-2 family, and Caspase 9, two main regulators of the mitochondrial apoptotic pathway. It also inhibits the cyclin-dependent kinases (CDKs) inhibitors p21CIP1 and p27KIP, directly related with the inhibition of cell cycle progression. Moreover, AKT can also inhibit the forkehead transcription factors FOXO1, 3, 4 and 6, involved in the transcriptional regulation of several genes including the proapoptotic CD95L, BCL2L11 (BIM), BBC3 (PUMA), CDKN2A (p21CIP1) and CDKN2B (p27KIP) [32]. In addition, AKT can phosphorylate PRAS40 and TSC2, two negative regulators of mTORC1 activity [33, 34].
Similar to mTORC2, mTORC1 is a large protein complex containing the kinase mTOR. However, in this case the complex is associated to the protein RAPTOR, which dictates substrate specificity [35]. mTORC1 senses and responds to environmental cues such as nutrient availability, stress, and mitogens to regulate protein synthesis through a highly orchestrated and complex mechanism. mTOR was originally identified in the early 1990s as a mutated protein that can confer resistance to the growth inhibitory effects of rapamycin in yeast and it was later considered a master regulator of cell growth and metabolism that signals to 4E-binding protein 1 (4EBP1) and 40S ribosomal protein S6 kinase (S6K), which are both important in the physiological control of mRNA translation. In fact, mTORC1 promotes protein synthesis by phosphorylating 4EBP1, which in turn prevents 4EBP1 binding to the eukaryotic initiation factor 4E (eIF4E) , enabling eIF4E to initiate cap-dependent translation. On the other hand, the activation of S6K1 by mTORC1 leads to an increase in mRNA biogenesis and cap-dependent translation. mTORC1 has also been demonstrated to activate RNA Pol I transcription and thus rRNA synthesis through a process involving the protein phosphatase 2A (PP2A) and the transcription initiation factor IA (TIF-IA) [36].
The AKT-mediated phosphorylations of PRAS40 and TSC2 inhibit their activity, leading to an increased mTORC1 signaling [33, 37]. Mechanistically, it has been suggested that phosphorylation of TSC2 by AKT promotes the translocation of the TSC complex away from the lysosomes, where the small GTPase RHEB is found, to activate mTORC1. In the absence of AKT activity, the Tuberous Sclerosis Complex (TSC) complex translocates to the lysosome, where TSC2 acts as a GTPase Activating Protein (GAP) towards RHEB. The resulting GDP-loaded RHEB is unable to activate mTORC1 leading to the inhibition of this complex [38].
6.2 PI3K/AKT Activation in Cancer
Many tumor types are characterized by constitutive activation of the PI3K/AKT pathway; however, here we will focus only on those malignancies for which the blockade of this signaling cascade may be a valid (and rational) therapeutic strategy . The net output of this pathway can be roughly defined as the algebraic sum of several alterations that contribute to activate the downstream effectors of PI3K/AKT/mTOR. Thus, any step from RTKs aberrant phopshorylation to sustained mTORC1 activity may be responsible for increased signaling and, possibly, represents an actionable therapeutic vulnerability.
HER2 overexpression (mainly by gene amplification) is a classical example of upstream activation of the PI3K pathway. It occurs in about 15–20% of breast cancer and, in lesser percentages, in other malignancies such as gastric, endometrial, ovary, salivary, colon. The same may apply for EGFR overexpression in triple negative breast cancer (lacking the expression of hormonal receptors and ERBB2 amplification, TNBC), lung and colon, MET in gastric and lung and FGFR1 in breast.
More downstream, activating mutations of PI3K (either in the regulatory subunit p85 or, more frequently, in the catalytic subunit p110) are responsible for the aberrant PIP3 production and consequent increase of AKT/mTOR signaling . Data from the breast tumor samples analyzed by the TCGA and our internal cohort of patients at Memorial Sloan Kettering Cancer Center show that about 25% of breast cancers exhibit mutations in PIK3CA, the gene encoding the p110α subunit of PI3K [39, 40]. These frequently involve hotspots that are characterized by mutations on the helical (E545K, E542K) and kinase (H1047R) domains of the p110α catalytic subunit of PI3K. Besides breast cancer, PIK3CA is frequently mutated also in head and neck cancer, endometrial cancer, ovarian cancer and hematological diseases.
Loss of expression/function of PTEN and/or INPP4B lipid phosphatases can also occur, also resulting in increase PIP3 production and activation of the pathway [41, 42]. PTEN loss, by genomic deletion of epigenetic silencing, is frequent in TNBC, prostate cancer, glioblastoma, endometrial cancer and stomach cancer.
AKT is also activated directly via mutations and copy-number alterations of the AKT isoforms. The most frequent AKT mutation is found in the PH domain of AKT1 where a glutamic acid is substituted with a lysine residue at amino acid 17 (E17K) [43], resulting in enhanced activity of the kinase. This mutation leads to a constitutive membrane localization of the kinase and increased phosphorylation on T308 and S473 in a PI3K-independent manner [43,44,45]. AKT1E17K mutation is present in several tumor types, but is more frequently detected in invasive breast carcinoma with an overall somatic mutation rate of 2.5% (TCGA results from 1098 patients).
Less common non-hotspot mutations in AKT1 with varying transforming potential have been reported in human breast cancers [46]. AKT3 is the most frequently amplified AKT isoform in breast cancer, and has been mostly studied in the triple-negative subtype in the context of resistance to therapy [47].
Activating mutations have been also reported to occur in mTOR. These alterations can induce resistance to either rapalogs (see below) or mTOR catalytic inhibitors [48,49,50] and can be present in both therapy naïve patients or emerge as a consequence of mTOR blockade.
Canonically, it is thought that activating mutations in different effectors of the PI3K/AKT/mTOR pathway are mutually exclusive. This assertion is, however, based mainly on primary untreated tumors. It is in fact still early to know whether this scenario could change in tumors undergoing pharmacological pressure.
6.3 Inhibitors Targeting the PI3K-AKT Pathway
The first inhibitors of this pathway were isolated more than two decades ago and targeted other kinases such as mTOR and DNA-PK. These agents included wortmannin and LY294002 and, due to the lack of therapeutic windows, their use was confined to the laboratory as tool compounds [51, 52]. The first “modern” inhibitor of the PI3K pathway was BEZ235 (Novartis Pharmaceuticals ). Initially thought to be a pan-PI3K inhibitor, they later discovered it targets also mTOR with equal or higher potency [53, 54]. Because of its strong antitumor activity in several preclinical models, either used as single agent or in combination with other targeted agents, BEZ235 was tested in a number of clinical trials. Unfortunately, despite anecdotal responses in a variety of tumor types, the toxicity profile and poor pharmacokinetic properties lowered the enthusiasm about this molecule [55]. Additional dual PI3K/mTOR inhibitors were successively developed and are now under clinical investigation.
Other molecules more selective for the PI3K enzyme were isolated shortly thereafter. GDC-0941 (Genentech Inc. [56]) and BKM120 (Novartis Pharmaceuticals [57]) target all the isoforms of PI3K (pan-PI3K) and are likely the most studied of this class. GDC-0941 showed good pharmacokinetic properties and has been tested in a variety of solid cancers. Despite anecdotal responses across many tumor types, its future clinical development is currently uncertain due to generally modest antitumor activity [58]. BKM120 has been extensively studied in many preclinical models and clinical settings, spanning from breast to glioblastoma, head and neck, lung and other cancers. A peculiar characteristic of this agent is the blood-brain barrier permeability [59], which on one hand renders it suitable for the treatment of brain tumors or brain metastases, but on the other can cause moderate to severe mood disorders [60]. BKM120 has recently shown interesting antitumor activity in both TNBC and estrogen receptor (ER)-positive breast tumors [60, 61]. Specifically, in TNBC BKM120 seems to induce DNA damage and sensitizes these tumors to the Poly (ADP-ribose) polymerase (PARP) inhibitor olaparib, independently of the BRCA status [62], whereas in ER-positive tumors BKM120 can synergize with the ER degrader fulvestrant. These combinatorial strategies have been tested in the clinic with promising preliminary results and, in the ER-positive setting, BKM120 showed a significantly increase activity in tumors bearing PIK3CA mutations.
Despite these encouraging evidences, the therapeutic window of these compounds is still a major limitation for their clinical development. In the attempt to obviate this bottleneck, pharmaceutical companies engaged in the development of isoform-specific PI3K inhibitors that, if given to the appropriate patients, can elicit strong antitumor activity with tolerable on-target toxicity. The two most promising compounds targeting specifically p110α are BYL719 (Novartis Pharmaceuticals [63]) and GDC-0032 (Genentech Inc. [64]). The latter also targets p110γδ and is more potent against the H1047R and E545K p110α mutants compared to the wild-type isoform. Used as monotherapy, both agents resulted in convincing clinical responses in a number of solid tumors bearing PIK3CA mutations, with particular activity in breast and head and neck cancers [65,66,67]. Not surprisingly, most of the efforts for the clinical advancement of these compounds are indeed focused in the treatment of these two tumor types. In breast cancer patients, both BYL719 and GDC-0032 are being tested in combination with anti-hormonal therapy [68] whereas head and neck patients are treated with single agent or in combination with the anti-EGFR antibody cetuxumab or with radiation.
Other isoform-specific PI3K inhibitors targeting p110β or p110δ have been developed for different indications. One of the most successful is CAL-101 , which was approved by the Food and Drug Administration (FDA) for the treatment of chronic lymphocytic leukemia, small lymphocytic lymphoma and follicular lymphoma [69].
Downstream PI3K, catalytic (e.g. AZD5363, GDC-0068) and allosteric (MK-2206) AKT inhibitors have also been investigated in both preclinical and clinical settings. Although these agents have shown antitumor activity in several PIK3CA-mutant and PTEN-deficient experimental models [70,71,72], their clinical efficacy in non-selected patients have been anecdotal [73,74,75,76]. In patients with tumors bearing the AKT1 E17K mutation, however, the activity of AZD5363 has been remarkable [77].
Finally, the allosteric inhibitor of mTORC1 and the catalytic inhibitor of mTOR have been extensively tested in numerous laboratory models and in clinical trials. Everolimus is certainly the most known and clinically successful allosteric mTORC1 inhibitor, having achieved the approval by the FDA for several solid tumors, including ER-positive breast cancer when combined with aromatase inhibitors [78]. Catalytic mTOR inhibitors have been developed to obviate the paradoxical increase in AKT caused by anti-mTORC1 allosteric agents (see below) and because they can inhibit directly mTORC2 and, as a consequence, AKT. These compounds are being studied in the clinic [79, 80], but their relatively narrow therapeutic window will likely require an accurate patient selection and/or combinations with other therapeutic agents.
6.4 Mechanisms of Resistance to PI3K/AKT/mTOR Inhibitors
Similar to virtually all anticancer drugs, resistance to PI3K/AKT/mTOR inhibitors can be de novo, when cells are intrinsically refractory to the antitumor activity of these agents, or acquired, when tumors initially respond but eventually escape therapy over time.
Typically, intrinsically resistant tumors either carry genomic alterations that prevent or nullify the inhibition of the target or are capable to adapt to the pharmacological stress by triggering the activation of compensatory pathways. Acquisition of resistance, instead, usually occurs via positive selection of tumor clones that are (or become) genetically or epigenetically predisposed to survive even in the presence of PI3K/AKT/mTOR suppression.
In this part, we will discuss the most common mechanisms of resistance to PI3K inhibition described to date.
6.4.1 Resistance Mediated by RTK Activation
Pharmacological inhibition of the PI3K/AKT/mTOR signaling cascade can induce a rapid overexpression/activation of RTKs that, in turn, can fuel downstream signaling pathways and limit the effectiveness of this therapy (Fig. 6.2). More than a decade ago, it was reported that inhibition of mTOR can release a negative feedback phosphorylation of AKT mediated by insulin receptor substrate-1 and result in the activation of the PI3K/AKT pathway [81]. This work, very provocative at that time, pioneered the field in what it turned out to be a common occurrence in targeted therapy: activation of RTKs in response to downstream effectors inhibition. A few years later, three independent investigations converged to the same conclusion that inhibition of the PI3K/AKT pathway leads to overexpression and activation of HER3, HER2 and other RTKs [82,83,84]. The relevance of this cellular adaptation was underscored by the fact that the concomitant inhibition of both PI3K/AKT and the upstream RTKs resulted in superior antitumor effects. As a matter of fact, a number of subsequent studies confirmed the validity of this therapeutic strategy in different preclinical models [72, 85,86,87].

Mechanisms of resistance mediated by kinases. Upon inhibition of PI3K or AKT the pro-survival mechanisms of the cancer cell are challenged, leading in multiple cases to cell death or cell cycle arrest. However, cancer cells can overcome these pharmacological stresses by relying on parallel signaling pathways that lead to the pro-survival phenotype. For instance, upregulation of RTKs is a common effect resulting from the transcriptional activity of FOXO transcription factors upon PI3K/AKT inhibition. Increased RTK signaling has been shown to activate the RAS/RAF/MEK/ERK mitogenic pathway. Two major signaling nodes, TSC2 and FOXO, contain several AKT phosphorylation consensus motifs that can be also phosphorylated by other kinases upon inhibition of PI3K/AKT. Some examples discussed depending on the tumor type include PIM2, SGK1/3, RSK3/4, ERK, and other putative kinases such as the PKC family. Importantly, most of these kinases could also be inhibited by selective pharmacologic inhibitors
Typically, RTK overexpression occurs rapidly in response to PI3K/AKT inhibition. However, in some instances this can also be the result of continuous suppression of the pathway. It is the case of AXL overexpression in response to acquired resistance to the PI3K inhibitor BYL719 in head and neck cancer models [88]. In this work, we describe that the increase expression of this RTK is sufficient to limit the sensitivity to PI3K inhibition by interacting with EGFR and circumventing PI3K pathway blockade. Although AXL expression is likely a multi-resistance mechanism [89], at least another study identified AXL as a causative player for inducing resistance to PI3K inhibition [90].
More than one mechanism triggering RTK overexpression upon PI3K/AKT inhibition is likely to be at play; none of which, however, seems to be attributable to stable genomic amplification of the genes coding these receptors (Fig. 6.2). Chandarlapaty et al. reported that FOXOs transcription factors shuttle to the nucleus of the cells as a result of AKT inhibition and promote RTK expression [82]. Another group reported that the PIM-1 kinase regulates the increase expression of RTKs in response to AKT inhibition in prostate cancer [91]. In any case, the overall output is the activation of downstream signaling that compensates for the pharmacological PI3K/AKT blockade.
6.4.2 Dependency on Other PI3K Isoforms
As mentioned above, current pharmacological approaches in the field of PI3K appear to move towards the inhibition of specific isoforms of this enzyme. For example, inhibitors targeting the PI3Kα have been shown to be more effective in malignancies harboring mutations in PIK3CA [65,66,67], while inhibitors targeting the PI3Kδ isoform have been approved for the treatment of relapsed chronic lymphocytic leukemia [92]. However, a clear disadvantage for the use and development of this class of compounds is that other isoforms could participate in the re-activation of the pathway because of the differential regulation of the PI3K isoforms. In fact, several reports indicate that a crosstalk among the different isoforms occurs in the context of PI3K inhibition resistance.
For instance, it has been shown that the pharmacologic effect of PI3Kα inhibitors is diminished as a result of increased PIP3 accumulation over time. This rebound is particularly evident in HER2-positive cells and is the result of an increased dependency on the p110β isoform, since both inhibition and knockdown of such isoform reduce the levels of PIP3 [93]. Moreover, the combination of p110α and p110β inhibitors exhibits greater anti-tumor effects than single agent treatment in BT474 xenografts. Although the mechanism is not well-understood, some evidences suggest that both RTKs and GPCRs could participate in this phenotype [93]. In line with these findings, a similar phenotype has been reported for PTEN-deficient prostate tumors. Loss of PTEN is linked to increased dependency on different PI3K isoforms in a tissue-specific manner. In the case of prostate malignancies, p110β appears to be the major player when PTEN is lost [94]. On the contrary, thyroid tumors, glomerulonephritis, and hamartoma syndrome appear to depend on both the p110α and p110β isoforms [95].
There is high prevalence of PTEN loss in prostate cancer, hence treatment with PI3K p110β inhibitors has been considered as a possible therapeutic strategy. However, there are compensatory mechanisms as a result of increased p110α signaling. In LNCAP prostate cancer cells, the increased p110α signaling is mediated by IGFR1 [96]. A plausible explanation is that the feedback mediated by S6K phosphorylation of IRS-1 would be critical to re-activate the PI3K pathway through an alternative PI3K isoform.
We have described a genetic mechanism of resistance to p110α inhibitors identified in the metastatic lesions from a patient that relapsed to the treatment with the p110α inhibitor BYL719 [42]. In this case, the pharmacological pressure upon treatment with BYL719 selected for tumor cell populations carrying inactivating mutations and deletion in PTEN. Similar to the observations in prostate cancer, loss of PTEN expression results in increased dependency on the p110β isoform, bypassing the therapeutic effects of BYL719 . Continuous efforts by us and others in genotyping resistant lesions have evidenced that this is a fairly common mechanism of resistance also observed in other p110α inhibitors such as GDC0032 (unpublished results). The combination of isoforms specific inhibitors is efficacious in treating patient-derived xenografts from resistant metastatic lesions and cell lines engineered to express shRNA against PTEN and also pan-PI3K inhibitors have been shown to be active in such context, because of their ability to target both isoforms [42]. The lack of predilection towards a specific isoform could also shed light on the fact that pan-PI3K inhibitors efficacy is not associated with the PIK3CA status in tumors and cell lines. In PTEN-negative breast cancer cell lines, a report has also shown that the mutations D1067Y/A/V in PIK3CB can drive resistance to the pan-PI3K inhibitor GDC0091 as a result of increased affinity to the lipid substrate PIP2 [97].
The strong dependency on the different PI3K isoforms, highlighted by the different mechanisms of resistance described above, suggest that these tumors are particularly addicted to this oncogenic pathway.
6.4.3 Resistance to PI3K/AKT Inhibitors by Ser/Thr Kinases
Because the PI3K pathway activates several Ser/Thr kinases, including AKT and S6K, to propagate the downstream signaling, it is plausible that other related kinases can compensate the inhibitory effects of targeting PI3K by phosphorylating overlapping substrates (Fig. 6.2 and reviewed in [22]). This effect is not exclusive of PI3K and AKT inhibitors, but common in most targeted therapies that block kinases involved in essential cellular processes , such as RAF and MEK, among others [98].
Different experimental approaches have been undertaken in order to identify alternative kinases that drive resistance to PI3K/AKT inhibitors. In general, screening technologies are useful and can address comprehensively the effect of every single kinase of the human kinome in the resistant phenotype. For instance, using open reading frame (ORF) gain-of-function screenings, sensitive cells are transfected or infected with libraries containing the cDNA of different kinases. Resistant clones are then selected upon exposure to therapeutic doses of the drug of interest and finally sequenced to identify the cDNA that drives resistance. Using this approach, it has been shown that the ribosomal protein kinases RSK3 and RSK4 have the ability to drive resistance to the pan-PI3K inhibitors BKM120 and GDC0941, the dual PI3K/mTOR inhibitor BEZ235, and the AKT inhibitor MK2206 [99]. Although the mechanism was not clearly elucidated, it appears that RSK3/4 overexpression could rescue the cap-dependent translation activity even upon mTORC1 inhibition. Because RSK kinases require ERK1/2 phosphorylation at the hydrophobic motif for its maximal activity, it has been suggested that combination with inhibitors of the MEK/ERK pathway would revert the resistant phenotype [100].
Another recent large-scale ORF screening has addressed the role of some kinases in the resistance to BYL719, identifying PIM and PKC kinase isoforms and AKT as putative mediators of resistance to this drug. Overexpression of PIM1 appears to induce resistance, not only to BYL719, but also to the pan-PI3K inhibitor GDC0941 and the AKT inhibitors MK2206 and GDC0068 [101]. Although the effects of the overexpression in driving resistance to these agents is clear, the effects of inhibiting PIM kinases in resistant cell lines are somehow mild, indicating that additional mechanisms of resistance could co-exist. In hematological cancers, the levels of the PIM2 isoform are elevated probably as a result of the activation of the upstream transcription factors STAT, which act as effectors of multiple cytokine receptors commonly hyperactivated in liquid malignancies [102]. This high expression of PIM2 is particularly evident in multiple myeloma and has been suggested to lead to resistance to PI3K inhibitors in these cells. Mechanistically, PIM2 was shown to phosphorylate TSC2 and PRAS40 and activate mTORC1 [103]. Other well-known substrates of PIM kinases are the eIF4E binding protein 1 4EBP1 that would engage into protein synthesis independently of mTORC1, the FOXO transcription factors, and the apoptosis-related protein BAD [104].
Activation of mTORC1 is a key event in the resistance to PI3K inhibitors in many tumors types, probably because of its role downstream of PI3K [105]. Activation of mTORC1 predicts sensitivity to such inhibitors, as tumors that display residual mTORC1 activity upon acute PI3K blockade will not respond significantly to the therapy [106]. The concomitant inhibition of PI3K and mTORC1 has been proven to sensitize resistant cell lines in breast and head and neck cancer [88], proving that mTORC1 plays a causative role in limiting the sensitivity to PI3K inhibitors.
Many Ser/Thr kinases have the ability to regulate the activity of the mTORC1, by either activating or inhibiting different regulators of the complex. Perhaps, the most important negative regulator of mTORC1 is TSC2, which is part of the trimeric TSC complex [107,108,109]. Lack of TSC2 has been shown to activate mTORC1 independently of the PI3K/AKT axis and, most likely, tumors that exhibit downregulation of this protein are refractory to PI3K inhibitors.
TSC2 contains several phosphorylation sites with consensus motifs for kinases involved in the regulation of cell growth and survival. Despite the lack of a crystal structure, it has been speculated that most of the TSC2 phosphorylations would prompt an electrostatic repulsion with the lysosomal membrane due to the negative charges [38].
In general, kinases that phosphorylate and inactivate TSC2 have been linked with resistance to PI3K inhibitors. We have previously discussed RSK and PIM kinases, which phosphorylate TSC2 at highly conserved sites present in residues S939 and T1462, among others that contain the consensus motif RXRXX(S/T), where X is any amino acid [110]. These sites have also been reported to be phosphorylated by the Serum Glucocorticoid-induced kinase (SGK) [111]. We reported that in cell lines that are intrinsically resistant to PI3Kα inhibitors SGK1 is elevated at both protein and mRNA levels, as a result of promoter de-methylation [111]. As expected, these same cell lines were shown to be correlated with resistance to AKT inhibitors. Mass spectrometry analysis of SGK1 kinase assays using TSC2 as a substrate revealed increased phosphorylation sites in the same sites as those previously reported to be phosphorylated by AKT and RSK.
Additionally, the exposure of sensitive cell lines to PI3K and AKT inhibitors results in increased expression of SGK3 at both the mRNA and protein levels by a mechanism that is yet unknown. At the same time, these cells exhibit re-activation of the mTORC1 signaling as a result of TSC2 phosphorylation, measured by using an antibody against the phospho-RXRXX(S/T) motif [112]. SGKs share many other substrates involved in cell survival with AKT, such as the FOXO transcription factors. This may explain the ability of these kinases to promote survival upon PI3K/AKT inhibition [113].
Other kinases that have been proposed to mediate resistance to PI3K and AKT inhibitors in head and neck cancers are PKC’s, a complex family of kinases that are classified between conventional (PKCα, βI,βII, and γ), novel (PKCδ, ε, θ, and η), and atypical (PKCζ, and ι/λ) according to their cofactor requirements (Fig. 6.2). Despite the lack of a precise biochemical mechanism leading to mTORC1 activity, it has been shown that these enzymes are responsible to regulate such complex downstream of EGFR signaling [88]. A plausible explanation is that PKC isoenzymes are able to phosphorylate TSC2, since the consensus motif for the PKC substrates partially overlaps with those described for AKT, SGK, and RSK [22]. This is explained by the fact that these kinases are structurally similar in their kinase domains and belong to the AGC family of kinases, a highly conserved group of enzymes involved in cell growth, survival, and proliferation. The regulation and activation of AGC kinases require three critical phosphorylation events that take place in the turn motif, activation loop, and hydrophobic motif [22]. Phosphorylation at the hydrophobic motif is carried out by different kinases present in the cell. For instance, the hydrophobic motif kinases for all the RSK isoforms is ERK1/2, while for AKT, PKC, and SGK is mTORC2. This phosphorylation is considered to be a priming event, because once phosphorylated it serves as a docking site for PDK1 to phosphorylate the activation loop. PDK1 is a constitutively active kinase because it has the ability to auto-phosphorylate its activation loop at S241 and lacks a hydrophobic motif . In contrast, PDK1 contains a hydrophobic pocket termed the PIF-interacting pocket that serves as a docking site for phosphorylated AGC kinases hydrophobic motif [114]. Inhibition of PDK1 results on the inhibition of most of the AGC kinases, because in the absence of activation motif phosphorylation, these kinases are inactive [115]. Therefore, inhibition of PDK1 could be considered as a strategy to target all these kinases that drive resistance to PI3K and AKT inhibitors, such as RSK, SGK, and PKC. Consistently, we have found that in breast cancer cell lines intrinsically resistant to PI3Kα inhibitors, PDK1 inhibition sensitizes to these therapeutic agents, as a result of SGK1 inhibition (Fig. 6.2 and [111]). Because PDK1 also regulates RSK and PKC, it is tempting to speculate that this therapy would be highly beneficial in cases where resistance is driven by such kinases. Small molecule inhibitors of PDK1 have been reported in the literature, however their efficacy did not match the expectations and, in most cases, phosphorylation of AKT at T308 was used to read-out PDK1 inhibition. However, AKT is the only PDK1 substrate that does not require hydrophobic motif phosphorylation as a priming event and its interaction with PDK1 is the result of a translocation to the plasma membrane upon PIP3 synthesis. When PDK1 is inhibited using small molecule drugs, AKT still has the ability to be phosphorylated by mTORC2 at the hydrophobic motif and uses the high affinity interaction between this phosphorylation and the PDK1 PIF-binding pocket as a mechanism to secure its proper activation. Consistent with this mechanism, inhibition of mTOR or mTORC2 deletion increases the sensitivity to PDK1 inhibitors and it also explains why the combination between PDK1 and PI3K/AKT inhibitors is effective [111].
Based on the current knowledge, other mechanisms of resistance mediated by Ser/Thr kinases could also take place in some contexts. For instance, the 5′ adenosine monophosphate-activated (AMPK) kinase has been shown to phosphorylate TSC2 at S1387 and T1271 leading to an increased GAP activity towards Rheb [116]. In this case, the activation of AMPK would lead to the inactivation of mTORC1, an expected outcome since AMPK is a sensor of low nutrients and high AMP/ATP ratio. AMPK is a trimeric complex formed by the catalytic core (α) and two regulatory subunits (β and γ) and is phosphorylated at the catalytic core by the upstream kinase LKB1 in the presence of AMP [117]. Since LKB1 loss is a fairly common event in cancer, as LKB1 acts as a tumor suppressor, it would be plausible that loss of LKB1 is a biomarker of resistance to PI3K and AKT inhibitors.
Finally, another group of kinases that is becoming attractive as a target in breast cancer is the cyclin-dependent kinase (CDK) family. The CDK4/6 inhibitor palbociclib has been recently approved for the treatment of metastatic ER-positive breast cancer in combination with anti-estrogen therapy [118]. Using a chemical library against PI3K inhibitor resistant cell lines, a study found that the inhibition of CDK4/6 sensitizes PIK3CA-mutated resistant cell lines in vitro and in vivo [119]. Although the exact mechanism by which this combination is beneficial has not been elucidated, it remains possible that different members of the PI3K/AKT/mTOR pathway regulate key players of the cell cycle, such as p16INK4A, p27KIP1, p21CIP1, or CDK4/6 directly.
6.4.4 Hormone Receptor-Dependent Resistance
Mutations in PIK3CA are enriched in breast cancers that express the ER [120]. Therefore, it has been hypothesized that there is an important crosstalk between the PI3K and ER pathways in luminal breast cancers [121]. Several studies have shown that the PI3K pathway is a mechanism of resistance to anti-estrogen therapy, used in the treatment of hormone-dependent breast cancers [122]. Consistent with these observations, the upstream receptor HER2 is also known to drive resistance to these agents and it is clinically considered as a biomarker of resistance to such inhibitors [123]. Consistently, clinical responses are observed when the combination of antiestrogen therapy and the mTORC1 inhibitor everolimus was given to patients whose disease was refractory to previous treatment with the aromatase inhibitors letrozole or anastrozole [78]. Although the addition of everolimus prolongs progression free-survival, the adverse effects observed are considerable. A similar problem has been recently observed when combining anti-estrogen therapy with pan-PI3K inhibitors such as NVP-BKM120 [124], urging the development of selective p110α inhibitors in the clinical setting.
Perhaps, for the relevance of this chapter, the opposite situation, in which the activation of ER signaling drives resistance to PI3K inhibitor, is more relevant. Our laboratory has previously demonstrated that cultured ER-positive breast cancer cell lines exhibit an increased luminal gene expression signature when exposed to therapeutic doses of PI3K and AKT inhibitors [125]. This signature is highly enriched in transcripts that are canonical targets of the ER transcription factor and, consistently, ER activity is increased upon PI3K inhibition. Chromatin-immunoprecipitation followed by high-throughput sequencing (ChIP-Seq) have demonstrated increased binding of ER in a large proportion of genes and have revealed the presence of consensus binding motifs for FOXA1 and PBX1, two cooperative transcription factors previously found to be critical in the estrogen-dependent activation of ER [126]. Although increased levels of ER mRNA and protein have been found, it is plausible that this is the results of a positive feedback loop, since ER expression is known to be regulated by ER itself.
Despite being termed pioneer factors, FOXA1 and PBX1 require the presence of active methylated histone marks (specifically H3K4me1/2) in order to bind DNA. This is particularly interesting because it suggests that the activity of methylases/demethyases can actively modify the accessibility of the ER complex to the chromatin and, in agreement with this hypothesis, KMT2D was found to play a key role in the regulation of this process. Mechanistically, the kinase AKT phosphorylates KMT2D at S1331 inhibiting the methyl-transferase activity of the enzyme, suggesting that AKT activation negatively affects ER transcription (Fig. 6.3). In the presence of PI3K or AKT inhibitors, KMT2D S1331 phosphorylation is lost and the enzymatic activity increased, priming the recruitment of FOXA1, PBX1, and consequently ER, into the designated loci [126]. These studies add supporting evidences for the combination of agents that degrade ER with PI3K inhibitors and open a new avenue for the design of small molecules that target the epigenome.

Epigenetic mechanisms of resistance to PI3K/AKT inhibitors. Resistance to PI3K and AKT inhibitors can also be regulated transcriptionally by several transcription factors. Hormone receptors, such as ER and AR, have been shown to drive resistance to PI3K/AKT inhibitors in breast and prostate luminal cancers. In the case of ER, the regulation of the methylase KMT2D (MLL2) by AKT is required to allow the recruitment of ER cofactors to the chromatin. It is possible that some similarity might exist with the mechanism regulating AR activation upon PI3K inhibition. Other transcription factors involved in the resistance to these inhibitors include Notch, which appears to counteract the inhibitory effects of mTORC1 through the expression of MYC. FOXO has also been shown to interact with β-catenin and promote a gene expression output leading to cell survival and metastasis. The case of BRD4 is less studied, but appears that would regulate the expression of RTK and MYC
In the context of prostate cancer, elegant studies using genetically-engineered mouse models of the disease have proven that PI3K inhibitors also result in the upregulation of the androgen receptor (AR) signaling [127]. As previously discussed, in this malignancy the p110β isoform is responsible for the downstream signaling [94]. The inhibition of PI3K with pan-PI3K inhibitors has an important effect in the activity of AR, a parallelism with ER in breast cancer. However, it remains to be elucidated whether the mechanism is the same. Interestingly, several reports have shown the presence of FOXA1 mutations in this cancer, suggesting that these could cooperate with androgen signaling [128].
6.4.5 Resistance to PI3K/AKT Inhibitors by Transcription Factors
Cancer cells can also become resistant to targeted therapies such as PI3K and AKT inhibitors by changing their transcriptional landscape, a process that is generally mediated by the activity of transcription factors that either activate or repress the expression of target genes (Fig. 6.3).
One of the first studies that systematically addressed resistance to anti-cancer agents demonstrated that both Notch and C-MYC transcription factors are markers of resistance when activated. In the case of Notch signaling, it was shown that overexpression of the intracellular active domain of NOTCH1 (ICN1) was sufficient to cause resistance in different breast cancer cells to BEZ235 [129]. ICN also caused resistance to the PI3K inhibitor PIK90, the mTOR inhibitor PP242 and mTORC1 inhibitor Everolimus, suggesting that the effect of this transcription factor was not specific to PI3K but it was rather driving resistance to the entire PI3K/mTOR pathway. In fact, cells overexpressing ICN have been shown to have similar levels of pS6K and p4EBP1, markers of activation of mTORC1, implying that the resistance is the result of an alternative pathway or a downstream effector, in this case, C-MYC. Knockdown of C-MYC in cells overexpressing ICN results in re-sensitization to these therapeutic agents, thus suggesting that C-MYC is the main downstream effector driving resistance. This is also consistent with the fact that the cap-dependent translation of C-MYC is dependent on mTORC1 [129]. Interestingly, in mouse models of T-cell leukemia, loss of Notch signaling was associated with resistance to the pan-PI3K inhibitor GDC-0941 [130]. Despite the involvement of NOTCH and C-MYC in PI3K/mTORC1 resistance has not been validated in clinical samples, there is a strong rationale to accept these transcription factors as putative modulators of resistance to such therapies.
Among the different transcription factors involved in the PI3K pathway, FOXOs is perhaps the better characterized due to its direct regulation by AKT and its role in cell survival [32]. Upon PI3K signaling, AKT phosphorylates FOXO at several residues and causes the binding with the 14-3-3 proteins in the cytoplasm, releasing it from their DNA-binding sites. Inhibition of AKT promotes a rapid de-phosphorylation and translocation to the nucleus, where FOXO’s engage into their transcriptional program. There are different transcription factors that have been shown to interact with and inhibit FOXO transcription factors, such as SMAD, FOXG1, PGC-1, and β-catenin, and could be potential mediators of resistance to PI3K inhibitors by blocking the cell death and cell cycle arrest mediated by FOXO’s. As a matter of fact, β-catenin drives resistance to PI3K and AKT inhibitors in colorectal cancers by modulating the transcriptional output of FOXO into driving metastasis [131].
6.4.6 Other Mechanisms Involved in the Resistance to PI3K/AKT Inhibitors
There are a number of novel topics that have become interesting for the treatment of cancer such as epigenetic inhibitors and nanoparticles in drug delivery . Although these fields are still recent, they have an interesting potential in the field of PI3K inhibition. For example, in order to decrease the systemic exposure of PI3K or AKT inhibitors (and therefore increase the therapeutic window of these agents), it has been show that the tumor-specific delivery of PI3K inhibitors can be achieved using nanoparticles. The advantages of this method are the reduction of secondary effects, such as hyperglycemia, and as a consequence the ability to combine other drugs that, in a systemic regime, would have severe adverse effects [132].
In the field of epigenetic inhibitors, two main targeting strategies have been explored in the context of PI3K therapy. For example, the use of histone deacetylase (HDAC) inhibitors has been demonstrated to be effective in preclinical models of medulloblastoma when combined with the PI3K inhibitor BKM120 [133]. Moreover, inhibitors of the bromodomain and extra terminal domain (BET) proteins also synergize with PI3K inhibitors when combined in breast cancer cells and transgenic mouse models [134]. Mechanistically, BRD4 appears to be involved in the transcriptional machinery required to upregulate RTKs upon PI3K/AKT inhibition, hence treatment with BRD4 inhibitors such as JQ1 would abrogate this effect.
Additional work will be required to identify the critical nodes of the epigenome that are required to target in order to modulate the response to PI3K and AKT inhibitors and pinpoint the specific tumor types that would benefit from such combinations.
6.5 Conclusions
The cumulative evidences regarding the role of the PI3K/AKT pathway in human cancers have prompted the development of inhibitors that specifically target this key signaling node. Despite the importance of PI3K and AKT in tumor biology, the clinical results have been less promising than initially anticipated. This is in part due to the multiple mechanisms of resistance that tumors exhibit to overcome these therapeutic agents. Both clinical and preclinical data suggest that pharmacologic combinations are required to increase the effectiveness of such compounds and, accordingly, clinical trials testing these combinations are undergoing. In general, it appears that the resistance to PI3K and AKT inhibitors is mediated by alternative kinase signaling that leads to the activation of downstream effectors, the most important of which is mTORC1. This signaling compensation stresses the importance of PI3K for the cells and could be explained from an evolutionary point of view as an attempt to maintain active a major pathway that regulates cell growth and survival. In fact, it is not surprising that many of the kinases involved in PI3K and AKT inhibitor resistance are part of the same family, the AGC kinases. Additional data has also revealed the importance of ER signaling in the resistance to these agents in breast cancer and clinical data could be supporting the combination with hormonal therapy soon. It will also be interesting to characterize novel mechanisms of resistance and targets such as epigenetic modulators and transcriptional regulators.
In summary, inhibitors of the PI3K/AKT pathway have a great potential in the clinical setting, but only when administered to the appropriate patients and in the right combination.
Abbreviations
- AGC:
-
Protein Kinase A, G, And C Kinase Family
- AKT:
-
RAC-Alpha Serine/Threonine-Protein Kinase
- AMP:
-
Adenosine Monophosphate
- AMPK:
-
AMP-Dependent Protein Kinase
- ARF:
-
ADP Ribosylation Factors
- ATP:
-
Adenosine Triphosphate
- BAD:
-
BCL2 Associated Agonist of Cell Death
- BCL2:
-
B-Cell Lymphoma 2
- BRD4:
-
Bromodomain And Extra Terminal Domain 4
- Cdc42:
-
Cell Division Cycle 42
- DEPTOR:
-
DEP Domain-Containing Mtor-Interacting Protein
- Eif4e:
-
Eukaryotic Translation Initiation Factor 4E
- ER:
-
Estrogen Receptor
- ERK:
-
Extracellular Signal–Regulated Kinase
- FOXA1:
-
Forkhead Box A1
- FOXG1:
-
Forkhead Box G1
- FOXO:
-
Forkhead Box O
- GAP:
-
GTP-Ase Activating Protein
- GDP:
-
Guanosine Diphosphate
- GTP:
-
Guanosine Triphosphate
- H3k4me1/2:
-
Histone 3 Lysine 4 Mono−/Di-Methylated
- HER2:
-
Human Epidermal Growth Factor Receptor 2
- IGFR1:
-
Insulin-Like Growth Factor 1 Receptor 1
- IRS1:
-
Insulin Receptor Substrate 1
- KMT2D:
-
Histone-Lysine N-Methyltransferase 2D
- LKB1:
-
Liver Kinase B1
- MAPK:
-
Mitogen-Activated Protein Kinases
- MEK:
-
MAPK/ERK Kinase
- MLST8:
-
Mammalian Lethal with SEC13 Protein 8
- MYC:
-
V-Myc Avian Myelocytomatosis Viral Oncogene Homolog
- P16ink4a:
-
16 kDa Inhibitor of Cyclin-Dependent Kinase Type 4A
- P21CIP1:
-
21 kDa CDK-Interacting Protein 1
- P27KIP:
-
27 kDa Kinase Inhibitor Protein
- PBX1:
-
Pre-B-Cell Leukemia Transcription Factor 1
- PDK1:
-
3-Phosphoinositide Dependent Protein Kinase-1
- PGC-1:
-
Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1
- PIF:
-
PDK1-Interacting Fragment
- PIM:
-
Proviral Integration Site for Moloney Murine Leukemia Virus-1
- PKC:
-
Protein Kinase C
- PRAS40:
-
Proline-Rich Akt Substrate of 40 kDa
- PROTOR:
-
Protein Observed with Rictor-1
- PTEN:
-
Phosphatase and Tensin Homolog
- Rac1:
-
Ras-Related C3 Botulinum Toxin Substrate 1
- RAF:
-
Rapidly Accelerated Fibrosarcoma
- RAPTOR:
-
Regulatory Associated Protein of MTOR Complex 1
- RHEB:
-
Ras Homolog Enriched in Brain
- RICTOR:
-
Rapamycin-Insensitive Companion of MTOR
- RSK:
-
90 kDa Ribosomal S6 Kinase
- SH2:
-
Src Homology 2
- SIN1:
-
Stress-Activated Map Kinase-Interacting Protein 1
- SMAD:
-
Mothers Against Decapentaplegic Homolog
- TSC2:
-
Tuberous Sclerosis Complex Protein 2
- VPS15:
-
Vacuolar Protein Sorting 15
- VPS34:
-
Vacuolar Protein Sorting 34
References
Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–34.
Casaletto JB, McClatchey AI. Spatial regulation of receptor tyrosine kinases in development and cancer. Nat Rev Cancer. 2012;12:387–400.
Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15:7–24.
Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–62.
Okkenhaug K, Graupera M, Vanhaesebroeck B. Targeting PI3K in cancer: impact on tumor cells, their protective stroma, angiogenesis, and immunotherapy. Cancer Discov. 2016;6:1090–105.
Vadas O, Burke JE, Zhang X, Berndt A, Williams RL. Structural basis for activation and inhibition of class I phosphoinositide 3-kinases. Sci Signal. 2011;4:re2.
Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13:195–203.
Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140–56.
Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, Vogelstein B, Gabelli SB, Amzel LM. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318:1744–8.
Burke JE, Williams RL. Synergy in activating class I PI3Ks. Trends Biochem Sci. 2015;40:88–100.
Renner O, Carnero A. Mouse models to decipher the PI3K signaling network in human cancer. Curr Mol Med. 2009;9:612–25.
Graupera M, Guillermet-Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J, Cutillas PR, Smith AJ, Ridley AJ, Ruhrberg C, Gerhardt H, Vanhaesebroeck B. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature. 2008;453:662–6.
Foukas LC, Claret M, Pearce W, Okkenhaug K, Meek S, Peskett E, Sancho S, Smith AJ, Withers DJ, Vanhaesebroeck B. Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature. 2006;441:366–70.
Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11:329–41.
Clayton E, Bardi G, Bell SE, Chantry D, Downes CP, Gray A, Humphries LA, Rawlings D, Reynolds H, Vigorito E, Turner M. A crucial role for the p110delta subunit of phosphatidylinositol 3-kinase in B cell development and activation. J Exp Med. 2002;196:753–63.
Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, Pearce W, Meek SE, Salpekar A, Waterfield MD, Smith AJ, Vanhaesebroeck B. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science. 2002;297:1031–4.
Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. 2000;287:1049–53.
Patrucco E, Notte A, Barberis L, Selvetella G, Maffei A, Brancaccio M, Marengo S, Russo G, Azzolino O, Rybalkin SD, Silengo L, Altruda F, Wetzker R, Wymann MP, Lembo G, Hirsch E. PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell. 2004;118:375–87.
Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, Bouabe H, Scudamore CL, Hancox T, Maecker H, Friedman L, Turner M, Okkenhaug K, Vanhaesebroeck B. Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature. 2014;510:407–11.
Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008;9:99–111.
Bayascas JR, Leslie NR, Parsons R, Fleming S, Alessi DR. Hypomorphic mutation of PDK1 suppresses tumorigenesis in PTEN(+/−) mice. Curr Biol. 2005;15:1839–46.
Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9–22.
Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–96.
Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–9.
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101.
Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–76.
Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008;27:1919–31.
Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, Lowry C, Newton AC, Mao Y, Miao RQ, Sessa WC, Qin J, Zhang P, Su B, Jacinto E. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008;27:1932–43.
Liu P, Gan W, Chin YR, Ogura K, Guo J, Zhang J, Wang B, Blenis J, Cantley LC, Toker A, Su B, Wei W. PtdIns(3,4,5)P3-Dependent activation of the mTORC2 kinase complex. Cancer Discov. 2015;5:1194–209.
Clark AR, Toker A. Signalling specificity in the Akt pathway in breast cancer. Biochem Soc Trans. 2014;42:1349–55.
Gonzalez E, McGraw TE. The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle. 2009;8:2502–8.
Webb AE, Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci. 2014;39:159–69.
Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–15.
Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–62.
Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–75.
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93.
Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37:217–22.
Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015;25:545–55.
Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, Butler A, Cheverton A, Gamble J, Hinton J, Jia M, Jayakumar A, Jones D, Latimer C, Lau KW, McLaren S, McBride DJ, Menzies A, Mudie L, Raine K, Rad R, Chapman MS, Teague J, Easton D, Langerod A, Oslo Breast Cancer C, Lee MT, Shen CY, Tee BT, Huimin BW, Broeks A, Vargas AC, Turashvili G, Martens J, Fatima A, Miron P, Chin SF, Thomas G, Boyault S, Mariani O, Lakhani SR, van de Vijver M, van ’t Veer L, Foekens J, Desmedt C, Sotiriou C, Tutt A, Caldas C, Reis-Filho JS, Aparicio SA, Salomon AV, Borresen-Dale AL, Richardson AL, Campbell PJ, Futreal PA, Stratton MR. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–4.
Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70.
Gewinner C, Wang ZC, Richardson A, Teruya-Feldstein J, Etemadmoghadam D, Bowtell D, Barretina J, Lin WM, Rameh L, Salmena L, Pandolfi PP, Cantley LC. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16:115–25.
Juric D, Castel P, Griffith M, Griffith OL, Won HH, Ellis H, Ebbesen SH, Ainscough BJ, Ramu A, Iyer G, Shah RH, Huynh T, Mino-Kenudson M, Sgroi D, Isakoff S, Thabet A, Elamine L, Solit DB, Lowe SW, Quadt C, Peters M, Derti A, Schegel R, Huang A, Mardis ER, Berger MF, Baselga J, Scaltriti M. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor. Nature. 2015;518:240–4.
Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellogg G, Touchman J, Patel K, Mousses S, Bittner M, Schevitz R, Lai MH, Blanchard KL, Thomas JE. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–44.
Kumar A, Purohit R. Cancer associated E17K mutation causes rapid conformational drift in AKT1 pleckstrin homology (PH) domain. PLoS One. 2013;8:e64364.
Landgraf KE, Pilling C, Falke JJ. Molecular mechanism of an oncogenic mutation that alters membrane targeting: Glu17Lys modifies the PIP lipid specificity of the AKT1 PH domain. Biochemistry. 2008;47:12260–9.
Yi KH, Axtmayer J, Gustin JP, Rajpurohit A, Lauring J. Functional analysis of non-hotspot AKT1 mutants found in human breast cancers identifies novel driver mutations: implications for personalized medicine. Oncotarget. 2013;4:29–34.
Chin YR, Yoshida T, Marusyk A, Beck AH, Polyak K, Toker A. Targeting Akt3 signaling in triple-negative breast cancer. Cancer Res. 2014;74:964–73.
Wagle N, Grabiner BC, Van Allen EM, Hodis E, Jacobus S, Supko JG, Stewart M, Choueiri TK, Gandhi L, Cleary JM, Elfiky AA, Taplin ME, Stack EC, Signoretti S, Loda M, Shapiro GI, Sabatini DM, Lander ES, Gabriel SB, Kantoff PW, Garraway LA, Rosenberg JE. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discov. 2014;4:546–53.
Wagle N, Grabiner BC, Van Allen EM, Amin-Mansour A, Taylor-Weiner A, Rosenberg M, Gray N, Barletta JA, Guo Y, Swanson SJ, Ruan DT, Hanna GJ, Haddad RI, Getz G, Kwiatkowski DJ, Carter SL, Sabatini DM, Janne PA, Garraway LA, Lorch JH. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N Engl J Med. 2014;371:1426–33.
Rodrik-Outmezguine VS, Okaniwa M, Yao Z, Novotny CJ, McWhirter C, Banaji A, Won H, Wong W, Berger M, de Stanchina E, Barratt DG, Cosulich S, Klinowska T, Rosen N, Shokat KM. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature. 2016;534:272–6.
Norman BH, Shih C, Toth JE, Ray JE, Dodge JA, Johnson DW, Rutherford PG, Schultz RM, Worzalla JF, Vlahos CJ. Studies on the mechanism of phosphatidylinositol 3-kinase inhibition by wortmannin and related analogs. J Med Chem. 1996;39:1106–11.
Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem. 1994;269:5241–8.
Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chene P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, Garcia-Echeverria C. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–63.
Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, Maira M, Garcia-Echeverria C, Parra JL, Arribas J, Baselga J. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68:8022–30.
Pongas G, Fojo T. BEZ235: when promising science meets clinical reality. Oncologist. 2016;21:1033–4.
Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, Friedman LS, Hayes A, Hancox TC, Kugendradas A, Lensun L, Moore P, Olivero AG, Pang J, Patel S, Pergl-Wilson GH, Raynaud FI, Robson A, Saghir N, Salphati L, Sohal S, Ultsch MH, Valenti M, Wallweber HJ, Wan NC, Wiesmann C, Workman P, Zhyvoloup A, Zvelebil MJ, Shuttleworth SJ. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-t hieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51:5522–32.
Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, Schnell C, Guthy D, Nagel T, Wiesmann M, Brachmann S, Fritsch C, Dorsch M, Chene P, Shoemaker K, De Pover A, Menezes D, Martiny-Baron G, Fabbro D, Wilson CJ, Schlegel R, Hofmann F, Garcia-Echeverria C, Sellers WR, Voliva CF. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012;11:317–28.
Sarker D, Ang JE, Baird R, Kristeleit R, Shah K, Moreno V, Clarke PA, Raynaud FI, Levy G, Ware JA, Mazina K, Lin R, Wu J, Fredrickson J, Spoerke JM, Lackner MR, Yan Y, Friedman LS, Kaye SB, Derynck MK, Workman P, de Bono JS. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2015;21:77–86.
Liu X, Ide JL, Norton I, Marchionni MA, Ebling MC, Wang LY, Davis E, Sauvageot CM, Kesari S, Kellersberger KA, Easterling ML, Santagata S, Stuart DD, Alberta J, Agar JN, Stiles CD, Agar NY. Molecular imaging of drug transit through the blood-brain barrier with MALDI mass spectrometry imaging. Sci Rep. 2013;3:2859.
Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, Demanse D, De Buck SS, Ru QC, Peters M, Goldbrunner M, Baselga J. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30:282–90.
Rodon J, Brana I, Siu LL, De Jonge MJ, Homji N, Mills D, Di Tomaso E, Sarr C, Trandafir L, Massacesi C, Eskens F, Bendell JC. Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Investig New Drugs. 2014;32:670–81.
Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzman M, Grueso J, Rodriguez O, Calvo MT, Aura C, Diez O, Rubio IT, Perez J, Rodon J, Cortes J, Ellisen LW, Scaltriti M, Baselga J. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012;2:1036–47.
Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, Kauffmann A, Guthy D, Erdmann D, De Pover A, Furet P, Gao H, Ferretti S, Wang Y, Trappe J, Brachmann SM, Maira SM, Wilson C, Boehm M, Garcia-Echeverria C, Chene P, Wiesmann M, Cozens R, Lehar J, Schlegel R, Caravatti G, Hofmann F, Sellers WR. Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014;13:1117–29.
Ndubaku CO, Heffron TP, Staben ST, Baumgardner M, Blaquiere N, Bradley E, Bull R, Do S, Dotson J, Dudley D, Edgar KA, Friedman LS, Goldsmith R, Heald RA, Kolesnikov A, Lee L, Lewis C, Nannini M, Nonomiya J, Pang J, Price S, Prior WW, Salphati L, Sideris S, Wallin JJ, Wang L, Wei B, Sampath D, Olivero AG. Discovery of 2-{3-[2-(1-isopropyl-3-methyl-1H-1,2-4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1 ,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): a beta-sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor activity. J Med Chem. 2013;56:4597–610.
Juric D, Rodon J, Gonzalez-Angulo AM, Burris HA, Bendell J, Berlin JD, Middleton MR, Bootle D, Boehm M, Schmitt A, Rouyrre N, Quadt C, Baselga J. Abstract CT-01: BYL719, a next generation PI3K alpha specific inhibitor: preliminary safety, PK, and efficacy results from the first-in-human study. Cancer Res. 2012;72(8). https://doi.org/10.1158/1538-7445.AM2012-CT-01.
Juric D, Krop I, Ramanathan RK, Xiao J, Sanabria S, Wilson TR, Choi Y, Parmar H, Hsu J, Baselga J, Von Hoff DD. Abstract LB-64: GDC-0032, a beta isoform-sparing PI3K inhibitor: results of a first-in-human phase Ia dose escalation study. Cancer Res. 2013;73(8). https://doi.org/10.1158/1538-7445.AM2013-LB-64.
Juric D, Krop I, Ramanathan RK, Wilson TR, Ware JA, Sanabria Bohorquez S, Savage H, Sampath D, Salphati L, Lin R, Jin H, Parmar H, Hsu JY, Von Hoff DD, Baselga J. Phase I dose escalation study of taselisib (GDC-0032), an oral PI3K Inhibitor, in patients with advanced solid tumors. Cancer Discov. 2017;7(7):704–15.
Mayer IA, Abramson V, Formisano L, Balko JM, Estrada MV, Sanders M, Juric D, Solit D, Berger MF, Won H, Li Y, Cantley LC, Winer EP, Arteaga CL. A Phase Ib Study of Alpelisib (BYL719), a PI3Kalpha-specific Inhibitor, with Letrozole in ER+/HER2- Negative Metastatic Breast Cancer. Clin Cancer Res. 2017;23(1):26–34.
Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, Byrd JC, Tyner JW, Loriaux MM, Deininger M, Druker BJ, Puri KD, Ulrich RG, Giese NA. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117:591–4.
Davies BR, Greenwood H, Dudley P, Crafter C, Yu DH, Zhang J, Li J, Gao B, Ji Q, Maynard J, Ricketts SA, Cross D, Cosulich S, Chresta CC, Page K, Yates J, Lane C, Watson R, Luke R, Ogilvie D, Pass M. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther. 2012;11:873–87.
Sangai T, Akcakanat A, Chen H, Tarco E, Wu Y, Do KA, Miller TW, Arteaga CL, Mills GB, Gonzalez-Angulo AM, Meric-Bernstam F. Biomarkers of response to Akt inhibitor MK-2206 in breast cancer. Clin Cancer Res. 2012;18:5816–28.
Tao JJ, Castel P, Radosevic-Robin N, Elkabets M, Auricchio N, Aceto N, Weitsman G, Barber P, Vojnovic B, Ellis H, Morse N, Viola-Villegas NT, Bosch A, Juric D, Hazra S, Singh S, Kim P, Bergamaschi A, Maheswaran S, Ng T, Penault-Llorca F, Lewis JS, Carey LA, Perou CM, Baselga J, Scaltriti M. Antagonism of EGFR and HER3 enhances the response to inhibitors of the PI3K-Akt pathway in triple-negative breast cancer. Sci Signal. 2014;7:ra29.
Yan Y, Serra V, Prudkin L, Scaltriti M, Murli S, Rodriguez O, Guzman M, Sampath D, Nannini M, Xiao Y, Wagle MC, Wu JQ, Wongchenko M, Hampton G, Ramakrishnan V, Lackner MR, Saura C, Roda D, Cervantes A, Tabernero J, Patel P, Baselga J. Evaluation and clinical analyses of downstream targets of the Akt inhibitor GDC-0068. Clin Cancer Res. 2013;19:6976–86.
Banerji U. Results of two phase I multicenter trials of AZD5363, an inhibitor of AKT1, 2 and 3: Biomarker and early clinical evaluation in Western and Japanese patients with advanced solid tumors. AACR 104th Annual Meeting. Cancer Res. 2013;73(8 Suppl). https://doi.org/10.1158/1538-7445.AM2013-LB-66.
Han HS. A phase I study of the AKT inhibitor (MK-2206) with concurrent trastuzumab and lapatinib in patients with HER2-positive solid tumors. ASCO Annual Meeting. J Clin Oncol. 2011;29(15 Suppl):3028. https://doi.org/10.1200/jco.2011.29.15_suppl.3028.
Michalarea, V. (2015) “BEECH”, a phase I/II study of the AKT inhibitor AZD5363 combined with paclitaxel in patients with advanced or metastatic breast cancer: results from the dose-finding study, including quantitative assessment of circulating tumor DNA as a surrogate for response/resistance. AACR. In: Proceedings of the 106th Annual meeting of the american association for cancer research (Abstract No. CT331).
Hyman DM, Smyth LM, Donoghue MTA, Westin SN, Bedard PL, Dean EJ, Bando H, El-Khoueiry AB, Perez-Fidalgo JA, Mita A, Schellens JHM, Chang MT, Reichel JB, Bouvier N, Selcuklu SD, Soumerai TE, Torrisi J, Erinjeri JP, Ambrose H, Barrett J-C, Dougherty B, Foxley A, Lindemann JPO, McEwen R, Pass M, Schiavon G, Berger MF, Chandarlapaty S, Solit DB, Banerji U, Baselga J, Taylor BS. AKT inhibition in solid tumors with AKT1 mutations. J Clin Oncol. 2017;35(20):2251–9. https://doi.org/10.1200/JCO.2017.73.0143.
Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, Deleu I, Perez A, Bachelot T, Vittori L, Xu Z, Mukhopadhyay P, Lebwohl D, Hortobagyi GN. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520–9.
Banerji U. First-in-human phase I trial of the dual mTORC1 and mTORC2 inhibitor AZD2014 in solid tumors. 2012 ASCO Annual Meeting. J Clin Oncol. 2012;30(15 Suppl):3004.
Varga A. Phase I expansion trial of an oral TORC1/TORC2 inhibitor (CC-223) in advanced solid tumors. 2013 ASCO Annual Meeting. J Clin Oncol. 2013;31(15):2606.
O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8.
Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71.
Serra V, Scaltriti M, Prudkin L, Eichhorn PJ, Ibrahim YH, Chandarlapaty S, Markman B, Rodriguez O, Guzman M, Rodriguez S, Gili M, Russillo M, Parra JL, Singh S, Arribas J, Rosen N, Baselga J. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011;30:2547–57.
Chakrabarty A, Sanchez V, Kuba MG, Rinehart C, Arteaga CL. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc Natl Acad Sci U S A. 2012;109:2718–23.
Wong MH, Xue A, Julovi SM, Pavlakis N, Samra JS, Hugh TJ, Gill AJ, Peters L, Baxter RC, Smith RC. Cotargeting of epidermal growth factor receptor and PI3K overcomes PI3K-Akt oncogenic dependence in pancreatic ductal adenocarcinoma. Clin Cancer Res. 2014;20:4047–58.
Garcia-Garcia C, Ibrahim YH, Serra V, Calvo MT, Guzman M, Grueso J, Aura C, Perez J, Jessen K, Liu Y, Rommel C, Tabernero J, Baselga J, Scaltriti M. Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to anti-HER2 therapy. Clin Cancer Res. 2012;18:2603–12.
Rexer BN, Chanthaphaychith S, Dahlman K, Arteaga CL. Direct inhibition of PI3K in combination with dual HER2 inhibitors is required for optimal antitumor activity in HER2+ breast cancer cells. Breast Cancer Res. 2014;16:R9.
Elkabets M, Pazarentzos E, Juric D, Sheng Q, Pelossof RA, Brook S, Benzaken AO, Rodon J, Morse N, Yan JJ, Liu M, Das R, Chen Y, Tam A, Wang H, Liang J, Gurski JM, Kerr DA, Rosell R, Teixido C, Huang A, Ghossein RA, Rosen N, Bivona TG, Scaltriti M, Baselga J. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell. 2015;27:533–46.
Scaltriti M, Elkabets M, Baselga J. Molecular pathways: AXL, a membrane receptor mediator of resistance to therapy. Clin Cancer Res. 2016;22:1313–7.
Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, Nilsson MB, Gudikote J, Tran H, Cardnell RJ, Bearss DJ, Warner SL, Foulks JM, Kanner SB, Gandhi V, Krett N, Rosen ST, Kim ES, Herbst RS, Blumenschein GR, Lee JJ, Lippman SM, Ang KK, Mills GB, Hong WK, Weinstein JN, Wistuba II, Coombes KR, Minna JD, Heymach JV. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19:279–90.
Cen B, Mahajan S, Wang W, Kraft AS. Elevation of receptor tyrosine kinases by small molecule AKT inhibitors in prostate cancer is mediated by Pim-1. Cancer Res. 2013;73:3402–11.
Fruman DA, Rommel C. PI3Kdelta inhibitors in cancer: rationale and serendipity merge in the clinic. Cancer Discov. 2011;1:562–72.
Costa C, Ebi H, Martini M, Beausoleil SA, Faber AC, Jakubik CT, Huang A, Wang Y, Nishtala M, Hall B, Rikova K, Zhao J, Hirsch E, Benes CH, Engelman JA. Measurement of PIP3 levels reveals an unexpected role for p110beta in early adaptive responses to p110alpha-specific inhibitors in luminal breast cancer. Cancer Cell. 2015;27:97–108.
Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM, Zhao JJ. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776–9.
Berenjeno IM, Guillermet-Guibert J, Pearce W, Gray A, Fleming S, Vanhaesebroeck B. Both p110alpha and p110beta isoforms of PI3K can modulate the impact of loss-of-function of the PTEN tumour suppressor. Biochem J. 2012;442:151–9.
Schwartz S, Wongvipat J, Trigwell CB, Hancox U, Carver BS, Rodrik-Outmezguine V, Will M, Yellen P, de Stanchina E, Baselga J, Scher HI, Barry ST, Sawyers CL, Chandarlapaty S, Rosen N. Feedback suppression of PI3Kalpha signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kbeta. Cancer Cell. 2015;27:109–22.
Nakanishi Y, Walter K, Spoerke JM, O’Brien C, Huw LY, Hampton GM, Lackner MR. Activating mutations in PIK3CB confer resistance to PI3K inhibition and define a novel oncogenic role for p110beta. Cancer Res. 2016;76:1193–203.
Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13:928–42.
Serra V, Eichhorn PJ, Garcia-Garcia C, Ibrahim YH, Prudkin L, Sanchez G, Rodriguez O, Anton P, Parra JL, Marlow S, Scaltriti M, Perez-Garcia J, Prat A, Arribas J, Hahn WC, Kim SY, Baselga J. RSK3/4 mediate resistance to PI3K pathway inhibitors in breast cancer. J Clin Invest. 2013;123:2551–63.
Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol. 2008;9:747–58.
Le X, Antony R, Razavi P, Treacy DJ, Luo F, Ghandi M, Castel P, Scaltriti M, Baselga J, Garraway LA. Systematic functional characterization of resistance to PI3K inhibition in breast cancer. Cancer Discov. 2016;6:1134–47.
Nawijn MC, Alendar A, Berns A. For better or for worse: the role of Pim oncogenes in tumorigenesis. Nat Rev Cancer. 2011;11:23–34.
Lu J, Zavorotinskaya T, Dai Y, Niu XH, Castillo J, Sim J, Yu J, Wang Y, Langowski JL, Holash J, Shannon K, Garcia PD. Pim2 is required for maintaining multiple myeloma cell growth through modulating TSC2 phosphorylation. Blood. 2013;122:1610–20.
Keane NA, Reidy M, Natoni A, Raab MS, O’Dwyer M. Targeting the Pim kinases in multiple myeloma. Blood Cancer J. 2015;5:e325.
Ilagan E, Manning BD. Emerging role of mTOR in the response to cancer therapeutics. Trends Cancer. 2016;2:241–51.
Elkabets M, Vora S, Juric D, Morse N, Mino-Kenudson M, Muranen T, Tao J, Campos AB, Rodon J, Ibrahim YH, Serra V, Rodrik-Outmezguine V, Hazra S, Singh S, Kim P, Quadt C, Liu M, Huang A, Rosen N, Engelman JA, Scaltriti M, Baselga J. mTORC1 inhibition is required for sensitivity to PI3K p110alpha inhibitors in PIK3CA-mutant breast cancer. Sci Transl Med. 2013;5:196ra199.
Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–34.
Li Y, Corradetti MN, Inoki K, Guan KL. TSC2: filling the GAP in the mTOR signaling pathway. Trends Biochem Sci. 2004;29:32–8.
Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–90.
Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A. 2004;101:13489–94.
Castel P, Ellis H, Bago R, Toska E, Razavi P, Carmona FJ, Kannan S, Verma CS, Dickler M, Chandarlapaty S, Brogi E, Alessi DR, Baselga J, Scaltriti M. PDK1-SGK1 signaling sustains AKT-independent mTORC1 activation and confers resistance to PI3Kalpha inhibition. Cancer Cell. 2016;30:229–42.
Bago R, Sommer E, Castel P, Crafter C, Bailey FP, Shpiro N, Baselga J, Cross D, Eyers PA, Alessi DR. The hVps34-SGK3 pathway alleviates sustained PI3K/Akt inhibition by stimulating mTORC1 and tumour growth. EMBO J. 2016;35:1902–22.
Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol Cell Biol. 2001;21:952–65.
Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15:161–70.
Najafov A, Sommer EM, Axten JM, Deyoung MP, Alessi DR. Characterization of GSK2334470, a novel and highly specific inhibitor of PDK1. Biochem J. 2011;433:357–69.
Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90.
Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–62.
Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, Harbeck N, Lipatov ON, Walshe JM, Moulder S, Gauthier E, Lu DR, Randolph S, Dieras V, Slamon DJ. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. 2016;375:1925–36.
Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, Lockerman EL, Pollack SF, Liu M, Li X, Lehar J, Wiesmann M, Wartmann M, Chen Y, Cao ZA, Pinzon-Ortiz M, Kim S, Schlegel R, Huang A, Engelman JA. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. 2014;26:136–49.
Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, Bowlby R, Shen H, Hayat S, Fieldhouse R, Lester SC, Tse GM, Factor RE, Collins LC, Allison KH, Chen YY, Jensen K, Johnson NB, Oesterreich S, Mills GB, Cherniack AD, Robertson G, Benz C, Sander C, Laird PW, Hoadley KA, King TA, Network TR, Perou CM. Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015;163:506–19.
Zhou W, Slingerland JM. Links between oestrogen receptor activation and proteolysis: relevance to hormone-regulated cancer therapy. Nat Rev Cancer. 2014;14:26–38.
Van Tine BA, Crowder RJ, Ellis MJ. ER and PI3K independently modulate endocrine resistance in ER-positive breast cancer. Cancer Discov. 2011;1:287–8.
Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, Beijersbergen RL, Valero V, Seoane J, Bernards R, Baselga J. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008;68:9221–30.
Mayer IA, Abramson VG, Isakoff SJ, Forero A, Balko JM, Kuba MG, Sanders ME, Yap JT, Van den Abbeele AD, Li Y, Cantley LC, Winer E, Arteaga CL. Stand up to cancer phase Ib study of pan-phosphoinositide-3-kinase inhibitor buparlisib with letrozole in estrogen receptor-positive/human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol. 2014;32:1202–9.
Bosch A, Li Z, Bergamaschi A, Ellis H, Toska E, Prat A, Tao JJ, Spratt DE, Viola-Villegas NT, Castel P, Minuesa G, Morse N, Rodon J, Ibrahim Y, Cortes J, Perez-Garcia J, Galvan P, Grueso J, Guzman M, Katzenellenbogen JA, Kharas M, Lewis JS, Dickler M, Serra V, Rosen N, Chandarlapaty S, Scaltriti M, Baselga J. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci Transl Med. 2015;7:283ra251.
Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, Dickler MN, Scaltriti M, Leslie CS, Armstrong SA, Baselga J. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science. 2017;355:1324–30.
Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, Scardino PT, Rosen N, Sawyers CL. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–86.
Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, Nickerson E, Chae SS, Boysen G, Auclair D, Onofrio RC, Park K, Kitabayashi N, MacDonald TY, Sheikh K, Vuong T, Guiducci C, Cibulskis K, Sivachenko A, Carter SL, Saksena G, Voet D, Hussain WM, Ramos AH, Winckler W, Redman MC, Ardlie K, Tewari AK, Mosquera JM, Rupp N, Wild PJ, Moch H, Morrissey C, Nelson PS, Kantoff PW, Gabriel SB, Golub TR, Meyerson M, Lander ES, Getz G, Rubin MA, Garraway LA. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–9.
Muellner MK, Uras IZ, Gapp BV, Kerzendorfer C, Smida M, Lechtermann H, Craig-Mueller N, Colinge J, Duernberger G, Nijman SM. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat Chem Biol. 2011;7:787–93.
Dail M, Wong J, Lawrence J, O’Connor D, Nakitandwe J, Chen SC, Xu J, Lee LB, Akagi K, Li Q, Aster JC, Pear WS, Downing JR, Sampath D, Shannon K. Loss of oncogenic Notch1 with resistance to a PI3K inhibitor in T-cell leukaemia. Nature. 2014;513:512–6.
Tenbaum SP, Ordonez-Moran P, Puig I, Chicote I, Arques O, Landolfi S, Fernandez Y, Herance JR, Gispert JD, Mendizabal L, Aguilar S, Ramon y Cajal S, Schwartz S Jr, Vivancos A, Espin E, Rojas S, Baselga J, Tabernero J, Munoz A, Palmer HG. Beta-catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat Med. 2012;18:892–901.
Mizrachi A, Shamay Y, Shah J, Brook S, Soong J, Rajasekhar VK, Humm JL, Healey JH, Powell SN, Baselga J, Heller DA, Haimovitz-Friedman A, Scaltriti M. Tumour-specific PI3K inhibition via nanoparticle-targeted delivery in head and neck squamous cell carcinoma. Nat Commun. 2017;8:14292.
Pei Y, Liu KW, Wang J, Garancher A, Tao R, Esparza LA, Maier DL, Udaka YT, Murad N, Morrissy S, Seker-Cin H, Brabetz S, Qi L, Kogiso M, Schubert S, Olson JM, Cho YJ, Li XN, Crawford JR, Levy ML, Kool M, Pfister SM, Taylor MD, Wechsler-Reya RJ. HDAC and PI3K Antagonists cooperate to inhibit growth of MYC-driven medulloblastoma. Cancer Cell. 2016;29:311–23.
Stratikopoulos EE, Dendy M, Szabolcs M, Khaykin AJ, Lefebvre C, Zhou MM, Parsons R. Kinase and BET inhibitors together clamp inhibition of PI3K signaling and overcome resistance to therapy. Cancer Cell. 2015;27:837–51.
Acknowledgments
We would like to thank the Breast Cancer Research Foundation and the Geoffrey Beene Cancer Research Center. Pau Castel is a Fellow of the Jane Coffin Childs Memorial Fund. We apologize for the impossibility to cite every author who has contributed to this field of inquiry.
Conflict of Interest
No potential conflicts of interest were disclosed.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Castel, P., Scaltriti, M. (2018). Mechanisms of Resistance to PI3K and AKT Inhibitors. In: Yarden, Y., Elkabets, M. (eds) Resistance to Anti-Cancer Therapeutics Targeting Receptor Tyrosine Kinases and Downstream Pathways. Resistance to Targeted Anti-Cancer Therapeutics, vol 15. Springer, Cham. https://doi.org/10.1007/978-3-319-67932-7_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-67932-7_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-67930-3
Online ISBN: 978-3-319-67932-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)