
Abstract
Archaea employ a eukaryote-like transcription apparatus to transcribe a bacteria-like genome; while the RNA polymerase, basal factors and promoter elements mirror the eukaryotic RNA polymerase II system, archaeal genomes are densely packed with genes organised into multicistronic transcription units. The molecular mechanisms of archaeal transcription have been studied and characterised in great detail in vitro, but until recently relatively little was known about its global characteristics. In this chapter we discuss an integrated view of transcription from the molecular to the global level. Systems biology approaches have provided compelling insights into promoter and terminator DNA elements, the genome-wide distribution of transcription initiation- and elongation factors and RNA polymerase, the archaeal transcriptome and chromatin organisation. Overall these analyses illuminate transcription from a genome-wide perspective and serve as a resource for the community. In addition, Big Data can often validate mechanistic models based on biochemical and structural information, and generate new working hypotheses that can be thoroughly tested and dissected in vitro. This is an exciting time to study gene expression in the archaea since we are at the brink of a comprehensive yet detailed understanding of transcription.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

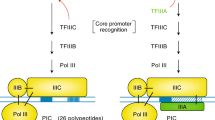

1.1 Introduction
Archaea are prokaryotes and as such share many properties with bacteria including circular genomes, densely packed with genes organised into operons. However, their transcription machinery is closely related to that of RNA polymerase II, the enzyme responsible for mRNA transcription in eukaryotes (Fig. 1.1). This similarity extends from the RNA polymerase (RNAP) subunit composition, via general transcription factors required for initiation, to their cognate promoter elements (Fig. 1.1a, b) (Werner and Grohmann 2011). In essence, archaeal transcription involves a eukaryotic-like machinery acting upon a bacterial-like template, making it an interesting and important subject to study. Archaeal transcription can be considered a simpler, stripped-down version of the RNAPII system, generally consisting of fewer and smaller components that facilitate the basic mechanisms of transcription. These are often obscured by the baroque complexity in eukaryotes—making archaea invaluable tools to dissect them. In vitro studies of archaeal transcription have focused on hyperthermophilic archaea due to their high biochemical tractability including the in vitro assembly of RNAPs from Methanocaldococcus jannaschii and Pyrococcus furiosus from individual recombinant subunits under defined conditions in the test tube (Naji et al. 2007; Smollett et al. 2015; Werner and Weinzierl 2002). This approach has not been successful with any eukaryotic RNAP thus far, and archaea have therefore provided invaluable model systems to elucidate the molecular mechanisms of RNAPII transcription (Fouqueau et al. 2013; Grohmann et al. 2011; Hirtreiter et al. 2010a, b; Kostrewa et al. 2009; Tan et al. 2008; Werner and Weinzierl 2005). Whilst such recombinant systems are required to carry out a definitive functional dissection of transcription, less attention has been paid to the systems level properties of the basic transcription machinery in archaea, including whole genome occupancy, transcription start site- and transcriptome mapping. As high-throughput sequencing technologies have become more accessible new avenues of research have become possible. In this chapter, we outline how systems biology can complement classical biochemistry/structural biology, and how this enhances our understanding of the different stages of the transcription cycle and the structure and function of chromatin (Fig. 1.2).

Evolution of the basal transcription machinery in the three domains of life. (a) Table of RNAP subunits and general transcription factors in the three domains of life. The columns represent the single RNAP transcription systems in bacteria, eury- and crenarchaea, and the three orthodox RNAPI, II and III systems in eukaryotes. The rows depict homologous factors, while functionally analogous but evolutionarily unrelated factors are shown as separated fields with dashed borders. Factors are colour-coded according to their function in transcription initiation (green), elongation (blue) and transcript cleavage (red). Subunits and factors that are not conserved in all domains are indicated with asterisks. (b) Schematic representation of key events in the evolution of the basal transcription machinery. General transcription factors are colored as in panel a, with chromatin proteins added in purple

The Archaeal transcription cycle. Transcription initiation is a recruitment cascade, the BRE and TATA promoter motifs sequester TBP and TFB, which in turn recruits RNAP to form the PIC. TFE stimulates DNA strand separation of the promoter IMR region, which stabilises the PIC. The later stages of initiation involved the synthesis of abortive transcripts, and promoter escape, which is likely facilitated by the swapping of TFE for Spt4/5, forming a processive transcription elongation complex. During elongation additional factors including transcript cleavage factors and Spt4/5 ensure highly processive transcription. At the 3′ end of the gene transcription is terminated by short poly-U signatures, and likely by hitherto uncharacterised termination factors
1.2 The Basal Transcription Machinery and the Archaeal Transcription Cycle
1.2.1 Promoter Recognition and Recruitment of the RNAP
In all domains of life transcription is initiated by the recruitment of basal, or general, transcription initiation factors to the promoter. Most archaeal promoters rely on three elements: the TATA box, B-recognition element (BRE) and the Initiator (Inr). TATA box and BRE are DNA sequence recognition motifs of the two general transcription factors TBP and TFB, respectively (Bell et al. 1999; Qureshi et al. 1995; Rowlands et al. 1994), both TBP and TFB are necessary and sufficient to facilitate promoter-directed transcription in vitro (Werner and Weinzierl 2002). TBP and TFB are homologous to eukaryotic TBP and TFIIB (Fig. 1.1), respectively, and have identical functions, albeit with a faster DNA-binding dynamics (Gietl et al. 2014) that may reflect different mechanisms of regulation (Blombach and Grohmann 2017). Global mapping of transcription start sites (TSSs) and subsequent promoter sequence analysis confirm in vitro observations in as much as TATA and BRE motifs are dominant elements in most archaeal promoters, with a few notable exceptions including the M. jannaschii ribosomal RNA promoter (Figs. 1.3 and 1.4) (Babski et al. 2016; Cho et al. 2017; Jäger et al. 2009, 2014; Li et al. 2015; Smollett et al. 2017; Wurtzel et al. 2010). This is in contrast to eukaryotes where strong TATA motifs (i.e., close to consensus sequence) are absent from the majority of promoters (Yang et al. 2007). Recently we have used Chromatin Immunoprecipitation followed by high-throughput sequencing (ChIP-seq) to characterise how promoter elements direct the recruitment of TBP, TFB and RNAP in vivo in the euryarchaeon M. jannaschii (Smollett et al. 2017). While BRE and TATA elements are the main contributors to promoter strength in vitro. There is only a weak correlation between BRE/TATA consensus score and TBP/TFB ChIP signals, and RNA steady-state levels in vivo. However, TBP/TFB binding does correlate with RNAP occupancy, which in turn correlates moderately well with RNA levels (Smollett et al. 2017). This shows that TBP and TFB direct pre-initiation complex (PIC) formation, and RNAP recruitment and loading into the transcription unit (TU) (Fig. 1.4). Yeast promoters show likewise little correlation between TATA box motif and TBP binding, with RNA levels being proportional to TBP occupancy (Kim and Iyer 2004). There could be several reasons for the discord between promoter motif strength and the binding of initiation factors in archaea and eukaryotes. In particular, the availability of the DNA template to the TBP, TFB and RNAP can be regulated by alternative chromatin structures, and gene-specific regulators may either enhance or inhibit PIC assembly (see Sect. 1.3). Several archaea encode multiple variants of TBP and TFB, in particular halophilic species such as Halobacterium NRC-1 contain 6 TBP and 7 TFB variants (Baliga et al. 2000); it has been proposed that the combination of TBP and TFB variants can direct a degree of promoter-specific regulation of transcription akin to bacterial sigma factors (Facciotti et al. 2007). A combination of different TBP/TFB deletion strains and ChIP analyses has revealed that only some TBP and TFB variants are essential and that different combinations of TBP/TFB bind to distinct promoters in vivo. Many promoters were associated with multiple TFB variants demonstrating a significant degree of redundancy (Facciotti et al. 2007), while subtle sequence biases in the BREs account for preferential binding of the different TFB variants (Seitzer et al. 2012).

Comparison of promoter consensus motifs in different archaea. Alignment of the DNA sequences upstream of TSS identified on a genome-wide scale identifies individual promoter elements including BRE, TATA box, IMR and Inr elements surrounding the TSS. Alignment of primary TSSs identified by whole genome sequencing of M. jannaschii (Smollett et al. 2017), Methanosarcina mazei (Jäger et al. 2009), Methanolobus psychrophilus (Li et al. 2015), Thermococcus kodakarensis (Jäger et al. 2014), T. onnurineus (Cho et al. 2017), Haloferax volcanii (Babski et al. 2016) and Solfolobus solfataricus (Wurtzel et al. 2010). Alignment visualised using WebLogo 3 adjusting to the background GC content for each organism (31.3% M. jannaschii, 41.5% M. mazei, 44.6% M. psychrophilus, 52% T. kodakarensis, 51.3% T. onnurineus, 65.5% H. volcanii, 35.8% S. solfataricus, http://weblogo.threeplusone.com/). Inset shows TATA box motif determined from same DNA sequences using MEME (http://meme-suite.org/tools/meme-chip). Adapted from Smollett et al. (2017)

Archaeal promoter elements govern transcription initiation. The interactions between promoter motifs (BRE/TATA), and sequence specific DNA-binding initiation factors (TBP/TFB) recruit RNAP to the promoter. During open complex formation the DNA strands of the promoter are separated within the IMR, an AT-rich region spanning from −12 to +2 relative to the TSS. TFE aids this process and stabilises the open PIC. The Inr surrounding the TSS plays an important role for the precise selection of the transcription start site
1.2.2 Stabilisation of the PIC by Open Complex Formation
The transcription initiation factor TFE (homologous to eukaryotic TFIIE) enhances the stability of the PIC by aiding DNA strand separation and loading of the template strand into the active site of RNAP (Blombach et al. 2015, 2016; Grohmann et al. 2011). This process is referred to as ‘open complex’ formation. The regulation of open complex formation is a crucial step in defining transcription output across all domains of life (reviewed in Blombach et al. 2016). The region of DNA to be separated, the initially melted region (IMR), extends from position −12 to +2 relative to the TSS (Bell et al. 1998; Blombach et al. 2015; Nagy et al. 2015). Global sequence analysis reveals that the IMR does not contain a specific sequence motif, but throughout the archaea have a significantly higher A and T content compared to the genome average, particularly at the upstream edge (Fig. 1.3) (Smollett et al. 2017). As A-T basepairs require less energy for DNA strand separation compared to G-C basepairs the AT-bias may have been selected to facilitate open complex formation (Fig. 1.4), while there is no correlation between AT content and promoter strength. This is similar to the bacterial −10 element, which is also AT-rich and forms the upstream edge of the transcription bubble (Sasse-Dwight and Gralla 1989; Zuo and Steitz 2015). Short AT-rich DNA motifs (IMR, −10 element) and factors (TFE, sigma, CarD, TFIIH) that contribute to open complex formation and stability have coevolved in all domains of life (Fig. 1.1). The eukaryotic counterpart of TFE, TFIIE, is a dimeric factor consisting of subunits TFIIEα and TFIIEβ. Many archaea employ monomeric TFE variants (homologous to TFIIEα), whereas crenarchaeal TFE variants are α/β heterodimers (Blombach et al. 2009, 2015, 2016).
1.2.3 Selection of the Transcription Start Site
Genome-wide studies show that mammalian genes can be transcribed from multiple promoters using multiple TSSs (Sandelin et al. 2007). In bacteria, the discriminator promoter element is important for genome-wide start site selection (Winkelman et al. 2016), and the bacterial core recognition element has been shown to influence TSS selection by interactions between a G nucleotide at register +2 in the non-template strand and the core RNAP (Vvedenskaya et al. 2016). The archaeal Inr is comprised of a dinucleotide motif ‘−1T+1[A/G]’ which directs precise start site selection in vivo (Figs. 1.3, 1.4 and 1.5) (Smollett et al. 2017). The Inr is a common feature in archaeal promoters, although the prevalence can vary between closely related species, e.g., it is present in the promoters of Thermococcus onnurineus, but not in T. kodakarensis (Fig. 1.3) (Cho et al. 2017; Jäger et al. 2014; Smollett et al. 2017). This suggests that TSS precision is not selected for some organisms, or may be compensated for by other factors. The archaeal Inr—essentially a preference for purine at the +1 and pyrimidine at the −1 position—is conserved in bacterial and eukaryotic promoters (Kadonaga 2012; Shultzaberger et al. 2007). Structural analyses suggests that base stacking interactions between the −1 nucleotide of the template strand and the initiating NTP plays a role in template DNA strand stabilisation within the PIC (Basu et al. 2014).

The Inr motif influences TSS selection. Many archaeal promoters include an Inr motif (−1T/+1[A/G]) and utilise single or multiple TSSs (+1). (a) A strong Inr motif will direct one specific TSS resulting in transcripts with identical 5′-termini. (b) Promoters with a weaker Inr motif will direct transcription from several TSS leading to RNA species with heterogenous 5′-termini (Smollett et al. 2017)
1.2.4 Promoter Escape Facilitated by Factor Swapping
All RNAP face a similar mechanical engineering challenge; while a network of high affinity interactions between promoter-bound initiation factors and RNAP is essential to facilitate efficient recruitment and PIC formation, the escape of RNAP from the promoter (i.e., productive transcription) requires that this network is dismantled (Werner 2012). Spt4/5 is homologous to DSIF in humans and NusG in bacteria, it is the only RNAP-associated transcription factor that is universally conserved in all domains of life (Fig. 1.1) (Werner 2012). Spt4/5 is not essential for transcription in vitro, but ChIP-seq profiles demonstrate that it associates with elongating RNAPs throughout the genome, on coding as well as noncoding TUs. As Spt4/5 and the initiation factor TFE bind to the RNAP clamp in a mutually exclusive manner in vitro, we have proposed that this exchange, or swap, between TFE and Spt4/5 occurs every time the RNAP progresses through the transcription cycle, and that the swap could enhance promoter escape (Grohmann et al. 2011; Werner 2012). Spt4/5 is recruited proximal to the promoter in vivo, in agreement with facilitating the transition from initiation to elongation (Figs. 1.6 and 1.7a) (Smollett et al. 2017). This is different from bacterial NusG, which is recruited to TEC in a stochastic fashion, and it is similar to the early recruitment of Spt4/5 in yeast (Mayer et al. 2010). In addition, a similar exchange between TFIIE and Spt4/5 has been shown at RNAPII promoters (Diamant et al. 2016; Larochelle et al. 2012).

An integrated view of transcription in archaea. ChIP occupancy profiles of the basal transcription machinery reflect the binding of PICs to promoters, and the distribution of RNAP-Spt4/5 TECs within the coding region as shown for the M. jannashii hsp60 TU. Both plus- and minus-strand RNA steady-state levels serve as proxy for transcription output of RNAP. Interestingly RNAPs do not strictly require Spt4/5 for transcription elongation in vitro, yet Spt4/5 closely follows RNAP in a genome-wide fashion, behaving as an ‘honorary’ RNAP subunit (Blombach et al. 2016; Smollett et al. 2017)

Two modes of Spt4/5 recruitment to RNAP. Global occupancy profiling of M. jannaschii RNAP and Spt4/5 reveals two patterns of recruitment. (a) Spt4/5 is recruited to RNAP proximal to the promoter at the majority of transcription units. This recruitment profile supports the theory that swapping between initiation factor TFE and elongation factor Spt4/5 aids promoter escape. (b) At a small subset of genes including the rRNA and CRISPR loci Spt4/5 is recruited hundreds of base pairs downstream of the transcription start site, during the early elongation phase of the transcription cycle (Smollett et al. 2017)
1.2.5 An Alternative Mode of Spt4/5 Recruitment
Genome-wide occupancy analysis allows us to not only define the ‘norm’ but also identify notable exceptions to the promoter-proximal Spt4/5 recruitment model (Fig. 1.7) (Mooney et al. 2009; Smollett et al. 2017). These exceptions include the ribosomal RNA operons and the abundant CRISPR loci where Spt4/5 is recruited during transcription elongation hundreds of base pairs downstream of the TSS. The underlying mechanisms behind this ‘delayed’ recruitment is currently not known, but likely includes novel gene-specific transcription factors, strong RNA secondary-structure or co-transcriptional processing—all of which are relevant for rRNA and CRISPR transcripts. The Sulfolobus solfataricus and Pyrococcus furiosus rRNA promoters have well defined BRE/TATA motifs and are very strong in vitro (Blombach et al. 2015; Micorescu et al. 2008; Qureshi et al. 1997), however, the M. jannaschii rRNA promoter shows surprisingly poor promoter motifs, and performs weakly in vitro, in apparent contrast with the high RNA levels and RNAP occupancy on the rRNA operons in vivo (Smollett et al. 2017). The lack of strong promoter motifs is akin to bacterial rRNA promoters, which tend to form unstable PICs, making them more amenable to regulation (Jensen and Pedersen 1990). It is possible that unknown transcription factors mask the Spt4/5 binding site on RNAP (clamp coiled coil) and activate M. jannaschii rRNA promoters. Alternatively, efficient promoter escape may occur at the weak rRNA promoter without Spt4/5. This is not the case for the CRISPR promoters, which have multiple promoters with strong matches to the consensus sequence (Smollett et al. 2017).
1.2.6 Termination of Transcription
Transcription termination remains one of the least understood mechanisms of gene expression in archaea. Specific DNA sequences and auxiliary factors can slow down the TEC and trigger dissociation of the TEC into RNAP, transcript and template, but the precise mechanisms and order of events is unclear. While the fundamental process appears conserved in all multisubunit RNAPs, the requirements for DNA sequence motifs and exogenous termination factors differs substantially (Epshtein et al. 2007, 2010; Porrua et al. 2016; Proudfoot 2016). Bacterial intrinsic terminators consist of a short RNA hairpin structure and a poly-U stretch; these terminators induce pausing and enable RNAP to undergo conformational changes such as an opening of the RNAP clamp (Hein et al. 2014), a process likely facilitated by the RNA hairpin that invades the DNA binding channel of RNAP (Epshtein et al. 2007). These allosteric changes lead to the dissociation of the TEC with the last residue of the poly-U stretch forming the RNA 3′ terminus (Ray-Soni et al. 2016). The limited number of archaeal terminators that have been studied in vitro share the requirement for a poly-U stretch (5–8 U-residues), but are not dependent on any RNA secondary structure elements, reminiscent of the eukaryotic RNAPIII system (Hirtreiter et al. 2010a; Santangelo et al. 2009; Santangelo and Reeve 2006; Spitalny and Thomm 2008) (Fig. 1.8a). This suggests that the termination mechanism is conserved across all domains of life, but that the archaeal and RNAPIII TECs dissociate more readily than the bacterial TEC—without the intervention of exogenous factors or RNA hairpins. It is noteworthy that one of the key differences between bacterial and archaeal RNAPs is the Rpo4/7 stalk domain, which enhances transcription termination and has been likened to an ‘inbuilt’ NusA elongation factor (Belogurov and Artsimovitch 2015; Hirtreiter et al. 2010a).

Transcription termination in archaea. (a) Short poly-U stretches implicated in triggering transcription termination in vitro and in vivo, and global RNA 3′ mapping demonstrates that transcript 3′ termini consist of U-residues for 30–40% of TU genome-wide. (b) Leaky termination can lead to alternative and extended 3′-UTRs, which can result in antisense transcript overlap between two genes organised in a convergent orientation, or read through into downstream TUs. (c) Archaeal genomes encode putative termination factors including 5′→3′ RNases and RNA helicases. In eukaryotes and bacteria factors with these activities facilitate transcription termination by ‘torpedo’ mechanisms
The genome-wide RNA 3′ termini of a euryarchaeon (Methanosarcina mazei) and a crenarchaeon (Sulfolobus acidocaldarius) have been mapped at base pair resolution using a systems biology approach (Term-seq) (Dar et al. 2016a, b). In agreement with the mechanisms characterised in vitro, the Term-seq dataset revealed that termination occurred in vivo immediately downstream of a poly-U motif without the need for RNA secondary structure elements (Dar et al. 2016a). In approximately half of convergent (i.e., head-to-head oriented) genes in S. acidocalaricus the terminator signal of a given TU was located in the coding region of the other TU, resulting in a potential antisense transcript overlap. This could be due to the high coding density of archaeal genomes and the resulting short intergenic regions, or have regulatory significance. Many TUs were associated with multiple RNA 3′ termini likely due to inefficient termination. Such ‘leaky’ termination could direct the synthesis of RNA isoforms that differ in the 3′-untranslated region (3′-UTR) targeted by small regulatory RNAs (Fig. 1.8b) (Dar et al. 2016a). However, Term-seq results have two principal caveats. Firstly, Term-seq cannot discriminate between ‘native’ RNA 3′ ends generated by transcription termination and ‘processed’ RNA 3′ ends resulting from nucleolytic digestion, either RNA-processing or -degradation. Secondly, termination motifs and RNA 3′ ends could only be identified in 30–39% of TUs, which suggests that alternative- or additional termination mechanisms are at work including template topology (positive supercoiling in hyperthermophiles) and hitherto unidentified termination factors. Strong terminator (poly-U) signals are present in intragenic regions but only 25% of these led to transcription termination. This could be due to transcription antitermination, a well-described phenomenon in bacteria that relies on factors that are conserved between bacteria and archaea including NusG (Spt4/5), NusA, NusE, and co-translating ribosomes (Santangelo and Artsimovitch 2011; Santangelo et al. 2008). Little is known about archaeal termination factors, but we can speculate about their properties. Both bacterial and eukaryotic termination factors use a ‘torpedo’ mechanism, i.e. they engage with the nascent transcript, translocate along the RNA in the 5′→3′ direction, and ultimately dissociate the TEC upon impact. 5′→3′ RNases (Xrn2 in mammals, Rat1 in yeast) and RNA-helicases (Rho factor in bacteria, Sen1 in eukaryotes) facilitate transcription termination in this fashion (Fig. 1.8c) (Han et al. 2016; Kim et al. 2004; El Hage et al. 2008; West et al. 2004; Epshtein et al. 2010; Porrua and Libri 2013). Archaeal genomes encode several candidates for torpedo-factors but none have been experimentally tested yet (Phung et al. 2013).
1.3 Additional Factors Affecting Transcriptional Output
1.3.1 Gene-Specific Transcription Regulators
The lack of a strong correlation between BRE/TATA promoter motifs and RNA levels genome wide (Kim and Iyer 2004; Smollett et al. 2017) suggests that additional forces are at work, including gene-specific regulators. The molecular mechanisms of several archaeal metabolic and stress response regulators have been elucidated in vitro, and their regulons characterised by ChIP-chip and ChIP-seq methods (Liu et al. 2016; Nguyen-Duc et al. 2013; Reichelt et al. 2016; Rudrappa et al. 2015; Tonner et al. 2015; Wilbanks et al. 2012). Archaeal regulators operate by a range of different mechanisms including repression by promoter occlusion and activation by enhancing the recruitment of the PIC; the mode of action of the same factor can depend on the location of the binding site relative to the promoter (Aravind and Koonin 1999; Charoensawan et al. 2010; Dahlke and Thomm 2002; Geiduschek and Ouhammouch 2005; Kanai et al. 2007; Lee et al. 2008; Lipscomb et al. 2009; Ochs et al. 2012; Peeters et al. 2013, 2015; Perez-Rueda and Janga 2010). Transcription regulators are described in greater detail in another chapter of this tome, we will only briefly mention example below.
The M. jannaschii Lrp-type regulator Ptr2 is an excellent example of how in vitro and in vivo approaches can complement each other. Ptr2 activates transcription from the rb2 promoter by recruiting TBP to the TATA box, a mechanism that was elucidated by elegant in vitro transcription experiments in the Geiduschek laboratory (Ouhammouch and Geiduschek 2001; Ouhammouch et al. 2003, 2005). Whole genome occupancy studies of M. jannaschii TBP validated this mechanism in vivo. By analysis of promoter sequences genome-wide, each TATA motif could be assigned a score that quantified its similarity to the global TATA consensus, i.e. the ideal TBP binding site. Subsequently, TBP binding to specific promoters could be predicted using a linear regression model, and compared to the actual occupancy of TBP experimentally determined by ChIP-seq. In case of the rb2 promoter the actual TBP occupancy far exceeded the predicted one (0.1 vs. 1 Log2[IP/input]), which is congruent with the notion that TBP-recruitment in vivo is strongly enhanced by Ptr2 (Smollett et al. 2017).
1.3.2 The Impact of Chromatin Structure on Transcription
All cellular genomes are organised and compacted by DNA-binding proteins that protect the DNA while still allowing the access of molecular machines that facilitate DNA replication, repair and recombination and last but not least transcription (Ammar et al. 2012; Cubonovaa et al. 2012; Peeters et al. 2015; Visone et al. 2014; Xie and Reeve 2004). In eukaryotes, histone-based chromatin has evolved into a major regulatory mechanism, with hundreds of post-translational modifications and remodelling complexes facilitating the precise execution of the genetic programme. Many archaea encode histone homologues, but it remains to be proven to which extent histone-based chromatin regulates gene expression in archaea. In addition to regulatory functions, histones are likely to protect the genomes of hyperthermophiles from thermal denaturation (Visone et al. 2014). Archaea vary in their repertoire of histones and other chromatin proteins (Peeters et al. 2015). Small archaeal chromatin proteins with the ability to bind and condense DNA were first described in Thermoplasma acidophilum (DeLange et al. 1981a, b; Searcy 1975; Searcy and Delange 1980), but archaeal histones were first characterised in the hyperthermophile Methanothermus fervidus (Sandman et al. 1990). In vitro experiments using a limited number of factors (TBP, TFB and RNAP) have shown that histones inhibit transcription under these conditions, but it remains unknown how additional general factors such as TFE, Spt4/5 and TFS assist RNAP transcribing through chromatin (Wilkinson et al. 2010; Xie and Reeve 2004).
While histones are not essential for cell viability in some archaea, deletion of histones changes the transcriptome by both up- and downregulating genes (Cubonovaa et al. 2012; Heinicke et al. 2004; Nalabothula et al. 2013). In eukaryotes this regulation chiefly occurs via post translational modifications of the histone tails (Bannister and Kouzarides 2011). Archaeal histones generally encompass only the histone fold and lack the tails of their eukaryotic counterparts. In M. jannaschii, no histone modifications could be identified in a top-down mass spectrometry approach (Forbes et al. 2004). However, most archaea with histones encode multiple paralogues, enabling different combinations of histone homo- and heterodimers to form alternative chromatin structures, either at specific regulatory sequences, different genomic loci or TU, or under different growth conditions. For example, in M. fervidus the expression levels of histone HMfA are higher than HMfB during exponential growth but decrease in stationary phase, a change which may result in more compact chromatin (Sandman et al. 1994).
High-throughput sequencing approaches including nucleosome sequencing have mapped the genome-wide histone occupancy, and identified the optimal archaeal histone DNA binding site, which is near-identical to eukaryotes and reflects a basepair sequence that enables DNA curvature/bending (Ammar et al. 2012; Maruyama et al. 2013; Nalabothula et al. 2013). Generally, archaeal histones dimerise in solution and interact with 30 bp of DNA. Limited MNase digestion of chromatin isolated from Haloferax volcanii resulted in nucleosome ladder with 60 bp steps corresponding to histone tetramers (Ammar et al. 2012), while Thermococcus kodakarensis and Methanothermobacter thermautophicus resulted in a pattern with 30 bp steps, indicative of histone dimers (Maruyama et al. 2013; Nalabothula et al. 2013). Both observations are congruent with a chromatin model where histones polymerise upon DNA binding (Fig. 1.9a). As is seen in eukaryotes, promoter regions, and specific genomic loci including the highly transcribed rRNA operons are apparently devoid of histone binding (nucleosome-free regions or NFR). Moreover, MNase digestion of in vitro reconstituted chromatin reproduces this pattern (Maruyama et al. 2013; Nalabothula et al. 2013). This not only suggests that the DNA sequence alone is sufficient to organise chromatin structure, but also implies that on-going transcription has little influence on the deposition of histones across the genome—and altogether emphasises a possible role of histones in transcription regulation (potential mechanisms are shown in Fig. 1.9b, c).

Interference of chromatin and transcription in archaea. (a) In euryarchaea histones compact and organise the genome into dynamic chromatin fibres that grow and shrink by association or dissociation of histone dimers at each end. These chromatin structures can interfere with transcription in a number of ways: (b) by denying access of initiation factors to promoter or (c) by providing a barrier to TEC
1.4 The Output of the Transcription System
1.4.1 The Archaeal Transcriptomes
The introduction of high-throughput sequencing approaches to determine global RNA levels provide a significant improvement compared to hybridisation-based approaches such as microarrays in terms of the dynamic range and the detection of low abundance transcripts (Zhao et al. 2014). RNA-seq data for several euryarchaeal species (Babski et al. 2016; Cho et al. 2017; Jäger et al. 2009, 2014; Li et al. 2015; Smollett et al. 2017) and the crenarcheaon S. solfataricus (Wurtzel et al. 2010) have provided new insights into archaeal transcriptomes, while other archaeal phyla remain unexplored. Because the RNA-seq approach is independent of prior knowledge about the coding regions and predicted TUs, these data sets can provide us with a wealth of novel non-coding transcripts including small regulator RNAs (discussed in Chap. 10), anti-sense RNA, and newly discovered mRNAs (Fig. 1.10) (Babski et al. 2016; Cho et al. 2017; Jäger et al. 2009, 2014; Li et al. 2015; Smollett et al. 2017; Straub et al. 2009; Tang et al. 2005; Toffano-Nioche et al. 2013; Wurtzel et al. 2010; Dar et al. 2016a). Archaeal ncRNA species with uncharacterized functions include processed fragments of mRNA UTRs. Methanogens (M. jannaschii, M. mazei and Methanolobus psychrophilus) and Thermococcales (T. kodakarensis, T. onnurineus and P. furiosus) all contain long 5′UTRs, including ribosome binding sites and potential sites of regulation by riboregulators and riboswitches (Cho et al. 2017; Jäger et al. 2009, 2014; Li et al. 2015; Smollett et al. 2017; Toffano-Nioche et al. 2013). In contrast, Sulfolobus and halophilic archaea are characterised by leaderless mRNAs where translation is initiated directly from the mRNA 5′-end (Babski et al. 2016; Brenneis et al. 2007; Koide et al. 2009; Torarinsson et al. 2005; Wurtzel et al. 2010). Term-seq has revealed the abundance of 3′-UTRs in archaea, which similar to the 5′-ends are longer in methanogens than in Sulfolobus (Dar et al. 2016a). Genes encoding ribosomal proteins tend to have long 5′UTRs in all archaea, even in species predominantly using leaderless mRNAs such as Sulfolobus (Li et al. 2015; Toffano-Nioche et al. 2013; Wurtzel et al. 2010), which suggests a common regulatory mechanism for these genes.

Features of the archaeal transcriptome. Global TSS mapping and RNA-seq highlight the diversity of archaeal transcripts as shown in M. jannaschii. (a) Transcription initiation using alternative promoters leads to the synthesis of distinct mRNA species with different 5′-UTRs, which provide opportunities for riboregulation by e.g., riboswitches. (b) and (c) These methods also lead to the discovery of novel transcripts including antisense RNAs (b) and small ORFs (c) missed in genome sequence-based annotations (Smollett et al. 2017)
1.4.2 Evidence for Pervasive Transcription in Archaea
Pervasive transcription describes the phenomenon of non-coding, often anti-sense transcripts that are not restricted by gene boundaries; it has been implicated in transcription regulation, transcription-coupled repair and genome evolution. RNA-seq demonstrates that pervasive transcription occurs in all domains of life (Clark et al. 2011; Smollett et al. 2017; Wade and Grainger 2014). In E. coli the comparison of transcriptome data obtained under different growth conditions, and library preparation techniques has yielded a more genuine and comprehensive map of TSSs. Furthermore the detection of novel transcripts in E. coli was aided by the deletion of nucleases including RNase E and RNase III that are involved in RNA turnover (Thomason et al. 2015; Wade 2015). The same approaches will likely enable a more accurate estimation as to the amount of pervasive transcription in archaea.
1.4.3 Deconvoluting RNA Synthesis and RNA Steady-State Levels
There are several limitations one needs to be aware of when analysing archaeal transcriptomes, in particular when attempting to correlate genome occupancy profiles of basal transcription factors and RNAP with RNA levels. Due to its high abundance rRNA is often depleted using standard procedures of RNA isolation and subsequent transcriptomics analyses. In addition RNA isolation methods and library preparation techniques tend to include size selection steps that introduce bias against small RNAs. Most importantly, RNA-seq data represent steady-state RNA levels that reflect RNA synthesis and degradation, and not nascent RNA synthesis. Attempts to determine mRNA half-lives haven been made for S. solfataricus and S. acidocaldarius. These studies revealed important differences in RNA stability depending on functional category of genes and RNA expression levels (Andersson et al. 2006). Several techniques have been developed to map the nascent transcriptome to obtain a more accurate global picture of ongoing RNA synthesis. In a NET-seq (Native elongating transcript sequencing) approach TEC are purified from biomass, the RNA associated with RNAPs is isolated and sequenced, which provides a snapshot of active transcription at a single-nucleotide resolution (Churchman and Weissman 2012). In transient transcriptome sequencing (TT-seq) approaches nascent RNA is metabolically labeled with uridine base analogues that allow the specific purification of the nascent RNA prior to sequencing (Schwalb et al. 2016). A caveat from an archaeal perspective is that the narrow phylogenetic distribution of the required uridine kinase activity would require the introduction of this enzyme by genetic manipulation to adapt such methods for archaea. A slightly different approach has been recently adapted for in vivo labeling of RNA in archaea for the first time using 4-Thiouracil rather than uridine analogues involving a different biochemical pathway from 4-Thiouracil to UMP via uracil phosphoribosyltransferase (Knüppel et al. 2017). Finally, approaches such as Gro-seq (Global run-on sequencing) that isolate TECs and carry out the metabolic labeling of nascent RNA by transcription elongation in vitro can be adapted to archaeal transcriptomics in a reasonably straightforward fashion (Core et al. 2008).
1.5 Future Directions
High-throughput sequencing approaches have greatly improved our understanding of the mechanisms of transcription in archaea. We can now begin to unravel connections between perturbations at the molecular level and changes of the entire system, between in vitro and in vivo data, aiming to understand transcription in a multiscalar fashion. The current experimental portfolio at our fingertips needs to be expanded by mapping the transcriptome-wide occupancy of RNA-binding transcription factors by techniques such as iCLIP (Konig et al. 2011), mapping of TECs by NET-seq and Gro-seq (see above), and genome-wide mapping of evolutionary pervasive DNA- and RNA-modifications (Huber et al. 2015). Once these aspects of archaeal transcription are described on a systems level, it will be possible to characterise the transcription apparatus in flux—as it changes in response to external stimuli and environmental insults. Future research would benefit from being expanded to include little characterised phyla including Nano- Thaum- and Lokiarchaeota to provide further insights into the evolution of transcription regulation in archaea. Last but not least, the ability to examine features of macromolecular metabolism genome-wide will allow us to correlate transcription with DNA replication, -recombination and -repair, and protein translation.
References
Ammar R, Torti D, Tsui K, Gebbia M, Durbic T, Bader GD, Giaever G, Nislow C (2012) Chromatin is an ancient innovation conserved between archaea and eukarya. Elife 1:e00078. doi:10.7554/eLife.00078
Andersson AF, Lundgren M, Eriksson S, Rosenlund M, Bernander R, Nilsson P (2006) Global analysis of mRNA stability in the archaeon Sulfolobus. Genome Biol 7(10):R99. doi:10.1186/gb-2006-7-10-r99
Aravind L, Koonin EV (1999) DNA-binding proteins and evolution of transcription regulation in the archaea. Nucleic Acids Res 27(23):4658–4670
Babski J, Haas KA, Nather-Schindler D, Pfeiffer F, Forstner KU, Hammelmann M, Hilker R, Becker A, Sharma CM, Marchfelder A, Soppa J (2016) Genome-wide identification of transcriptional start sites in the haloarchaeon Haloferax volcanii based on differential RNA-Seq (dRNA-Seq). BMC Genomics 17(1):629. doi:10.1186/s12864-016-2920-y
Baliga NS, Goo YA, Ng WV, Hood L, Daniels CJ, DasSarma S (2000) Is gene expression in Halobacterium NRC-1 regulated by multiple TBP and TFB transcription factors? Mol Microbiol 36(5):1184–1185
Bannister AJ, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21(3):381–395. doi:10.1038/cr.2011.22
Basu RS, Warner BA, Molodtsov V, Pupov D, Esyunina D, Fernandez-Tornero C, Kulbachinskiy A, Murakami KS (2014) Structural basis of transcription initiation by bacterial RNA polymerase holoenzyme. J Biol Chem 289(35):24549–24559. doi:10.1074/jbc.M114.584037
Bell SD, Jaxel C, Nadal M, Kosa PF, Jackson SP (1998) Temperature, template topology, and factor requirements of archaeal transcription. Proc Natl Acad Sci U S A 95(26):15218–15222
Bell SD, Kosa PL, Sigler PB, Jackson SP (1999) Orientation of the transcription preinitiation complex in archaea. Proc Natl Acad Sci U S A 96(24):13662–13667. doi:10.1073/pnas.96.24.13662
Belogurov GA, Artsimovitch I (2015) Regulation of transcript elongation. Annu Rev Microbiol 69:49–69. doi:10.1146/annurev-micro-091014-104047
Blombach F, Grohmann D (2017) Same same but different: the evolution of TBP in archaea and their eukaryotic offspring. Transcription 8(3):162–168. doi:10.1080/21541264.2017.1289879
Blombach F, Makarova KS, Marrero J, Siebers B, Koonin EV, van der Oost J (2009) Identification of an ortholog of the eukaryotic RNA polymerase III subunit RPC34 in crenarchaeota and thaumarchaeota suggests specialization of RNA polymerases for coding and non-coding RNAs in archaea. Biol Direct 4:39. doi:10.1186/1745-6150-4-39
Blombach F, Salvadori E, Fouqueau T, Yan J, Reimann J, Sheppard C, Smollett KL, Albers SV, Kay CW, Thalassinos K, Werner F (2015) Archaeal TFEalpha/beta is a hybrid of TFIIE and the RNA polymerase III subcomplex hRPC62/39. Elife 4:e08378. doi:10.7554/eLife.08378
Blombach F, Smollett KL, Grohmann D, Werner F (2016) Molecular mechanisms of transcription initiation-structure, function, and evolution of TFE/TFIIE-like factors and open complex formation. J Mol Biol 428(12):2592–2606. doi:10.1016/j.jmb.2016.04.016
Brenneis M, Hering O, Lange C, Soppa J (2007) Experimental characterization of cis-acting elements important for translation and transcription in halophilic archaea. PLoS Genet 3(12):e229. doi:10.1371/journal.pgen.0030229
Charoensawan V, Wilson D, Teichmann SA (2010) Genomic repertoires of DNA-binding transcription factors across the tree of life. Nucleic Acids Res 38(21):7364–7377. doi:10.1093/nar/gkq617
Cho S, Kim MS, Jeong Y, Lee BR, Lee JH, Kang SG, Cho BK (2017) Genome-wide primary transcriptome analysis of H2-producing archaeon Thermococcus onnurineus NA1. Sci Rep 7:43044. doi:10.1038/srep43044
Churchman LS, Weissman JS (2012) Native elongating transcript sequencing (NET-seq). Curr Protoc Mol Biol Chapter 4:Unit 4 14 11–17. doi:10.1002/0471142727.mb0414s98
Clark MB, Amaral PP, Schlesinger FJ, Dinger ME, Taft RJ, Rinn JL, Ponting CP, Stadler PF, Morris KV, Morillon A, Rozowsky JS, Gerstein MB, Wahlestedt C, Hayashizaki Y, Carninci P, Gingeras TR, Mattick JS (2011) The reality of pervasive transcription. PLoS Biol 9(7):e1000625; discussion e1001102. doi:10.1371/journal.pbio.1000625
Core LJ, Waterfall JJ, Lis JT (2008) Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322(5909):1845–1848. doi:10.1126/science.1162228
Cubonovaa L, Katano M, Kanai T, Atomi H, Reeve JN, Santangelo TJ (2012) An archaeal histone is required for transformation of Thermococcus kodakarensis. J Bacteriol 194(24):6864–6874. doi:10.1128/JB.01523-12
Dahlke I, Thomm M (2002) A Pyrococcus homolog of the leucine-responsive regulatory protein, LrpA, inhibits transcription by abrogating RNA polymerase recruitment. Nucleic Acids Res 30(3):701–710
Dar D, Prasse D, Schmitz RA, Sorek R (2016a) Widespread formation of alternative 3′ UTR isoforms via transcription termination in archaea. Nat Microbiol 1(10):16143. doi:10.1038/nmicrobiol.2016.143
Dar D, Shamir M, Mellin JR, Koutero M, Stern-Ginossar N, Cossart P, Sorek R (2016b) Term-seq reveals abundant ribo-regulation of antibiotics resistance in bacteria. Science 352(6282):aad9822. doi:10.1126/science.aad9822
DeLange RJ, Green GR, Searcy DG (1981a) A histone-like protein (HTa) from Thermoplasma acidophilum. I. Purification and properties. J Biol Chem 256(2):900–904
DeLange RJ, Williams LC, Searcy DG (1981b) A histone-like protein (HTa) from Thermoplasma acidophilum. II. Complete amino acid sequence. J Biol Chem 256(2):905–911
Diamant G, Bahat A, Dikstein R (2016) The elongation factor Spt5 facilitates transcription initiation for rapid induction of inflammatory-response genes. Nat Commun 7:11547. doi:10.1038/ncomms11547
El Hage A, Koper M, Kufel J, Tollervey D (2008) Efficient termination of transcription by RNA polymerase I requires the 5′ exonuclease Rat1 in yeast. Genes Dev 22(8):1069–1081. doi:10.1101/gad.463708
Epshtein V, Cardinale CJ, Ruckenstein AE, Borukhov S, Nudler E (2007) An allosteric path to transcription termination. Mol Cell 28(6):991–1001. doi:10.1016/j.molcel.2007.10.011
Epshtein V, Dutta D, Wade J, Nudler E (2010) An allosteric mechanism of Rho-dependent transcription termination. Nature 463(7278):245–249. doi:10.1038/nature08669
Facciotti MT, Reiss DJ, Pan M, Kaur A, Vuthoori M, Bonneau R, Shannon P, Srivastava A, Donohoe SM, Hood LE, Baliga NS (2007) General transcription factor specified global gene regulation in archaea. Proc Natl Acad Sci U S A 104(11):4630–4635. doi:10.1073/pnas.0611663104
Forbes AJ, Patrie SM, Taylor GK, Kim YB, Jiang L, Kelleher NL (2004) Targeted analysis and discovery of posttranslational modifications in proteins from methanogenic archaea by top-down MS. Proc Natl Acad Sci U S A 101(9):2678–2683. doi:10.1073/pnas.0306575101
Fouqueau T, Zeller ME, Cheung AC, Cramer P, Thomm M (2013) The RNA polymerase trigger loop functions in all three phases of the transcription cycle. Nucleic Acids Res 41(14):7048–7059. doi:10.1093/nar/gkt433
Geiduschek EP, Ouhammouch M (2005) Archaeal transcription and its regulators. Mol Microbiol 56(6):1397–1407. doi:10.1111/j.1365-2958.2005.04627.x
Gietl A, Holzmeister P, Blombach F, Schulz S, von Voithenberg LV, Lamb DC, Werner F, Tinnefeld P, Grohmann D (2014) Eukaryotic and archaeal TBP and TFB/TF(II)B follow different promoter DNA bending pathways. Nucleic Acids Res 42(10):6219–6231. doi:10.1093/nar/gku273
Grohmann D, Nagy J, Chakraborty A, Klose D, Fielden D, Ebright RH, Michaelis J, Werner F (2011) The initiation factor TFE and the elongation factor Spt4/5 compete for the RNAP clamp during transcription initiation and elongation. Mol Cell 43(2):263–274. doi:10.1016/j.molcel.2011.05.030
Han Z, Libri D, Porrua O (2016) Biochemical characterization of the helicase Sen1 provides new insights into the mechanisms of non-coding transcription termination. Nucleic Acids Res 45(3):1355–1370. doi:10.1093/nar/gkw1230
Hein PP, Kolb KE, Windgassen T, Bellecourt MJ, Darst SA, Mooney RA, Landick R (2014) RNA polymerase pausing and nascent-RNA structure formation are linked through clamp-domain movement. Nat Struct Mol Biol 21(9):794–802. doi:10.1038/nsmb.2867
Heinicke I, Muller J, Pittelkow M, Klein A (2004) Mutational analysis of genes encoding chromatin proteins in the archaeon Methanococcus voltae indicates their involvement in the regulation of gene expression. Mol Genet Genomics 272(1):76–87. doi:10.1007/s00438-004-1033-5
Hirtreiter A, Damsma GE, Cheung AC, Klose D, Grohmann D, Vojnic E, Martin AC, Cramer P, Werner F (2010a) Spt4/5 stimulates transcription elongation through the RNA polymerase clamp coiled-coil motif. Nucleic Acids Res 38(12):4040–4051. doi:10.1093/nar/gkq135
Hirtreiter A, Grohmann D, Werner F (2010b) Molecular mechanisms of RNA polymerase - the F/E (RPB4/7) complex is required for high processivity in vitro. Nucleic Acids Res 38(2):585–596. doi:10.1093/nar/gkp928
Huber SM, van Delft P, Mendil L, Bachman M, Smollett K, Werner F, Miska EA, Balasubramanian S (2015) Formation and abundance of 5-hydroxymethylcytosine in RNA. Chembiochem 16(5):752–755. doi:10.1002/cbic.201500013
Jäger D, Förstner KU, Sharma CM, Santangelo TJ, Reeve JN (2014) Primary transcriptome map of the hyperthermophilic archaeon Thermococcus kodakarensis. BMC Genomics 15:684. doi:10.1186/1471-2164-15-684
Jäger D, Sharma CM, Thomsen J, Ehlers C, Vogel J, Schmitz RA (2009) Deep sequencing analysis of the Methanosarcina mazei Go1 transcriptome in response to nitrogen availability. Proc Natl Acad Sci U S A 106(51):21878–21882. doi:10.1073/pnas.0909051106
Jensen KF, Pedersen S (1990) Metabolic growth rate control in Escherichia coli may be a consequence of subsaturation of the macromolecular biosynthetic apparatus with substrates and catalytic components. Microbiol Rev 54(2):89–100
Kadonaga JT (2012) Perspectives on the RNA polymerase II core promoter. Wiley Interdiscip Rev Dev Biol 1(1):40–51. doi:10.1002/wdev.21
Kanai T, Akerboom J, Takedomi S, van de Werken HJ, Blombach F, van der Oost J, Murakami T, Atomi H, Imanaka T (2007) A global transcriptional regulator in Thermococcus kodakaraensis controls the expression levels of both glycolytic and gluconeogenic enzyme-encoding genes. J Biol Chem 282(46):33659–33670. doi:10.1074/jbc.M703424200
Kim J, Iyer VR (2004) Global role of TATA box-binding protein recruitment to promoters in mediating gene expression profiles. Mol Cell Biol 24(18):8104–8112. doi:10.1128/MCB.24.18.8104-8112.2004
Kim M, Krogan NJ, Vasiljeva L, Rando OJ, Nedea E, Greenblatt JF, Buratowski S (2004) The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature 432(7016):517–522. doi:10.1038/nature03041
Knüppel R, Kuttenberger C, Ferreira-Cerca S (2017) Toward time-resolved analysis of RNA metabolism in archaea using 4-thiouracil. Front Microbiol 8:286. doi:10.3389/fmicb.2017.00286
Koide T, Reiss DJ, Bare JC, Pang WL, Facciotti MT, Schmid AK, Pan M, Marzolf B, Van PT, Lo FY, Pratap A, Deutsch EW, Peterson A, Martin D, Baliga NS (2009) Prevalence of transcription promoters within archaeal operons and coding sequences. Mol Syst Biol 5:285. doi:10.1038/msb.2009.42
Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J (2011) iCLIP–transcriptome-wide mapping of protein-RNA interactions with individual nucleotide resolution. J Vis Exp 50:2638. doi:10.3791/2638
Kostrewa D, Zeller ME, Armache KJ, Seizl M, Leike K, Thomm M, Cramer P (2009) RNA polymerase II-TFIIB structure and mechanism of transcription initiation. Nature 462(7271):323–330. doi:10.1038/nature08548
Larochelle S, Amat R, Glover-Cutter K, Sanso M, Zhang C, Allen JJ, Shokat KM, Bentley DL, Fisher RP (2012) Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol 19(11):1108–1115. doi:10.1038/nsmb.2399
Lee SJ, Surma M, Hausner W, Thomm M, Boos W (2008) The role of TrmB and TrmB-like transcriptional regulators for sugar transport and metabolism in the hyperthermophilic archaeon Pyrococcus furiosus. Arch Microbiol 190(3):247–256. doi:10.1007/s00203-008-0378-2
Li J, Qi L, Guo Y, Yue L, Li Y, Ge W, Wu J, Shi W, Dong X (2015) Global mapping transcriptional start sites revealed both transcriptional and post-transcriptional regulation of cold adaptation in the methanogenic archaeon Methanolobus psychrophilus. Sci Rep 5:9209. doi:10.1038/srep09209
Lipscomb GL, Keese AM, Cowart DM, Schut GJ, Thomm M, Adams MW, Scott RA (2009) SurR: a transcriptional activator and repressor controlling hydrogen and elemental sulphur metabolism in Pyrococcus furiosus. Mol Microbiol 71(2):332–349. doi:10.1111/j.1365-2958.2008.06525.x
Liu H, Wang K, Lindas AC, Peeters E (2016) The genome-scale DNA-binding profile of BarR, a beta-alanine responsive transcription factor in the archaeon Sulfolobus acidocaldarius. BMC Genomics 17:569. doi:10.1186/s12864-016-2890-0
Maruyama H, Harwood JC, Moore KM, Paszkiewicz K, Durley SC, Fukushima H, Atomi H, Takeyasu K, Kent NA (2013) An alternative beads-on-a-string chromatin architecture in Thermococcus kodakarensis. EMBO Rep 14(8):711–717. doi:10.1038/embor.2013.94
Mayer A, Lidschreiber M, Siebert M, Leike K, Soding J, Cramer P (2010) Uniform transitions of the general RNA polymerase II transcription complex. Nat Struct Mol Biol 17(10):1272–1278. doi:10.1038/nsmb.1903
Micorescu M, Grunberg S, Franke A, Cramer P, Thomm M, Bartlett M (2008) Archaeal transcription: function of an alternative transcription factor B from Pyrococcus furiosus. J Bacteriol 190(1):157–167. doi:10.1128/JB.01498-07
Mooney RA, Davis SE, Peters JM, Rowland JL, Ansari AZ, Landick R (2009) Regulator trafficking on bacterial transcription units in vivo. Mol Cell 33(1):97–108. doi:10.1016/j.molcel.2008.12.021
Nagy J, Grohmann D, Cheung AC, Schulz S, Smollett K, Werner F, Michaelis J (2015) Complete architecture of the archaeal RNA polymerase open complex from single-molecule FRET and NPS. Nat Commun 6:6161. doi:10.1038/ncomms7161
Naji S, Grünberg S, Thomm M (2007) The RPB7 orthologue E’ is required for transcriptional activity of a reconstituted archaeal core enzyme at low temperatures and stimulates open complex formation. J Biol Chem 282(15):11047–11057. doi:10.1074/jbc.M611674200
Nalabothula N, Xi L, Bhattacharyya S, Widom J, Wang JP, Reeve JN, Santangelo TJ, Fondufe-Mittendorf YN (2013) Archaeal nucleosome positioning in vivo and in vitro is directed by primary sequence motifs. BMC Genomics 14:391. doi:10.1186/1471-2164-14-391
Nguyen-Duc T, van Oeffelen L, Song N, Hassanzadeh-Ghassabeh G, Muyldermans S, Charlier D, Peeters E (2013) The genome-wide binding profile of the Sulfolobus solfataricus transcription factor Ss-LrpB shows binding events beyond direct transcription regulation. BMC Genomics 14:828. doi:10.1186/1471-2164-14-828
Ochs SM, Thumann S, Richau R, Weirauch MT, Lowe TM, Thomm M, Hausner W (2012) Activation of archaeal transcription mediated by recruitment of transcription factor B. J Biol Chem 287(22):18863–18871. doi:10.1074/jbc.M112.365742
Ouhammouch M, Dewhurst RE, Hausner W, Thomm M, Geiduschek EP (2003) Activation of archaeal transcription by recruitment of the TATA-binding protein. Proc Natl Acad Sci U S A 100(9):5097–5102. doi:10.1073/pnas.0837150100
Ouhammouch M, Geiduschek EP (2001) A thermostable platform for transcriptional regulation: the DNA-binding properties of two Lrp homologs from the hyperthermophilic archaeon Methanococcus jannaschii. EMBO J 20(1–2):146–156. doi:10.1093/emboj/20.1.146
Ouhammouch M, Langham GE, Hausner W, Simpson AJ, El-Sayed NM, Geiduschek EP (2005) Promoter architecture and response to a positive regulator of archaeal transcription. Mol Microbiol 56(3):625–637. doi:10.1111/j.1365-2958.2005.04563.x
Peeters E, Driessen RP, Werner F, Dame RT (2015) The interplay between nucleoid organization and transcription in archaeal genomes. Nat Rev Microbiol 13(6):333–341. doi:10.1038/nrmicro3467
Peeters E, Peixeiro N, Sezonov G (2013) Cis-regulatory logic in archaeal transcription. Biochem Soc Trans 41(1):326–331. doi:10.1042/BST20120312
Perez-Rueda E, Janga SC (2010) Identification and genomic analysis of transcription factors in archaeal genomes exemplifies their functional architecture and evolutionary origin. Mol Biol Evol 27(6):1449–1459. doi:10.1093/molbev/msq033
Phung DK, Rinaldi D, Langendijk-Genevaux PS, Quentin Y, Carpousis AJ, Clouet-d’Orval B (2013) Archaeal beta-CASP ribonucleases of the aCPSF1 family are orthologs of the eukaryal CPSF-73 factor. Nucleic Acids Res 41(2):1091–1103. doi:10.1093/nar/gks1237
Porrua O, Boudvillain M, Libri D (2016) Transcription termination: variations on common themes. Trends Genet 32(8):508–522. doi:10.1016/j.tig.2016.05.007
Porrua O, Libri D (2013) A bacterial-like mechanism for transcription termination by the Sen1p helicase in budding yeast. Nat Struct Mol Biol 20(7):884–891. doi:10.1038/nsmb.2592
Proudfoot NJ (2016) Transcriptional termination in mammals: stopping the RNA polymerase II juggernaut. Science 352(6291):aad9926. doi:10.1126/science.aad9926
Qureshi SA, Baumann P, Rowlands T, Khoo B, Jackson SP (1995) Cloning and functional analysis of the TATA-binding protein from Sulfolobus shibatae. Nucleic Acids Res 23(10):1775–1781. doi:10.1093/nar/23.10.1775
Qureshi SA, Bell SD, Jackson SP (1997) Factor requirements for transcription in the archaeon Sulfolobus shibatae. EMBO J 16(10):2927–2936. doi:10.1093/emboj/16.10.2927
Ray-Soni A, Bellecourt MJ, Landick R (2016) Mechanisms of bacterial transcription termination: all good things must end. Annu Rev Biochem 85:319–347. doi:10.1146/annurev-biochem-060815-014844
Reichelt R, Gindner A, Thomm M, Hausner W (2016) Genome-wide binding analysis of the transcriptional regulator TrmBL1 in Pyrococcus furiosus. BMC Genomics 17:40. doi:10.1186/s12864-015-2360-0
Rowlands T, Baumann P, Jackson SP (1994) The TATA-binding protein - a general transcription factor in eukaryotes and archaebacteria. Science 264(5163):1326–1329. doi:10.1126/science.8191287
Rudrappa D, Yao AI, White D, Pavlik BJ, Singh R, Facciotti MT, Blum P (2015) Identification of an archaeal mercury regulon by chromatin immunoprecipitation. Microbiology 161(12):2423–2433. doi:10.1099/mic.0.000189
Sandelin A, Carninci P, Lenhard B, Ponjavic J, Hayashizaki Y, Hume DA (2007) Mammalian RNA polymerase II core promoters: insights from genome-wide studies. Nat Rev Genet 8(6):424–436. doi:10.1038/nrg2026
Sandman K, Grayling RA, Dobrinski B, Lurz R, Reeve JN (1994) Growth-phase-dependent synthesis of histones in the archaeon Methanothermus fervidus. Proc Natl Acad Sci U S A 91(26):12624–12628
Sandman K, Krzycki JA, Dobrinski B, Lurz R, Reeve JN (1990) HMf, a DNA-binding protein isolated from the hyperthermophilic archaeon Methanothermus fervidus, is most closely related to histones. Proc Natl Acad Sci U S A 87(15):5788–5791
Santangelo TJ, Artsimovitch I (2011) Termination and antitermination: RNA polymerase runs a stop sign. Nat Rev Microbiol 9(5):319–329. doi:10.1038/nrmicro2560
Santangelo TJ, Cubonova L, Matsumi R, Atomi H, Imanaka T, Reeve JN (2008) Polarity in archaeal operon transcription in Thermococcus kodakaraensis. J Bacteriol 190(6):2244–2248. doi:10.1128/JB.01811-07
Santangelo TJ, Cubonova L, Skinner KM, Reeve JN (2009) Archaeal intrinsic transcription termination in vivo. J Bacteriol 191(22):7102–7108. doi:10.1128/JB.00982-09
Santangelo TJ, Reeve JN (2006) Archaeal RNA polymerase is sensitive to intrinsic termination directed by transcribed and remote sequences. J Mol Biol 355(2):196–210. doi:10.1016/j.jmb.2005.10.062
Sasse-Dwight S, Gralla JD (1989) KMnO4 as a probe for lac promoter DNA melting and mechanism in vivo. J Biol Chem 264(14):8074–8081
Schwalb B, Michel M, Zacher B, Fruhauf K, Demel C, Tresch A, Gagneur J, Cramer P (2016) TT-seq maps the human transient transcriptome. Science 352(6290):1225–1228. doi:10.1126/science.aad9841
Searcy DG (1975) Histone-like protein in the prokaryote Thermoplasma acidophilum. Biochim Biophys Acta 395(4):535–547
Searcy DG, Delange RJ (1980) Thermoplasma acidophilum histone-like protein. Partial amino acid sequence suggestive of homology to eukaryotic histones. Biochim Biophys Acta 609(1):197–200
Seitzer P, Wilbanks EG, Larsen DJ, Facciotti MT (2012) A Monte Carlo-based framework enhances the discovery and interpretation of regulatory sequence motifs. BMC Bioinf 13:317. doi:10.1186/1471-2105-13-317
Shultzaberger RK, Chen Z, Lewis KA, Schneider TD (2007) Anatomy of Escherichia coli sigma70 promoters. Nucleic Acids Res 35(3):771–788. doi:10.1093/nar/gkl956
Smollett K, Blombach F, Reichelt R, Thomm M, Werner F (2017) A global analysis of transcription reveals two modes of Spt4/5 recruitment to archaeal RNA polymerase. Nat Microbiol 2:17021. doi:10.1038/nmicrobiol.2017.21
Smollett K, Blombach F, Werner F (2015) Transcription in archaea: preparation of Methanocaldococcus jannaschii transcription machinery. Methods Mol Biol 1276:291–303. doi:10.1007/978-1-4939-2392-2_17
Spitalny P, Thomm M (2008) A polymerase III-like reinitiation mechanism is operating in regulation of histone expression in archaea. Mol Microbiol 67(5):958–970. doi:10.1111/j.1365-2958.2007.06084.x
Straub J, Brenneis M, Jellen-Ritter A, Heyer R, Soppa J, Marchfelder A (2009) Small RNAs in haloarchaea: identification, differential expression and biological function. RNA Biol 6(3):281–292
Tan L, Wiesler S, Trzaska D, Carney HC, Weinzierl RO (2008) Bridge helix and trigger loop perturbations generate superactive RNA polymerases. J Biol 7(10):40. doi:10.1186/jbiol98
Tang TH, Polacek N, Zywicki M, Huber H, Brugger K, Garrett R, Bachellerie JP, Huttenhofer A (2005) Identification of novel non-coding RNAs as potential antisense regulators in the archaeon Sulfolobus solfataricus. Mol Microbiol 55(2):469–481. doi:10.1111/j.1365-2958.2004.04428.x
Thomason MK, Bischler T, Eisenbart SK, Forstner KU, Zhang A, Herbig A, Nieselt K, Sharma CM, Storz G (2015) Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J Bacteriol 197(1):18–28. doi:10.1128/JB.02096-14
Toffano-Nioche C, Ott A, Crozat E, Nguyen AN, Zytnicki M, Leclerc F, Forterre P, Bouloc P, Gautheret D (2013) RNA at 92 degrees C: the non-coding transcriptome of the hyperthermophilic archaeon Pyrococcus abyssi. RNA Biol 10(7):1211–1220. doi:10.4161/rna.25567
Tonner PD, Pittman AM, Gulli JG, Sharma K, Schmid AK (2015) A regulatory hierarchy controls the dynamic transcriptional response to extreme oxidative stress in archaea. PLoS Genet 11(1):e1004912. doi:10.1371/journal.pgen.1004912
Torarinsson E, Klenk HP, Garrett RA (2005) Divergent transcriptional and translational signals in archaea. Environ Microbiol 7(1):47–54. doi:10.1111/j.1462-2920.2004.00674.x
Visone V, Vettone A, Serpe M, Valenti A, Perugino G, Rossi M, Ciaramella M (2014) Chromatin structure and dynamics in hot environments: architectural proteins and DNA topoisomerases of thermophilic archaea. Int J Mol Sci 15(9):17162–17187. doi:10.3390/ijms150917162
Vvedenskaya IO, Vahedian-Movahed H, Zhang Y, Taylor DM, Ebright RH, Nickels BE (2016) Interactions between RNA polymerase and the core recognition element are a determinant of transcription start site selection. Proc Natl Acad Sci U S A 113(21):E2899–E2905. doi:10.1073/pnas.1603271113
Wade JT (2015) Where to begin? Mapping transcription start sites genome-wide in escherichia coli. J Bacteriol 197(1):4–6. doi:10.1128/JB.02410-14
Wade JT, Grainger DC (2014) Pervasive transcription: illuminating the dark matter of bacterial transcriptomes. Nat Rev Microbiol 12(9):647–653. doi:10.1038/nrmicro3316
Werner F (2012) A nexus for gene expression-molecular mechanisms of Spt5 and NusG in the three domains of life. J Mol Biol 417(1-2):13–27. doi:10.1016/j.jmb.2012.01.031
Werner F, Grohmann D (2011) Evolution of multisubunit RNA polymerases in the three domains of life. Nat Rev Microbiol 9(2):85–98. doi:10.1038/nrmicro2507
Werner F, Weinzierl RO (2002) A recombinant RNA polymerase II-like enzyme capable of promoter-specific transcription. Mol Cell 10(3):635–646
Werner F, Weinzierl RO (2005) Direct modulation of RNA polymerase core functions by basal transcription factors. Mol Cell Biol 25(18):8344–8355. doi:10.1128/MCB.25.18.8344-8355.2005
West S, Gromak N, Proudfoot NJ (2004) Human 5′→3′ exonuclease Xrn2 promotes transcription termination at co-transcriptional cleavage sites. Nature 432(7016):522–525. doi:10.1038/nature03035
Wilbanks EG, Larsen DJ, Neches RY, Yao AI, Wu CY, Kjolby RA, Facciotti MT (2012) A workflow for genome-wide mapping of archaeal transcription factors with ChIP-seq. Nucleic Acids Res 40(10):e74. doi:10.1093/nar/gks063
Wilkinson SP, Ouhammouch M, Geiduschek EP (2010) Transcriptional activation in the context of repression mediated by archaeal histones. Proc Natl Acad Sci U S A 107(15):6777–6781. doi:10.1073/pnas.1002360107
Winkelman JT, Vvedenskaya IO, Zhang YC, Zhang Y, Bird JG, Taylor DM, Gourse RL, Ebright RH, Nickels BE (2016) Multiplexed protein-DNA cross-linking: scrunching in transcription start site selection. Science 351(6277):1090–1093. doi:10.1126/science.aad6881
Wurtzel O, Sapra R, Chen F, Zhu Y, Simmons BA, Sorek R (2010) A single-base resolution map of an archaeal transcriptome. Genome Res 20(1):133–141. doi:10.1101/gr.100396.109
Xie Y, Reeve JN (2004) Transcription by an archaeal RNA polymerase is slowed but not blocked by an archaeal nucleosome. J Bacteriol 186(11):3492–3498. doi:10.1128/JB.186.11.3492-3498.2004
Yang C, Bolotin E, Jiang T, Sladek FM, Martinez E (2007) Prevalence of the initiator over the TATA box in human and yeast genes and identification of DNA motifs enriched in human TATA-less core promoters. Gene 389(1):52–65. doi:10.1016/j.gene.2006.09.029
Zhao S, Fung-Leung WP, Bittner A, Ngo K, Liu X (2014) Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS One 9(1):e78644. doi:10.1371/journal.pone.0078644
Zuo Y, Steitz TA (2015) Crystal structures of the E. coli transcription initiation complexes with a complete bubble. Mol Cell 58(3):534–540. doi:10.1016/j.molcel.2015.03.010
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter

Cite this chapter
Smollett, K., Blombach, F., Fouqueau, T., Werner, F. (2017). A Global Characterisation of the Archaeal Transcription Machinery. In: Clouet-d'Orval, B. (eds) RNA Metabolism and Gene Expression in Archaea. Nucleic Acids and Molecular Biology, vol 32. Springer, Cham. https://doi.org/10.1007/978-3-319-65795-0_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-65795-0_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-65794-3
Online ISBN: 978-3-319-65795-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)