
Abstract
Over the last decade, tryptophan catabolism has been firmly established as a powerful mechanism of innate and adaptive immune tolerance. The catabolism of tryptophan is a central pathway maintaining homeostasis by preventing autoimmunity or immunopathology that would result from uncontrolled and overreacting immune responses. This is driven by the key and rate-limiting enzymes indoleamine-2,3-dioxygenase 1 (IDO1) and tryptophan-2,3-dioxygenase 2 (TDO), resulting in local depletion of tryptophan, while tryptophan catabolites accumulate, including kynurenine and its derivatives, depending on the presence of downstream enzymes in the kynurenine pathway. These metabolic modifications result in a local microenvironment that is profoundly immunosuppressive, as a result of various mechanisms whose respective role remains incompletely characterized. Drugs targeting this pathway, specifically IDO1, are already in clinical trials with the aim at reverting cancer-induced immunosuppression. Recent studies have demonstrated favorable pharmacokinetics profiles for first-generation (Indoximod NLG8189) and second-generation IDO1 inhibitors (INCB024360 and NLG919). Targeting tryptophan catabolism in combination with additional methods of therapy may improve efficacy of cancer immunotherapy. These methods include, but are not limited to vaccination, adoptive cellular therapy, checkpoint inhibitor blockade, and cyclooxygenase-2 (COX2) inhibition. Over the last decade, there has been a considerable increase in our understanding of the regulation and downstream mediators of tryptophan metabolism. This detailed understanding will expand opportunities to interfere with the pathway therapeutically on multiple levels. The object of this chapter is to highlight current and past key findings that implicate tryptophan catabolism as an important mediator of cancer immunity and discuss the development of multiple therapeutic targets.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Indoleamine 2,3-dioxyenase (IDO)
- Tryptophan
- Kynurenine
- Clinical trial
- Immune tolerance
- Cancer
- Immunotherapy
9.1 Introduction
Over the last decade, tryptophan catabolism has been firmly established as a powerful mechanism of innate and adaptive immune tolerance. The catabolism of tryptophan is a central pathway maintaining homeostasis by preventing autoimmunity or immunopathology that would result from uncontrolled and overreacting immune responses. This is driven by the key and rate-limiting enzymes indoleamine-2,3-dioxygenase 1 (IDO1) and tryptophan-2,3-dioxygenase 2 (TDO), resulting in local depletion of tryptophan, while tryptophan catabolites accumulate, including kynurenine and its derivatives, depending on the presence of downstream enzymes in the kynurenine pathway. These metabolic modifications result in a local microenvironment that is profoundly immunosuppressive, as a result of various mechanisms whose respective role remains incompletely characterized. Drugs targeting this pathway, specifically IDO1, are already in clinical trials with the aim at reverting cancer-induced immunosuppression. Recent studies have demonstrated favorable pharmacokinetics profiles for first-generation (Indoximod NLG8189) and second-generation IDO1 inhibitors (INCB024360 and NLG919). Targeting tryptophan catabolism in combination with additional methods of therapy may improve efficacy of cancer immunotherapy. These methods include, but are not limited to vaccination, adoptive cellular therapy, checkpoint inhibitor blockade, and cyclooxygenase-2 (COX2) inhibition. Over the last decade, there has been a considerable increase in our understanding of the regulation and downstream mediators of tryptophan metabolism. This detailed understanding will expand opportunities to interfere with the pathway therapeutically on multiple levels. The object of this chapter is to highlight current and past key findings that implicate tryptophan catabolism as an important mediator of cancer immunity and discuss the development of multiple therapeutic targets.
9.2 Tryptophan Catabolism and the Role of IDO
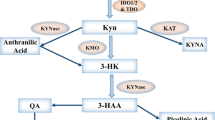
Indoleamine 2,3 dioxygenase has be identified as a major source of immune regulation. The enzymatic activity of IDO is mediated by catabolism of the essential amino L-tryptophan (Trp) into L-kynurenine (Kyn) via the kynurenine pathway metabolic cascade [1, 2] (Fig. 9.1). The amino acid tryptophan is required by all forms of life for protein synthesis and additional significant metabolic functions [2]. The first and rate-limiting step of kynurenine catabolism is facilitated by three distinct enzymes: indoleamine 2,3-dioxygenase (IDO1), indoleamine 2,3-dioxygenase 2 (IDO2), and tryptophan 2,3-dioxygenase 2 (TDO) [1,2,3,4,5,6,7,8]. Of this family of genes, IDO1 is a potent immunoregulatory enzyme. Gene expression of the non-reductant IDO2 is not induced by interferons that regulate IDO1 gene expression [4, 9, 10]. Moreover, the structurally distinct enzyme, TDO is a hepatic enzyme that catabolizes tryptophan selectively expressed in the liver [6, 11]. Stress-related glucocorticoids, as opposed to inflammatory signals induce the TDO gene [12]. These data suggest that unlike IDO1, IDO2 and TDO may have distinct functional roles [13]. Therefore, IDO1 is the major focus of this editorial, and will be referred to as “IDO”, unless otherwise specified in this review.

Metabolic modulation result in a local microenvironment becoming profoundly immunosuppressive. Indoleamine 2,3-dioxygenase (IDO1) is an enzyme that catalyzes the essential amino acid, tryptophan, in the initial rate-limiting step along the kynurenine pathway. IDO is expressed by tumor cells, tolerogenic dendritic cells (DCs) and myeloid derived suppresser cells (MDSCs) mediate tryptophan deprivation leading to T cell cycle arrest; and the immunosuppressive catabolite, kynurenine mediates differentiation of CD4+ T cells into regulatory T cells, as well as T cell cycle arrest
IDO is encoded by the INDO gene and has a molecular weight of 45 kDa. Additionally, the gene contains 10 exons and is located on chromosome 8 in human and mice [14, 15]. The IDO gene is tightly regulated by inflammatory mediators such as type I and type II interferons, lipopolysaccharide (LPS), and tumor necrosis factor-alpha (TNF-α). Likewise, the IDO gene promoter region is comprised of the interferon stimulated response element (ISRE) and interferon-gamma activated sequence (GAS) element. Upregulation of the IDO gene is expressed in a wide range of tissues and cellular subsets, such as in the placenta during pregnancy, in a variety of tissues during an infection, transplantation [16], autoimmunity, and cancer [16,17,18,19]. In regulatory dendritic cells, IDO expression is upregulated by negative signaling from cytotoxic T lymphocyte protein 4 (CTLA-4) and the glucocorticoid induced tumor necrosis factor receptor (GITR) ligand [20, 21]. This reverse negative signaling was described for the CTLA-4 receptor present on T regulatory cells that binds to its ligands, CD80 and CD86 on dendritic cells and mediated induction of IDO in an interferon-gamma (IFN-γ) dependent manner [20, 22]. Likewise, the toll-like receptor 9 (TLR9) ligand, CpG oligodeoxynucleotides (ODNs) mediate the induction of IDO expression in dendritic cell subsets via type I interferon signaling pathway [23]. Moreover, T regulatory cells are generated by human plasmacytoid dendritic cells via IDO catabolism of tryptophan [24, 25]. These signaling pathways are critical for establishing tolerance to tumor antigens. Additionally, inflammatory stimuli from the signal transducer and activator of transcription 3 (STAT3) stimulated IDO upregulation in myeloid-derived suppressor cells [26]. The initial indication of a role for IDO in immune tolerance was demonstrated when it was ascertained that IDO enzyme activity mediated immune privilege and was required to prevent T cell-driven rejection of allogeneic fetuses during pregnancy in mice [27]. Inhibition of the enzymatic activity of IDO by 1-methyl-tryptophan resulted in the rejection of allogeneic, as opposed to syngeneic fetuses. These findings proposed that IDO-medicated depletion of local tryptophan prevented T cell responses to the fetus by limiting the amino acid to proliferating T cells [28]. Likewise, studies performed in the context of IDO genetic knockout mouse models or IDO pharmacological inhibitors have confirmed that the enzyme plays a crucial role in immune tolerance and inflammatory tumorigenesis [27, 29, 30].
IDO modulates of immune responses by two distinct mechanisms. These mechanisms include immune cells deprivation of tryptophan which leads to activation of stress response pathways [19, 28, 31], and through the generation of kynurenine pathway suppressive metabolites [32]. T cells are highly sensitive to tryptophan depletion [33]. A decreased in local tryptophan availability leads to the accumulation of uncharged tryptophan transfer ribonucleic acids (tRNAs). Subsequently, an increase in uncharged tRNAs results in the induction of an integrated stress response kinase general control non-derepressible 2 (GCN2) in T cells. GCN2 kinase acts as a molecular sensor in T cells, and upon activation triggers a stress response program that can result in cell cycle arrest, T cell differentiation, or apoptosis [34]. Amino acid withdrawal modifies the phenotype of dendritic cells and macrophages in GCN2 kinase-dependent manner [35,36,37]. IDO expressing macrophages acquire the capacity to suppress natural killer (NK) cells and CD8 T cells proliferation. Moreover, GCN2 activation in CD8 effector T cells has been proved to inhibit cell proliferation and induce anergy following T cell receptor stimulation by down regulating the TCR zeta-chain [38, 39]. Likewise, naïve CD4+ T cells GCN2 appears to be important for the differentiation and activation of regulatory T cells (Tregs) [40]. We show that these IDO+ pDCs directly activate resting CD4+CD25+Foxp3+ Tregs for potent suppressor activity. Additionally, data from in vitro studies suggest that tryptophan-derived catabolites also inhibit the ability of T cell and natural killer cell proliferation [41].
A second mechanism of IDO-mediated immune suppression is dependent on the accumulation of the tryptophan catabolites, kynurenine. The kynurenine pathway generates several metabolites, including L-kynurenine, kynurenic acid, anthranilic acid, 3 hydroxykynurenine, 3-hydroxyanthranilic acid, quinolinic acid, picolinic acid and nicotinamide adenine dinucleotide [42]. IDO expressing cells release L-kynurenine, 3-hydroxyanthranilic acid (3-HAA) and other tryptophan metabolites into circulation. As a result, innate and adaptive immune responses are modulated in the cells that sense these amino acid catabolites. Moreover, kynurenine is an endogenous ligand of the aryl hydrocarbon receptor (AHR) [43]. The AHR is a ligand-activated member of the basic helix-loop-helix (bHLH) family of transcription factors, originally identified as a receptor for environmental xenobiotic toxin 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Upon ligation, the inactive form of AHR dissociates from cytoplasmic chaperone molecules, associates with the AHR nuclear translocator and binds to the target gene in the nucleus to modulate gene transcription [43, 44]. Tumors select for IDO-mediated tryptophan catabolism that produces kynurenine that binds to AHR and assist the tumor to evade immune surveillance. Moreover, an elevated level of AHR present in tumors is a poor prognostic factor in patients [43, 45]. The role of AHR in immune regulation, inflammation and tumorigenesis genetic studies was defined in genetic mice studies. These experiments confirmed that regulatory T cells (Tregs) generation differentiation from naïve CD4+CD25− T cells is dependent on kynurenine activation of the AHR in the present of transforming growth factor beta (TGF-β) [46]. Activation of AHR mediated immune suppression, by promoting FoxP3 differentiation, suppression effector T cell anti-tumor immunity, and decreasing immunogenicity of dendritic cells [11, 43, 47]. However, effect is not seen in AHR null T cells. Specifically, Mezrich et al. demonstrated that naïve T cells exposed to KYN leads to mRNA transcription of the downstream targets of AHR activation, cytochrome P450 family 1 subfamily A polypeptide 1 (CYP1a1), and cytochrome P450 family 1 subfamily B polypeptide 1 (CYP1b1) [46]. AHR plays a role in the generation of Th17 cells in vitro and in vivo [48, 49]. Kynurenine metabolites, via the IDO pathway, also tip the balance of Th17 cell to Treg cells, in favor of Treg cells by suppression of pro-inflammatory Th17 pathway and promoting Treg differentiation [50]. In addition, activation by endogenous AHR ligands, exogenous ligand, 6-formlindolo [3,2-b] carbazole (FICZ), mediates Th17 cell formation upon AHR activation [51]. Further work is needed to understand the mechanisms by which AHR ligation by KYN regulation transcriptional regulation of immune suppressive factors (Fig. 9.2).

Ligation and activation of the aryl hydrocarbon receptor regulate gene transcription. IDO catabolism of tryptophan leads to the generation of kynurenine (Kyn), an endogenous ligand of the aryl hydrocarbon receptor (AhR). The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that modulates gene transcription. The AhR is present in its inactive form accompanied by Heat Shock Protein 90 chaperones in the cytoplasm of the cell. Following ligation, the cytosolic Kyn-AhR complex undergoes transformation and translocates to the nucleus. In the nucleus the AhR forms a heterodimer with the AhR nuclear translocator (ARNT) which interacts with the core-binding motif of the responsive elements located in regulatory regions of AhR target genes and gene transcription occurs
IDO is a potent immunoregulatory enzyme [17, 33, 39], and a major source of immune suppression within the tumor microenvironment of ovarian cancer and other tumor types [19, 52] (Fig. 9.3). Constitutive IDO expression in human tumors creates a suppressive microenvironment due to the depletion of typtophan and the synthesis of immune suppressive kynurenine metabolites [53, 54]. Elevated levels of IDO enzyme activity correlates with reduced frequency of tumor infiltrating T lymphocytes in murine cancer models [19]. Studies investigating the role of TILs in ovarian cancer, demonstrated that the presence of intra-epithelial CD8+ infiltrating lymphocytes favor overall survival in epithelial ovarian cancer (EOC) patients [52]. A meta-analysis study confirmed a significant survival advantage associated with tumor-infiltrating lymphocytes (TILs) in several tumor types [55,56,57,58,59]. Additionally, these studies indicate that: (1) the beneficial prognostic effect of CD8+ tumor infiltrating lymphocytes in human EOC is negatively affected by CD4+CD25+FOXP3+ Treg cells; (2) IDO expression in human ovarian cancer correlates with poor prognosis and decreases overall survival; (3) IDO mediated depletion of tryptophan results in suppression of T cell responses in ovarian cancer patients; and (4) tumor-derived antigen specific CD8+ T lymphocytes from human ovarian cancer demonstrate impaired effector function and enriched expression of the inhibitory molecule, programmed cell death–1 (PD-1), a marker of T cell exhaustion [60]. These results suggest that the regulatory influences of IDO via arrest of T cell proliferation, expansion of CD4+ Tregs, or promotion of T cell exhaustion may dampen the efficacy of tumor reactive effector CD8+ T lymphocytes. Collectively, these findings provide impetus to characterize the molecular and cellular basis of how IDO induced tryptophan catabolism leads to T cell exhaustion; and test novel strategies for overcoming IDO mediated immune tolerance.

Indoleamine 2,3-dioxyenase is a potent immunoregulatory enzyme within the tumor microenvironment. Immune suppression within the tumor microenvironment is orchestrated by the functions of the cells present that promote tumor growth. Tumors have diverted an immune suppressive role of IDO1 to their own benefit in order to continue to evade immune attack. IDO1 expression by the tumor and tolerogenic dendritic cells (DCs) is associated with a reduced effector T cell function, and an increased frequency of regulatory T cells (Tregs) at the tumor site. Additional immunosuppressive mechanisms within the tumor microenvironment are mediated by: myeloid derived suppressor cells (MDSC), macrophages with an ‘M2’ phenotype, arginase (Arg1), inducible nitric oxide synthase (iNOS), nitric oxide (NO), prostaglandin E2 (PGE2), interleukin-10 (IL-10), transforming growth factor βeta (TGF-β), and programmed cell death ligand 1 (PD-L1)
9.3 Cancer Immunotherapy Targeting Tryptophan Catabolism
Several tumor types express IDO, providing them the ability to evade anti-tumor immune surveillance and facilitate immune escape. Consequently, this has motivated the development of IDO inhibitors. Preclinical data demonstrated that a competitive inhibitor of IDO, 1-methyl-tryptophan [61] prevented anergic T cells and suppressed tumor growth in murine carcinoma models. There are multiple approaches targeting tryptophan catabolism in cancer across different levels: (1) blocking IDO expression utilizing strategies which interfere with upstream pathways that regulation IDO transcription and/or translation; (2) enzymatic inhibition by suppressing IDO and/or TDO enzyme activity; (3) and combinatory therapies which target inhibition of tryptophan catabolism with other therapies.
9.4 Inhibitors of IDO Expression
A recent study has linked the therapeutic effects of imatinib, a tyrosine-kinase inhibitor drug, in gastrointestinal stromal tumors (GIST) to inhibition of IDO1 expression, which is driven by the oncogenic KIT signaling. Treatment of experimental tumors with imatinib suppressed IDO expression, resulting in the reversal of IDO1-mediated immunosuppression and thus activation of T effector cells and suppression of Tregs [62]. This finding is striking, because it lends credence to the notion that IDO1 targeting may already be providing a benefit in the context of imatinib treatment of GIST and advocate for conduction preclinical studies in tumor models involving immunocompetent hosts. Based on the association between CTLA-4 and IDO, a rational therapeutic consequence of this observation is to combine KIT blockade with an anti-CTLA-4 approach in GIST, which is currently tested in a clinical trial NCT01643278 (source http://clinicaltrials.gov). Based on COX2-IDO regulatory connection, antitumor implications have been analyzed in a preclinical model of breast cancer treatment with cyclooxygenase-2 (COX2) inhibitors [63], illustrating its therapeutic relevance in principle. T regulatory cell functions disrupted by COX2 inhibition have also been found to be mediated by IDO1 inhibition, possibly contributing to the anticancer properties of COX2 inhibitors [64]. One interesting aspect of IDO is that its enzyme appears to be spontaneously recognized by specific CD8+ T cells that are present in humans [65]. Along this line are preclinical and clinical studies targeting IDO-expressing cells by a peptide vaccine, where early evidence was obtained of long-lasting disease stabilization and a partial response against liver metastasis in metastatic lung cancer patients vaccinated with an IDO-derived peptide, in the absence of notable toxicity [66].
9.5 IDO1 Enzyme Inhibitors
The IDO1 blocking agent 1-methyl-tryptophan (1-MT) has been shown to have significant ability to inhibit IDO1 activity, and co-operate with chemotherapy in mediating regression of established tumors in murine models [30]. In the murine models, the effect of 1-MT was lost in immunodeficient Rag1-knockout (Rag1-KO) hosts, indicating that the antitumor effect of 1-MT was immune mediated. The overall effects of 1-MT include enhanced T cell responses against tumor antigen, allograft antigen, and autoantigens in vivo [67, 68]. Additionally, by blocking IDO, 1-MT inhibits the production of tryptophan catabolites such as kynurenine that have been shown to directly reduce T cell and NK cell proliferation [31, 69,70,71]. 1-MT is a mixture of the two racemic isoforms 1-methyl-D-tryptophan (D-1-MT) and 1-methyl-L-tryptophan (L-1-MT). In previous studies, while the L stereoisomer of 1-MT was a more potent inhibitor of IDO, the D stereoisomer was shown to be less active in inhibiting IDO1, but to have superior antitumor activity and to be more effective in inhibiting IDO-expressing tolerogenic DCs in pre-clinical models [72]. In addition, IDO2 was reported to be preferentially targeted by D-1-MT [73]. These findings have been challenged by other groups [7, 74,75,76]. Nevertheless, D-1-MT is being developed clinically as an IDO-inhibitor (indoximod, NLG8189) for the treatment of several cancers with the aim at reversing cancer-associated immunosuppression.
It is reported that in addition or alternatively to direct IDO1 inhibition, D-1-MT may interfere with transcellular tryptophan transport [77]. Tryptophan transport system L is commonly overexpressed in tumor cells and seems to be the main route for transcellular tryptophan transport in T cells [78]. Myeloid antigen presenting cells have been shown to express an additional high-affinity tryptophan transport mechanism [78] thus being able to take up tryptophan efficiently in a low tryptophan containing microenvironment. Hence, under tryptophan depleting conditions, such as cancer, it seems likely that T cells are more affected by tryptophan starvation and tryptophan is efficiently being shifted toward tryptophan consuming cells. D-1-MT may act as a tryptophan mimetic, provide via mammalian target of rapamycin (mTOR), an intracellular tryptophan sufficiency signal to the cell, maintain mTOR activity also in T cells, thus restore their activity [79]. These findings identify mTOR suppression as an IDO1 effector mechanism and D-1-MT acts as a high-potency tryptophan mimetic in reversing mTOR inhibition and autophagic induction by IDO1 through tryptophan transport interfering. Since L-1-MT is principally capable of exerting the same effects, it is not yet entirely clear why D-1-MT is more effective in restoring T cell activity under physiological conditions [79].
In addition to directly inhibiting IDO enzymatic activity, second-generation IDO1 inhibitors such as INCB024360 and NLG919 have entered clinical trials. These new inhibitors may have a more favorable pharmacokinetic profile. Phase I clinical trials with these orally available compounds have demonstrated safety and indicated biological efficacy based on serum parameters demonstrating reversal of tryptophan depletion and kynurenine accumulation, complementing in vitro experiments data [80].
In order to improve the efficacy of cancer immunotherapy, it has become clear that clinical studies targeting tryptophan catabolism should combine with other anti-cancer therapies, based on preclinical animal works. For instance, in spontaneously arising aggressive mammary tumors in the MMTV-neu/HER2 transgenic mouse model of breast cancer, 1-MT had little effect on tumor outgrowth but it could dramatically empower the efficacy of a variety of chemotherapeutic agents, triggering stable regressions of otherwise mainly recalcitrant tumors [30]. As a result, clinical phase I trials have combined indoximod with chemotherapy agents, including the first-in-man phase I trial of indoximod and docetaxel therapy for solid metastaic tumors (NCT02835729; NCT01792050; NCT02077881; NCT01191216; NCT01042535) [81, 82]. A recent preclinical study suggested that IDO1 is a critical resistance mechanism attenuating the efficacy of immunotherapies by antibodies disrupting CTLA-4, PD-1 or GITR, and that 1-MT can safely leverage the antitumor properties of these antibodies [22]. Trials combining indoximod (NCT02073123) or INCB024360 (NCT01604889) with the anti-CTLA-4 antibody ipilimumab in patients with melanoma are underway. Conceptually, and supported by preclinical studies, IDO1 inhibition may enhance the efficacy of active cancer vaccines as it may break cancer-induced tolerance. Two phase II studies are currently evaluating this combination approach (NCT01560923; NCT01042535 and NCT01302821) [83].
Additionally, in an on-going trial by the Cancer Immunotherapy Trials Network (NCT02042430), to determine the magnitude by which INCB024360 alters the frequency of tumor-infiltrating CD8+ T cells once IDO blockade is administered prior to surgery in patients whom are newly diagnosed Stage III-IV with epithelial ovarian, fallopian tube, or primary peritoneal cancer (Fig. 9.4). In this study, ovarian cancer patients receive INCB024360 orally for up to 3 weeks and undergo surgery at the completion of treatment with the IDO1 inhibitor. In another approach, IDO inhibitors are combined with other immunotherapeutic strategies. In one example, the Roswell Park Cancer Institute-University of Pittsburgh Cancer Institute Ovarian Cancer SPORE investigators testing whether concomitant inhibition of IDO-mediated immune tolerance and vaccination against NY-ESO-1 will enhance the generation of durable anti-tumor CD8+ T cells in patients (Fig. 9.5). The successful completion of these studies will result in the generation of critical data that will bring about further evaluation of IDO blockade to relieve Treg cell-mediated immune tolerance, promote conditions that favor durable host immunity, and prolong disease free survival in ovarian cancer patients (http://trp.cancer.gov/spores/abstracts/roswell_ovarian.htm). An overview of IDO1 inhibitors in clinical trial is described in (Table 9.1).

Clinical Trial, NCT02042430, study scheme utilizing an IDO1 inhibitor. This is a pilot clinical trial which studies indoleamine 2,3-dioxygenase (IDO1) inhibitor, INCB024360, before surgery in newly diagnosed stage III-IV epithelial ovarian, fallopian tube, or primary peritoneal cancer patients. Presented here is the clinical trial scheme which includes the patient eligibility criteria, number of enrollment, IDO1 inhibitor treatment dosage and schedule, and tissue samples that are collected pre- and post IDO1 inhibitor treatment, as well as after surgery

NCT02166905 study scheme. This clinical trial is designed to test whether inhibition of IDO will augment vaccine induced immune responses in patients with ovarian cancer in remission. Presented here is the clinical trial scheme which includes the patient eligibility criteria, the study size, the treatment cycles schedule, and the tissue samples that are collected pre- and post treatment
9.6 Conclusions
The tryptophan catabolism is a central driver of malignant development and progression. It acts in tumor, stromal and immune cells to support pathogenic inflammatory processes that engender immune tolerance to tumor antigens. Mechanistic investigations have defined the aryl hydrocarbon receptor, the master metabolic regulator mTOR1 and the stress kinase GCN2 as key effector signaling elements for tryptophan metabolism. The opportunity to interfere with tryptophan metabolism have expanded well beyond inhibiting IDO1 due to the advances in understanding the regulation, as well as the cellular and molecular targets of this pathway. There is an interest in pharmacological targeting of TDO for cancer immunotherapy. Downstream effectors such as AHR and tryptophan transport mechanisms will have to be taken into consideration for future therapeutic strategies. Since it is questionable whether these IDO1/TDO inhibitors will be effective by themselves, rational combinations with already available and/or yet to be identified immunomodulators, such as cancer vaccines or checkpoint inhibitors, deserve thorough basic research and preclinical studies.
References
Lob S, et al. Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: can we see the wood for the trees? Nat Rev Cancer. 2009;9(6):445–52.
Moffett JR, Namboodiri MA. Tryptophan and the immune response. Immunol Cell Biol. 2003;81(4):247–65.
Ball HJ, et al. Characterization of an indoleamine 2,3-dioxygenase-like protein found in humans and mice. Gene. 2007;396(1):203–13.
Metz R, et al. Novel tryptophan catabolic enzyme IDO2 is the preferred biochemical target of the antitumor indoleamine 2,3-dioxygenase inhibitory compound D-1-methyl-tryptophan. Cancer Res. 2007;67(15):7082–7.
Mehler AH, Knox WE. The conversion of tryptophan to kynurenine in liver. II. The enzymatic hydrolysis of formylkynurenine. J Biol Chem. 1950;187(1):431–8.
Knox WE, Mehler AH. The conversion of tryptophan to kynurenine in liver. I. The coupled tryptophan peroxidase-oxidase system forming formylkynurenine. J Biol Chem. 1950;187(1):419–30.
Lob S, et al. IDO1 and IDO2 are expressed in human tumors: levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol Immunother. 2009;58(1):153–7.
Thackray SJ, Mowat CG, Chapman SK. Exploring the mechanism of tryptophan 2,3-dioxygenase. Biochem Soc Trans. 2008;36(Pt 6):1120–3.
Ball HJ, et al. Indoleamine 2,3-dioxygenase-2; a new enzyme in the kynurenine pathway. Int J Biochem Cell Biol. 2009;41(3):467–71.
Fatokun AA, Hunt NH, Ball HJ. Indoleamine 2,3-dioxygenase 2 (IDO2) and the kynurenine pathway: characteristics and potential roles in health and disease. Amino Acids. 2013;45(6):1319–29.
Platten M, et al. Cancer Immunotherapy by Targeting IDO1/TDO and Their Downstream Effectors. Front Immunol. 2014;5:673.
Kanai M, et al. Tryptophan 2,3-dioxygenase is a key modulator of physiological neurogenesis and anxiety-related behavior in mice. Mol Brain. 2009;2:8.
Croitoru-Lamoury J, et al. Interferon-gamma regulates the proliferation and differentiation of mesenchymal stem cells via activation of indoleamine 2,3 dioxygenase (IDO). PLoS One. 2011;6(2):e14698.
Kadoya A, et al. Gene structure of human indoleamine 2,3-dioxygenase. Biochem Biophys Res Commun. 1992;189(1):530–6.
Najfeld V, et al. Localization of indoleamine 2,3-dioxygenase gene (INDO) to chromosome 8p12→p11 by fluorescent in situ hybridization. Cytogenet Cell Genet. 1993;64(3–4):231–2.
Brandacher G, Margreiter R, Fuchs D. Implications of IFN-gamma-mediated tryptophan catabolism on solid organ transplantation. Curr Drug Metab. 2007;8(3):273–82.
Mellor AL, Munn DH. Tryptophan catabolism and regulation of adaptive immunity. J Immunol. 2003;170(12):5809–13.
Curti A, et al. The role of indoleamine 2,3-dioxygenase in the induction of immune tolerance: focus on hematology. Blood. 2009;113(11):2394–401.
Uyttenhove C, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9(10):1269–74.
Grohmann U, et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol. 2002;3(11):1097–101.
Grohmann U, et al. Reverse signaling through GITR ligand enables dexamethasone to activate IDO in allergy. Nat Med. 2007;13(5):579–86.
Holmgaard RB, et al. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. 2013;210(7):1389–402.
Mellor AL, et al. Cutting edge: CpG oligonucleotides induce splenic CD19+ dendritic cells to acquire potent indoleamine 2,3-dioxygenase-dependent T cell regulatory functions via IFN Type 1 signaling. J Immunol. 2005;175(9):5601–5.
Chen W, et al. The indoleamine 2,3-dioxygenase pathway is essential for human plasmacytoid dendritic cell-induced adaptive T regulatory cell generation. J Immunol. 2008;181(8):5396–404.
Fallarino F, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. 2003;4(12):1206–12.
Yu J, et al. Noncanonical NF-kappaB activation mediates STAT3-stimulated IDO upregulation in myeloid-derived suppressor cells in breast cancer. J Immunol. 2014;193(5):2574–86.
Munn DH, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281(5380):1191–3.
Mellor AL, Munn DH. Tryptophan catabolism and T-cell tolerance: immunosuppression by starvation? Immunol Today. 1999;20(10):469–73.
Mellor AL, et al. Prevention of T cell-driven complement activation and inflammation by tryptophan catabolism during pregnancy. Nat Immunol. 2001;2(1):64–8.
Muller AJ, et al. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11(3):312–9.
Munn DH, et al. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189(9):1363–72.
Fallarino F, et al. T cell apoptosis by kynurenines. Adv Exp Med Biol. 2003;527:183–90.
Mellor AL, et al. Cells expressing indoleamine 2,3-dioxygenase inhibit T cell responses. J Immunol. 2002;168(8):3771–6.
McGaha TL, et al. Amino acid catabolism: a pivotal regulator of innate and adaptive immunity. Immunol Rev. 2012;249(1):135–57.
Ravishankar B, et al. Tolerance to apoptotic cells is regulated by indoleamine 2,3-dioxygenase. Proc Natl Acad Sci U S A. 2012;109(10):3909–14.
Manlapat AK, et al. Cell-autonomous control of interferon type I expression by indoleamine 2,3-dioxygenase in regulatory CD19+ dendritic cells. Eur J Immunol. 2007;37(4):1064–71.
Liu H, et al. GCN2-dependent metabolic stress is essential for endotoxemic cytokine induction and pathology. Mol Cell Biol. 2014;34(3):428–38.
Fallarino F, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol. 2006;176(11):6752–61.
Munn DH, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22(5):633–42.
Sharma MD, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117(9):2570–82.
Frumento G, et al. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. 2002;196(4):459–68.
Hayashi T, et al. 3-Hydroxyanthranilic acid inhibits PDK1 activation and suppresses experimental asthma by inducing T cell apoptosis. Proc Natl Acad Sci U S A. 2007;104(47):18619–24.
Opitz CA, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478(7368):197–203.
Mimura J, Fujii-Kuriyama Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim Biophys Acta. 2003;1619(3):263–8.
Prendergast GC. Cancer: Why tumours eat tryptophan. Nature. 2011;478(7368):192–4.
Mezrich JD, et al. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185(6):3190–8.
Pilotte L, et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci U S A. 2012;109(7):2497–502.
Kimura A, et al. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci U S A. 2008;105(28):9721–6.
Veldhoen M, et al. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med. 2009;206(1):43–9.
Quintana FJ, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453(7191):65–71.
Veldhoen M, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453(7191):106–9.
Sato E, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102(51):18538–43.
Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117(5):1147–54.
Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 2012;72(21):5435–40.
Zhang L, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348(3):203–13.
Callahan MJ, et al. Increased HLA-DMB expression in the tumor epithelium is associated with increased CTL infiltration and improved prognosis in advanced-stage serous ovarian cancer. Clin Cancer Res. 2008;14(23):7667–73.
Hwang WT, et al. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol Oncol. 2012;124(2):192–8.
Hamanishi J, et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A. 2007;104(9):3360–5.
Tomsova M, et al. Prognostic significance of CD3+ tumor-infiltrating lymphocytes in ovarian carcinoma. Gynecol Oncol. 2008;108(2):415–20.
Matsuzaki J, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A. 2010;107(17):7875–80.
Cady SG, Sono M. 1-Methyl-DL-tryptophan, beta-(3-benzofuranyl)-DL-alanine (the oxygen analog of tryptophan), and beta-[3-benzo(b)thienyl]-DL-alanine (the sulfur analog of tryptophan) are competitive inhibitors for indoleamine 2,3-dioxygenase. Arch Biochem Biophys. 1991;291(2):326–33.
Balachandran VP, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011;17(9):1094–100.
Basu GD, et al. Cyclooxygenase-2 inhibitor enhances the efficacy of a breast cancer vaccine: role of IDO. J Immunol. 2006;177(4):2391–402.
Lee SY, et al. The immune tolerance of cancer is mediated by IDO that is inhibited by COX-2 inhibitors through regulatory T cells. J Immunother. 2009;32(1):22–8.
Sorensen RB, et al. The immune system strikes back: cellular immune responses against indoleamine 2,3-dioxygenase. PLoS One. 2009;4(9):e6910.
Iversen TZ, et al. Long-lasting disease stabilization in the absence of toxicity in metastatic lung cancer patients vaccinated with an epitope derived from indoleamine 2,3 dioxygenase. Clin Cancer Res. 2014;20(1):221–32.
Grohmann U, et al. IL-6 inhibits the tolerogenic function of CD8 alpha+ dendritic cells expressing indoleamine 2,3-dioxygenase. J Immunol. 2001;167(2):708–14.
Grohmann U, et al. CD40 ligation ablates the tolerogenic potential of lymphoid dendritic cells. J Immunol. 2001;166(1):277–83.
Hwu P, et al. Indoleamine 2,3-dioxygenase production by human dendritic cells results in the inhibition of T cell proliferation. J Immunol. 2000;164(7):3596–9.
Munn DH, et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science. 2002;297(5588):1867–70.
Mellor AL, et al. Cutting edge: induced indoleamine 2,3 dioxygenase expression in dendritic cell subsets suppresses T cell clonal expansion. J Immunol. 2003;171(4):1652–5.
Munn DH. Indoleamine 2,3-dioxygenase, tumor-induced tolerance and counter-regulation. Curr Opin Immunol. 2006;18(2):220–5.
Lob S, et al. Levo- but not dextro-1-methyl tryptophan abrogates the IDO activity of human dendritic cells. Blood. 2008;111(4):2152–4.
Qian F, et al. Efficacy of levo-1-methyl tryptophan and dextro-1-methyl tryptophan in reversing indoleamine-2,3-dioxygenase-mediated arrest of T-cell proliferation in human epithelial ovarian cancer. Cancer Res. 2009;69(13):5498–504.
Qian F, et al. Effects of 1-methyltryptophan stereoisomers on IDO2 enzyme activity and IDO2-mediated arrest of human T cell proliferation. Cancer Immunol Immunother. 2012;61(11):2013–20.
Yuasa HJ, et al. 1-L-methyltryptophan is a more effective inhibitor of vertebrate IDO2 enzymes than 1-D-methyltryptophan. Comp Biochem Physiol B Biochem Mol Biol. 2010;157(1):10–5.
Travers MT, et al. Indoleamine 2,3-dioxygenase activity and L-tryptophan transport in human breast cancer cells. Biochim Biophys Acta. 2004;1661(1):106–12.
Seymour RL, et al. A high-affinity, tryptophan-selective amino acid transport system in human macrophages. J Leukoc Biol. 2006;80(6):1320–7.
Metz R, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology. 2012;1(9):1460–8.
Liu X, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. 2010;115(17):3520–30.
Soliman HH, et al. A first in man phase I trial of the oral immunomodulator, indoximod, combined with docetaxel in patients with metastatic solid tumors. Oncotarget. 2014;5(18):8136–46.
Soliman HH, et al. A phase I study of indoximod in patients with advanced malignancies. Oncotarget. 2016;7(16):22928–38.
Vacchelli E, et al. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology. 2014;3(10):e957994.
Funding
This work was supported by the Roswell Park Alliance Foundation, NIH 1R01CA158318-01A1 and RPCI-UPCI Ovarian Cancer SPORE P50CA159981-01A1.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter

Cite this chapter
Amobi, A., Qian, F., Lugade, A.A., Odunsi, K. (2017). Tryptophan Catabolism and Cancer Immunotherapy Targeting IDO Mediated Immune Suppression. In: Kalinski, P. (eds) Tumor Immune Microenvironment in Cancer Progression and Cancer Therapy. Advances in Experimental Medicine and Biology, vol 1036. Springer, Cham. https://doi.org/10.1007/978-3-319-67577-0_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-67577-0_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-67575-6
Online ISBN: 978-3-319-67577-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)