
Abstract
Passive transport across lipid bilayers is a significant, if not dominant, route for the permeation of biologically active amphiphiles through cell membranes. Often, the quantitative description of the rate of permeation is based on a single kinetic parameter, the permeability coefficient. However, the nature of the interactions between amphiphilic molecules and lipid bilayers is complex and involves different steps (insertion, translocation and desorption), which affect both the extent of partition and the rate of permeation. Quantitative knowledge of the rate constants associated with each individual step is required for proper understanding of the whole process, and certainly useful in prediction of the ability of new drug compounds to access the interior of their cell targets. This chapter reviews the formalisms applicable to the kinetics of interaction of small solutes with lipid bilayers. Several important limiting cases, corresponding to different ranges of aqueous solubility and membrane partition, are considered, and selected examples of applications of fluorescence spectroscopy to quantitative description of solute/bilayer interaction are presented. We also address the state of the art regarding methods for calculation of rate constants of solute/lipid interaction and permeability coefficients from molecular dynamics simulations. These methods rely on accurate computation of free energy profiles of solutes across lipid bilayers, and strategies to this purpose, namely employing enhanced sampling of improbable states with the so-called umbrella sampling method, are discussed.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

The interaction of small molecules with biological membranes is of fundamental importance in organelle, cell and whole organism homeostasis. Most biologically active small molecules such as metabolites and pharmaceuticals are amphiphilic and as a consequence they interact to some extent with hydrophobic assemblies such as lipid bilayers. This is actually a requirement for their efficient distribution between the distinct aqueous compartments in the living being, all delimited by biomembranes. Most biologically active amphiphiles are not recognized by transporters in the biological membrane and they cross those barriers by passive mechanisms. The rate of equilibration between the distinct compartments depends on the permeability coefficient through the lipid bilayer of biomembranes, which in turn is a function of the rate of translocation between the bilayer leaflets, as well as on the rates of insertion and desorption into/from each leaflet. In addition, a large fraction of cellular enzymes are associated with membranes (either permanently or transiently due to electrostatic interactions and/or regulated acylation), and the effective concentration of the bioactive compound depends on its partition between the aqueous medium and the membrane as well as on the rates of interaction with the membrane.
Given its importance, it is to some extent surprising that so little information is available on the rates of interaction of small amphiphilic molecules with lipid bilayers and biological membranes. This is justified by the extreme difficulty on obtaining experimental data of high quality due to the limited availability of techniques with the required sensitivity and time resolution. An example that illustrates well this difficulty may be found in the effort dedicated by several authors to characterize the rates of interaction of fatty acids with lipid bilayers. The kinetics of the interaction is somewhat easier to characterize for the case of fluorescent molecules, but nevertheless the available information is still quite limited. Molecular Dynamics (MD) simulations is a well-established methodology to obtain molecular details on the interaction between amphiphilic molecules and lipid bilayers. More recently, this methodology has been applied to obtain information regarding the kinetics of the interaction.
In this chapter we will review the methodologies available to characterize the kinetics of interaction between small molecules and lipid bilayers. The distinct methodologies commonly used, and the required mathematical formalisms, will be presented with a critical evaluation of their strengths and limitations. The list of references given is by no mean extensive, the objective being simply to illustrate the distinct methodologies available.
4.1 Experimental Approaches to Characterize the Interaction Between Small Molecules and Lipid Bilayers
To characterize quantitatively the kinetics of interaction between a solute and lipid bilayers it is necessary to quantify the concentration of solute associated with the bilayer. Fluorescence based methodologies are by far the most convenient because the fluorescence properties of the solute (quantum yield, lifetime and/or anisotropy) are strongly dependent on the environment. This permits following the transfer of a fluorescent molecule between media with distinct properties without the need to physically separate them. In addition, fluorimeters are inexpensive and easy to work with, display a high sensitivity (allowing the use of small concentrations that do not perturb the lipid bilayer), and little interference from other molecules present in the system as most molecules do not fluoresce.
The kinetic models required to characterize the kinetics of interaction of small molecules with liposomes will be presented and discussed in the first part of this section. A focus on fluorescent molecules will be given, with the discussion of common difficulties and possible artifacts. The models presented are also valid for the case of non-fluorescent molecules, provided that their concentration in the distinct compartments may be quantified. Some common methods to follow non-fluorescent molecules, both directly and using fluorescent probes, will also be described.
We have opted to present the kinetic models organized according to solute solubility in the aqueous media, considering separately very high, moderate and low aqueous solubility, because the experimental approaches and the mathematical formalisms depend strongly on this solute property.
4.1.1 Solutes with High Solubility in the Aqueous Media and Insoluble in the Lipid Bilayer
Usually, when the solubility of the molecule of interest in the aqueous media is very high it does not associate significantly with the lipid bilayer. The solute molecules in the vicinity of the lipid bilayer on one side, will permeate directly into the other side of the bilayer with a given rate constant κ, scheme (4.1):
where \( {S}_{\mathrm{W}}^{\mathrm{o}} \) and \( {S}_{\mathrm{W}}^{\mathrm{i}} \) represent the solute in the aqueous media outside and inside the liposomes, respectively. It is assumed that the rate constant for crossing the bilayer in both directions is the same, which should hold for symmetric bilayers.
The differential equations that relate the time dependence of the amount of solute on both sides of the bilayer with the rate constants for crossing the bilayer are given below.
Note that the total amount of solute in a given aqueous compartment is represented by an uppercase S, whereas a lowercase s represents the solute in the immediate vicinity of the bilayer. The rate for crossing the bilayer is proportional to the difference in the amount of solute in the immediate vicinity of the bilayer (and not to the difference in the total amount of solute in the two aqueous compartments), because only those solute molecules are able to cross the bilayer with the rate constant κ. Solute molecules further away have first to diffuse into the region near the bilayer, and after crossing the barrier the solutes will equilibrate with the respective aqueous compartment by diffusion.
For small and polar molecules the diffusion in the aqueous media is much faster than crossing the lipid bilayer barrier. Therefore, the solute molecules in the immediate vicinity of the bilayer are in equilibrium with the solute in the bulk aqueous phase. If the solute does not interact with the surface of the lipid bilayer, the concentration in the volume of aqueous phase in the immediate vicinity of the lipid bilayer is the same as the concentration in the respective bulk aqueous phase. In this case, the fraction of solute in the immediate vicinity of the lipid bilayer is equal to the ratio between the volume of the aqueous phase with a thickness that depends on the solute dimensions, and the area of the bilayer in direct contact with it, K = 1 in Eq. (4.3). For the case of ions and charged membranes, the solute may be enriched or depleted at the surface of the bilayer due to electrostatic attraction or repulsion; in this case the distribution coefficient between the bulk aqueous phase and the aqueous layer in the immediate vicinity of the bilayer (K) will be larger or smaller than 1, respectively. The product of the distribution coefficient (K) and the characteristic length (λ) converts the solute concentration in bulk aqueous phase (in units of mol dm−3) into the surface concentration of solute near the permeation barrier (in units of mol dm−2).
See Fig. 4.1 for the definition of the distinct variables and parameters in Eq. (4.3).

Schematic drawing with the kinetic scheme considered for the transport of very polar solutes across a lipid membrane. The lipid bilayer is represented by the medium intensity gray bar ( ), the aqueous layer in the immediate vicinity of the bilayer by the light gray bar (
), the aqueous layer in the immediate vicinity of the bilayer by the light gray bar ( ), and the bulk aqueous phase by very light grey (
), and the bulk aqueous phase by very light grey ( ). The volumes of the aqueous phases outside and inside the liposomes are represented by \( {V}_{\mathrm{w}}^{\mathrm{o}} \) and \( {V}_{\mathrm{w}}^{\mathrm{i}} \), respectively; the volumes of aqueous phase immediately in the vicinity of the bilayer are represented by \( {v}_{\mathrm{w}}^{\mathrm{o}} \) and \( {v}_{\mathrm{w}}^{\mathrm{i}} \); and the total area of the bilayer surface in direct contact with the aqueous media is represented by A
o and A
i (
). The volumes of the aqueous phases outside and inside the liposomes are represented by \( {V}_{\mathrm{w}}^{\mathrm{o}} \) and \( {V}_{\mathrm{w}}^{\mathrm{i}} \), respectively; the volumes of aqueous phase immediately in the vicinity of the bilayer are represented by \( {v}_{\mathrm{w}}^{\mathrm{o}} \) and \( {v}_{\mathrm{w}}^{\mathrm{i}} \); and the total area of the bilayer surface in direct contact with the aqueous media is represented by A
o and A
i ( ). The solute in the bulk aqueous phase (\( {S}_{\mathrm{w}}^{\mathrm{o}} \) and \( {S}_{\mathrm{w}}^{\mathrm{i}} \)) equilibrates rapidly with the solute in the immediate vicinity of the lipid bilayer (\( {s}_{\mathrm{w}}^{\mathrm{o}} \) and \( {s}_{\mathrm{w}}^{\mathrm{i}} \)) and slowly permeates the bilayer with the rate constant κ
). The solute in the bulk aqueous phase (\( {S}_{\mathrm{w}}^{\mathrm{o}} \) and \( {S}_{\mathrm{w}}^{\mathrm{i}} \)) equilibrates rapidly with the solute in the immediate vicinity of the lipid bilayer (\( {s}_{\mathrm{w}}^{\mathrm{o}} \) and \( {s}_{\mathrm{w}}^{\mathrm{i}} \)) and slowly permeates the bilayer with the rate constant κ
Using the relations given in Eq. (4.3), the differential equations for the total amount of solute in each aqueous compartment as a function of the total amount of solute in that compartment may be found, Eq. (4.4). Note that for large unilamelar vesicles (LUVs) and giant unilamelar vesicles (GUVs), the area of the bilayer in contact with the aqueous media outside and inside the liposomes is the same, that means A o = A i = A.
When the concentration of solute in a given aqueous compartment with respect to the volume of that aqueous compartment is the property being followed (as is the case for permeation through cell monolayers), it is convenient to describe the time evolution of the solute in terms of its concentration with respect to the volume of the aqueous compartment where it is dissolved \( \left({\left[{S}_{\mathrm{w}}^{\mathrm{x}}\right]}_{V_{\mathrm{w}}^{\mathrm{x}}}={nS}_{\mathrm{w}}^{\mathrm{x}}/{V}_{\mathrm{w}}^{\mathrm{x}}\right) \), Eq. (4.5).
The differential equations that describe the time evolution of the concentration of solute in the two aqueous compartments with respect to the volume of the specific compartment are no longer symmetric. This is because the incremental concentration in each aqueous compartment due to the transfer of a given amount of solute depends on the volume of the respective compartment.
When using the fluorescence of the whole solution to follow the permeation of a solute through the lipid bilayer, it is not usually possible to quantify the concentration of solute in a given compartment with respect to the volume of that compartment, but rather the corresponding concentration with regard to the total volume of the solution, \( {\left[{S}_{\mathrm{w}}^{\mathrm{x}}\right]}_{V_{\mathrm{T}}}={nS}_{\mathrm{w}}^{\mathrm{x}}/{V}_{\mathrm{T}} \). The differential equations obtained for the time variation in the concentration of solute in each aqueous compartment, with respect to the total volume of the system, are given by:
Using the molar conservation equation for the total amount of solute \( \left({nS}^{\mathrm{T}}={nS}_{\mathrm{w}}^{\mathrm{o}}+{nS}_{\mathrm{w}}^{\mathrm{i}}\right) \), one can obtain the differential equation for each of the species (namely \( {S}_{\mathrm{w}}^{\mathrm{o}} \)) where its own concentration is the only time dependent variable:
Equation (4.7) has the form:
and its integration gives:
where x 0 is the value of variable x at time equal to zero and x ∞ is its value at the end of the time window considered in the experiment.
The rate constant for transfer of solute from the aqueous media outside the liposomes to the aqueous media inside the liposomes (k) is therefore given by:
The above expression may be simplified because several of the variables depend on common parameters. The total area of lipid bilayer in contact with each aqueous compartment is given by:
where \( {N}_{{\mathrm{L}}_{\mathrm{V}}} \) is the number of liposomes in the total volume of solution considered, and r the radius of the liposomes. On the other hand, the total volume of aqueous media inside the liposomes is given by:
If the aqueous volume inside the liposomes is much smaller than the total volume of the solution, and because the volume of the lipid bilayer is negligible, the aqueous volume outside the liposomes may be considered equal to the total volume of solution. In that case, the rate constant for transfer of solute through the lipid bilayer of the liposomes depends on the size of the liposomes, being given by:
The rate constant of transfer of solute is therefore inversely proportional to the size of the liposomes. Due to this dependence, the kinetic parameter that is usually reported is the permeability coefficient (P). This parameter may be calculated from the rate of accumulation of solute in the acceptor compartment, which is described by Eq. (4.14) for the case where no significant back transfer of solute occurs:
where the superscripts D and A denote the donor and acceptor compartments, and \( {nS_{\mathrm{w}}^{\mathrm{D}}}_{(0)} \) is the amount of solute in the donor compartment at t = 0. The differential equation for the amount of S in the acceptor compartment is given by,
At the beginning of transfer from D to A, when \( {nS}_{\mathrm{w}}^{\mathrm{A}}=0 \) and \( {nS}_{\mathrm{w}}^{\mathrm{D}}={nS}^{\mathrm{T}} \), the above equation simplifies to:
Substituting Eq. (4.16) in Eq. (4.14) leads to the relation between the permeability coefficient and the transfer rate constant:
If the donor compartment is the aqueous medium inside the liposomes, \( {V}_{\mathrm{w}}^{\mathrm{A}}\kern0.5em \gg {V}_{\mathrm{w}}^{\mathrm{D}} \) and the above equation simplifies to:
Substituting in the expression the Eqs. (4.11), (4.12) and (4.13), for A, \( {V}_{\mathrm{w}}^{\mathrm{D}} \) and k, respectively, one obtains the following equation:
which relates the permeability coefficient with the parameters initially considered in the kinetic model, κ and λ.
In this mechanism of permeation it is assumed that the solutes do not interact with the bilayer (except for some eventual electrostatic interaction). Therefore, the intrinsic rate constant for crossing the barrier (κ) is related with the formation of transient hydrated defects due to thermal fluctuations, which depend on the bilayer thickness and lipid-lipid interactions. The rate constant is also affected by the size of the solute which will have to diffuse through the transient pores. For a given ion, the rate of permeation is expected to decrease exponentially with the increase in the thickness of the bilayer. This has been observed for the permeation of small ions through thin lipid bilayers [1].
The mathematical formalism above has been used to characterize the rate of permeation of the anion dithionite through LUVs of distinct lipid composition [2]. Dithionite is not fluorescent but its concentration in the aqueous media inside the LUVs was quantitatively followed via its reaction with the fluorescent group nitrobenzoxadiazole (NBD) covalently linked to the head group of 1,2-dimiristoyl-sn-glycero-3-phosphoethanolamine (DMPE). The same formalism has been used to calculate the permeability coefficient of several non-fluorescent small molecules (water, urea and glycerol) [1, 3] and ions (such as amino acids, peptides, thyroid hormones, phosphate and H+/OH−) [1, 4,5,6]. The time evolution of the concentration of the relevant solute in the inner or outer aqueous media was followed by changes in the liposome volume (therefore leading to the permeability coefficient under an osmotic gradient) [1, 3, 4], using specific electrodes [1], through the selective reaction of the solute in the inner or outer aqueous phase [4, 5], or by solute quantification after physical separation of the two aqueous compartments [6].
To characterize the rate of permeation of fluorescent molecules through lipid bilayers, the most common approach is to encapsulate the fluorescent molecule at high concentrations (leading to efficient fluorescence quenching) inside liposomes. The fluorescent molecules outside the liposomes are then removed (usually by size exclusion chromatography), and the permeation through the lipid bilayer is followed through the time dependent fluorescent increase due to dilution of the fluorophore when going from the inner to the outer aqueous media [7]. This approach can only be used for molecules with low and very low permeability, because it requires previous removal of the probe located in the aqueous media outside the liposomes. Additionally, the fluorescent molecules must have a very high solubility in the aqueous phase to achieve self-quenching concentrations, and they cannot interact efficiently with the membranes to ensure slow permeability. The method cannot therefore be used to characterize amphiphilic molecules. It has been used mostly to characterize the effect of several membrane perturbing molecules, such as peptides and surfactants, on the barrier properties of liposomes [7,8,9,10,11,12]. The fluorescent probe used to report on the bilayer properties is most commonly carboxyfluorescein (CBF), but calcein has also been employed [8]. To follow quantitatively the permeation, attention should be given to the fact that fluorescence intensity may have a non-linear relation with the extent of transfer [7, 13].
4.1.2 Solutes with a Moderate Solubility in the Aqueous Media and in the Lipid Bilayer
The overall permeation of solutes with moderate solubility in both the aqueous and the lipidic phases is usually described by the partition-diffusion mechanism. Early descriptions of permeation following this mechanism considered the presence of several diffusion barriers in the system water/bilayer/water [14]. However, the approximation of a single rate limiting step, the diffusion through the bilayer non-polar core, became the prevailing model. According to this formalism, the overall permeability coefficient (P) is related with the microscopic parameters: partition coefficient between the water and the bilayer (K P), diffusion coefficient of the solute through the bilayer (D), and thickness of the barrier (h), by:
This model for permeation through membranes is based on the work done by Overton more than a century ago [15], and Eq. (4.20) is commonly referred to as Overton’s rule [16, 17].
Equation (4.20) is formally equivalent to the equation derived in the previous section for solutes that do not partition into the lipid bilayer, Eq. (4.19), with the equilibrium distribution between the bulk aqueous phase and the immediate vicinity of the lipid bilayer (K) replaced by the partition coefficient between the aqueous and lipidic phases (K P). The first order rate constant associated with transport with the diffusion coefficient D is D/ℓ 2, where ℓ is the distance between the two equilibrium positions that is being crossed by diffusion [14]. Therefore, Eq. (4.20) is equivalent to Eq. (4.19) with the solute characteristic length (λ) equal to the thickness of the diffusion barrier. The length parameter in Eq. (4.20), h, is usually considered as the thickness of the bilayer, although it is in fact the distance between the equilibrium positions of the solute center of mass on both sides of the bilayer. The assumptions in the derivation of Eq. (4.20) are very similar to those considered in the previous section: (1) negligible accumulation of solute in the bilayer; (2) rapid transport of solute from the bulk aqueous media to the bilayer; and (3) transport through the lipid bilayer as a single step.
The major difference between the partition/diffusion model and the model presented in the last section is the nature of the intrinsic rate constant for transport through the barrier; diffusion of solute dissolved in the nonpolar portion of the lipid bilayer, and diffusion through transient hydrated defects, respectively. The distinction between the two permeation mechanisms may be done through the dependence of the overall permeability coefficient on the thickness of the bilayer. Assuming an invariant partition coefficient and diffusion coefficient; P is predicted to depend inversely on the thickness of the bilayer for the partition/diffusion mechanism of permeation, while an exponential dependence is predicted for the pore mechanism [1]. The permeation of small neutral molecules such as water, urea and glycerol, has been shown to follow the predictions from the partition/diffusion mechanism of permeation through lipid bilayers. This same mechanism is observed for the permeation of large ions, due to their significant solubility in the lipid bilayer, the small probability of pore formation with the appropriate size and the slow diffusion of the large ion through the transient pore.
There are several reports on the overall permeability coefficient of small molecules through liposomes considering the partition/diffusion mechanism. The methods used are essentially equal to those described in the previous section for solutes very soluble in the aqueous phase, except that permeation may be too fast to allow the physical separation of the inner and outer aqueous compartments. The permeation of fluorescent amphiphiles into GUVs has also been followed directly using fluorescence microscopy [18], and the permeation of weak acids and bases has been characterized through the pH variation in the aqueous compartment inside the LUVs, as measured by the fluorescence probes carboxyfluorescein or pyranin [19].
When comparing distinct solutes along a homologous series, Eq. (4.20) provides a good description for the dependence of the rate of overall permeation coefficient with the solute hydrophobicity. However, for structurally unrelated solutes the correlation between P and K P is poor and no clear relation is obtained with the diffusion coefficient as predicted by the size of the solutes. This is due to the assumptions considered in the development of the partition/diffusion mechanism which are not valid for the case of medium size and amphiphilic molecules.
In the partition/diffusion mechanism of permeation, the barrier region of the lipid bilayer is treated as a homogeneous medium through which the solute diffuses due to the concentration gradient on both sides of the barrier. There are several difficulties associated with this assumption, namely the high transversal heterogeneity of the lipid bilayer (with density, viscosity and polarity gradients), which is not compatible with the assumption of a smooth continuous resistance offered by the media on the diffusing molecules required to treat transport as diffusion [14]. It is therefore challenging, if not impossible, to know what would be the diffusion coefficient to use in Eq. (4.20) in order to predict the permeability coefficient from the structure of a given molecule; the more general inhomogeneous solubility-diffusion model, which can accommodate this heterogeneity, will be described below in the context of MD simulations. Also, for amphiphilic molecules (with well-defined polar and non-polar regions) the transport through the bilayer center cannot be considered as random diffusion because the energy of the solute in the bilayer does not depend only on the position of its center of mass, but also on the orientation of the polar and non-polar regions. The high Gibbs energy state in the transport of amphiphilic molecules through the bilayer usually corresponds to the solubilization of the polar region in the non-polar center of the bilayer (transition state) and is more conveniently treated as a single step corresponding to translocation from the equilibrium position in one side of the bilayer to the other. The rate constant of translocation depends on the activation energy barrier, which is a function of the interactions that the solute establishes with the lipids and the hydration shell at the equilibrium positions and with the non-polar portion of the lipids when in the transition state. The prediction of this activation energy, and therefore the rate constant for translocation, from the structure of the amphiphile and the properties of the bilayer is a feasible task, allowing the prediction of the overall permeation from the structure of the permeating solute.
Another important limitation of the partition/diffusion model is the assumption that transport through the non-polar region of the bilayer is the rate limiting step. In the overall process of entering an aqueous compartment delimited by a lipid membrane (cell, organelle or liposome) the amphiphile first interacts with the outer leaflet (rate of insertion), followed by equilibration with the inner leaflet (rate of translocation, or diffusion, through the non-polar part of the bilayer), and then it equilibrates with the inner aqueous compartment (rate of desorption). For amphiphiles with a high Hydrophilic/Lipophilic Balance (HLB), the rate limiting step in the overall process is usually translocation through the non-polar center of the bilayer. In this case, the permeability coefficient is directly proportional to the rate of translocation and to the partition coefficient between the aqueous media and the lipid bilayer as predicted by Eq. (4.20). However, in the last years several exceptions to this behavior have been observed, and it is now well established that for amphiphiles with intermediate and low HLB, Eq. (4.20) does not adequately predict the rate of overall permeation through lipid bilayers [20,21,22,23], as the rate limiting step is the desorption from the bilayer (and not translocation) [20].
The overall rate of permeation is the relevant parameter to evaluate how fast the amphiphile crosses the biological membranes. However, a rational interpretation of the dependence of this parameter on the structure and properties of the permeating amphiphile is not straightforward, because it depends on several steps, each being affected differently by the amphiphile properties [20]. To rationalize and gain predictive value on the dependence of the overall permeation with the molecular properties of the amphiphile, it is necessary to obtain all the relevant rate constants (insertion, desorption and translocation) for a large set of structurally unrelated molecules.
To obtain the rate constants of insertion and desorption from the lipid bilayer it is necessary to consider those steps explicitly in the kinetic scheme. Therefore, the association between the amphiphile and the lipid bilayer is not assumed to be in fast equilibrium. The resulting kinetic scheme is given below.
where S W o represents the amphiphile (solute) in the aqueous media outside the liposomes; L V the liposomes; S L o and S L i represent the amphiphile in the outer and inner leaflet of the liposomes, respectively; k f is the rate constant for translocation between the leaflets; \( {k}_{+}^{{\mathrm{L}}_{\mathrm{V}}} \) is the rate constant for insertion of the amphiphile in the lipid bilayer of the liposomes; and k − is the rate constant for desorption from the lipid bilayer into the aqueous media.
We call the reader’s attention to the fact that the notation used for the rate constants of desorption and translocation does not include reference to the topology of the lipid phase, while this is included in the notation used for the rate constant of insertion. This is because the rate of insertion requires the encounter between the amphiphile in the aqueous media and the lipid phase, which depends on the size of the lipid assemblies for the case of processes near or at the diffusion limit. In accordance, the lipid phase is represented with the topology present in the solution (liposomes, L V). This allows the comparison between the obtained rate constant of insertion and the diffusion limited rate constant. Additionally, this formalism permits the comparison between the rates of insertion in lipid aggregates of distinct sizes with the uncoupling between the contributions from size and other properties [24]. It should however be noted that the usual equations to calculate the diffusion limited rate constants are only valid in the absence of electrostatic interaction between the reactants [25]. Also, the model considers that all the volume occupied by the reactants is active, which is only an approximation for the case of liposomes and leads to significant deviations for the case of very large liposomes and cells [26].
Although the interaction is considered to take place with the individual liposomes, equilibration of the amphiphile between the aqueous phase and the lipid leaflet in direct contact is considered to occur via partition and not binding to well defined binding sites. In addition, the capacity of the liposomes to interact with the amphiphile is considered as independent of the presence of amphiphile already associated with the liposomes; this being valid only for small local concentrations of solute. As a consequence, the concentration of liposomes (binding agent available for the interaction) remains constant throughout the equilibration process, which significantly simplifies the mathematical description of the interaction between small molecules and liposomes.
In kinetic scheme (4.21), the equilibration of the amphiphile with the aqueous media inside the liposomes is not considered. Although this approximation is valid for LUVs because the volume of the encapsulated aqueous media is negligible, it does not hold for GUVs. Another simplification considered is that the rate constant for translocation is the same in both directions. This is valid for liposomes with a small curvature (diameter equal to or larger than 100 nm) with the same lipid composition in both leaflets.
The rate constant for equilibration between the aqueous media and the liposome leaflet in direct contact is given by,Footnote 1
If this process occurs at least one order of magnitude faster than translocation into the inner leaflet, the two processes are uncoupled and the fluorescence variation that follows the addition of liposomes to the fluorescent amphiphile is a single-exponential function, from which the rate constant k is directly obtained. Performing the experiment at different liposome concentrations permits obtaining the rate constant of insertion and the rate constant of desorption, [27] Eq. (4.22).
When the rate of translocation is much faster than the rate of equilibration between the aqueous phase and the exposed liposome leaflet, the fluorescence variation observed is also a single-exponential function but the relation between the transfer rate constant with the intrinsic rate constants for insertion and desorption is given by,
The rate of desorption is proportional to half the value of the respective rate constant because only the amphiphile in the outer leaflet of the bilayer (half of the total amphiphile) is able to desorb into the aqueous phase outside the liposomes.
Both situations described above allow the characterization of the rate constants of insertion and desorption, but not the rate constant of translocation. An important difficulty is whether Eqs. (4.22) or (4.23) should be used if no independent information is available regarding the relative rate of translocation.
For some amphiphiles, the two distinct processes occur on similar time scales and the fluorescence variation observed does not follow a single-exponential function. In this situation the time dependence of the fluorescence variation must be described by the integration of the full set of differential equations obtained from the kinetic scheme, Eq. (4.24), and all the rate constants may in principle be obtained.
The relative weights of the fast and slow steps reflect the equilibrium association of the amphiphile with the lipid in the outer leaflet and with the total lipid (outer and inner leaflet). For very small lipid concentrations, doubling the amount of lipid leads to a proportional increase in the amount of amphiphile associated with the membrane. In this case equal weight is expected for the fluorescence increase in the fast and slow processes of interaction with the LUVs. On the other hand, for very large concentrations of lipid all the amphiphile associates with the outer leaflet of the liposomes and the equilibration with the inner leaflet does not lead to any further fluorescence variation. The optimal range of lipid concentrations to characterize the rate constants for interaction with the outer leaflet and the rate constant for translocation, depends on the fraction of amphiphile associated with the lipid bilayer at equilibrium, which in turn depends on the liposome concentration and equilibrium constant for association with the liposome:
where [S L](fast) is the concentration of amphiphile associated with the outer leaflet of the liposomes before equilibration with the inner leaflet, and [S L](∞) is the concentration of amphiphile associated with the liposomes after equilibration with both the outer and inner leaflet; \( {K}_{{\mathrm{L}}_{\mathrm{V}}} \) is the equilibrium association constant with the outer leaflet \( \left({K}_{{\mathrm{L}}_{\mathrm{V}}}={k}_{+}^{{\mathrm{L}}_{\mathrm{V}}}/{k}_{-}\right) \), and Δ (slow) is the amplitude of the slow process relative to the total fluorescence variation.
The effect of the rate of translocation and liposome concentration on the time evolution of the concentration of solute associated with the liposome is shown in Fig. 4.2. When the lipid concentration is high (panels A and B) all the solute interacts with the liposomes, even when the inner leaflet is not accessible (slow translocation, panel A). At long times, the solute in the outer leaflet equilibrates with the inner leaflet but without any effect in the total amount of solute associated with the liposome. The two situations (A and B) cannot be distinguished simply by the analysis of the fluorescence variation (proportional to the total amount of solute associated with the liposome, S L) because both the time dependence and the amplitude of the variation are the same. On the other hand, for sufficiently small liposome concentrations (panels C and D), the initial accumulation of solute in the outer leaflet is smaller than the equilibrium value when both the outer and inner leaflets are accessible. In this case, a slow process is observed in the case of slow translocation (panel C), with a relative amplitude Δ (slow) equal to 0.33 for the parameters considered in this simulation.

Kinetics of equilibration of an amphiphile from the aqueous phase to liposomes. The rate constants for insertion and desorption are the same in all panels \( \left({k}_{+}^{{\mathrm{L}}_{\mathrm{V}}}=5\times {10}^8{\mathrm{M}}^{-1}{\mathrm{s}}^{-1},k\_={10}^{-1}\kern0.24em {\mathrm{s}}^{-1}\right) \) while translocation is slow in panels A/C (k
f = 10−2 s−1) and fast in panels B/D (k
f = 10 s−1). The liposome concentration is 10−8 and 10−10 M (corresponding to a lipid concentration of 1 mM and 10 μM for 100 nm LUVs) in panels A/B and C/D, respectively. The concentration of solute in the distinct compartments (\( {S}_{\mathrm{W}}^{\mathrm{o}} \)  , \( {S}_{\mathrm{L}}^{\mathrm{o}} \)
, \( {S}_{\mathrm{L}}^{\mathrm{o}} \)  , \( {S}_{\mathrm{L}}^{\mathrm{i}} \)
, \( {S}_{\mathrm{L}}^{\mathrm{i}} \)  , and the total amphiphile associated with the liposome S
L
, and the total amphiphile associated with the liposome S
L  ) have been calculated by numerical integration of Eq. (4.24)
) have been calculated by numerical integration of Eq. (4.24)
When both a fast and a slow process are identified in the fluorescence variation associated with the equilibration of an amphiphile with liposomes, special attention should be given to investigate the possibility of amphiphile aggregation in the aqueous phase, as this may be the origin of the fluorescence variation not following a single-exponential function. This may be done through the dependence of the relative amplitude of the slow process with the liposome concentration, Eq. (4.25), and also through its dependence on the total amphiphile concentration while maintaining the liposome concentration (the rate and weight of the slow step are expected to be independent on the total amphiphile concentration if it reflects translocation). A small local concentration of amphiphile in the lipid bilayer should be used when performing this evaluation because high local concentrations may affect its rate of translocation [28, 29].
The independent evaluation of the rate of translocation would simplify significantly the assignment of the kinetic steps to the distinct rates observed in the fluorescence variation. This is the case for NBD-labelled amphiphiles due to their fast and irreversible reaction with dithionite [2, 27, 30]. The comparison between the rate of fluorescence decrease when dithionite is added to pre-equilibrated liposomes containing the NBD-amphiphile and the fluorescence variation observed when liposomes are added to the amphiphile in the aqueous media, allows the unequivocal identification of the kinetic steps involved [2, 27]. For this goal it is mandatory that dithionite can only react with the NBD-amphiphile located in the outer leaflet. The amount of dithionite that has permeated to the aqueous media inside the liposomes may be evaluated from its rate of permeation [2], leading to less than 1% of the concentration outside the liposomes 1 h after addition to 100 nm LUVs prepared from POPC, 2 h for membranes prepared from POPC/Chol 1:1, and 20 h for the case of liposomes prepared from SM/Chol 6:4, at 35 °C [2]. Whether this small dithionite concentration is negligible or not depends on the rate of its reaction with the NBD amphiphile, because what is important is that the NBD amphiphile does not react with dithionite while it is located in the inner leaflet of the liposomes.
This methodology has been followed to obtain all the rate constants for the interaction of fluorescent amphiphiles with lipid bilayers; namely for NBD-labelled fatty amines with a short alkyl chain [27], lyso-phospholipids [31], short acyl chain phospholipids [32], and deuteroporphyrin [33]. The rate constants for insertion and desorption have also been characterized for the interaction of a quaternary alkyl amine (labelled with 7-hydroxycoumarin) with lipid bilayers with several lipid compositions and in distinct phases [34, 35].
Some of the kinetic parameters for the interaction of non-fluorescent amphiphiles have also been characterized. For this purpose, most approaches are still based on fluorescence [21, 36], although other methodologies such as isothermal titration calorimetry [28] and nuclear magnetic resonance [37] have also been used.
4.1.3 Solutes with Very Low Solubility in the Aqueous Media and a High Partition into the Lipid Bilayer
The section above describes methodologies to characterize the kinetics of association of amphiphiles with liposomes where the amphiphile is initially in the aqueous phase in the monomeric state. This requires that the solubility of the amphiphile in the aqueous media is significant. Usually near 1 μM is required although concentrations as small as a few nM have been used for amphiphiles with a high fluorescent quantum yield when associated with the lipid bilayer and a low fluorescence in the aqueous phase, as is the case for NBD-labelled amphiphiles [27]. The kinetic parameters for the interaction of amphiphiles with very low solubility in the aqueous phase must be characterized through their exchange between distinct binding agents. The only requirement for the donor and acceptor binding agents is that at least one fluorescence parameter of the amphiphile (fluorescence intensity, spectrum, lifetime and/or anisotropy) changes when the latter is associated with one or the other binding agents. Countless variations may be encountered on this approach.
The kinetic scheme that describes the equilibration of an amphiphile between donor and acceptor LUVs is given below:
where the superscript D/A represents the donor and acceptor vesicles, respectively.
The time variations in the concentration of amphiphile in the distinct compartments may be obtained from the numerical integration of the differential equations obtained from the above kinetic scheme, Eq. (4.27):
In some situations approximations may be assumed which greatly simplify the set of differential equations and may lead to a simple analytical solution. When the solubility in the aqueous phase is very low, the steady-state approximation for the amphiphile in this compartment may be assumed because it corresponds to a negligible fraction of the total amphiphile.
If the rate of solute translocation in both the donor and acceptor liposomes is much smaller than the rate of exchange between the liposomes, only the solute in the outer leaflet of the donor liposome is able to equilibrate with the outer leaflet of the acceptor liposome. In this case, the exchange between the donor and acceptor liposomes follows a single-exponential function and the rate constant for exchange (k) is given by:
It should be recalled that the above derivation assumed a negligible amount of solute in the aqueous phase at all times, that means \( {K}_{{\mathrm{L}}_{\mathrm{V}}}^{{\mathrm{L}}_{\mathrm{V}}^{\mathrm{D}}}\left[{L}_{\mathrm{V}}^{\mathrm{D}}\right]\gg 1 \) where \( {K}_{{\mathrm{L}}_{\mathrm{V}}}^{{\mathrm{L}}_{\mathrm{V}}^{\mathrm{D}}} \) is the equilibrium constant for the solute between the aqueous phase and the outer leaflet of the donor liposomes, \( {K}_{{\mathrm{L}}_{\mathrm{V}}}^{{\mathrm{L}}_{\mathrm{V}}^{\mathrm{D}}}={k}_{+}^{{\mathrm{L}}_{\mathrm{V}}^{\mathrm{D}}}/{k}_{-}^{\mathrm{D}} \).
When the rate constants for interaction of the amphiphile with the donor and acceptor liposomes are the same, Eq. (4.29) simplifies to \( k={k}_{-}^{\mathrm{D}}={k}_{-}^{\mathrm{A}} \), that is the rate constant for exchange becomes equal to the rate constant of desorption. Therefore, when the lipid composition of the donor and acceptor liposomes is very similar, it is not possible to obtain the rate constant for insertion. Additionally, changing the concentration of acceptor liposomes does not affect the rate constant of exchange. The case of exchange between liposomes with the same properties will be further analyzed and discussed below in relation with Fig. 4.3.

Effect of the rate of translocation and acceptor concentration on the kinetics of solute exchange between LUVs with the same lipid composition. The rate constants of insertion and desorption are the same in all panels, \( {k}_{+}^{{\mathrm{L}}_{\mathrm{V}}^{\mathrm{D}}}={k}_{+}^{{\mathrm{L}}_{\mathrm{V}}^{\mathrm{A}}}=5\times {10}^9\kern0.24em {\mathrm{M}}^{-1}\;{\mathrm{s}}^{-1} \); \( {k}_{-}^{\mathrm{D}}={k}_{-}^{\mathrm{A}}=0.1\kern0.24em {\mathrm{s}}^{-1} \), as well as the concentration of donor liposomes, 5 × 10−10 M (corresponding to 50 μM lipid for 100 nm LUVs) and solute (10−6 M). The concentration of acceptor liposomes is the same as that of donor liposomes in panels A, B and C, and is 10 times larger in panels D, E and F. The rate constant for translocation is 10−3 s−1 (panel A and D), 10−2 s−1 (panel B and E) and 1 s−1 (panel C and F). The concentrations of solute in the distinct compartments (\( {S}_{\mathrm{L}}^{\mathrm{Xo}} \)  , \( {S}_{\mathrm{L}}^{\mathrm{Xi}} \)
, \( {S}_{\mathrm{L}}^{\mathrm{Xi}} \)  , \( {S}_{\mathrm{L}}^{\mathrm{X}} \)
, \( {S}_{\mathrm{L}}^{\mathrm{X}} \)  ; with the concentrations in the donor compartment in thinner lines and those in the acceptor compartment in thicker lines) were calculated through the numerical integration of Eq. (4.27)
; with the concentrations in the donor compartment in thinner lines and those in the acceptor compartment in thicker lines) were calculated through the numerical integration of Eq. (4.27)
If translocation through both the donor and acceptor liposomes is much faster than the rate of exchange, the expression for the rate constant of exchange is half the value obtained by Eq. (4.29) because only half of the total amphiphile exchanging between the two liposome populations is directly accessible to the aqueous phase that mediates the exchange process (the solute in the outer leaflet). Relevant variations in those exchange experiments may involve lipoproteins or small unilamelar vesicles (SUVs). In this case the fraction of solute directly accessible to the aqueous phase may be different from one half, and the equations derived for the rate constant of exchange will be different [26, 27].
The expression obtained for the rate constant of solute exchange between two populations of LUVs, Eq. (4.29), depends on four unknown parameters: the rate constants for insertion into the outer leaflet of the donor and acceptor liposomes \( \left({k}_{+}^{{\mathrm{L}}_{\mathrm{V}}^{\mathrm{D}}}\kern0.5em \mathrm{and}\kern0.5em {k}_{+}^{{\mathrm{L}}_{\mathrm{V}}^{\mathrm{A}}}\right) \), and the rate constants for desorption from the outer leaflet of the liposomes \( \left({k}_{-}^{\mathrm{D}}\kern0.5em \mathrm{and}\kern0.5em {k}_{-}^{\mathrm{A}}\right) \). By performing the exchange experiments at distinct concentration ratios of donor and acceptor liposomes it is possible to obtain the rate constants for desorption from the donor and acceptor liposomes (see Fig. 4.4 and discussion below). When the concentration of acceptor liposomes is much larger than that of donors, the rate constant for exchange approaches the rate constant for desorption from the donor liposomes; while at very small concentrations of acceptor liposomes, the rate constant for exchange tends towards the rate constant for desorption from the acceptor liposomes. The total concentration of liposomes should be high enough to guarantee the validity of the steady-state approximation, but at relatively low values to ensure that exchange occurs through the aqueous compartment and not due to collisions between the liposomes [38, 39]. Using this methodology, it is also possible to obtain the ratio between the rate constants for insertion in the donor and acceptor liposomes. To characterize the rate constants for insertion in each liposome, it is necessary to obtain independently the equilibrium constant for association with at least one liposome population.

Effect of the concentration of acceptor liposomes in the rate and extent of exchange for donor and acceptor liposomes with distinct properties. The rate constants of insertion and desorption are the same in all panels: \( {k}_{+}^{{\mathrm{L}}_{\mathrm{V}}^{\mathrm{D}}}=5\times {10}^9\;{\mathrm{M}}^{-1}{\mathrm{s}}^{-1} \), \( {k}_{+}^{{\mathrm{L}}_{\mathrm{V}}^{\mathrm{A}}}=5\times {10}^{10}\;{\mathrm{M}}^{-1}\;{\mathrm{s}}^{-1} \), \( {k}_{-}^{\mathrm{D}}=0.1\;{\mathrm{s}}^{-1} \), \( {k}_{-}^{\mathrm{A}}=1\kern0.24em {\mathrm{s}}^{-1} \), as well as the concentration of donor liposomes, 1 × 10−9 M (corresponding to 0.1 mM lipid for 100 nm LUVs) and total solute (10−6 M). The top panels (A to C) illustrate the case of slow translocation (\( {k}_{\mathrm{f}}^{\mathrm{D}}={k}_{\mathrm{f}}^{\mathrm{A}}={10}^{-4}\;{\mathrm{s}}^{-1} \)) while in panels D to F the translocation is faster than exchange (\( {k}_{\mathrm{f}}^{\mathrm{D}}={k}_{\mathrm{f}}^{\mathrm{A}}=10\kern0.24em {\mathrm{s}}^{-1} \)). Note the different scales in the upper and lower panels. The concentration of acceptor LUVs is equal to 1 × 10−10 ( ), 2 × 10−10 (
), 2 × 10−10 ( ), 5 × 10−10 (
), 5 × 10−10 ( ), 1 × 10−9 (
), 1 × 10−9 ( ), 3 × 10−9 (
), 3 × 10−9 ( ) and 1 × 10−8 M (
) and 1 × 10−8 M ( ). The data in plots A, B, D and E was obtained through the numerical integration of Eq. (4.27), in the central panels the concentration in the acceptor liposomes was normalized to its value at 30 min to highlight the different kinetics. The rate constants of exchange shown in plots C and F were obtained from the best fit of a single-exponential function to the time dependent concentration of solute in the acceptor liposomes (\( {S}_{\mathrm{L}}^{\mathrm{A}} \)) shown in plots A and D respectively; the lines in plots C and F are the best fit of Eq. (4.31) with the parameters: a
0 = 1.0, a
∞ = 0.10 and b = 9.9 for plot C and a
0 = 0.49, a
∞ = 0.052 and b = 9.6 for plot F
). The data in plots A, B, D and E was obtained through the numerical integration of Eq. (4.27), in the central panels the concentration in the acceptor liposomes was normalized to its value at 30 min to highlight the different kinetics. The rate constants of exchange shown in plots C and F were obtained from the best fit of a single-exponential function to the time dependent concentration of solute in the acceptor liposomes (\( {S}_{\mathrm{L}}^{\mathrm{A}} \)) shown in plots A and D respectively; the lines in plots C and F are the best fit of Eq. (4.31) with the parameters: a
0 = 1.0, a
∞ = 0.10 and b = 9.9 for plot C and a
0 = 0.49, a
∞ = 0.052 and b = 9.6 for plot F
The above models predict a single-exponential function for the time variation of the signal due to exchange between the donor and the acceptor binding agents, this reflecting a very slow or very fast translocation. To characterize the rate of translocation using exchange experiments, it is necessary to change the conditions in order to alter the relative rates of translocation and insertion/desorption so as to occur in similar time scales. This may be achieved through variations in the solution pH and/or temperature. If the signal variation due to exchange does not follow a single-exponential, there is no simple analytical expression to allow the calculation of the intrinsic rate constants from the observed rate of exchange. In this case it is advisable to perform the numerical integration of the differential equations, although important information may be obtained from the best fit with a two-exponential function. The case of exchange between liposomes with distinct properties will be further analyzed and discussed below in relation with Fig. 4.4.
The simulation of the time variation in the solute concentration for donor and acceptor liposomes with the same lipid composition is shown in Fig. 4.3 for different rate constants and liposome concentrations. In panels A and D, translocation is much slower than exchange between the liposomes and only the solute in the outer leaflet of the donor liposomes equilibrates with the outer leaflet of the acceptor liposomes. The amount of solute that is transferred to the acceptor liposomes is dependent on the relative concentrations of donor and acceptor liposomes, being larger for higher ratios of acceptor/donor liposomes (in panel A the liposome concentration is 5 × 10−10 M for both donor and acceptor while in panel D there is a tenfold excess of acceptor liposomes). However, the rate constant for the exchange is the same in both situations, and equal to the rate of desorption (which is the same for both donor and acceptor liposomes). The effect of the concentration of acceptor liposomes for the case of fast translocation is shown in panels C and F with a larger fraction of solute exchanged when the concentration of acceptor liposomes is increased, while keeping the same time dependence. The exchange rate constant obtained in panels C and F is half the value of the rate constant for desorption (observed in panels A and D), due to the fact that all solute is exchanging but only that in the outer leaflets is in contact with the aqueous phase that mediates the exchange process. The case of translocation slower than exchange between the outer leaflets, but occurring in similar time scales, is shown in panels B and E. The total amount of solute transferring towards the acceptor vesicles is affected by the relative concentration of donor and acceptor, but the relative amplitudes of the fast and slow processes are unchanged. The correct description of the concentrations time dependence (amplitude and rate constants) requires the numerical integration of the differential equations. However, a good approximation is obtained for the rate constants (desorption and translocation) through the best fit of a bi-exponential equation such as Eq. (4.30) for the concentration of solute in the acceptor liposomes.
The main disadvantage of this approach is that the relative amplitudes of the fast and slow steps are not simply the fractions of solute in the outer and inner leaflets, precluding the validation of the slow step as being translocation (and not an artifact such as amphiphile aggregation).
Simulations of the time variation in the concentration of solute in the acceptor liposomes for the case of donor and acceptor liposomes with distinct properties are shown in Fig. 4.4. Transfer is considered at distinct relative concentrations of donor and acceptor liposomes (R A/D), for the two limit situations of slow (panels A to C) and fast translocation (panels D to F).
As the concentration of acceptor liposomes increases, the amount of solute that exchanges from the donor to the acceptor liposomes increases (panels A and D). The initial rate of transfer is independent on the concentration of acceptor. However, the rate constant for the exchange process becomes lower as the concentration of acceptor liposomes is increased, because the solute transfer proceeds during a longer time interval (panels B and E) [20]. Both sets of simulations lead to a single-exponential function for the time dependence of the concentration of solute in the acceptor liposomes. The rate constant of exchange may be obtained from the best fit of a single-exponential function to the time variation in the property being observed (fluorescence from the solute or any other property proportional to the concentration of the solute in the acceptor liposomes). The dependence of the exchange rate constant on the ratio between the concentrations of donor and acceptor liposomes is shown in plot C and F, together with the best fit of the general function shown in Eq. (4.31) which has the same dependence on R A/D as Eq. (4.29) but is valid for any fraction of exchangeable solute.
For the case of exchange between LUVs, b gives the ratio between the rate constants for insertion in the acceptor and donor liposomes (\( {k}_{+}^{{\mathrm{L}}_{\mathrm{V}}^{\mathrm{A}}}/{k}_{+}^{{\mathrm{L}}_{\mathrm{V}}^{\mathrm{D}}} \)), whereas the parameters a 0 and a ∞ are related with the rate constants of desorption from the acceptor and donor binding agents, respectively. If the approximation of slow translocation is valid, \( {a}_0={k}_{-}^{\mathrm{A}} \) and \( {a}_{\infty }={k}_{-}^{\mathrm{D}} \); while if translocation is fast \( {a}_0=\frac{k_{-}^{\mathrm{A}}}{2} \) and \( {a}_{\infty }=\frac{k_{-}^{\mathrm{D}}}{2} \). The values obtained for the kinetic parameters using this methodology are essentially equal to the values considered in the simulations (see Fig. 4.4).
As was discussed, to characterize the kinetics of interaction of poorly water soluble solutes with lipid membranes, an exchange protocol between two binding agents must necessarily be used. However, the donor and acceptor binding agents do not need to be both liposomes. An approach frequently used is to first equilibrate the solute with an aqueous soluble protein (such as serum albumin) and follow the kinetics of equilibration with the acceptor liposomes. In this case the interaction between the solute and the protein is usually faster than transfer to the liposomes. This situation greatly simplifies the equations that describe the exchange process because the fast equilibration approximation may be used. Additionally, all solute bound to the protein (binding agent, B) is accessible to the aqueous media, kinetic scheme below.
The fast equilibrium approximation for the intermediate (solute in the aqueous phase) is given by:
and the rate constant for exchange is given by one of the two Eqs. (4.34), depending on whether translocation into the inner leaflet of the acceptor liposome is much slower or much faster than interaction with the outer leaflet:
As a final remark on those exchange protocols one should note that to obtain the rate constant of transfer the property being followed must be proportional to the concentration of solute in the compartment of interest. Additionally, to avoid the physical separation between the solute associated with the donor and acceptor compartments, the relation between the property and solute concentration must be different for the distinct compartments. Furthermore, the relations must be quantitatively known if the equilibrium constants are to be obtained, and/or, if the identity of the slow step is to be validated from the relative amplitude of signal variation due to this process.
There are several examples in the literature with the characterization of the exchange rate for fluorescent amphiphiles. In some studies, a clear minority, it was possible to obtain all the rate constants involved. It is worth mentioning the early work by Nichols and co-workers with the characterization of the rate constants of insertion and desorption of phospholipids with the fluorescent group NBD attached to the acyl chain of phosphatidylcholines (NBD-PC) [32, 40]. The transfer of NBD-PC was followed through quenching of its fluorescence in one of the compartments due to self-quenching or to fluorescence resonance energy transfer (FRET) to rhodamine labelled phospholipid. All the parameters were characterized for the transfer between bovine serum albumin (BSA) and LUVs, for alkyl amines with different length (labeled with NBD in the amine group, NBD-Cn) [27], phosphatidylethanolamine with NBD in the polar head group (NBD-DMPE) [2, 41] and dehydroergosterol; [42] the exchange being followed via the different fluorescence quantum yield of the fluorescent amphiphiles in the donor and acceptor binding agents. The rate of desorption from LUVs for several pyrene labelled phospholipids has also been characterized [43, 44], as well as the rate of translocation; [45] transfer being followed via the decrease in the formation of pyrene excimers. The rate constants for desorption and translocation of fatty acids labelled with the fluorescent group 9-anthroyloxy has also been characterized through the exchange between BSA and liposomes (based on the distinct quantum yield of the fluorophore) [46], or exchange between liposomes (with the incorporation of a FRET acceptor in the donor or acceptor liposomes) [47].
Fluorescence based methods have also been used to characterize the exchange of non-fluorescent amphiphiles, such as fluorescence quenching of the protein used as donor or acceptor [48,49,50], pH and electrostatic potential variations at the surface of the liposomes and/or in the bulk aqueous compartments for the case of exchange of weak acids and bases or charged amphiphiles [49, 50].
4.2 Molecular Dynamics Simulations to Characterize the Interaction and Permeation of Small Molecules Through Lipid Bilayers
Molecular dynamics (MD) simulations are a powerful tool to study the interaction of amphiphiles with lipid membranes, as they can give atomistic insight into processes and phenomena that often cannot be considered experimentally in sufficient detail [51, 52]. Additionally, the field of MD simulations in biological sciences has developed to a level where predictions of new phenomena are frequently being made, thereby generating quite important added value to complement experiments. Indeed, currently there is a variety of software applications and methodologies that makes the MD simulations easily accessed by the scientific community [53]. In the context of the interaction of amphiphilic molecules with lipid membranes, one of the central simulation techniques involves the calculation of free energies [54]. Most commonly, sampling strategies are used, notably the use of biased simulations through the Umbrella Sampling (US) [55, 56] method to calculate the Potential of Mean Force (PMF) profiles for the amphiphiles interacting with a lipid bilayer. Previous work has resulted in PMF profiles for the interaction of a variety of solutes with different bilayer compositions [57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77]. In addition to disclosing mechanistic details, these data are highly useful since they may depict the free energy barriers associated with insertion, translocation and desorption, and hence allow the computation of their rates as well as equilibrium constants [78, 79]. In this context, the systematic comparison and validation between experimental and simulation data is imperative. However, this may not be a straightforward challenge.
4.2.1 General Description on the Generation of the PMF Profile Through Lipid Bilayers
The concept of PMF was originally introduced by Kirkwood [80]. Regarding the interaction of solutes with lipid bilayers, the energy profile across the bilayer normal allows the calculation of the free energy barriers for the processes of insertion, desorption and translocation, which are the individual microscopic steps for membrane permeation. US [55, 56] is probably the most popular technique to compute the PMF along a given reaction coordinate. This technique aims to overcome limited sampling at energetically unfavorable configurations by restraining the simulation system with an additional (typically harmonic) potential [55]. Conjugated with the explicit umbrella potential, stratification strategies are used [81], whereby the reaction pathway is divided into a large number of small overlapping windows.
Generally, to calculate the PMF of the interaction of amphiphiles with lipid bilayers using US, a set of initial structures is first generated along the reaction coordinate. Although the choice of the reaction coordinate is a highly non-trivial matter, especially with complex molecules, the most immediate and simplest choice is usually the distance from the molecule to the membrane’s center of mass (COM) along the bilayer normal direction. Then, a set of production runs is carried out, applying a biasing harmonic potential between the molecule and the bilayer, relative to a reference position. From these simulations, distance distributions of the molecule’s selected coordinate around the reference position are obtained. Finally, the PMF is generated, correcting for the contribution of the biasing potential [81, 82].
Accordingly, a set of Nw separate umbrella simulations, corresponding to each umbrella window, are carried out, with an umbrella potential,
which restrains the system at the position \( {\zeta}_i^c\left(i=1,\dots, {\mathrm{N}}_{\mathrm{w}}\right) \) with a force constant K i . From each of the Nw umbrella simulations an umbrella histogram h i (ζ) is recorded, representing the probability distribution \( {\mathrm{P}}_i^b\left(\zeta \right) \) along the reaction coordinate biased by the umbrella potential w i (ζ).
After running the simulations, the data of each umbrella window are subsequently pasted together using histogram based algorithms. The most widely used technique to compute the PMF from the umbrella histograms, that is, to unbias the distributions \( {\mathrm{P}}_i^b\left(\zeta \right) \), is probably the weighted histogram analysis method (WHAM) [83]. The purpose of WHAM is to estimate the smallest statistical uncertainty of the unbiased probability distribution from the umbrella histograms, and compute the PMF [81,82,83]. The unbiased probability distribution, P(ζ), is related to the PMF by
where, k B is the Boltzmann constant, T is the temperature and ζ 0 is an arbitrary reference point where the PMF W(ζ 0) is defined to be zero. Following this procedure, an energy profile over a reaction coordinate is obtained, e.g. the energy profile of a molecule through the direction normal to a lipid bilayer.
4.2.2 The General Simulation Protocol
The number of umbrella sampling windows needed to generate a PMF profile makes such kind of procedures computationally demanding. For symmetric lipid bilayers the free energy profile is usually calculated for one leaflet, and the other leaflet is represented symmetrically. For a complete definition of the PMF, adjacent umbrella windows should span the space between the membrane center (z = 0) and the bulk water region (z ≈ 4 nm), usually separated by 1–2 Å. Since the distribution histograms should overlap properly, the harmonic umbrella potential used to restrain the position of the amphiphile should be adapted to the umbrella spacing [81, 82]. For an asymmetric bilayer, sampling of the reaction path must be done through the entire membrane, at least doubling the computational cost. From the performed simulations, the unbiased PMF is then obtained using WHAM [81, 82].
For a given molecule, different choices of variable complexity may be adopted to define the reaction coordinate—the key parameter in free energy profile computations—of a PMF profile. Usually, the distance z of the molecule’s COM, or a chemically significant atom/set of atoms (e.g. the most polar group for the case of relatively large and amphiphilic molecules), in respect to the membrane COM along the normal coordinate, is chosen as the reaction coordinate. With this definition, the location z = 0 nm represents thus the COM of the lipid molecules [60, 61, 84]. This choice results in the so called 1D-PMF. In order to improve sampling, the position and orientation of the molecules may be simultaneously restrained, resulting in a 2D-PMF. This type of reaction coordinate has been important to differentiate between distinct translocation mechanisms [85,86,87].
To obtain a PMF profile, the production simulations must start from several system configurations with the molecule at different positions of the reaction coordinate. Different procedures may be used to generate these initial structures [53]. Generation of initial structures from unrestrained simulations would be the most adequate procedure to minimize artifacts [68, 75]. However, this may be not possible for the majority of the systems where an enhanced sampling technique is used. Therefore, the most popular strategy to generate sets of initial structures for US simulations is to artificially pull the amphiphile along the reaction coordinate. In this process, slow pulling rates (e.g., ∼0.005 nm/ps) and low force constants (e.g., ∼500 kJ/mol/nm2) are used in order to avoid artificial deformations of the lipid bilayer. Even with such a careful procedure, different directions of pulling (either starting at the water, WC, or at the bilayer center, CW) may not give the same results [84], as shown in Fig. 4.5. Plotting the PMF profiles obtained from the CW and WC initial pulling directions simultaneously and considering different reference positions (bilayer center used as reference in Fig. 4.5A and the water in Fig. 4.5B) shows that this difference is caused by poor sampling at the lipid/water interface, as highlighted by the dashed boxes in the Fig. 4.5.

PMF profiles of NBD-C16 in a POPC bilayer, calculated with the CW (black) and WC (red) schemes. In CW, the data used for analysis covered a period from 20 to 110 ns, and in WC a period from 120 ns until the end of the simulation. In (A) the PMF is defined to be zero in the center of the bilayer, and in (B) the PMF is defined to be zero in the water region. Reprinted with permission from reference [84]. Copyright 2014 American Chemical Society
4.2.3 Sampling Issues
While US is a seemingly simple technique, there are several potential problems that may compromise the quality of the results. Sampling issues may be critical in the determination of energy profiles from MD simulations. Sampling problems have been addressed by some research groups, being actively discussed in the literature [66,67,68, 84, 88]. If the sampling is not sufficiently extensive, then the condition of ergodicity is broken and the free energy values found through the analysis are not accurate enough. The sources of sampling problems are several, stemming from solute size, bilayer size, bilayer defects, initial conformations or choice of the reaction coordinate, usually being expressed as orthogonal degrees of freedom separated by hidden energy barriers. Strategies to overcome these problems have been reviewed [88].
Recently, the use of advanced simulation techniques has been proposed to improve sampling in the study of solute permeation through lipid membranes, focusing on the importance of orientation and conformational motions [67, 77, 85, 87, 89, 90]. A related problem regarding sampling issues concerns equilibration. For each simulation window, the system should be properly equilibrated before adequate sampling of the reaction coordinate (for WHAM analysis) can take place [66, 67]. If these issues are not taken care of, the simulation data may include artifacts. For instance, as was shown in Fig. 4.5, when the amphiphile is pulled from the water phase to the interior of the lipid bilayer (WC), different results may be obtained compared to when the amphiphile is pulled in the opposite direction (CW). This is induced by artifacts which were shown to arise from sampling problems at the membrane-water interface, causing the simulation results to not converge despite extensive simulation times [84], as shown in Fig. 4.6. Systematic variation of PMF profiles and energy barriers, when considering different simulation times for analysis, is indicative of non-converged data.

Convergence of the PMF calculated for NBD-C16 in a POPC bilayer, with WC: (A, B) increasing the total simulation time by 5 ns intervals until the maximum simulation time of 130 ns; (C, D) discarding successive 5 ns intervals for equilibration with a maximum simulation time of 130 ns. The profiles shown in panels (A, C), and (B, D), have the reference position defined to be at the center of the bilayer and in the water region, respectively. The arrows show how the PMF profile evolves. The time dependence of the free energy barriers for translocation (black) and desorption (red) are shown as insets in panels (A, C). Reprinted with permission from reference [84]. Copyright 2014 American Chemical Society
Additionally, an appropriate choice of the reaction coordinate is decisive for finding physically correct results [84]. It has been shown that the definition of the reaction coordinate may influence the results through generation of membrane deformations during the production runs [66]. Therefore, in each particular case, one must understand how PMF calculations should be performed to avoid any problems that would result in unphysical data. For example, the distance between the solute and a locally defined center of the bilayer (only taking into account the lipid within a cylinder centered at the solute and aligned along the z-axis) may be advantageously employed to minimize membrane deformation artifacts [84]. Alternatively, as mentioned above, in some literature reports, free energy surfaces are computed, characterized by two reaction coordinates. In these works, one of the reaction coordinates is defined as the distance to the bilayer center, and the other is an angular coordinate which accounts for solute orientation [85,86,87].
Despite the simulation being sampling the true energy minima, the convergence of free energy profiles should be always carefully assessed. The total simulation time for a given umbrella window includes an equilibration of the system, followed by fluctuations around equilibrium. Preferably, the PMF should be computed using only the simulation times after equilibration. The decision on whether or not the system has reached equilibrium after a given simulation time is not trivial. The final PMF (hopefully corresponding to an equilibrated system) may be evaluated in three ways:
-
1.
assume (perhaps incorrectly) that no equilibration is needed, and in each sampling window use simulation data from increasingly long times to generate consecutive PMFs.
-
2.
systematically increase the slice of the simulation time used for equilibration in each sampling window, and use the rest of the simulation data for analysis.
-
3.
systematically increase the amount of data used for equilibration in each sampling window, and analyze the PMF profiles carried out over a fixed time interval (for instance tens of nanoseconds).
In all three analysis schemes, not only the values of the barriers but also the shapes of the profiles should be compared to each other [84], as exemplified in Fig. 4.7. The achievement of small and non-systematic variations of the PMF profiles and energy barriers is indicative of good convergence.

Convergence of the PMF of NBD-C16 in a POPC bilayer, calculated in the CW case: (A) Increasing the total simulation time by 5 ns intervals until the maximum of 110 ns. The arrows indicate the evolution of the PMF profiles (black line stands for a 5 ns simulation, red being 10 ns, etc.). (B) Discarding successive 5 ns intervals from the simulation data (for equilibration) with a maximum simulation time of 110 ns. (C) Data analyzed over 20 ns intervals. The time dependence of the free energy barriers for translocation/flip-flop (black) and desorption (red) are shown as insets in each plot. In the middle panel, Δ‡Gt o and Δ‡Gd o indicate the free energy barriers for the translocation and desorption processes, respectively. Reprinted with permission from reference [84]. Copyright 2014 American Chemical Society
4.2.4 The General Description of the PMF Profile
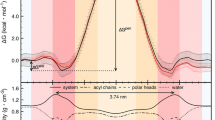
The PMF profile defines the variation in the Gibbs free energy of the system as a function of the reaction coordinate, solute position relative to the center of the bilayer for the case of permeation through lipid bilayers. The energy minimum gives the equilibrium location of the solute in the hydrated bilayer, and the energy maxima correspond to transition states in the reaction pathway. For amphiphilic molecules, an energy maximum is commonly encountered at the bilayer center due to the energetically unfavorable solvation of the polar portion of the molecule by this nonpolar environment [91, 92]. An energy maximum is also frequently observed at the bilayer/water interface due to both the hydrophobic effect (as the nonpolar portion of the molecule becomes in contact with water), and to the high density of the system at this region [61, 63, 64, 84]. For amphiphiles with long and/or bulky nonpolar groups, a decrease in the system Gibbs energy when the amphiphile leaves the membrane would be expected. This is because when the nonpolar portion of the amphiphile is partially in the aqueous media and partially inserted in the bilayer, the energetic penalty arising from the hydrophobic effect is almost complete and an extra energetic penalty is observed due to the formation of a cavity in the lipid bilayer beneath the amphiphile (with the consequent loss of lipid/lipid interactions) [32, 41, 61, 64, 93, 94]. However, for most amphiphiles, the PMF obtained does not show a decrease in energy as the amphiphile moves from its most external position at the bilayer/water interface into the bulk water, see Figs. 4.5, 4.6 and 4.7 for the case of NBD-C16 in POPC bilayers. When the PMF profile is analyzed from the amphiphile in bulk water towards its equilibrium position in the bilayer, the absence of an energy barrier at the bilayer/water interface indicates that insertion is a diffusion controlled process. This is in contradiction with the experimental results obtained for this system [27] and may result from poor sampling at this location in the reaction coordinate [66, 84, 88].
The energy barrier obtained from PMF profiles at the bilayer center may also not correspond to the energy required to place the polar portion of the amphiphile in the nonpolar environment of the bilayer center. This is because this configuration is not necessarily involved in the most probable translocation pathway followed by the amphiphile. However, there is not enough information available to evaluate whether this corresponds or not to the pathway observed in the real system.
A review on computational studies of translocation (flip-flop) of phospholipids, sterols and fatty acids has been recently published [95]. While in some systems the results are compatible with the polar portion of the amphiphile in the center of the lipid bilayer as the transition state [2, 84], more complex descriptions have been raised, from different transition paths [85,86,87, 96,97,98], to the formation of large pores in the lipid bilayer [61, 64, 99,100,101,102,103]. In the cases without pore formation, three main translocation paths have been proposed [104]. In a “push-in flip-flop”, a transverse motion occurs first, followed by the rotation of the molecule; in a “sliding flip-flop”, the transverse and rotational motions occur simultaneously; and in a “rotation flip-flop”, the rotation motion occurs first followed by the transverse motion of the molecule. The most probable translocation pathway certainly depends on the properties of the amphiphile and lipid bilayer, as well as on the local concentration of amphiphile. The confirmation of several local minima along the translocation path will represent a challenge to the development of new mechanistically meaningful kinetic models for the analysis of experimental data.
The formation of large pores in the lipid bilayer has been implicated in the translocation of phospholipids [61, 64, 99,100,101,102]. However, the pore mediated mechanism for the translocation of phospholipid should also not be taken for granted. Lipid translocation has been observed by US simulations [61, 64] or in unrestrained simulations where pores were induced [101, 102]. It should be noted that the formation of a pore implies a considerable change in the structure of the membrane that has not been considered in the kinetic models discussed above in this chapter. It should be worthy to know whether the formation of pores is being induced by the process of restraining the molecules in the membrane, without avoiding the deformation of the latter. For the case of cholesterol (which is not reported to translocate through membrane pores), once initiated, the translocation process occurs on average in 73 ns [97], a time comparable to the duration of each single restricted US simulation in recent studies [63, 66, 84, 105]. Although it may be argued that the minimum free energy path implies the formation of a pore [100, 104], with too long simulation times the membrane has the opportunity to “over-equilibrate” and deform around the restrained molecule. For solutes with strong electrostatic interactions this may result in the artificial formation of a pore. Spontaneous pore formation in a lipid bilayer has only been observed in long MD simulations for the short acyl chain phospholipid DLPC [100]. The most representative lipids of biological membranes have a longer acyl chain and the spontaneous formation of pores have a much higher energetic cost being an extremely rare event [1, 100].
For the case of the NBD-Cn series, as shown in Fig. 4.8, there is no clear tendency regarding the dependence of the translocation energy barrier on the number of carbons in the alkyl chain [27, 84]. The dependence of the desorption and translocation energy barriers on the number of carbons of the amphiphile’s alkyl chain is in qualitative agreement with experimental results for the interaction of these amphiphiles with POPC bilayers [27], and for the transfer of labeled phospholipids between vesicles [45].

PMF profiles of the NBD-Cn molecules in a POPC bilayer. PMFs of NBD-C4, -C8, -C12, and -C16 are depicted as blue, red, green, and black lines, respectively. Reprinted with permission from reference [84]. Copyright 2014 American Chemical Society
4.2.5 How to Obtain Kinetics from the PMF
Clearly, the ultimate goal of calculation of PMF profiles is to be able to estimate rate constants for the lipid/amphiphile interaction processes and permeability coefficients that can be used to calculate the rate of permeation through the bilayer. For this purpose, the Transition State Theory (TST), also known as absolute-rate theory, may be used [106,107,108]. This theory is usually applied to obtain all thermodynamic parameters for the transition states of each process involved in amphiphile permeation across bilayers from the experimental rate constants [27, 41, 109, 110].
The thermodynamic formulation of the transition state theory describes the rate constant of reaction (k) as
where k B T/h is a frequency factor, associated to the conversion from the transition state to the products, being k B, h and R the Boltzmann, Planck and gas constants, respectively; T is the absolute temperature; Δ ‡ G o is the Gibbs energy variation from the reactants to the transition state; and κ, the so-called transmission coefficient, is an efficiency parameter.
The value of Δ ‡ G o may be directly obtained from the PMF profile. To calculate the rate constant associated with crossing each energy barrier in the PMF, it is also necessary to know the transmission coefficient. A value κ = 1 is expected for elementary chemical reactions in which every vibration of the mode responsible for converting the activated complex (corresponding to the transition state of the system) to the product is effective. However, in solution, solvent cage effects are present, which delay conversion between the activated complex and the products. Moreover, the processes involved in solute permeation across the membrane are of a physical nature, and such a vibrational mode cannot be identified.
One possible strategy to overcome this problem involves additional (unrestrained) MD simulations. Regarding the translocation step, one may carry out several simulations where the amphiphile is allowed to relax from the transition state (assumed as z = 0) towards its equilibrium position. Typical relaxation curves for the NBD-Cn amphiphiles are shown in Fig. 4.9. It should be noted that, in order to achieve a statistically significant value using this strategy, an extreme large number of simulations is implied. This is particularly problematic for highly ordered systems, namely in cholesterol-containing membranes [63]. In a simple analysis, the resulting average of relaxation curves may be analyzed with a single-exponential function. For amphiphilic molecules with the size of a lipid, the characteristic relaxation time is on the order of a few to a few tens of nanoseconds [105]. Therefore, this procedure allows the calculation of the pre-exponential factor, \( \left(\kappa \frac{k_{\mathrm{B}}T}{h}\right) \), which is half the value of the relaxation rate constant. This statistical factor stems from the equal probability of going from the transition state back to the reactant (equilibrium position in the initial leaflet) or towards the product (equilibrium position in opposite leaflet) [108]. The pre-exponential factor obtained, in the order of 108 s−1, is considerably lower than the value used when applying TST with κ = 1 (1012 s−1). This suggests that the experimentally derived values for the thermodynamic activation functions should be obtained with much lower pre-exponential factors, as already used in several literature works [64, 105, 112]. For the case of the insertion/desorption steps, a similar strategy can in principle be used. Simulations where the amphiphiles are allowed to relax from the insertion/desorption transition state until the equilibrium position may be performed. However, the complexity of this membrane region may require a multi-exponential function fit to the relaxation data. It is expected that these simulations show high variability, with the amphiphile desorbing to water in some cases and inserting towards its membrane equilibrium position in others. In general, the main disadvantages of this method are the large number of unrestrained simulations required to obtain a statistically significant average relaxation curve; and the possibility of complex relaxation kinetics, of unclear interpretation. In any case, transmission coefficients may be still estimated, and used for improved calculation of the rate constants.

Transfer of the amphiphiles from the center of the bilayer to the equilibrium position evaluated as the z(t) distance from the NBD COM to the bilayer center, in POPC bilayers: (A) NBD-C4, (B) NBD-C8, (C) NBD-C12, (D) NBD-C16. NBD equilibrium positions determined from unrestrained simulations [111] (blue) and from PMF minima [84] (green) are shown for comparison. This results are an extension of the work of reference [84]
If translocation of the solute occurs through pores, the observed rate constant of translocation depends on both the rate at which the solute translocates through the pore, and on the density of pores in the bilayer. The mathematical formalism that should be used in this situation has been developed by Tieleman and co-workers and applied to predict the rate of translocation of phospholipids and the permeability coefficient of very polar solutes [61].
The conversion between calculated free energy barriers and experimental rate constants for the interaction of amphiphiles with lipid bilayers may also be conducted following the Kramers theory of reaction rates [113], applied to the theoretical description of amphiphile monomer-micelle dissociation developed by Aniansson et al. [93]. Characteristic free energies for the desorption of amphiphiles from lipid bilayers have been obtained from the experimentally observed rate constants following this model [32, 94]. The relation between the rate constant of desorption and the Gibbs energy variation between the equilibrium position in the bilayer and the transition state (Δ ‡ G o) is:
where D m is the diffusion constant for the exiting amphiphile, l b is the width of the barrier that is RT energy units below its maximum. This expression can be understood intuitively, since l b 2/D m is the time for diffusional motion over the distance l b and exp[−Δ ‡ G o/(RT)] is the relative probability that a monomer resides in the region of length l b, or within RT energy units of Δ ‡ G o. The value of l b considered in the experimental reports was around 0.1 nm being independent on the length of the alkyl chain in homologous series of amphiphiles [32, 94]. Also, in experiments D m is assumed to be close to the diffusion constant of the free amphiphile in solution. Considering l b = 0.1 nm and D m = 5 × 10−6 cm2/s, a pre-exponential factor of 5 × 1010 s−1 is obtained.
The length parameter (l b) may be easily obtained from the PMF profile. Additionally, the diffusion coefficient at the reaction coordinates near the transition state may be estimated using the force autocorrelation at each sampling window, as will be further addressed in the next section. For example, regarding the above mentioned NBD-Cn series, l b ∼ 0.2–0.3 nm and D ∼ 3–4 × 10−7cm2/s are obtained for the translocation barrier, leading to pre-exponential factors of the order of ∼5 × 108 s−1. On the other hand, for the desorption barrier, typical values l b ∼ 0.2–0.4 nm and D ∼ 5 × 10−6 cm2/s are found, leading to pre-exponential factors near ∼5 × 109 s−1.
Regarding the example of the NBD labeled amphiphiles described above, the simulated free energy barriers are clearly lower than the values estimated from the experimental rate constants, assuming the absolute rate theory (i.e., a frequency of conversion from the transition to the final states of k B T/h) [106], for both translocation and desorption processes [27, 84]. In any case, a linear dependence of the desorption free energy barrier on the number of carbons n is observed in both approaches. The slope based on simulations is 4.4 kJ/CH2, compared to 3.5 kJ/CH2 based on experiments. The quantitative disagreement between experimental and simulation results is in part a consequence of the direct application of this theory, as the actual reaction frequency is likely much lower than this limiting value [114]. However, the value of this reaction frequency is expected to depend more on the dynamics of the bilayer than on the solute itself, therefore conserving the linear dependence of the energy barriers, and giving good qualitative results.
4.2.6 Calculation of Permeability Coefficients from MD Simulations
The overall permeation through the lipid bilayer and the permeability coefficient P may be calculated from the rate constants of each step in the process [20]. MD simulations also allow calculation of permeability coefficients, which are defined according to Eq. (4.14). In silico estimation of P could provide a means to reduce costs involved in drug design and development.
By keeping track of the motions of all individual molecules in the system, MD simulations have the potential for calculation of solute permeation across membrane systems. However, on the time scales available to atomistic MD, permeation events are exceedingly rare in unbiased simulations. This precludes direct calculation of permeability coefficients from MD using Eq. (4.14). Despite this setback, there are indirect methods that can be utilized to this purpose [92]. In this section, we focus on the most used approach, the so-called inhomogeneous solubility-diffusion model [115, 116], which leads to the following expression for P:
Here, ΔG o(z) is the Gibbs free energy across the bilayer (i.e., PMF profile, see above subsections), and D z is the solute diffusion coefficient along the z-direction. z 1 and z 2 represent locations in the water phase on either side of the membrane, and therefore the integration is carried out across the whole bilayer. D z(z) may be obtained using the Einstein relation in short time ranges, or, more commonly, using the force autocorrelation method, described below in brief.
For a given z value, a constrained MD run is carried out, in which the solute depth is fixed to the intended value. The local time dependent friction coefficient of the diffusing molecule ξ(t) is related to the time autocorrelation function of the fluctuations of the instantaneous force acting on the solute when located at z, F(z, t), relative to its time average 〈F(z)〉 t , by:
where,
Assuming that ξ(t) is large and decays rapidly compared to other time scales in the system, a satisfactory description of the full dynamics is provided by the static friction coefficient ξ:
When studying diffusion across a free energy barrier, the above condition is met if the slope of the free energy barrier over a distance covered by the particle during the decay time of its friction coefficient is lower than the thermal fluctuation, RT. In this case, ξ is related to the local diffusion coefficient D z (z) of the permeating solute at depth z by:
The inhomogeneous solubility-diffusion model has been applied to several small hydrophilic and hydrophobic molecules calculating the local resistance of the membrane to the permeation as the inverse of the local permeability coefficient. It was found that for hydrophilic molecules the main barrier is represented by the hydrocarbon core, while for the hydrophobic molecules the main barrier to permeation is offered by the head group region [91]. More recent applications include a number of studies on the permeation of larger compounds, such as drugs and hormones [117, 118].
A simpler alternative to the integration procedure of Eq. (4.39) may be obtained in the framework of Kramers reaction rate theory, assuming that ΔG o(z) is characterized by a large barrier value at some value of z = z*, and that D z (z) is constant (≅ D) in that region. Under these hypotheses, one may approximate the permeability coefficient in a transition-state-like approximation according to [119, 120]
where
and ΔΔG o refers to the difference between ΔG o(z) at the maximum and the bottom of the energy barrier, that means, equal to Δ ‡ G o.
Although this approach only considers a single energy barrier, an extension to a multibarrier situation could be envisaged in the framework of the inhomogeneous solubility-diffusion model as proposed originally by Diamond and Katz [119]. As derived by these authors, the reciprocal of the permeability coefficient has the significance of a resistance, which consists of three resistances in series, r i (corresponding to insertion, translocation and desorption, respectively). Each of these terms can be obtained by taking the reciprocal of a hypothetical single-barrier P i value, obtained from Eq. (4.44), using the pertaining ΔΔG o and D. Finally, they are combined to obtain the global permeation coefficient through
Generally, the calculated permeability coefficients are qualitatively consistent with experiments, in that ranking orders are well reproduced. However, P values from simulations are typically several orders of magnitude larger than those from experiments. While several reasons could be invoked to account for this disagreement (including the diversity of experimental approaches and measured values, the much simpler systems used in simulations, unsolved force field and/or sampling issues), it is noteworthy that the ranking order among a set of compounds constitutes, by itself, valuable information for drug design [121]. Identically to the calculation of rate constants for individual processes, consideration of enhanced sampling strategies and/or multi-dimensional free energy surfaces (including orientational degrees of freedom) to identify hidden energy barriers may lead to improved quantitative accordance with experimental permeability coefficients, especially for relatively large molecules (in which reorientations can take place on the same time scale of displacements along the bilayer normal) [122].
4.3 Concluding Remarks
In this chapter, we address both experimental and simulation-based strategies to obtain kinetic information regarding the processes of interaction (insertion, desorption, translocation) between an amphiphilic solute and lipid membranes. The problem is formulated in general terms, from which limiting cases of practical importance are derived. Different experimental approaches are described, with an emphasis on fluorescence spectroscopy. On the other hand, MD simulations have evolved to the point where, in addition to detailed mechanistic information, free energy profiles and permeability coefficient values across the bilayer can be obtained for small- and medium-sized solutes.
Clearly, there is still plenty of room for improvement regarding quantitative agreement between calculated and experimental energy barriers and corresponding equilibrium and rate constants, due to both MD (important sampling issues), theory (uncertainty in frequency factor for TST) and experiment related (variety of approaches leading to scattered reported values) issues. However, experiment and simulation are most often in good qualitative accordance, and future advances in sampling strategies and identification of hidden energy barriers [77], may lead to an effective utility of MD simulations in prediction of permeation and in rational drug design.
Notes
- 1.
In this and in the next equations, the concentrations are calculated with respect to the total volume of the solution except when explicitly indicated.
References
Paula S, Volkov AG, VanHoek AN, Haines TH, Deamer DW. Biophys J. 1996;70:339.
Moreno MJ, Estronca L, Vaz WLC. Biophys J. 2006;91:873.
Verkman AS, Dix JA, Seifter JL. Am J Physiol. 1985;248:F650.
Chakrabarti AC, Deamer DW. Biochim Biophys Acta. 1992;1111:171.
Chehin RN, Isse BG, Rintoul MR, Farias RN. J Membrane Biol. 1999;167:251.
Mayer PT, Xiang TX, Niemi R, Anderson BD. Biochemistry. 2003;42:1624.
Barbet J, Machy P, Truneh A, Leserman LD. Biochim Biophys Acta. 1984;772:347.
Allen TM, Cleland LG. Biochim Biophys Acta. 1980;597:418.
Lasch J. Biochim Biophys Acta. 1995;1241:269.
Loura LMS, de Almeida RFM, Coutinho A, Prieto M. Chem Phys Lipids. 2003;122:77.
Pokorny A, Almeida PFF. Biochemistry. 2004;43:8846.
Ahyayauch H, Goni FM, Bennouna M. J Liposome Res. 2003;13:147.
Chen RF, Knutson JR. Anal Biochem. 1988;172:61.
Zwolinski BJ, Eyring H, Reese CE. J Phys Colloid Chem. 1949;53:1426.
Overton E. Vierteljschr d Naturforsch Ges Zurich. 1899;44:88.
Deamer DW, Kleinzeller A, Fambrough DM. Membrane permeability: 100 years since Ernest Overton, vol. 48. San Diego: Academic Press; 1999.
Missner A, Pohl P. Chemphyschem. 2009;10:1405.
Li S, Hu PC, Malmstadt N. Anal Chem. 2010;82:7766.
Eyer K, Paech F, Schuler F, Kuhn P, Kissner R, Belli S, Dittrich PS, Kramer SD. J Control Release. 2014;173:102.
Filipe HAL, Salvador A, Silvestre JM, Vaz WLC, Moreno M. J Mol Pharm. 2014;11:3696.
Thomae AV, Koch T, Panse C, Wunderli-Allenspach H, Kramer SD. Pharm Res. 2007;24:1457.
Sawada GA, Barsuhn CL, Lutzke BS, Houghton ME, Padbury GE, Ho NFH, Raub TJ. J Pharmacol Exp Ther. 1999;288:1317.
Kramer SD, Lombardi D, Primorac A, Thomae AV, Wunderli-Allenspach H. Chem Biodivers. 2009;6:1900.
Estronca LMBB, Moreno MJ, Laranjinha JAN, Almeida LM, Vaz WLC. Biophys J. 2005;88:557.
Steinfeld JI, Francisco JS, Hase WL. Chemical kinetics and dynamics. 2nd ed. New Jersey: Prentice-Hall; 1999.
Estronca LMBB, Filipe HAL, Salvador A, Moreno MJ, Vaz WLC. J Lipid Res. 2014;55:1033.
Cardoso RMS, Martins PAT, Gomes F, Doktorovova S, Vaz WLC, Moreno MJ. J Phys Chem B. 2011;115:10098.
Martins PT, Velazquez-Campoy A, Vaz WLC, Cardoso RMS, Valerio J, Moreno MJ. J Am Chem Soc. 2012;134:4184.
Donovan JM, Jackson AA. Biochemistry. 1997;36:11444.
McIntyre JC, Sleight RG. Biochemistry. 1991;30:11819.
Sampaio JL, Moreno MJ, Vaz WLC. Biophys J. 2005;88:4064.
Nichols JW. Biochemistry. 1985;24:6390.
Kuzelova K, Brault D. Biochemistry. 1994;33:9447.
Pokorny A, Almeida PFF, Vaz WLC. Biophys J. 2001;80:1384.
Pokorny A, Almeida PFF, Melo ECC, Vaz WLC. Biophys J. 2000;78:267.
Luo M, Fadeev EA, Groves JT. J Am Chem Soc. 2005;127:1726.
Cabral DJ, Small DM, Lilly HS, Hamilton JA. Biochemistry. 1987;26:1801.
Jones JD, Thompson TE. Biochemistry. 1989;28:129.
Almeida PF. Biophys J. 1922;1999:76.
Nichols JW, Pagano RE. Biochemistry. 1981;20:2783.
Abreu MSC, Moreno MJ, Vaz WLC. Biophys J. 2004;87:353.
Estronca LMBB, Moreno MJ, Vaz WLC. Biophys J. 2007;93:4244.
Roseman MA, Thompson TE. Biochemistry. 1980;19:439.
Frank A, Barenholz Y, Lichtenberg D, Thompson TE. Biochemistry. 1983;22:5647.
Homan R, Pownall HJ. Biochim Biophys Acta Biomembr. 1988;938:155.
Kamp F, Westerhoff HV, Hamilton JA. Biochemistry. 1993;32:11074.
Storch J, Kleinfeld AM. Biochemistry. 1986;25:1717.
Richieri GV, Anel A, Kleinfeld AM. Biochemistry. 1993;32:7574.
Langner M, Isac T, Hui SW. Biochim Biophys Acta Biomembr. 1995;1236:73.
Cupp D, Kampf JP, Kleinfeld AM. Biochemistry. 2004;43:4473.
Loura LMS, Ramalho JPP. Molecules. 2011;16:5437.
Lyubartsev AP, Rabinovich AL. Soft Matter. 2011;7:25.
Javanainen M, Martinez-Seara H. Biochim Biophys Acta Biomembr. 2016;1858:2468.
Gumbart JC, Roux B, Chipot C. J Chem Theory Comput. 2012;9:794.
Torrie GM, Valleau JP. J Comput Phys. 1977;23:187.
Kästner J. Wiley Interdiscip Rev: Comput Mol Sci. 2011;1:932.
Wennberg CL, van der Spoel D, Hub JS. J Am Chem Soc. 2012;134:5351.
MacCallum JL, Bennett WFD, Tieleman DP. Biophys J. 2008;94:3393.
Sapay N, Bennett WFD, Tieleman DP. Biochemistry. 2010;49:7665.
Hinner MJ, Marrink SJ, de Vries AH. J Phys Chem B. 2009;113:15807.
Tieleman DP, Marrink S-J. J Am Chem Soc. 2006;128:12462.
Bennett WFD, Tieleman DP. J Lipid Res. 2012;53:421.
Bennett WFD, MacCallum JL, Tieleman DP. J Am Chem Soc. 2009;131:1972.
Sapay N, Bennett WFD, Tieleman DP. Soft Matter. 2009;5:3295.
MacCallum JL, Tieleman DP. J Am Chem Soc. 2006;128:125.
Neale C, Bennett WFD, Tieleman DP, Pomès R. J Chem Theory Comput. 2011;7:4175.
Neale C, Madill C, Rauscher S, Pomès R. J Chem Theory Comput. 2013;9:3686.
Paloncýová M, Berka K, Otyepka M. J Chem Theory Comput. 2012;8:1200.
Comer J, Schulten K, Chipot C. J Chem Theory Comput. 2014;10:554.
Peters GH, Werge M, Elf-Lind MN, Madsen JJ, Velardez GF, Westh P. Chem Phys Lipids. 2014;184:7.
Wang Y, Hu D, Wei D. J Chem Theory Comput. 2014;10:1717.
Martin LJ, Chao R, Corry B. Biophys Chem. 2014;185:98.
Jakobtorweihen S, Zuniga AC, Ingram T, Gerlach T, Keil FJ, Smirnova I. J Chem Phys. 2014;141:045102.
Ma J, Domicevica L, Schnell JR, Biggin PC. Phys Chem Chem Phys. 2015;17:19766.
Paloncýová M, Berka K, Otyepka M. J Phys Chem B. 2013;117:2403.
Paloncýová M, DeVane R, Murch B, Berka K, Otyepka M. J Phys Chem B. 2014;118:1030.
Jämbeck JPM, Lyubartsev AP. J Phys Chem Lett. 2013;4:1781.
Awoonor-Williams E, Rowley CN. Biochim Biophys Acta Biomembr. 2016;1858:1672.
Dickson CJ, Hornak V, Pearlstein RA, Duca JS. J Am Chem Soc. 2016;139:442–52.
Kirkwood JG. J Chem Phys. 1935;3:300.
Roux B. Comput Phys Commun. 1995;91:275.
Hub JS, de Groot BL, van der Spoel D. J Chem Theory Comput. 2010;6:3713.
Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA. J Comput Chem. 1992;13:1011.
Filipe HAL, Moreno MJ, Róg T, Vattulainen I, Loura LMS. J Phys Chem B. 2014;118:3572.
Jo S, Rui H, Lim JB, Klauda JB, Im W. J Phys Chem B. 2010;114:13342.
Wei C, Pohorille A. J Phys Chem B. 2014;118:12919.
Parisio G, Sperotto MM, Ferrarini A. J Am Chem Soc. 2012;134:12198.
Neale C, Pomès R. Biochim Biophys Acta Biomembr. 2016;1858:2539.
Ghaemi Z, Minozzi M, Carloni P, Laio A. J Phys Chem B. 2012;116:8714.
Cardenas AE, Elber R. Mol Phys. 2013;111:3565.
Bemporad D, Essex JW, Luttmann C. J Phys Chem B. 2004;108:4875.
Xiang T-X, Anderson BD. Adv Drug Deliv Rev. 2006;58:1357.
Aniansson EAG, Wall SN, Almgren M, Hoffmann H, Kielmann I, Ulbricht W, Zana R, Lang J, Tondre C. J Phys Chem. 1976;80:905.
Jones JD, Thompson TE. Biochemistry. 1990;29:1593.
Parisio G, Ferrarini A, Sperotto MM. Int J Adv Eng Sci Appl Math. 2016;8:134.
Ogushi F, Ishitsuka R, Kobayashi T, Sugita Y. Chem Phys Lett. 2012;522:96.
Choubey A, Kalia RK, Malmstadt N, Nakano A, Vashishta P. Biophys J. 2013;104:2429.
Róg T, Stimson LM, Pasenkiewicz-Gierula M, Vattulainen I, Karttunen M. J Phys Chem B. 2008;112:1946.
Bennett WFD, Tieleman DP. J Chem Theory Comput. 2011;7:2981.
Bennett WFD, Sapay N, Tieleman DP. Biophys J. 2014;106:210.
Gurtovenko AA, Onike OI, Anwar J. Langmuir. 2008;24:9656.
Gurtovenko AA, Vattulainen I. J Phys Chem B. 2007;111:13554.
Huang K, García AE. Biophys J. 2013;104:412.
Arai N, Akimoto T, Yamamoto E, Yasui M, Yasuoka K. J Chem Phys. 2014;140:064901.
Bennett WFD, MacCallum JL, Hinner MJ, Marrink SJ, Tieleman DP. J Am Chem Soc. 2009;131:12714.
Eyring H. Chem Rev. 1935;17:65.
Wynne-Jones WFK, Eyring H. J Chem Phys. 1935;3:492.
Evans MG, Polanyi M. Trans Faraday Soc. 1935;31:0875.
McConnell HM, Kornberg RD. Biochemistry. 1971;10:1111.
McLean LR, Phillips MC. Biochemistry. 1981;20:2893.
Filipe HAL, Moreno MJ, Loura LMS. J Phys Chem B. 2011;115:10109.
Neuvonen M, Manna M, Mokkila S, Javanainen M, Rog T, Liu Z, Bittman R, Vattulainen I, Ikonen E. PLos One. 2014;9:e103743.
Kramers HA. Physica. 1940;7:284.
Roux B. J Gen Physiol. 1999;114:605.
Diamond JM, Katz Y. J Membrane Biol. 1974;17:121.
Marrink SJ, Berendsen HJC. J Phys Chem. 1994;98:4155.
Orsi M, Essex JW. Soft Matter. 2010;6:3797.
Carpenter TS, Kirshner DA, Lau EY, Wong SE, Nilmeier JP, Lightstone FC. Biophys J. 2014;107:630.
Wilson MA, Pohorille A. J Am Chem Soc. 1996;118:6580.
Ulander J, Haymet AD. J Biophys J. 2003;85:3475.
Orsi M, Essex JW, editors. Molecular simulations and biomembranes: from biophysics to function. Cambridge: The Royal Society of Chemistry; 2010. p. 76.
Parisio G, Stocchero M, Ferrarini A. J Chem Theory Comput. 2013;9:5236.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter

Cite this chapter
Filipe, H.A.L., Cardoso, R.M.S., Loura, L.M.S., Moreno, M.J. (2017). Interaction of Amphiphilic Molecules with Lipid Bilayers: Kinetics of Insertion, Desorption and Translocation. In: Chattopadhyay, A. (eds) Membrane Organization and Dynamics . Springer Series in Biophysics, vol 20. Springer, Cham. https://doi.org/10.1007/978-3-319-66601-3_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-66601-3_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-66600-6
Online ISBN: 978-3-319-66601-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)