
Abstract
Transmembrane proteins inhabit a highly asymmetric environment that is, to a first approximation, two-dimensional. Many of them serve to transmit information between aqueous compartments, while others serve as conduits for the transport of material between compartments. In serving these functions they have to adopt at least two stable structures and rapidly interconvert between them. The paucity of atomic resolution structures has been limiting in elucidating the mechanisms by which these proteins carry out their functions. However, this century has seen the determination of the three-dimensional structures of a number of membrane proteins, leading to the start of an understanding of the dynamics displayed by them within the bilayer. Without attempting to be exhaustive, we provide illustrative examples of dynamics in membrane proteins and review their underlying mechanisms as they insert, fold and function in biological membranes.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


10.1 Introduction
Proteins can be broadly classified by the milieu they reside in. Those embedded in the non-polar environment of membranes and which have portions that traverse the membrane, are called integral membrane proteins. These proteins carry out a variety of functions ranging from providing a local structural framework at the membrane to the transmission of physiologically critical material and information across membranes. Inasmuch as they form the interface between the cell and its environment, plasma membrane resident proteins have been the subject of intense study. However, the detailed characterisation of these proteins and their mechanistic understanding has been impeded because of the challenge of obtaining purified protein in sufficient quantities. Nevertheless, there has been progress made into structure determination of membrane proteins over the past two decades. While only 1% or so of protein structures deposited in the Protein Data Bank (PDB) over this period are of membrane proteins, the number of membrane protein structures determined in the twenty-first century greatly exceed the total number determined prior to this period. Armed with these static pictures, it is now becoming possible to understand the functional dynamics of membrane proteins.
The α-helix, first recognized by Linus Pauling 65 years ago [1] and the β-sheet, Pauling’s “other” secondary structure [2], provided the only solutions to the problem of satisfying the hydrogen bonding requirements of the peptide bond within the bilayer (Fig. 10.1). The edges of a β-sheet would be unstable in a bilayer, hence it folds on itself to close and form a barrel in the membrane. β-barrels are seen primarily in proteins of the outer membranes of bacteria, mitochondria and plastids. While structures are available for a few β-barrel proteins (such as in Fig. 10.1), considerably more structural and biochemical literature is available for proteins composed of transmembrane helices (TMHs). There is also a substantial bias towards information about plasma membrane proteins compared to those residing on endomembranes.

Two major categories of membrane proteins. Integral Membrane Proteins are either all-helical as in (a) or β-barrel as in (b). (a) KcsA—the first ion channel to be crystallised is an all helix protein; shown here embedded in the lipid bilayer. It is representative of the closed state of almost all potassium channels. The pore has a selectivity filter at the level of lipid head-groups in the upper leaflet visible in the cut-away model in the left panel. The C-terminus of the helices obstruct the channel. The right panel highlights the constricted channel aperture. On the other hand, there is Voltage Dependent Anion Channel (VDAC) (b). It is an interesting β-barrel protein found on the outer membrane of mitochondria. It posesses an atypical odd number of β-strands unlike other β-barrels which have an even number. The right panel shows the barrel from the top highlighting the lone helix that is hypothesised to alter the conductivity of the protein. Both (a) and (b) are drawn surrounded by a homogenous dipalmitoylphosphatidylcholine (DPPC) lipid bilayer modelled around the crystal structures available in PDB; [PDB id: 1BL8, 2JK4]
Proteins that mediate transduction of information and exchange matter across the membrane have to adopt at least two forms: one that facilitates transmission and another that does not. The interconversion between these forms is controlled by an external signal such as a ligand, voltage, pressure, temperature, etc. However, the proteins first have to be made on ribosomes as linear polypeptides, folded into their final three-dimensional (3D) structure and then be transported to their location of action where they function. Hence, the dynamics of such membrane proteins can be studied broadly at three stages: (i) insertion into the membrane and folding therein, (ii) transport to the appropriate organelle and (iii) interconversion between a transmission competent state and the ground state.
10.2 Membrane Protein Translocation/Insertional Dynamics
Protein dynamics initiate right when a polypeptide starts emerging from the ribosomal exit tunnel. The highly hydrophobic nascent polypeptides have to be folded and transported to their site of function. While there has been considerable progress studying the folding of soluble proteins, membrane proteins pose major difficulties. Unfolding membrane proteins to structure-less entities generally results in irreversible denaturation, while refolding from partially folded states fails to capture the entire process (reviewed by Stanley and Fleming [3]). A further complication occurs due to the fact that the membrane ambient into which the protein has to fold is not homogenous, being polar at the head-groups and very non-polar at the mid-plane [4].
Khorana’s group showed that individual helices of bacteriorhodopsin (bR) can insert and fold independently, then assemble into a functional entity capable of binding retinal and pumping protons on absorbing light [5]. This led to the postulation of a two-stage folding process with independent insertion of individual helices, followed by their assembly into a higher order structure within the membrane [6]. Kaback’s group expressed lactose permease as contiguous polypeptides and if the breaks between fragments are introduced in loop regions, the peptides could associate to mediate active lactose transport [7]. Thus, the case for two-stage folding is strong, but the data is limited to very few membrane proteins.
Recent experiments have used cell free translation systems to extend this knowledge to insertion of freshly synthesised bacteriorhodopsin into nanodiscs. These data indicate that, in this system, retinal is required for formation of the fully folded and functional form of the protein [8]. However, for most membrane proteins, we are limited to the knowledge of the machinery that promotes insertion into the membrane.
10.2.1 Prokaryotic Translocation Machinery
10.2.1.1 α-Helical Proteins
10.2.1.1.1 SecYEG Translocase
α-helical proteins are predominantly found in the inner membrane of Gram negative bacteria. The growing hydrophobic chain emerging from the ribosome exit tunnel is identified by a Signal Recognition Particle (SRP) [9]. A groove within the SRP recognises the non-polar signal peptide that might be the first TM segment or a region distal to it [10, 11]. The signal peptide loaded SRP is then transferred to the SecYEG translocon by the SRP-FtsY composite. FtsY is a receptor for SRP and utilises energy from GTP hydrolysis to load the nascent polypeptide on the SecYEG complex. SecY is a 10 transmembrane helix protein that appears to have two exit points [12]. One is a channel with a 3–5 Å hydrophobic constriction and the other permits lateral entry to the membrane [13, 14].
An α-helix blocks the channel in SecY and the lateral entry port between TM2-TM7 is also gated. The former is opened by signal peptide binding and the latter by protein translocation through the channel [14]. This process is initiated when the nascent polypeptide inserts into the channel as a loop. Signal peptide gets intercalated into the walls of SecY while the distal region gets close to the pore [15]. This binding moves the pore blocking helix by about 20 Å [16]. Subsequently, the lateral entry gate opens into the membrane on protein translocation into the channel, enabling insertion into the membrane. The signal sequence remains bound till the entire protein has passed through [17].
Peptide propulsion through SecY requires the force generated either by the ribosome itself [18] or by an ATPase-SecA [19] and a proton motif force [20]. SecA has a two helix finger domain which is found near the entrance of the SecYEG channel [21]. It is believed that this two helix finger pushes substrates in to the channel [22]. Moreover, binding of SecA to SecY creates a 5 Å gap in the lateral gate manned by TM2 and TM7 [21]. Hence, SecY in conjunction with SecA or the ribosome can mediate co- or post-translational insertion, respectively, in the inner membrane of Gram negative bacteria.
10.2.1.1.2 YidC Insertase
The YidC Insertase enables integration of membrane proteins in a SecYEG-independent manner [23]. In contrast to SecYEG translocon that can mediate both co- and post-translational translocation, YidC facilitates only post-translational translocation [24]. How a substrate protein chooses between alternate pathways to integrate into the membrane is not currently known. But, it is worth noting that YidC can associate with SecYEG to form a supercomplex [25].
The YidC structure reveals a novel fold. It has a cytoplasmic helical hairpin and a central membrane embedded groove that opens towards the cytoplasm and the membrane. It is proposed that the cytoplasmic hairpin facilitates the entry of the substrate protein into the groove. The groove being blocked on the extracellular side, guides the processing TMH to slide into the bilayer core [26].
10.2.1.2 β-Barrel Proteins
β-barrel proteins are found in the outer membrane of Gram negative bacteria. The insertion of these outer-membrane proteins (OMPs) is mediated by a complex machinery composed of a Bam supercomplex and a few chaperones [27]. Upon synthesis in the cytoplasm, the polypeptide is carried across the inner membrane by the Sec translocon (Sect. 10.2.1.1). Once in the periplasmic space, it is bound by chaperones SurA and Skp which protect and escort the nascent OMP to the outer membrane [27]. A number of weak contacts mediate the transfer of the protein to the Bam complex where it is folded and passed into the membrane.
Multiple crystal structures are available for the 5 Bam(A–E) individual proteins that make up the supercomplex (reviewed by O’Neil et al. [28]). Moreover earlier this year, multiple groups solved the structure of the complete complex [29,30,31]. This has brought substantial advance into understanding the molecular architecture that inserts and aids the formation of tertiary structure of an OMP.
10.2.2 Eukaryotic Translocation Machinery
10.2.2.1 α-Helical Proteins
Despite the large number of membrane bound organelles, eukaryotes have only three sites of TMH insertion. Co-translational insertion occurs at the endoplasmic reticulum (ER), inner mitochondrial and the thylakoid membrane. Sec61 which is present on the ER membrane is a homologue of SecY [32]. Though absent in the ER, Oxa1 in the inner mitochondrial membrane and Alb3/Alb4 in chloroplasts appear to be YidC homologues in eukaryotes [24]. Post-translational translocation has also been observed at the ER mediated by Sec62 and Sec63. There are other proteins in the ER that aid the translocation in specific ways (reviewed in Denks et al. [33]) and include TRAM, TRAP, oligosaccharyl transferase, BiP, etc. The process of targeting proteins to different membranes is beyond the scope of this article but the reader is referred to an excellent review [34].
10.2.2.2 β-Barrel Proteins
In Eukaryotes, β-barrel proteins are found predominantly on the outer membrane of mitochondria and plastids. Homologues of the Prokaryotic OMP insertion and translocation machinery are found in the Eukaryotic organelles [35]. In the mitochondrion, polypeptides are imported from the cytoplasm through the TOM (Translocase of Outer Membrane) complex where TOM40, itself a β-barrel, forms the protein conduction channel. The imported protein is stabilised by small TIM (Translocase of Inner Membrane) chaperones—Tim9 & Tim10 and MIA (Mitochondrial Inner membrane Assembly) chaperones. The SAM (Sorting and Assembly Machinery) complex finishes the insertion of β-barrel precursors into the outer membrane. In the SAM complex, SAM50 is thought to perform the same role as the Prokaryotic protein—BamA [36]. The chloroplast membrane protein insertion pathway remains relatively obscure with only a few implicated proteins (Tic-Toc complex) that have been characterised [37].
10.3 Energetics Underlying Protein Dynamics in Lipid Bilayers
Integral membrane proteins, especially TM helices, upon reaching their destination are met with similar and dissimilar hydrophobic molecules. In other words, they reside in a complex milieu where lateral interactions between adjacent helices occur in competition with interactions with surrounding lipids and also with the encapsulating water. Furthermore, there are global bilayer effects which arise due to the electric field across the membrane and phase separation amongst lipids. Several studies have been undertaken to elucidate the forces stabilising the final 3D structures adopted by TMHs. Some understanding of the energetics of transitions between stable states has also been obtained in a few cases.
10.3.1 Van der Waal’s Interactions
Van der Waals (VdW) packing interactions contribute significantly to the integrity of the core of membrane proteins. This is in contrast to soluble proteins where the core is stabilised by hydrophobic interactions [38]. However, membrane proteins cannot utilise the entropic gain of partitioning away from water if their interactions occur in the interior of the bilayer. The first example that emphasized the importance of VdW interactions in membrane protein dynamics came from studying the transmembrane helix (TMH) of Glycophorin A (GpA) [39]. Interhelical packing in GpATM was more stabilising compared to helix-lipid interactions, thereby promoting its homo-dimerization. The dimer interface in GpA is formed by a GxxxG motif where the Glycines occur on the same side of the lone TMH of GpA [40]. GxxxG has become probably the most studied dimerization motif in membranes and we take it up in more detail in Sect. 10.4.1. In addition, the VdW packing in membrane proteins is optimized for function. For instance, bR has only a fourth of void space in the interior of the protein when compared to mechanosensitive channels like MscL (Mechanosensitive Channel of Large conductance) which are loosely packed [41]. The empty pockets in receptors and channels allows for conformational flexibility required to undergo gating transitions [41].
10.3.2 Hydrogen Bonding
Hydrogen bonding is widespread in membrane proteins. Apart from participating in secondary structure formation, hydrogen bonding is also critical for tertiary structure formation. However, experimental determinations of the contribution of hydrogen bonds to stability fall short by 3.5–4.5 kcal mol−1 of the expected value in several cases [42]. Nevertheless, a single polar residue has been shown sufficient to drive homo-dimerization of an otherwise hydrophobic model TMH [43, 44]. The contribution of Glycines is also particularly well studied in the context of hydrogen bonding in membranes. Especially, the GASRight/GxxxG/GG4 motif has been shown to be partly stabilised by hydrogen-bonding [45].
10.3.3 Salt Bridges
Salt bridge interactions are thought to contribute over long range [46]. Debye lengths in the interior of membranes can be very long compared to those in the aqueous phase. Further, the energy of all electrostatic interactions is enhanced in the low dielectric of the bilayer. Making a salt-bridge in a hydrophobic membrane is also energetically favoured when the contributing charged residues are in close proximity as seen in bacteriorhodopsin [47, 48]. Since burying a charged residue in the bilayer core is energetically costly, salt-bridges often contribute to function. There is experimental evidence supporting the role of specific salt bridges in altering the functional state of α-helical membrane proteins such as CFTR (Cystic Fibrosis Transmembrane conductance Regulator), Kcv (Potassium channel chlorella virus) [49], rhodopsin and TCR (T-cell receptor). In CFTR, a change from such coulombic interactions between R347-D924-D993 to R352-D993 can change its conductance [50]. Notably, rhodopsin becomes constitutively active upon removal of one salt-bridge [51] and salt bridge formation can promote homo/hetero-dimerization of TMH in TCR α, β and ζζ [52]. Interestingly, the dimerization in TCR occur through Aspartate and Threonine residues in preference to Glycines of GxxxG which are also present in the latter protein.
10.3.4 Aromatic–Aromatic Interactions
The WxxW, WxxY, YxxY motifs have also been shown to drive dimerization of TMHs through aromatic-aromatic interactions between Tryptophan and Tyrosine residues [53]. These are long range (approx. 7 Å) interactions, usually considered in the context of strengthening the tertiary folded structure of integral membrane proteins [54], e.g. there are seven aromatic pairs that form in KcsA. Also, a ZAX motif, where Z = Alanine, Tryptophan, Phenylalanine, or Tyrosine and X = Alanine, Histidine, Lysine, Arginine show very high dimerization affinity in TMHs. This motif is stabilised by cation-π interactions [55]. It is the same cation-π interactions between protein and lipids that preferentially stabilise Tryptophan and Tyrosine residues at the membrane-water interface [56] and sometimes promote tilts in TM helices [57].
It should be noted that lipids in the bilayer have also been implicated in enabling interaction of TMHs and affecting function (reviewed in [58]). These interactions may be specific or non-specific and manifest themselves as change in helical tilts, perpendicular shifts of helices with respect to the membrane normal (reviewed by Lee [59]) or even inversion of helical topology post-insertion [60]. Lipids can also modulate the strength of existing dimers, shifting their dynamic equilibrium, like it is shown for the Glycophorin A TM dimer [61].
10.4 Dynamics of Helix Dimerization
The binding of ligands to signal transducing receptors on one side of the membrane, results in conformational rearrangements leading to changes in accessibility of critical residues on the other side. This alters protein-protein interactions with downstream binding partners that translates to a signal being transduced in the far compartment. Transmission of binding information across the membrane may be expected to implicate conformational rearrangements of transmembrane helices. The simplest case would involve proteins with a single transmembrane helix. We review instances of protein-protein interactions mediated by a single transmembrane helix (TMH) in different contexts focussing on well-characterised instances of bitopic proteins.
10.4.1 GxxxG Motif
The GxxxG/GG4 or the GASRight motif is a bona-fide motif that induces dimerization of α-helices both in bilayers and micelles. This motif was first observed in Glycophorin A (GpA) [39, 62] which itself was one of the first membrane proteins to be sequenced [63]. Shortly after the protein was sequenced, it was observed that the TM region of GpA (GpATM) is responsible for dimerization of the protein [64]. This was inferred from Dodecyl Polyacrylamide gels (SDS-PAGE) of erythrocyte ghosts and it wasn’t until a decade after, that dimerization in gels could be reproduced synthetically in liposomes [65]. On mapping the dimerization interface of GpATM, it was found that a minimal sequence of GxxxG, where the ‘x’ represent dimerization insensitive amino-acids, could cause dimerization of unrelated transmembrane helices [66]. This discovery marked GxxxG as a common dimerization motif. Subsequent work which produced a NMR (Nuclear Magnetic Resonance) structure of the GpATM validated the biochemically identified interface (Fig. 10.2) [40]. Also, a search through the sequences of transmembrane helices available at the time revealed an enrichment of the said motif pointing to its physiological importance [67].

Dimerization of Glycophorin A TM (GpATM) helix. GpATM is one of the most studied examples in the context of helical dimerization in membranes (see Sect. 10.4.1 in the main text for more details). The GxxxG motif (shown in pink) utilises hydrogen bonding and Van der Waals interactions to stabilise an SDS resistant helical dimer
Proteins apart from GpA, like EGFR [68], ErbB4 [69], BNIP3 [70], etc. carrying a GxxxG motif form similar right handed helical TM dimers with a crossing angle of –40°. Of all the 3D structures of TM helices—bitopic and polytopic—in membrane proteins, when clustered pairwise, 12.8% had similar parallel, right-handed geometry as dimeric GpATM [71]. Proteins like RTKs (Receptor Tyrosine Kinases—ErbBs), neuropilins [72], immunologically important receptors like TLRs (Toll-like Receptors) [73], MHC (Major Histocompatibility Complex) [74] and Integrins [75, 76] have all been shown to use the GxxxG motif to dimerise. This geometry is stabilised by Van der Waals packing and hydrogen bonding (Sect. 10.3) [45].
10.4.1.1 GxxxG Is Not Sufficient for Dimerization
The experimental tools developed during discovery of GxxxG as a dimerization motif spawned a great deal of research on finding other such motifs. These validation tools utilise a similar concept of expressing TMH of interest fused with a protein that is active only as a dimer. First of these was ToxR, where the TMH dimerization induces ToxR dimerization leading to controlled expression of β-galactosidase enzyme which can be monitored [77]. Variations of this method include TOXCAT [78], POSSYCCAT [79], GALLEX [80], BACTH [81], AraTM [82] and MaMTH [83]. As a result, polar motifs (SxxxSSxxT and SxxxSSxxT) [84], glycine zipper (GxxxGxxxG) and its variants with SAT substitutions [85], WxxW, WxxY and YxxY [53] have been shown to cause association of monomeric TMHs. Still, these dimer interfaces have limited associated literature which has been mentioned at the appropriate places.
These observations lead to the idea that any small-xxx-small motif could cause dimerization. However, this assumption was proved incorrect and believably so, as more than 55% of all predicted TM helices have a small-xxx-small motif [86]. On similar lines, it was also found that GxxxG in GpATM is also sensitive to the sequence context in which it occurs [87, 88]. Neither artificially enriching transmembrane sequences from a randomised pool nor searching the entire sequence space of naturally occurring sequences carrying the dimerization motif, reveals any common contextual scaffolding pattern for GxxxG [86].
Studying GxxxG on more proteins revealed that the presence of GxxxG by itself does not cause dimerization. When recording dimerization status of peptides in denaturing polyacrylamide gels, unlike GpATM, a vast majority do not show association [89]. Even a high sequence conservation does not ensure that the GASRight motif will convey association of the TMHs [90, 91]. In the same vein, its corollary is also true; not every TM helical interaction relies on GxxxG or small-xxx-small motif. For instance, Dap12 which is an immunologically important, signalling competent receptor that associates with other proteins such as the T-cell receptor was shown to use a polar residue to dimerise in spite of the presence of a GxxxG motif [92] (See also Sect. 10.3.3). Another example illustrating the same is discussed in Sect. 10.4.2.1.
10.4.1.2 Ab Initio Prediction of Dimerization
Despite the advancement of technology, most simulations of transbilayer segments are computationally limiting. The application of force-field based modelling has remained the only option for membrane biologists trying to study helical interactions until recently. Nevertheless, simplified approaches are being developed to circumvent the issue. PREDDIMER is one such algorithm that is available through a web server [93]. Another method—CATM, is available as an open-source compilable C++ library on the internet [45]. CATM screens the helical interface with respective to a set of 463 geometries that any GxxxG motif can afford and then optimises the resulting structure using Monte Carlo simulations. While CATM presently only computes homo-dimeric interactions, PREDDIMER can be used for both homo- and hetero-dimeric structures. Both these solutions have stood validation against the experimentally solved available 3D structures for interacting TM helices.
In summary, we can explain some of the available structures containing the GASRight motif invoking hydrogen bonding and Van der Waals forces which has enabled respectable prediction of helical dimerization in membranes. However, more work is needed to have a unifying model to understand and predict the mechanism of GxxxG driven dynamics in natural membranes.
10.4.2 Beyond a Passive GxxxG Motif
A high resolution 3D structure alone isn’t enough to furnish mechanistic information about a protein. Nevertheless, it allows for more guided experiments to be performed. More importantly, it provides researchers with a starting point for simulating the molecules. Once, arguably the largest barrier for computational biologists—de novo tertiary structure prediction is surpassed, molecular dynamics (MD) simulations can help in understanding their dynamics in nature [94]. This approach has helped gain deeper insights into GxxxG driven dimerization and glean more general principles which can be applied to complex polytopic transmembrane proteins such as ion channels (KcsA, MscL; Sect. 10.6.1) that have a glycine zipper motif.
10.4.2.1 GxxxG Motif as a TM Switch
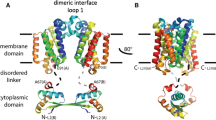
The signalling proteins mentioned in Sect. 10.4.1 use as diverse a range of domains on extracellular and intracellular side of the plasma membrane as the number of signals themselves. Despite the multitude of folds and structures on either side of the membrane, all these proteins traverse the plasma membrane using a single TM helix. The GxxxG motif has been implicated in stabilising the dimerization of such TMHs. But there are instances, where using two GxxxG motifs, a TMH could go from one dimeric conformation to another. If two GxxxG motifs occur at the N- and C-termini of the same helix, then it becomes straightforward to imagine how two motifs can enable two structurally distinct forms to exist (Fig. 10.3). Also, one could hypothesize the introduction of an energy barrier between the two, such that interconversion between these two forms can be coupled to another process. Thus, hinting at the potential of GxxxG as a naturally occurring intramembranous molecular switch.

Small-xxx-small motif in intra-membrane dynamics. A cartoon depiction of EGFR TM helices. The helices are proposed to switch from one conformation to another to shuttle between a signalling “On” state and an “Off” state. With both these interfaces stabilised by the small-xxx-small motif, the helices can lock-in either of them, affecting the interaction of the juxtamembrane region. This transduces the activation caused by extracellular ligand binding to kinase activity in the cytoplasm
It was in 2002 that the role of GxxxG as a switch was first formally hypothesised [95]. However, it was not until 11 years later that experimental evidence supporting the hypothesis was obtained in a plasma membrane bitopic receptor protein—EGFR (Epidermal Growth Factor Receptor) [68, 94]. EGFR or ErbB1 is one of the four Erb receptors of the Receptor Tyrosine Kinase family and being the cell surface receptor of the EGF peptide ligand, it has a crucial signalling role in cellular proliferation, migration and differentiation. But, its ligand-mediated activation was not clear until the TMH was studied. EGFR/ErbB1 was found to go from an active to inactive state utilising the two small-xxx-small motifs in its TMH (Fig. 10.3). Performing MD simulations on the structures of transmembrane and juxtamembrane segment obtained by NMR spectroscopy, it was demonstrated that the TMH of ErbB1/EGFR can indeed stabilise in either of the N- or C-terminal dimers. This change in the mode of dimerization controlled by EGF binding/unbinding modulates the activity of cytosolic kinase domains of EGFR [68] that has been implicated in certain cancers for over 30 years.
Another notable example that demands a mention here is that of ErbB2 or HER2 (Human EGF Receptor 2). ErbB2, like EGFR, has two small-xxx-small motifs in its TMH [96]. It was shown that mutating the C-terminal GxxxG motif did not affect the dimerization potential to any significant degree [96]. Moreover, from the NMR structure it was found that ErbB2 used the N-terminal motif for homo-dimerization [97] This observation would appear confounding to the argument to have two dimerization motifs on a transmembrane helix for switching between active and inactive states. However, earlier this year another NMR structure of ErbB2 was solved [98]. Surprisingly, it was found that the TM helix of ErbB2 can indeed form a C-terminal right-handed dimer but without using the GxxxG motif. Instead, it uses an unusual (Sect. 10.4.1.1), highly hydrophobic stretch of residues IxxxVxxLLxxVLxxVFxxL. Thus, even ErbB2 can form both N- and C-terminal dimers with their potential use as a switch. Also, the C-terminal GxxxG motif of ErbB2 is suggested to be involved in hetero-dimerization [99].
10.5 TM Helices in Endomembranes
There is some evidence that STIM1 (Stromal Interaction Molecule 1) on the metazoan endoplasmic reticulum (ER) membranes can also form dimers with the small-xxx-small motif in its single transmembrane helix [100]. This dimerization has implications for the conversion of STIM1 from inactive to active state. The inactive to active state conversion is initiated by depletion of Ca2+ in the lumen of the ER. This signal is translocated across the ER membrane to activate Orai1, a highly selective, STIM1 gated Ca2+ channel. STIM together with Orai orchestrates the process of Store Operated Calcium Entry (SOCE) which is crucial for immunological signalling and cellular Ca2+ homeostasis; reviewed by Prakriya and Lewis [101].
ADCK3 (AarF Domain Containing Kinase 3) is a mitochondrial inner membrane protein which has been categorically studied in the context of helical dimerization. It also utilises a Glycine zipper motif (Sect. 10.4.1.1) to form homodimers [102]. ADCK3 protein has a role in Coenzyme Q (Ubiquinone) biosynthesis and a naturally occurring mutation in ADCK3 has been correlated to respiratory phenotypes and cerebellar ataxia [103]. However, the physiological relevance of homo-dimerization of ADCK3 TMH remains a speculation. Thus, despite the unarguable biological importance of integral proteins on endomembrane organelles, the transmembrane region of these protein remain understudied. This is exemplified by almost no literature highlighting more than a passive role for the TM region of such proteins.
10.6 Dynamics of Ion Channels
The extension of dimerization studies to multi-pass membrane proteins has been limited, due in part to the relatively small number of well characterized instances where a reversible equilibrium association can be observed and manipulated in native membranes. One system that has been studied, is the homo-dimeric ClC-ec1 Cl−/H+ antiporter of Escherichia coli. This 50-kDa membrane protein dimerizes via a membrane embedded, non-polar interface lined mainly by Isoleucines and Leucines. Chadda et al. [104] have diluted this protein into liposomes to the point where monomers and dimers can both be observed by single molecule fluorescence. Such an approach has allowed the extraction of thermodynamic parameters, including the free energy of dimerization, but fails to provide atomic level information of the dimerization process [104].
There are relatively few multi-pass membrane proteins for which a mechanism of transition between stable states has been worked out at atomic resolution. Of these, ion transporters are probably the best understood and have been extensively studied for four decades. The first structure of a membrane protein [105] was of bacteriorhodopsin (bR)—a light-driven H+ pump—which is also the first membrane protein to have essentially all its characterised intermediates crystallised. These structures and a vast body of biophysical and mutagenesis studies have revealed individual steps of proton translocation through the protein coupled to movements of transmembrane helices. These include long range proton migration using a hydrogen-bonded chain, proton transfer between protonable groups within the membrane and also proton transfer utilising water molecules present between protein residues [106]. Consequently, it is the best understood membrane protein today. After bR, the protein that attracted concerted attention was the voltage gated potassium channel that is involved in the generation of action potentials in excitable tissues. Ion channels constitute a class of transporters that stabilize an aqueous pore across the membrane through which ions can diffuse down their electro-chemical gradient. These pores are responsible for electrical signalling in the nervous system, volume regulation and other critical physiological processes. The appearance of a series of high resolution structures of ion channels makes it possible to infer the stimulus-driven conformational changes underlying the transition from conducting to non-conducting states and vice-versa.
Ion channels stabilise columns of water that traverse the membrane and hence the functional channel has multiple pore-lining helices. For a given stoichiometry of ideal helices lining the conducting pore, the enclosed pore radius depends on the tilt of the helices with respect to the membrane normal. Conversely, for a given helical tilt, the pore radius increases with the number of constituent helices. Thus, gating transitions from conducting to non-conducting states can be achieved by altering helical tilt or by changing the number of constituent helices. The disposition of side chains that project into the pore lumen is another critical feature determining effective pore radius.
10.6.1 Voltage Gated K+ Channels
Voltage gated ion channels are tetrameric proteins contributing one helix per subunit to the aqueous pore. These channels are strongly ion-selective. Selectivity is conferred by a selectivity filter which is non-helical and tightly packed against the pore helix. This ensures that interatomic distances within the filter remain constant so as to achieve selectivity between ions as similar as Na+ and K+ (Fig. 10.4). Much of our knowledge of ion channel structure starts with a series of crystal structures of prokaryotic channels solved by Rod MacKinnon. The first such structure was that of the KcsA, a homo-tetrameric K+ channel that crystallised in the Closed State [107] (Figs. 10.1 and 10.4). The protein has two transmembrane helices, one of which contributes to the lining of the aqueous pore. The re-entrant loop connecting these helices contains the selectivity filter. The pore-lining helices are at an angle to the membrane normal, and the point of closest approach or the “bundle crossing” point between them leaves insufficient room for a hydrated K+ ion to pass. The MacKinnon group subsequently solved the structure of the MthK channel, which is also K+ conducting, but crystallised in the Open State [108]. A comparison of these structures revealed that channel opening and closing probably proceeds through an iris-type opening, involving bending of the four pore-lining helices so that the diameter at the narrowest point is about 12 Å (Fig. 10.4).

The Open ↔ Closed transition in ion channels. The bacterial potassium channels KcsA and MthK crystallised in the Closed and Open States respectively. (a) Views of the two tetrameric structures without lipid and with two chains omitted for clarity. Top: MthK which crystallised in the Open State. Highlighted in green is the conserved Glycine residue that allows a kink in the pore lining helix. Bottom: KcsA which crystallised in the Closed State. Note the bundle crossing of the helices highlighted by a rectangle in the view at left. The non-helical, tightly-packed selectivity filter is shown in magenta. The orange spheres represent the space between the helix backbones towards the cytosolic end of the pore as viewed from a cytosolic vantage point. The bend in the helix in MthK that occurs around the highlighted Glycine is responsible for opening of the pore by about 12 Å to allow for a hydrated K+ to pass. The pore lining helices splay about 30° to open the channel as represented in (b); [PDB id: 1BL8, 3LDC]
Voltage gated ion channels undergo a transition between the Open (or conducting) and Closed (or non-conducting) States in response to changes in transmembrane electric field. These two stable states differ by about 10 Kcal mol−1 in energy at resting membrane potentials. For simplicity, we will not consider other states such as an activated state or any of the inactivated states, all of which are non-conducting. Each subunit of voltage gated K+ channels (Kv channels) has six transmembrane helices with a re-entrant loop, containing the selectivity filter, connecting the last two helices. These last two helices and included loop are analogous to the KcsA and MthK channels. The last helix, S6, lines the pore while the positively charged fourth helix, S4, constitutes the voltage sensor. This basic architecture of the channel is shared by all voltage gated channels whose structure has been solved to date.
The determination of crystal structures of rat Kv1.2 in the Open State [109] provided a starting point to understand the dynamics of the voltage driven Open to Closed transition of the channel. Deducing a model for the Closed State starting from an open structure requires integrating the vast body of biochemical and electrophysiological work, coupled with data on the mutagenesis of a significant fraction of the residues to elucidate conformational switching in these proteins. This challenge has been approached by several groups [110,111,112]. Unfortunately, in the absence of an experimentally determined structure for the Closed State, there is no final answer at this time. Upadhyay et al. [111] started with the crystal structure of Kv1.2, which has a bent S6 as in MthK (Fig. 10.4) and modelled the Closed State of the pore based on the structure of KcsA [107]. The pore lining S6 helix had to be remodelled by the action of the electric field so as to achieve the narrow “bundle crossing” seen in the KcsA structure. This involves straightening of the S6 helix, resulting in closure of the channel inasmuch as the C-γ of V408 in diagonally opposite subunits are positioned around 5.3 Å apart. This spacing generates a constriction too small to be negotiated by a hydrated K+ ion (which is over 8 Å in diameter).
Like the potassium channels (Kv), voltage gated Na+ channel and Ca2+ channels, are Closed at the resting potentials of neuronal cells, around −60 mV. They open on depolarising the cells—i.e. going to less negative potentials. The early view of channel gating was that electric field actively forced the channels into opening from a “ground” Closed State. On the other hand, Upadhyay et al argued that since the crystals were formed in the absence of a bilayer, they are perforce zero-field structures. Which is to say that the Open State of the channel is the “ground state” of the channel. This is supported by the observation that all available structures of voltage gated channels are of the Open State. The application of an inside-negative electric field applies force on the voltage sensing helix, S4, moving it towards the cytoplasmic side of the plasma membrane. S4, in turn, tugs on the S4–S5 linker so that S5 prods S6, resulting in its “unkinking” and straightening, which constricts the spacing at the bundle crossing point. The Closed State is thus a “cocked gun” kept closed by action of the electric field. Once this is relieved by depolarising the membrane, S5 (by the action of S4) moves away from S6 allowing it to swing open rapidly resulting in channel opening. Since the electric field is used to constrain S6 into the Closed State, an S4 with significantly lower charge would require a greater electric field to apply the same pressure on S6 to keep it closed. The most dramatic instance of such a shift in the voltage of channel opening is probably that reported by Miller and Aldrich [113] for the double mutant R365N:R371I of the Shaker channel which has a mid-point of activation at −180 mV as compared to the wild type channel which opens at −20 mV.
10.6.2 Ligand Gated Ion Channels
Ligand gated channels can be broadly classified, for the purposes of this review, into two classes—those with four subunits and those with five. Glutamate receptor channels are the prototypic tetrameric neurotransmitter receptors while serotonin, GABA, glycine, and acetylcholine gate pentameric receptors. Though structures of ligand binding domains have been available for some time in both liganded and free states, an understanding of how ligand binding leads to channel opening requires the structure of the full length channel. These have been elucidated over the past few years allowing the examination of plausible pathways to ligand driven transitions in these proteins.
Glutamate receptors come in several flavours, named after ligands that can activate the channel (apart from glutamate, which opens all GluRs). Structures of full length AMPA channels have been solved with either an antagonist bound [114] or with an auxiliary subunit called TARP bound to it [115]. In both cases, the channel is in a closed state. Here the narrowest point of the channel is the bundle crossing point of the M2 helices from each of the subunits, which is structurally similar to the bundle crossing seen in the S6 helices of voltage gated K+ channels. Thus, channel opening is presumed to follow an iris opening transition as inferred for the K+ channels. The conformational changes in the ligand binding domain effected by binding of ligand have been observed in crystal structures of the isolated domain and the manner in which this is likely to affect the transmembrane helices is apparent [116].
The pentameric ligand gated receptor family includes receptors for nicotinic acetylcholine (nAChRs), serotonin (5-HT3Rs), glycine (GlyRs) and GABA (GABAARs). X-ray structures of the ancestral prokaryotic receptor channels were solved almost a decade ago [117,118,119]. These channels are symmetric homo-pentamers, whereas eukaryotic channels are often hetero-multimeric. Nonetheless, the prokaryotic channels probably represent a common minimal core. Indeed, more recent structures of a mouse serotonin receptor [120], a human GABA receptor [121] and glycine receptors from humans [122] and zebrafish [123] confirm this conservation.
The transition between Open and Closed States can be clearly seen in comparing crystal structures of the channels in the respective states. Fortunately, there are structures of the Closed State of unliganded channels (GLIC at pH 7 and the Glutamate gated Chloride channel [117, 119, 124]) as well as the strychnine-bound state of the Gly receptors [123]. In all of these structures, side chains of long, hydrophobic amino acid residues point into the lumen of the pore restricting it to under 2 Å radius precluding the transport of hydrated ions like Na+. The presence of the hydrophobic side chains prevents passage of desolvated ions, which would need compensatory ligands from the channel lining residues. Open State structures of GLIC at pH 4 [117, 119, 124], glutamate activated chloride channel in the presence of agonists [125, 126] and the glycine receptor bound to glycine [122] are also available. The pore-lining M2 helices move outwards towards the M3 helices of the same subunit and also undergo an anticlockwise rotation which move the constricting Isoleucine side chains away from the pore and towards the helical interface. This results in an opening of the pore to allow the passage of hydrated ions. Such a model of channel opening is consistent with a variety of mutagenesis studies which implicate an activation-gate hydrophobic block in the nAChR [127], as well as the finding that mutation of the critical hydrophobic resides to polar residues results in stabilisation of the Open State.
10.7 Concluding Remarks
There are several stages in the life of a membrane protein where dynamics play a critical role. We have very little information on the folding of membrane proteins, although the machinery involved has been identified. It may be, that the process will have to be studied with the full panoply of the cellular machinery intact. Trafficking of the membrane integrated protein to its target location is well studied. Operation of the protein in its native location requires the elucidation of structures of the protein in the various stable states that it adopts. The gradually increasing repository of atomic resolution structures provide a starting point for understanding how these remarkable proteins do what they do.
References
Pauling L, Corey RB, Branson HR. The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain. Proc Natl Acad Sci U S A. 1951;37:205–11.
Pauling L, Corey RB. The pleated sheet, a new layer configuration of polypeptide chains. Proc Natl Acad Sci U S A. 1951;37:251–6.
Stanley AM, Fleming KG. The process of folding proteins into membranes: challenges and progress. Arch Biochem Biophys. 2008;469:46–66.
White SH, Wimley WC. Hydrophobic interactions of peptides with membrane interfaces. Biochim Biophys Acta. 1998;1376:339–52.
Liao MJ, Huang KS, Khorana HG. Regeneration of native bacteriorhodopsin structure from fragments. J Biol Chem. 1984;259:4200–4.
Popot JL, Engelman DM. Membrane-protein folding and oligomerization—the 2-stage model. Biochemistry. 1990;29:4031–7.
Zen KH, Mckenna E, Bibi E, Hardy D, Kaback HR. Expression of lactose permease in contiguous fragments as a probe for membrane-spanning domains. Biochemistry. 1994;33:8198–206.
Baumann A, Kerruth S, Fitter J, Buldt G, Heberle J, Schlesinger R, Ataka K. In-situ observation of membrane protein folding during cell-free expression. PLoS One. 2016;11:e0151051.
Berndt U, Oellerer S, Zhang Y, Johnson AE, Rospert S. A signal-anchor sequence stimulates signal recognition particle binding to ribosomes from inside the exit tunnel. Proc Natl Acad Sci U S A. 2009;106:1398–403.
Batey RT, Rambo RP, Lucast L, Rha B, Doudna JA. Crystal structure of the ribonucleoprotein core of the signal recognition particle. Science. 2000;287:1232–9.
Janda CY, Li J, Oubridge C, Hernandez H, Robinson CV, Nagai K. Recognition of a signal peptide by the signal recognition particle. Nature. 2010;465:507–10.
Van den Berg B, Clemons WM Jr, Collinson I, Modis Y, Hartmann E, Harrison SC, Rapoport TA. X-ray structure of a protein-conducting channel. Nature. 2004;427:36–44.
Cannon KS, Or E, Clemons WM Jr, Shibata Y, Rapoport TA. Disulfide bridge formation between SecY and a translocating polypeptide localizes the translocation pore to the center of SecY. J Cell Biol. 2005;169:219–25.
du Plessis DJ, Berrelkamp G, Nouwen N, Driessen AJ. The lateral gate of SecYEG opens during protein translocation. J Biol Chem. 2009;284:15805–14.
Shaw AS, Rottier PJM, Rose JK. Evidence for the loop model of signal-sequence insertion into the endoplasmic-reticulum. Proc Nat Acad Sci U S A. 1988;85:7592–6.
Harris CR, Silhavy TJ. Mapping an interface of SecY (PrlA) and SecE (PrlG) by using synthetic phenotypes and in vivo cross-linking. J Bacteriol. 1999;181:3438–44.
Plath K, Mothes W, Wilkinson BM, Stirling CJ, Rapoport TA. Signal sequence recognition in posttranslational protein transport across the yeast ER membrane. Cell. 1998;94:795–807.
Connolly T, Gilmore R. Formation of a functional ribosome-membrane junction during translocation requires the participation of a GTP-binding protein. J Cell Biol. 1986;103:2253–61.
Osborne AR, Clemons WM Jr, Rapoport TA. A large conformational change of the translocation ATPase SecA. Proc Natl Acad Sci U S A. 2004;101:10937–42.
Schiebel E, Driessen AJM, Hartl F-U, Wickner W. ΔμH+ and ATP function at different steps of the catalytic cycle of preprotein translocase. Cell. 1991;64:927–39.
Zimmer J, Nam Y, Rapoport TA. Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature. 2008;455:936–43.
Erlandson KJ, Miller SB, Nam Y, Osborne AR, Zimmer J, Rapoport TA. A role for the two-helix finger of the SecA ATPase in protein translocation. Nature. 2008;455:984–7.
Samuelson JC, Chen M, Jiang F, Moller I, Wiedmann M, Kuhn A, Phillips GJ, Dalbey RE. YidC mediates membrane protein insertion in bacteria. Nature. 2000;406:637–41.
Dalbey RE, Kuhn A, Zhu L, Kiefer D. The membrane insertase YidC. Biochim Biophys Acta. 2014;1843:1489–96.
Houben EN, Ten Hagen-Jongman CM, Brunner J, Oudega B, Luirink J. The two membrane segments of leader peptidase partition one by one into the lipid bilayer via a Sec/YidC interface. EMBO Rep. 2004;5:970–5.
Kumazaki K, Chiba S, Takemoto M, Furukawa A, Nishiyama K, Sugano Y, Mori T, Dohmae N, Hirata K, Nakada-Nakura Y, Maturana AD, Tanaka Y, Mori H, Sugita Y, Arisaka F, Ito K, Ishitani R, Tsukazaki T, Nureki O. Structural basis of Sec-independent membrane protein insertion by YidC. Nature. 2014;509:516–20.
Hagan CL, Silhavy TJ, Kahne D. beta-Barrel membrane protein assembly by the Bam complex. Annu Rev Biochem. 2011;80:189–210.
O’neil PK, Rollaer SE, Noinaj N, Buchanan SK. Fitting the pieces of the beta-barrel assembly machinery complex. Biochemistry. 2015;54:6303–11.
Bakelar J, Buchanan SK, Noinaj N. The structure of the beta-barrel assembly machinery complex. Science. 2016;351:180–6.
Gu Y, Li H, Dong H, Zeng Y, Zhang Z, Paterson NG, Stansfeld PJ, Wang Z, Zhang Y, Wang W, Dong C. Structural basis of outer membrane protein insertion by the BAM complex. Nature. 2016;531:64–9.
Han L, Zheng J, Wang Y, Yang X, Liu Y, Sun C, Cao B, Zhou H, Ni D, Lou J, Zhao Y, Huang Y. Structure of the BAM complex and its implications for biogenesis of outer-membrane proteins. Nat Struct Mol Biol. 2016;23:192–6.
Voorhees RM, Fernandez IS, Scheres SH, Hegde RS. Structure of the mammalian ribosome-Sec61 complex to 3.4 A resolution. Cell. 2014;157:1632–43.
Denks K, Vogt A, Sachelaru I, Petriman NA, Kudva R, Koch HG. The Sec translocon mediated protein transport in prokaryotes and eukaryotes. Mol Membr Biol. 2014;31:58–84.
Mellman I, Nelson WJ. Coordinated protein sorting, targeting and distribution in polarized cells. Nat Rev Mol Cell Biol. 2008;9:833–45.
Dolezal P, Likic V, Tachezy J, Lithgow T. Evolution of the molecular machines for protein import into mitochondria. Science. 2006;313:314–8.
Dukanovic J, Rapaport D. Multiple pathways in the integration of proteins into the mitochondrial outer membrane. Biochim Biophys Acta. 2011;1808:971–80.
Soll J, Schleiff E. Protein import into chloroplasts. Nat Rev Mol Cell Biol. 2004;5:198–208.
Harpaz Y, Gerstein M, Chothia C. Volume changes on protein folding. Structure. 1994;2:641–9.
Lemmon MA, Flanagan JM, Treutlein HR, Zhang J, Engelman DM. Sequence specificity in the dimerization of transmembrane alpha-helices. Biochemistry. 1992;31:12719–25.
Mackenzie KR, Prestegard JH, Engelman DM. A transmembrane helix dimer: structure and implications. Science. 1997;276:131–3.
Adamian L, Liang J. Helix-helix packing and interfacial pairwise interactions of residues in membrane proteins. J Mol Biol. 2001;311:891–907.
Bowie JU. Membrane protein folding: how important are hydrogen bonds? Curr Opin Struct Biol. 2011;21:42–9.
Choma C, Gratkowski H, Lear JD, Degrado WF. Asparagine-mediated self-association of a model transmembrane helix. Nat Struct Biol. 2000;7:161–6.
Gratkowski H, Lear JD, Degrado WF. Polar side chains drive the association of model transmembrane peptides. Proc Natl Acad Sci U S A. 2001;98:880–5.
Mueller BK, Subramaniam S, Senes A. A frequent, GxxxG-mediated, transmembrane association motif is optimized for the formation of interhelical C alpha-H hydrogen bonds. Proc Nat Acad Sci U S A. 2014;111:E888–95.
Kumar S, Nussinov R. Salt bridge stability in monomeric proteins. J Mol Biol. 1999;293:1241–55.
Balashov SP, Imasheva ES, Govindjee R, Sheves M, Ebrey TG. Evidence that aspartate-85 has a higher pK(a) in all-trans than in 13-cis Bacteriorhodopsin. Biophys J. 1996;71:1973–84.
Eisenstein L, Lin SL, Dollinger G, Odashima K, Termini J, Konno K, Ding WD, Nakanishi K. Ftir difference studies on apoproteins - Protonation states of aspartic and glutamic-acid residues during the photocycle of bacteriorhodopsin. J Am Chem Soc. 1987;109:6860–2.
Hertel B, Tayefeh S, Kloss T, Hewing J, Gebhardt M, Baumeister D, Moroni A, Thiel G, Kast SM. Salt bridges in the miniature viral channel Kcv are important for function. Eur Biophys J. 2010;39:1057–68.
Cui G, Freeman CS, Knotts T, Prince CZ, Kuang C, Mccarty NA. Two salt bridges differentially contribute to the maintenance of cystic fibrosis transmembrane conductance regulator (CFTR) channel function. J Biol Chem. 2013;288:20758–67.
Kim JM, Altenbach C, Kono M, Oprian DD, Hubbell WL, Khorana HG. Structural origins of constitutive activation in rhodopsin: role of the K296/E113 salt bridge. Proc Natl Acad Sci U S A. 2004;101:12508–13.
Call ME, Pyrdol J, Wiedmann M, Wucherpfennig KW. The organizing principle in the formation of the T cell receptor-CD3 complex. Cell. 2002;111:967–79.
Sal-Man N, Gerber D, Bloch I, Shai Y. Specificity in transmembrane helix-helix interactions mediated by aromatic residues. J Biol Chem. 2007;282:19753–61.
Hong H, Park S, Jimenez RH, Rinehart D, Tamm LK. Role of aromatic side chains in the folding and thermodynamic stability of integral membrane proteins. J Am Chem Soc. 2007;129:8320–7.
Johnson RM, Hecht K, Deber CM. Aromatic and cation-pi interactions enhance helix-helix association in a membrane environment. Biochemistry. 2007;46:9208–14.
Yau WM, Wimley WC, Gawrisch K, White SH. The preference of tryptophan for membrane interfaces. Biochemistry. 1998;37:14713–8.
Van der Wel PCA, Strandberg E, Killian JA, Koeppe RE. Geometry and intrinsic tilt of a tryptophan-anchored transmembrane α-helix determined by 2H NMR. Biophys J. 2002;83:1479–88.
Mcintosh TJ, Simon SA. Roles of bilayer material properties in function and distribution of membrane proteins. Annu Rev Biophys Biomol Struct. 2006;35:177–98.
Lee AG. How lipids affect the activities of integral membrane proteins. Biochim Biophys Acta. 2004;1666:62–87.
Bogdanov M, Heacock PN, Dowhan W. A polytopic membrane protein displays a reversible topology dependent on membrane lipid composition. EMBO J. 2002;21:2107–16.
Anbazhagan V, Schneider D. The membrane environment modulates self-association of the human GpA TM domain—implications for membrane protein folding and transmembrane signaling. Biochim Biophys Acta. 2010;1798:1899–907.
Lemmon MA, Flanagan JM, Hunt JF, Adair BD, Bormann BJ, Dempsey CE, Engelman DM. Glycophorin-a dimerization is driven by specific interactions between transmembrane alpha-helices. J Biol Chem. 1992;267:7683–9.
Segrest JP, Jackson RL, Marchesi VT, Guyer RB, Terry W. Red cell membrane glycoprotein: amino acid sequence of an intramembranous region. Biochem Biophys Res Commun. 1972;49:964–9.
Furthmayr H, Marchesi VT. Subunit structure of human erythrocyte glycophorin A. Biochemistry. 1976;15:1137–44.
Bormann BJ, Knowles WJ, Marchesi VT. Synthetic peptides mimic the assembly of transmembrane glycoproteins. J Biol Chem. 1989;264:4033–7.
Lemmon MA, Treutlein HR, Adams PD, Brunger AT, Engelman DM. A dimerization motif for transmembrane alpha-helices. Nat Struct Biol. 1994;1:157–63.
Senes A, Gerstein M, Engelman DM. Statistical analysis of amino acid patterns in transmembrane helices: the GxxxG motif occurs frequently and in association with beta-branched residues at neighboring positions. J Mol Biol. 2000;296:921–36.
Endres NF, Das R, Smith AW, Arkhipov A, Kovacs E, Huang Y, Pelton JG, Shan Y, Shaw DE, Wemmer DE, Groves JT, Kuriyan J. Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell. 2013;152:543–56.
Bocharov EV, Mineev KS, Goncharuk MV, Arseniev AS. Structural and thermodynamic insight into the process of “weak” dimerization of the ErbB4 transmembrane domain by solution NMR. Biochim Biophys Acta. 2012;1818:2158–70.
Sulistijo ES, Mackenzie KR. Structural basis for dimerization of the BNIP3 transmembrane domain. Biochemistry. 2009;48:5106–20.
Walters RF, Degrado WF. Helix-packing motifs in membrane proteins. Proc Natl Acad Sci U S A. 2006;103:13658–63.
Sawma P, Roth L, Blanchard C, Bagnard D, Cremel G, Bouveret E, Duneau JP, Sturgis JN, Hubert P. Evidence for new homotypic and heterotypic interactions between transmembrane helices of proteins involved in receptor tyrosine kinase and neuropilin signaling. J Mol Biol. 2014;426:4099–111.
Godfroy JI 3rd, Roostan M, Moroz YS, Korendovych IV, Yin H. Isolated Toll-like receptor transmembrane domains are capable of oligomerization. PLoS One. 2012;7:e48875.
Dixon AM, Drake L, Hughes KT, Sargent E, Hunt D, Harton JA, Drake JR. Differential transmembrane domain GXXXG motif pairing impacts major histocompatibility complex (MHC) class II structure. J Biol Chem. 2014;289:11695–703.
Li R, Gorelik R, Nanda V, Law PB, Lear JD, Degrado WF, Bennett JS. Dimerization of the transmembrane domain of Integrin alphaIIb subunit in cell membranes. J Biol Chem. 2004;279:26666–73.
Schneider D, Engelman DM. Involvement of transmembrane domain interactions in signal transduction by alpha/beta integrins. J Biol Chem. 2004;279:9840–6.
Langosch D, Brosig B, Kolmar H, Fritz HJ. Dimerisation of the glycophorin A transmembrane segment in membranes probed with the ToxR transcription activator. J Mol Biol. 1996;263:525–30.
Russ WP, Engelman DM. The GxxxG motif: a framework for transmembrane helix-helix association. J Mol Biol. 2000;296:911–9.
Gurezka R, Langosch D. In vitro selection of membrane-spanning leucine zipper protein-protein interaction motifs using POSSYCCAT. J Biol Chem. 2001;276:45580–7.
Schneider D, Engelman DM. GALLEX, a measurement of heterologous association of transmembrane helices in a biological membrane. J Biol Chem. 2003;278:3105–11.
Karimova G, Pidoux J, Ullmann A, Ladant D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci U S A. 1998;95:5752–6.
Su PC, Berger BW. Identifying key juxtamembrane interactions in cell membranes using AraC-based transcriptional reporter assay (AraTM). J Biol Chem. 2012;287:31515–26.
Petschnigg J, Groisman B, Kotlyar M, Taipale M, Zheng Y, Kurat CF, Sayad A, Sierra JR, Mattiazzi Usaj M, Snider J, Nachman A, Krykbaeva I, Tsao MS, Moffat J, Pawson T, Lindquist S, Jurisica I, Stagljar I. The mammalian-membrane two-hybrid assay (MaMTH) for probing membrane-protein interactions in human cells. Nat Methods. 2014;11:585–92.
Dawson JP, Weinger JS, Engelman DM. Motifs of serine and threonine can drive association of transmembrane helices. J Mol Biol. 2002;316:799–805.
Kim S, Jeon TJ, Oberai A, Yang D, Schmidt JJ, Bowie JU. Transmembrane glycine zippers: physiological and pathological roles in membrane proteins. Proc Natl Acad Sci U S A. 2005;102:14278–83.
Teese MG, Langosch D. Role of GxxxG motifs in transmembrane domain interactions. Biochemistry. 2015;54:5125–35.
Doura AK, Fleming KG. Complex interactions at the helix-helix interface stabilize the glycophorin A transmembrane dimer. J Mol Biol. 2004;343:1487–97.
Doura AK, Kobus FJ, Dubrovsky L, Hibbard E, Fleming KG. Sequence context modulates the stability of a GxxxG-mediated transmembrane helix-helix dimer. J Mol Biol. 2004;341:991–8.
He L, Hoffmann AR, Serrano C, Hristova K, Wimley WC. High-throughput selection of transmembrane sequences that enhance receptor tyrosine kinase activation. J Mol Biol. 2011;412:43–54.
Domanska G, Motz C, Meinecke M, Harsman A, Papatheodorou P, Reljic B, Dian-Lothrop EA, Galmiche A, Kepp O, Becker L, Gunnewig K, Wagner R, Rassow J. Helicobacter pylori VacA toxin/subunit p34: targeting of an anion channel to the inner mitochondrial membrane. PLoS Pathog. 2010;6:e1000878.
Toutain CM, Clarke DJ, Leeds JA, Kuhn J, Beckwith J, Holland IB, JACQ A. The transmembrane domain of the DnaJ-like protein DjlA is a dimerisation domain. Mol Genet Genomics. 2003;268:761–70.
Call ME, Wucherpfennig KW, Chou JJ. The structural basis for intramembrane assembly of an activating immunoreceptor complex. Nat Immunol. 2010;11:1023–9.
Polyansky AA, Chugunov AO, Volynsky PE, Krylov NA, Nolde DE, Efremov RG. PREDDIMER: a web server for prediction of transmembrane helical dimers. Bioinformatics. 2014;30:889–90.
Arkhipov A, Shan Y, Das R, Endres NF, Eastwood MP, Wemmer DE, Kuriyan J, Shaw DE. Architecture and membrane interactions of the EGF receptor. Cell. 2013;152:557–69.
Fleishman SJ, Schlessinger J, Ben-Tal N. A putative molecular-activation switch in the transmembrane domain of erbB2. Proc Natl Acad Sci U S A. 2002;99:15937–40.
Mendrola JM, Berger MB, King MC, Lemmon MA. The single transmembrane domains of ErbB receptors self-associate in cell membranes. J Biol Chem. 2002;277:4704–12.
Bocharov EV, Mineev KS, Volynsky PE, Ermolyuk YS, Tkach EN, Sobol AG, Chupin VV, Kirpichnikov MP, Efremov RG, Arseniev AS. Spatial structure of the dimeric transmembrane domain of the growth factor receptor ErbB2 presumably corresponding to the receptor active state. J Biol Chem. 2008;283:6950–6.
Bragin PE, Mineev KS, Bocharova OV, Volynsky PE, Bocharov EV, Arseniev AS. HER2 transmembrane domain dimerization coupled with self-association of membrane-embedded cytoplasmic juxtamembrane regions. J Mol Biol. 2016;428:52–61.
Gerber D, Sal-Man N, Shai Y. Two motifs within a transmembrane domain, one for homodimerization and the other for heterodimerization. J Biol Chem. 2004;279:21177–82.
Ma G, Wei M, He L, Liu C, Wu B, Zhang SL, Jing J, Liang X, Senes A, Tan P, Li S, Sun A, Bi Y, Zhong L, Si H, Shen Y, Li M, Lee MS, Zhou W, Wang J, Wang Y, Zhou Y. Inside-out Ca(2+) signalling prompted by STIM1 conformational switch. Nat Commun. 2015;6:7826.
Prakriya M, Lewis RS. Store-operated calcium channels. Physiol Rev. 2015;95:1383–436.
Khadria AS, Mueller BK, Stefely JA, Tan CH, Pagliarini DJ, Senes A. A Gly-zipper motif mediates homodimerization of the transmembrane domain of the mitochondrial kinase ADCK3. J Am Chem Soc. 2014;136:14068–77.
Liu YT, Hersheson J, Plagnol V, Fawcett K, Duberley KE, Preza E, Hargreaves IP, Chalasani A, Laura M, Wood NW, Reilly MM, Houlden H. Autosomal-recessive cerebellar ataxia caused by a novel ADCK3 mutation that elongates the protein: clinical, genetic and biochemical characterisation. J Neurol Neurosurg Psychiatry. 2014;85:493–8.
Chadda R, Krishnamani V, Mersch K, Wong J, Brimberry M, Chadda A, Kolmakova-Partensky L, Friedman LJ, Gelles J, Robertson JL. The dimerization equilibrium of a ClC Cl(-)/H(+) antiporter in lipid bilayers. Elife. 2016;5:e17438.
Henderson R, Unwin PNT. Three-dimensional model of purple membrane obtained by electron microscopy. Nature. 1975;257:28–32.
Lanyi JK. Proton transfers in the bacteriorhodopsin photocycle. Biochim Biophys Acta Bioenergetics. 2006;1757:1012–8.
Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, Mackinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77.
Jiang Y, Lee A, Chen J, Cadene M, Chait BT, Mackinnon R. The open pore conformation of potassium channels. Nature. 2002;417:523–6.
Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903.
Grabe M, Lai HC, Jain M, Jan YN, Jan LY. Structure prediction for the down state of a potassium channel voltage sensor. Nature. 2007;445:550–3.
Upadhyay SK, Nagarajan P, Mathew MK. Potassium channel opening: a subtle two-step. J Physiol. 2009;587:3851–68.
Yarov-Yarovoy V, Baker D, Catterall WA. Voltage sensor conformations in the open and closed states in ROSETTA structural models of K(+) channels. Proc Natl Acad Sci U S A. 2006;103:7292–7.
Miller AG, Aldrich RW. Conversion of a delayed rectifier K+ channel to a voltage-gated inward rectifier K+ channel by three amino acid substitutions. Neuron. 1996;16:853–8.
Sobolevsky AI, Rosconi MP, Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–56.
Zhao Y, Chen S, Yoshioka C, Baconguis I, Gouaux E. Architecture of fully occupied GluA2 AMPA receptor-TARP complex elucidated by cryo-EM. Nature. 2016;536:108–11.
Sobolevsky AI. Structure and gating of tetrameric glutamate receptors. J Physiol. 2015;593:29–38.
Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux JP, Delarue M, Corringer PJ. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature. 2009;457:111–4.
Hilf RJ, Dutzler R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature. 2008;452:375–9.
Hilf RJ, Dutzler R. Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature. 2009;457:115–8.
Hassaine G, Deluz C, Grasso L, Wyss R, Tol MB, Hovius R, Graff A, Stahlberg H, Tomizaki T, Desmyter A, Moreau C, Li XD, Poitevin F, Vogel H, Nury H. X-ray structure of the mouse serotonin 5-HT3 receptor. Nature. 2014;512:276–81.
Miller PS, Aricescu AR. Crystal structure of a human GABAA receptor. Nature. 2014;512:270–5.
Huang X, Chen H, Michelsen K, Schneider S, Shaffer PL. Crystal structure of human glycine receptor-alpha3 bound to antagonist strychnine. Nature. 2015;526:277–80.
Du J, Lu W, Wu S, Cheng Y, Gouaux E. Glycine receptor mechanism elucidated by electron cryo-microscopy. Nature. 2015;526:224–9.
Sauguet L, Shahsavar A, Poitevin F, Huon C, Menny A, Nemecz A, Haouz A, Changeux JP, Corringer PJ, Delarue M. Crystal structures of a pentameric ligand-gated ion channel provide a mechanism for activation. Proc Natl Acad Sci U S A. 2014;111:966–71.
Althoff T, Hibbs RE, Banerjee S, Gouaux E. X-ray structures of GluCl in apo states reveal a gating mechanism of Cys-loop receptors. Nature. 2014;512:333–7.
Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011;474:54–60.
Purohit P, Gupta S, Jadey S, Auerbach A. Functional anatomy of an allosteric protein. Nat Commun. 2013;4:2984.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter

Cite this chapter
Lall, S., Mathew, M.K. (2017). Dynamics of Membrane Proteins. In: Chattopadhyay, A. (eds) Membrane Organization and Dynamics . Springer Series in Biophysics, vol 20. Springer, Cham. https://doi.org/10.1007/978-3-319-66601-3_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-66601-3_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-66600-6
Online ISBN: 978-3-319-66601-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)