
Abstract
Over the past several decades, numerous studies have shown that inflammatory diseases and infections trigger or promote the development and progression of many types of cancers, including hepatocellular carcinoma (HCC). Despite considerable progress in HCC diagnosis and improvement of the curative strategies, its incidence is still increasing in western countries and the prognosis of patients with advanced HCC remains in general very poor. Diverse immune cell types associated with the release of a large spectrum of inflammatory cytokines appeared to be a key component in HCC emergence, progression and in therapeutic failures. In addition to curative strategies for HCC, it is of interest to identify and block the immune cell types and their associated cytokines susceptible to trigger tumor initiation and progression
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Over the past several decades, numerous studies have shown that inflammatory diseases and infections trigger or promote the development and progression of many types of cancers [65], including hepatocellular carcinoma (HCC) which occurs in most cases at late stages of chronic liver disease associated with viral hepatitis B and C infection, metabolic disorders, and alcohol heavy consumption [41].
Chronic liver diseases, whatever their etiologies, are often associated with a sustained inflammatory response leading to repeated injury, fibrosis and at late stages, to cirrhosis. HCC arises in the setting of cirrhotic livers in 80 to 90% of cases and progresses in an inflammatory context. Despite significant progress in HCC diagnosis and improvement of the curative strategies, its incidence is still increasing in western countries and the prognosis of patients with advanced HCC remains in general very poor. Recurrence and non-response to the current anti-cancer treatment occur frequently. Diverse immune cell types associated with the release of a large spectrum of inflammatory cytokines appeared to be a key component in HCC emergence, progression and in therapeutic failures. Many factors produced by infiltrating immune cells such as chemokines, growth factors, cytokines and proangiogenic factors contribute to the promotion of cell survival, proliferation, epithelial-mesenchymal transition and genomic DNA instability [55].
Thus, a better characterization of the underlying molecular and cellular mechanisms by which HCC development and resistance occur may open new insights for the development of more effective anticancer therapies. In addition to curative strategies for HCC, it is of interest to identify and block the immune cell types and their associated cytokines susceptible to trigger tumor initiation and progression.
9.1 Cytokines in Chronic Liver Diseases: A Risk Factor for HCC Initiation
9.1.1 Chronic Alcohol Consumption is a Risk Factor for HCC Emergence
Chronic alcohol consumption is associated with hepatic inflammation leading to cirrhosis development, and constitute a risk factor for HCC initiation. Chronic liver diseases are mainly due to recurrent and excessive liver inflammatory processes, as observed in alcoholic liver disease (ALD). ALD ranks among the major causes of morbidity and mortality worldwide, with a mortality rate in the USA and in Western Europe estimated to be approximately 5–6%. In USA, ALD is responsible for up to 100,000 deaths per year. ALD can present as steatosis (fatty liver, i.e. accumulation of triglycerides in hepatocytes), the prevalent lesion found in excessive drinkers that is now recognized as harbinger of worse disease to follow when liver insult is sustained. Indeed, steatosis may progress towards alcoholic steatohepatitis (ASH), when accompanied with liver inflammation and hepatocyte injury, that promotes liver fibrogenesis with a 20% risk of cirrhosis after 10–20 years. Severe alcoholic hepatitis (AH) is a specific clinical form characterized by a prolonged and intense inflammatory reaction despite alcohol withdrawal, and associated with a spontaneous 50% mortality rate after 6 months. Current management of steatosis and mild to moderate forms of AH relies upon abstinence. In severe AH, corticosteroids reduce the mortality rate to 15–20% after 6 months. Nevertheless, outcome after 1 year remains grim with survival rates ranging between 50 to 60%. Overall, these figures underscore the urgent need for novel therapeutic approaches targeting inflammation in the management of ALD.
9.1.1.1 Role of Innate Immune Cytokines
Activation of hepatic innate immune cells is the first step that triggers inflammation in alcoholic patients. Indeed, dysregulated cytokine signaling, particularly of those released by the resident macrophages of the liver (Kupffer cells), plays a pivotal role in the pathogenesis of ALD. In particular, several clinical and experimental studies have shown that overproduction of TNF-α by activated Kupffer cells is central to the inflammatory process associated with ALD [95, 121, 126]. The mechanism leading to increased TNF-α production in response to ethanol involves enhanced intra-hepatic oxidative stress and altered gut permeability, thereby allowing enhanced translocation of endotoxin (LPS) into the portal blood, and activation of Kupffer cells following binding of LPS to its receptor Toll Like Receptor 4 (TLR4). TNF-α [141] induces hepatocyte cell death and inflammatory cell infiltration [12]. Furthermore, liver regeneration has also been shown to be impaired in ALD [60]. In addition, clinical treatments of patients with severe AH based on TNF-α neutralization with pentoxifilin (PTX) [1] or monoconal antibodies like infliximab [104] or etanercept [13] have been shown to prevent inflammation but were associated with severe side effects including a higher susceptibility to infection leading to increased mortality rate. In addition, above and beyond its detrimental role in liver inflammation, TNF-α is not only critical for the host defence, but it also induces IL-6 synthesis to initiate hepatocyte proliferation and liver regeneration. IL-6 and TNF-α are two cytokines mainly produced by Kupffer cells in the liver and markedly induced after partial hepatectomy. TNF-α is a major regulator of the initiation of liver regeneration. It is known that IL-6 and TNF-α can stimulate hepatocyte proliferation by activating intracellular signalling pathways such as signal transducer and activator of transcription 3 (STAT-3) and CCAAT/enhancer-binding protein B (C/EBP). Altogether, these data point out the importance of controlling the balance between the inflammatory immune response required for pathogen elimination and liver regeneration, and the exacerbated inflammatory processes leading to hepatocyte cell death.
The molecular mechanisms associated with Kupffer cell activation are linked to acquisition of an M1 phenotype characterized by the production of a storm of inflammatory cytokines including TNF-α, IL-6, IL-12 and IFN-γ, which supports resistance to extracellular bacterial infection. However, overwhelming production of those cytokines by Kupffer cells is responsible for the development of AH. Contrastingly, M2-polarized macrophages are defined by production of IL-10, IL-4, and IL-13. Although those macrophages cannot control bacterial infection, they are critical for tempering the triggered inflammatory process, and in promoting tissue repair. And recently an atypical M2 profile has been reported that combines M1 and M2 characteristics [11] producing IL-6, TGF-β and the chemokine CXCL8 (also known as IL-8) susceptible to promote liver fibrosis.
Dendritic cells (DCs) are professional antigen presenting cells. They have the unique capacity to catch, process and load all kinds of antigens and prime effectors immune cells namely CD4+ and CD8+ T cells. Basically, two types of DCs are described. The first DC type conventional DCs respond to lipopolysaccharide and lipoteichoic acid via TLR4 and TLR2, respectively, and produce TNF-α, IL-6, IL-12. The second DC type known as plasmacitoid DCs respond to TLR7 and TLR9 activation by producing IFN-α [102]. Impaired DC functions highly contribute to tumor escape from immune-surveillance in patients with cancer [44].
9.1.1.2 Impact of Adaptive Immune Response
Indeed, in addition to the crucial role of macrophages, increasing evidence also points out the crucial role of T lymphocytes in mediating hepatitis in ALD [8, 17, 77]. The decrease in peripheral lymphocyte number is associated with an increase in the ratio of T helper cells to suppressor cells in the liver. Today, four major distinct CD4+ T cell subtypes have been described: the Th1, Th2, T regulatory (Treg) and more recently, the Th17 phenotype.
-
Th1 cells that mainly produce IFN-γ, mediate immune response against intracellular pathogens, and are also involved in some autoimmune diseases [99, 107].
-
Th2 cells that produce principally IL-4, IL-10 and IL-13 are involved in host defence against extracellular parasites [99, 107] and suppress Th1 cell proliferation [38].
-
Th17 cells defined as producer of IL-17, IL-21 and IL-22, play a major role in immune response against bacteria and fungi and participate also to the induction of several autoimmune diseases [133], but could also protect hepatocytes in acute hepatitis through IL-22 production [142]. Recently, the deleterious role of T helper lymphocytes secreting IL-17 (Th17 cells) in recruiting neutrophils has been reported in ALD [88]. In addition, persistent IL-17 production has been identified in numerous other chronic liver diseases with deleterious functions leading to cirrhosis and HCC development [52, 84]. These findings strongly support the potent role of T cells in the progression of AH. Indeed, T helper differentiation into a specific phenotype is mainly controlled by innate immune cells that in turn could respond to the variety of produced cytokines by those differentiated T cells.
-
Treg cells regulate immune response by maintaining the self-tolerance and are beneficial for treating autoimmune diseases [118]. First described as suppressive T cells, [118] Treg lymphocytes are involved in immunosuppression and mainly contribute to tumor immune escape. CD4+ CD25high FoxP3+ Treg cells are induced by several microenvironmental factors including IL-10, TGF-β and VEGF which are overexpressed in HCC [10, 50, 140]. However, studies have reported a negative correlation between an increase in Treg infiltrating cells and clinical outcome in HCC patients [58]. In addition, it has been shown that in many types of liver disorders including chronic viral hepatitis, liver cirrhosis and HCC, Treg cell number increase favored HCC appearance and growth [74]. IL-10 mainly produced by Treg is the most studied anti-inflammatory cytokine regarding HCC. It has been shown that IL-10 is overexpressed in patients with HCC with less optimistic prognosis compared [10, 21]. Consistent with the previous studies, these reports strongly suggest that through IL-10 production, Treg lymphocytes promote HCC progression.
9.1.1.3 Inflammation-Associated Reactive Oxygen and Nitrogen Species (ROS and RNS)
Notable discoveries in mechanisms involved in ALD demonstrated the critical role of Kupffer cells in mediating AH through TNF-α overproduction, and led scientists to propose therapeutic strategies neutralizing TNF-α-mediated inflammation. In association with TNF-α, over-production of IL-6 was pointed out as driving factor of liver carcinogenesis [106]. The major mechanism that has been highlighted is the IL-6-induced reactive oxygen species (ROS) production and epigenetic changes triggering HCC development [123].
Due to ethanol oxidization by the cytochrome CYP2E1, acetaldehyde and reactive oxygen species (ROS) accumulate in the liver. This ROS accumulation promotes lipid peroxidation, DNA damage, chromosome instability and epigenetic disturbance. In an inflammatory context, epithelial and immune cell activation induce the production of reactive oxygen and nitrogen species (RONS) by NADPH oxidase, and nitric oxide synthase (NOS). Studies on chronic inflammatory diseases including inflammatory bowel diseases (IBD) and Helicobacter pylori-induced gastritis have reported increased level of RONS, suggesting a link between RONS production and cancer risk [111, 116]. RONS production induces cell damages including oxidative stress, lipids, proteins and DNA abnormalities through 8-oxodG and 8-nitrodeoxyguanosine accumulation [101] and therefore promote tumor initiation and malignancy. The discovery of RONS-induced DNA damages in chronic inflammatory responses including hepatitis is consistent with the involvement of RONS in diseases characterized by a higher cancer risk [101]. Furthermore, 8-oxodG and 8-nitrodeoxyguanine reactivity plays an important role in hepatitis C virus-induced chronic hepatitis [61]. All together these observations enhance the link between inflammation-induced RONS and carcinogenesis. Therapeutical use of antioxidants (e.g., S-adenosyl-l-methionine, polyunsaturated phosphatidylcholine) to restore alcohol-induced methionine and oxidative balance disruption, has shown promising effects but still needs to be further studied [115].
9.1.2 Cytokines in Fibrosis and Increased Risk of HCC Initiation
Tissue damage due to sustained inflammation leads in most cases to fibrosis development. Liver fibrosis is caused by a large variety of chronic liver diseases and represents an important cause of mortality in the world. The immune system protects the host from foreign pathogens without disrupting tolerance toward self-antigens but during fibrogenesis, inflammation contribute to the deposition and accumulation of collagen leading to an important modification of the physiological liver architecture.
Hepatic stellate cells represent the major cell population responsible for increased deposition of extracellular matrix proteins including collagen molecules. Collagens can also be produced to a lesser extent by other cell types including progenitor cells, portal fibroblasts, and cholangiocytes. Many immune cells including Kupffer cells, natural killer cells and dendritic cells have been shown to participate to liver fibrogenesis by releasing pro-inflammatory cytokines. These cytokines such as IFN-α and IFN-β, IL-6 and IL-22, activate the JAK-STAT signaling pathways by binding to their respective receptors.
9.1.2.1 Antifibrotic Cytokines
Interferon type 1, 2 and 3 were identified as cytokines that in general inhibit liver fibrosis development. For instance, IFN-α treatment significantly reduces the hepatic fibrosis in mice by blocking collagen gene transcription via the interaction of p300 transcription factor and phosphorylated STAT1 [66]. Similarly, IFN-γ deficient mice are more susceptible to develop liver fibrosis induced by CCl4 administration or under a 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) diet [67, 112]. The anti-fibrotic function of IFN-γ are more likely mediated through the induction of hepatic stellate cell-growth arrest and apoptosis [112].
It has been recently demonstrated that triple knockout mice for IL-10, IL-12/23 and IL-13Ra2 are more susceptible to many pathologies related to liver including S. mansoni-induced liver fibrosis model. Levels of liver enzymes, hepatosplenomegaly and ascites were increased, suggesting that IL-10, IL-12p40, and IL-13Rα2 contribute cooperatively to reduce liver fibrosis in this model of S. mansoni infected mice [97].
In the liver, IL-6 and IL-22 are mainly responsible for the activation of STAT3.
IL-6 knock-out mice seems more susceptible to liver injury and fibrosis after CCl4 treatment [79]. Furthermore, the lack of gp130/STAT3-mediated signaling in hepatocytes leads to worsened DDC diet feeding related chronic cholestatic liver injury and fibrosis progression [109]. Similarly, hepatocyte-specific STAT3 knockout mice displays a higher degree of liver fibrosis compared with wild-type mice in various models of liver fibrosis induced by CCl4 administration, feeding with a 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) diet [109], feeding with a choline-deficient, ethionine-supplemented (CDE) diet [80].
In hepatocytes, STAT5 activation is mainly induced by growth hormone (GH). Using the hepatocyte-specific STAT5 knockout mice developed in Dr. Lothar Hennighausen’s laboratory [26], STAT5 loss in hepatocytes has been shown to promote increased TGF-β levels and enhanced STAT3 activity induced by GH in the liver after CCl4 administration [6, 62]. Moreover, STAT5 deletion in the liver promoted hepatic tumorigenesis induced by CCl4 injection in wild-type mice [62] and was responsible for the development of spontaneous liver cancer in liver-specific glucocorticoid receptor knockout mice [100] or in GH transgenic mice [43]. Although the effects of STAT5 in hepatocytes have been widely investigated, little is known about the potential functions of STAT5 in the fibrogenic hepatic stellate cells (HSC).
9.1.2.2 Pro-fibrotic Cytokines
It is also known that IFN-α/β and IL-12 can activate STAT4 in immune cells and promote inflammation. In several animal models, treatment with IL-12 has been shown to induce liver inflammation and reduce liver tumor growth [18, 57]. This inflammatory response is characterized by the activation of NK and NKT cells which in turn produce IFN-γ [124]. Via the activation of STAT4 in immune cells, IL-12 seems to act as a pro-inflammatory cytokine promoting fibrosis and liver injury.
Th2 cytokines such as IL-4 and IL-13 are considered to be pro-inflammatory. Studies have shown that administration of IL-13 inhibitors or IL-13 gene blockage results in a significant reduction of liver fibrosis in the S. mansoni infection model. In humans, a correlation has been established between elevated levels of IL-13 and liver fibrosis in patients with chronic HBV or HCV infection, suggesting that IL-13 promotes liver fibrosis in an infection dependent or independent context [135]. These pro-fibrogenic effects can be explained by the fact that IL-13 induces HSC activation and promotes the production of fibrotic proteins by HSCs in a STAT6 dependent manner. Indeed, STAT6 blockage with siRNA inhibits HSC activation in vitro [3, 122]. Moreover, STAT6-deficient mice present smaller amounts of collagen deposition in the liver compared with wild- type mice after infection with S. mansoni [71]. Like TGF-β, IL-4 is known to have pro-fibrotic properties as it contributes to HSC activation and collagen production [3, 69]. Furthermore, IL-4 expression levels are higher in fibrotic liver from S. mansoni-infected baboons [37]. Although the role of IL-13 in liver fibrosis is reported in the S. mansoni infection model, studies to come need to clarify to what extent STAT6 in HSC is implicated in liver fibrogenesis in patients with chronic liver diseases.
IL-6 is a major pro-regenerative factor and induces the acute phase in the liver by stimulating hepatocytes to produce acute phase proteins such as C-reactive protein, serum amyloid A and complement C3 [114]. Clinical studies showed that IL-6 hepatic expression is increased and positively correlated with the degree of liver fibrosis [32, 136]. As IL-6 receptors are widely expressed on all types of cells in the liver, it can explain how IL-6 may have distinct roles in all these types of cells by regulating positively and negatively liver fibrosis. It has been shown that IL-6 can directly promote HSC survival and proliferation resulting in enhanced liver fibrosis. Collectively, the major effect of IL-6 on liver fibrosis is the result of the balance between on the one hand the inhibitory effect through STAT3 activation in hepatocytes and the stimulatory effects enhancing HSC survival, which depend on liver fibrosis stage and etiology.
9.1.3 Influence of Cytokines in Liver Progenitor/Stem Cell Activation: A Potent Mechanism of HCC Initiation
Liver progenitor cell (LPC) proliferation is reported in ductular reaction, often observed, in cirrhotic livers, hepatitis B and C viral infections, alcoholic or non-alcoholic steatohepatitis. Their appearance is associated with increased incidence of HCC. An interest in LPC biology emerged because of their stem-cell-like capacities to promote liver regeneration and to generate liver cancer. LPCs can differentiate into mature hepatocytes and biliary cells. Their capacity to restore injured hepatic tissue has been well documented [36]. However, LPCs were also defined as precursors for HCC and described as potent Cancer Stem Cells (CSCs) when they undergo “transformation” and generate heterogeneous lineage of cancer cells [29, 73, 94, 117]. Many transcription factors such as NANOG, cMYC, KLF-4, OCT4, SOX2 are stemness markers which have been reported to be increased in cancers [131].
The signaling pathways identified in HCC are also observed in isolated liver CSCs (eg, Wnt, Notch, TGF-β, Hedgehog, and PI3K/AKT/mTOR [86, 87]. Liver CSCs can be identified based on the expression of several cell markers such as CD90, CD44, CD24, CD13, epithelial cell adhesion molecule (EpCAM), CD133 (prominin-1), and oval cell marker OV6 as well as Hoechst dye efflux or aldehyde dehydrogenase expression and activities [86, 87]. Among those markers, double positive CD133+ and EpCAM+ cells display higher expression of stem-cell related genes and appearance of drug-resistance to chemotherapeutics. CSCs can initiate tumor in xenograft transplantation experiments. Moreover, the high capacity of resistance of CSC to sorafenib therapy suggests that CSCs could contribute to the poor prognosis [54] and participate to HCC recurrence. Several lines of evidence suggest a potential role of inflammatory microenvironment in CSC-initiation and progression towards HCC.
Recently, a correlation between IGF-1R and the expression of stemness markers in HBV-related HCC has been reported and suggests that inflammatory cytokines are involved in CSC development. The hepatic microenvironment is markedly disrupted in chronic liver diseases and characterized by infiltration of lymphocytes, activation of stellate cells and expansion of hepatic progenitor cells. One of the main axis involved in liver inflammation is IL-6/STAT3 signaling that in collaboration with TGF-β potentially promotes CSC survival and proliferation in the liver [98, 125].
9.2 Mechanisms of Cytokine Contribution to Tumor Growth
9.2.1 Role of Inflammation Related micro-RNA in HCC Development
In addition to its ability to modulate liver immune response, alcohol can also lead to epigenetic changes in inflammatory-associated genes via the multiple mechanisms [27]. These include (i) DNA-methyl transferases increasing methylation on gene promoters, (ii) an alteration of the physiological interaction between the transcriptional proteins to the DNA due to inappropriate methylation, acetylation phosphorylation and/or ubiquitination, and (iii) more recently, a transcriptional regulation mediated by micro-RNAs (miRNA) [102]. Epigenetic regulation of DNA methylation, phosphorylation, and ubiquitination by alcohol has been extensively reviewed in many previous articles. Here we mainly discuss the role of miRNAs in pathogenesis of HCC.
MiRNA are single-stranded non-coding RNAs composed of 20 nucleotides approximately. They are mostly responsible for the post-transcriptional epigenetic regulation of targeted gene expression. The interest for the miRNA raised when evidence of aberrant expression of several miRNAs was reported in many types of cancers [15]. Many pathways such as p53, RAS/MAPK, PI3K/ AKT/mTOR, WNT/β-catenin, and TGF-β are involved in HCC development. Abnormal expression of some miRNA has been observed in HCC compared to normal liver tissue [105]. It has been shown that miRNA-199a and miRNA-122 are highly expressed in healthy liver. Interestingly, the expression of these two miRNAs is markedly disrupted in HCC [63]. MiR-199 has been shown to stop cell cycle at G1 phase. A correlation has been reported between the downregulation of miR-199a (a member of miR-199 family) and increased recurrence rate with reduced laps before recurrence of HCC [68].
MiR-122 is only expressed in adult normal liver and seems to be a key factor in the regulation of hepatocyte differentiation by inhibiting genes not exclusive to the liver [139] which makes it a particular miRNA in liver physiopathology. Consistent with these findings, in up to 70% of HCC, miR-122 is downregulated indicating that this miRNA should have an antitumor activity. Furthermore, miR-122 is known to promote apoptosis, block the tumor cell cycle, reduce in vivo cancer cell malignancy and increase efficacy of drugs such as Sorafenib and also doxorubicin by inhibiting p53 activity [5, 40]. Interestingly, in liver cancer patients, miR-122 loss is correlated with the development of metastasis and a reduced period before recurrence [25, 127]. Thanks to a miR-122 KO mouse model, the role of this miR is now better defined [64, 128]. MiR-122KO developed chronic liver inflammation, fibrosis and HCC like spontaneous tumors comforting the antitumor potential of miR-122. Indeed, miR122 targets CULT-1 and reduces its activity which explain the undifferentiated phenotype of HCC cells. Similar to miR-199, miR-122 inhibits cyclin G1 leading to an upregulation of p53 which is increased in HCC [51]. The link between miRNA and the inflammatory response seems very strong suggesting that some cytokines could be involved in miRNA regulation. This mechanism by which cytokines could induce some miRNA targeting antitumor genes and thus promote tumor growth seems to be a very promising axis to explore in carcinogenesis.
In a study, up to 80% of HCC analyses showed a significant increase of miR-221 expression. This upregulation of miR-221 leads to increased tumor growth and cancer cell proliferation [47, 96]. Consistent with these observations, in transgenic mice overexpressing miR-221 in the liver, higher HCC tumorigenicity was reported and could be inhibited by administrating anti-miR-221 nucleotides, called antago-miR [16].
9.2.2 Cytokine-Induced Oncogenic Intracellular Pathways
Raf/MAPK/ERK
signaling pathway is involved in cell growth and differentiation. The extracellular signal is translated from tyrosine kinase receptors including VEGFR, IGFR, PDGFR, EGFR and MET, triggering a cascade of intracellular phosphorylations [4]. RAS, a GTPase protein, and Raf, a serine/threonine kinase regulate the signal transduction in this pathway [75]. A study has shown that in HCC Raf kinase inhibitor is down-regulated leading to an over-activity of the Raf/MAPK/ERK pathway [85]. New therapeutic approaches including Sorafenib aimed to target and inhibit Raf kinase, and consequently to limit tumor growth [137].
PI3K/Akt/mTOR
like the Raf/MAPK/ERK signaling pathway, controls proliferation, growth, motility and cell survival. HCC patients present an over-activation of this pathway. Indeed, it has been reported that in over 40% of HCC patients, Akt signaling and mTOR effector (p70s6k) were activated leading to increased cell survival and growth through an inhibition of TGF-β induced apoptosis [20]. These observations highlighted this signaling pathway as a potential target for therapeutic perspectives. Some strategies have been developed to block this pathway such as PI-103 inhibiting the phosphoinositide 3 Kinase (PI3K) and mTOR activation. These treatments showed efficacy in blocking Raf/MAP/ERK and PI3K/Akt/mTOR pathways leading to the reduction of EGF-induced proliferation of tumor cells [48].
Wnt/β-catenin
targets many processes including cell determination, stemness but also intercellular adhesion by interacting with E-cadherin and proliferative signal transmission through β-catenin activity [110]. Aberrant Beta-catenin activation is found in almost 40% of human HCC. B-catenin degradation is regulated by Adenomatous Polyposis Coli (APC) protein [22, 23]. Growth factors from the extracellular microenvironment bind to the Frizzled (Fzd) receptors expressed on the cell surface and activate this pathway. In murine and human HCC, an abnormal activity of this pathway has been reported [30, 56]. In absence of Wnt, the destruction complex formed by AXIN1, adenomatous polyposis coli (APC), glycogen synthase kinase-3β (GSK-3β), and casein kinase 1 (CK1) proteins drive the proteolysis of β-catenin through the ubiquitin/proteasome mechanism by phosphorylating the protein. Another protein has been reported to be increased in HCC; PRC1 controls cytoskeleton organization and increase Wnt signaling by contributing to the sequestration of the complex at the membrane, promoting tumorigenesis and metastasis development [19]. The role of Wnt/β-catenin in tumor growth is an attractive field to explore for paving the way to new therapeutic options. Indeed, antibody based therapies have already been developed. Blocking the β-catenin signaling reduced HCC tumor growth and increased apoptosis through the administration of anti-Wnt-1 antibodies [134].
NF-κB
plays a central role in liver injury, fibrosis and HCC development [91]. Its activation in macrophages lead to a large production of cytokines shaping an inflammatory tumor microenvironment of HCC [72]. In a DEN induced HCC murine model, Kupffer cell and hepatocyte specific blockade of IKK-β which is a major activator of NF-κB, leads to decrease tumor size and reduced the production of inflammatory cytokines including TNF-α and IL-6. However, an increase of hepato-tumorigenesis was reported with a deletion of IKK-β only in hepatocytes [92]. These paradoxical results highlight the double-edged sword role and complexity of NF-κB signaling but clearly show the key role played by immune cells in shaping a favorable pro-tumor microenvironment.
9.2.3 Cytokine Gene Polymorphisms Contribute to Altered Immune Response
In addition to the environmental factors responsible for cytokine release during chronic liver diseases, alteration in cytokine or cytokine receptor gene expressions can occur and chronically dysregulate the inflammatory response leading to cancer development. Such alteration results from single nucleotide polymorphisms (SNPs) in coding or non-coding regions of the genes. Several cytokine gene polymorphisms have been recently reported, including IL-1β [34], resulting in increased cytokine release. Furthermore, genetic polymorphisms of IL-6 are associated with fibrosis progression in chronic HCV infected patients [28]. Polymorphisms were identified in virtually all other cytokines such as TNF-α, IL-6, IL-10, TGF-β1 and IFN-γ [33].
9.3 Cytokines in HCC Progression with Metastasis
Tumor microenvironment is shaped by myeloid and lymphoid cells including tumor infiltrating lymphocytes (TILs) responsible for the control of tumor growth (Fig. 9.1). According to their phenotypes myeloid and lymphoid cells will inhibit or promote tumor growth. Myeloid cells basically play a major role in the immune response against tumor by recognizing these tumor antigens and generating humoral and cellular specific immune responses. However, their ability in tumor-associated antigen recognition is often altered and lead to pro-tumoral properties of those cells. Among lymphoid cells, two types of TILs can be distinguished: anti-tumoral effector cells such as CD8+ lymphocytes which are associated with a better prognosis when they are highly present in the tumor, that are opposed to pro-tumoral cells such as regulatory T cells (Treg) which are associated with a poor prognosis.

Immune cell microenvironment of HCC. The HCC microenvironment is composed of pro-tumoral immune cells including M2 macrophages, myeloid derived supressor cells, Treg and Th17 lymphocytes, and anti-tumoral cells gathering M1 macrophages, cytotoxic CD8 T cells, Th1 and NK cells
9.3.1 Myeloid Immune Response in HCC Progression
Tumor Associated Macrophages (TAMs)
also play a key regulatory role in tumor-related inflammation and angiogenesis. TAMs are also involved in tumor relapse by facilitating tumor regrowth, revascularization, and spread after anti-cancer therapies. TAMs are associated to tumor growth through the production of growth factors such as EGF, VEGF and bFGF. They contribute to the invasiveness of tumor cells and metastasis by favoring extracellular matrix remodeling via the release of metalloproteases 2 and 9 (MMP2 and MMP9). They also contribute to vascularize the tumor by increasing angiogenesis and lymphangiogenesis via MMP9, VEGF and PDGF synthesis [45].
Tumor-associated neutrophils (TAN)
similarly to TAMs, have been described [93] CXCL8 chemokine production by tumoral cells in HCC are responsible for the chemotaxis of neutrophils in the stroma surrounding the nodules [83]. More recently, an analysis of 919 HCC identified an overexpression of the CXCL5 chemokine that correlated with an increase in neutrophil-infiltrating cells in the liver and a poor prognosis of the diseases [144]. Those recruited neutrophils favors tumoral progression in part by increase in ROS production (as mentioned earlier in this chapter) [55]. Interestingly, neutrophil-derived ROS was associated with mutations and DNA damage [53] and activation of proteolytic enzymes including MMP-2, 7, 8, 9 and inactivation of the Tissue inhibitor metalloprotease-1 (TIMP-1) which consequently favour tumor invasiveness [31].
Myeloid Derived-Suppressor Cells (MDSCs)
represent a heterogeneous population of cells sharing similarities with TAMs and TANs. They are often observed in HCC and their presence is associated with a poor prognosis. This population of cells is basically composed of main sub-types: monocytic MDSCs and granulocytic MDSCs. Under certain conditions they can adopt several phenotypes that control their ability to promote or restrain tumor progression. For instance, in hypoxic conditions or in presence of tumor -derived factions, MDSC can differentiate into immunosuppressive TAMs [24]. Furthermore studies reported in vitro their capacity to differentiate in a macrophage, DC or granulocyte phenotype [103]. However, in general, MDSCs are described as suppressor of T cell activation and therefore can alter T-cell mediated anti-tumor function and favour tumor progression [130].
9.3.2 Lymphoid Immune Response in HCC Progression
9.3.2.1 Infiltrating CD8+ T Cells and Their Associated Cytokines
CD8+ T cells are major actors in antitumor immunity through their antigen specific cytotoxicity capacities targeting tumor antigens. These latter are ingested by host antigen presenting cell such as dendritic cells and processed into peptides which are presented via class I and class II MHC molecules respectively to CD8+ and CD4+T cells. In many cancer including colorectal and ovarian cancers, an increased number of tumor infiltrating CD8+ T lymphocytes (TIL) predicts a favorable prognosis. Regarding HCC, a correlation has also been reported between the presence of TIL and patient prognosis [46]. Indeed, the penetration of CD8+ T cells is correlated to an improved recurrence-free survival after liver resection [59]. These beneficial effects are explained by the inflammatory microenvironment generated by CD8+ effector T cells within the tumor leading to the establishment of an anti-tumor response. Studies have shown in murine models that through IL-12 stimulation, CD8+ T cells were activated which induce IFN-γ release leading to increased hepatoma cell apoptosis [76].
Recent findings [39] have highlighted CD8+ T specific responses targeting tumor-associated antigens (TAA) in HCC. It has been shown that TAA-specific CD8+ T cell immune response was visible in more than 1 out of 2 HCC patients and already detectable in early stages of the disease. Consistent with the correlation between improved progression-free survival TAA-specific CD8+ T responses these results comfort the major role played by these cells in anti-tumor immunity.
However, in some patients with HCC, impaired functions of CD8+ T cells have been reported [49]. Indeed, tumors develop mechanisms to escape to immune surveillance; one of them is the up-regulation of the ligand for PD-1 (PD-L1) responsible for addressing an inhibitory signal to PD-1 expressing cells namely CD8+ and CD4+ T cells [42]. This PD1/PD-L1 interaction leads to T cell inactivation and consequently, to the inhibition of their anti-tumor function and ultimately to the promotion of tumor aggressiveness. PD-L1 expression in HCC has recently been characterized and its crucial role in HCC progression has been strongly suggested [14].
Interestingly, it has been observed in HCC patient cohort that even with increased peripheral and intratumor PD-1 expression on CD8+ T cells, tumor cells were also rich in PD-L1 expression. These findings thus showed a correlation between a high PD-L1 expression within the tumor and a poorer outcome with early HCC recurrence after liver resection because of the induction of CD8+ T cell apoptosis [120]. The challenges of next studies will be to determine the mechanisms by which tumors promote PD-L1 expression on tumor cells and to find strategies to bypass the inhibitory signal delivered to PD-1 expressing cells including CD8+ T lymphocytes.
9.3.2.2 IL-17-producing Cells
IL-17 is in majority produced by Th17 lymphocytes and targets a large variety of cells through its ubiquitously expressed receptor IL-17RA. However, other IL-17-producing cell types have been identified including γδ T cells or neutrophils [113]. It has been reported that in HCC IL-17 levels were increased compared to non-tumor tissues [89]. Furthermore, a positive correlation has been established between high expression of IL-17 and microvessel density in tissues and poor survival in patients with HCC [143] suggesting that IL-17 may promote HCC growth by promoting angiogenesis.
In addition, neutrophils detected inside HCC tumors are associated with a poor recurrence-free survival for patients with HCC after liver resection. Peritumoral neutrophils promote angiogenesis leading therefore to stimulate tumor growth [81, 82]. More surprisingly, a study has shown that IL-17 can recruit neutrophils. Peritumoral tissue was also found enriched in Th17 lymphocytes which number is correlated with tumor activated monocytes that have been reported to induce IL-17 producing cells proliferation [81, 82]. Despite the positive correlation between IL-17 producing cells and poor survival in HCC patients, the underlying mechanisms by which these cells lead to HCC progression remain poorly defined.
9.3.2.3 Inflammation-Induced Epithelial Mesenchymal Transition (EMT)
Physiologically, EMT is a key step during embryogenesis, pathological events, inflammation but it can also trigger metastasis development in cancer context [9, 70]. During this process, morphological modifications occurs in epithelial cells which adopt a fibroblast-like phenotype. Through a complex cytoskeleton reorganization, many intercellular junctions characterizing epithelial cells are lost such as desmosomes, adherent junctions, tight junctions and gap junctions. EMT thus promotes epithelial markers loss including E-cadherin in favor of an induction of fibroblast markers such as fibronectin, matrix metalloproteinase. Such transformation allows those transformed cells to leave the original tissue and colonize other tissue through the blood circulation. TGF-β is one of the most relevant inflammatory mediator involved in EMT. It is considered as a key factor in embryogenesis but also in fibrosis and cancer development in many models [35, 138] through SMAD2, SMAD3 and SMAD4 [119, 129]. Studies have brought evidence that inflammation promotes EMT via the induction of pro-inflammatory cytokines. It has been shown that together TNF-α and IL-6 enhance TGF-β signaling pathways which stimulate EMT [7]. These two cytokines are known to trigger NF-κB which induces many factors implicated in EMT. Lastly, ROS synthesis has also been shown to induce EMT [131]. Exploring more deeply the involvement of cytokines in EMT in a context of CHC could open new therapeutic options.
9.4 HCC Therapeutic Failure and Cytokine-Based Therapy
At late stage, patients are not eligible for surgical resection of the tumor or for liver transplant, and the efficacy of classical radiotherapy and chimiotherapy is very poor. Since 2008, the SHARP (Sorafenib HCC Assessment Randomised Protocol) trials combining multikinase inhibitory and anti-angiogenic properties is considered as a standard for advanced HCC and showed an improved overall survival in Child–Pugh class A patients with advanced HCC upon treatment. HCC-patients given Sorafenib have a longer progression-free survival (PFS) with a median overall survival reaching 10.7 months in sorafenib treated patients vs 7.9 months in the placebo patients [90] Moreover, the high capacity of resistance of CSC to sorafenib therapy suggests that CSCs may contribute to the poor prognosis.
In HCC many cytokine levels are deregulated leading to promote or inhibit carcinogenesis. The development of combined therapy like IFN-α with ribavirin has markedly reduced HCC incidence. However antiviral therapies only help to delay the development of HCC. More and more anti-tumor therapeutic options use strategies to regulate cytokines levels or modulate immune cell activity. Loco-regional immune-chemotherapy based on lymphokine-activated killer cells (LAK) is a relevant approach in HCC treatment. LAKs release many cytokines including IFN-γ, IL-2 and IL-12 promoting cytolytic activity against tumor cells [78]. Many studies also proposed to enhance cytokine responses and more specifically to the liver. A murine HCC model was developed with adenoviral vector carrying IL-12 leading to reduce tumor growth and induce immune infiltration potentially responsible for the inhibition of angiogenesis [2]. Consistently, IL-12 intrahepatic administration in BALB/c has shown early infiltration of lymphocytes and macrophages resulting in a reduction of tumor progression [108]. Furthermore, combined immunotherapy in a murine model of HCC based on IL-12 and GM-CSF triggered a powerful antitumor response and avoid the side-effect of IL-12 treatment alone [132].
9.5 Conclusion
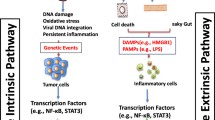
Although liver inflammation is critical for protection against infections and for triggering liver regeneration mechanisms, it must be finely tuned and “turned off” right after the clearance of the pathogens and the achievement of tissue repair. Indeed, excessive and recurrent liver inflammation is a common process observed in livers of alcoholic patients and in non-alcoholic steatohepatitis, in drug and chemical intoxication, during viral and bacterial infection, as well as in certain idiopathic liver pathologies such as autoimmune hepatitis. A high variety of immune cells can infiltrate the liver tissue (Fig. 9.2). Their quantity and their activity defined by their ability to produce a large spectrum of cytokines, depend on the underlying chronic liver disease. One of the major challenges in the liver field is to understand the cellular and molecular mechanisms underlying the chronic inflammatory processes associated with acute and chronic injury. The recent advances in immunology field sheds some light on the important role of cytokine milieu in which tumoral cells can emerge and proliferate. This also demonstrates how complex and heterogeneous is the liver inflammatory response according to the etiology leading to HCC. This strongly suggests that a better characterization of the inflammatory process would allow developing a personalized medicine for patients, and would constitute a promising strategy in HCC prevention and cure.

Influence of cytokines in HCC initiation and progression. Inflammatory cytokines are responsible for stellate cell activation and fibrosis development (IL-6, TGF-b, IL-1b), liver progenitor cell expansion (IL-6, TNF-a, IL-22) and DNA alteration via ROS production
References
Akriviadis E, Botla R, Briggs W, Han S, Reynolds T, Shakil O. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: a double-blind, placebo-controlled trial. Gastroenterology. 2000;119(6):1637–48.
Andrews KJ, Ribas A, Butterfield LH, Vollmer CM, Eilber FC, Dissette VB, Nelson SD, Shintaku P, Mekhoubad S, Nakayama T, Taniguchi M, Glaspy JA, McBride WH, Economou JS. Adenovirus-interleukin-12-mediated tumor regression in a murine hepatocellular carcinoma model is not dependent on CD1-restricted natural killer T cells. Cancer Res. 2000;60(22):6457–64.
Aoudjehane L, Pissaia A Jr, Scatton O, Podevin P, Massault PP, Chouzenoux S, Soubrane O, Calmus Y, Conti F. Interleukin-4 induces the activation and collagen production of cultured human intrahepatic fibroblasts via the STAT-6 pathway. Lab Investig. 2008;88(9):973–85.
Avila MA, Berasain C, Sangro B, Prieto J. New therapies for hepatocellular carcinoma. Oncogene. 2006;25(27):3866–84.
Bai S, Nasser MW, Wang B, Hsu SH, Datta J, Kutay H, Yadav A, Nuovo G, Kumar P, Ghoshal K. MicroRNA-122 inhibits tumorigenic properties of hepatocellular carcinoma cells and sensitizes these cells to sorafenib. J Biol Chem. 2009;284(46):32015–27.
Baik M, Yu JH, Hennighausen L. Growth hormone-STAT5 regulation of growth, hepatocellular carcinoma, and liver metabolism. Ann N Y Acad Sci. 2011;1229:29–37.
Bates RC, Mercurio AM. Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol Biol Cell. 2003;14(5):1790–800.
Batey RG, Wang J. Molecular pathogenesis of T lymphocyte-induced liver injury in alcoholic hepatitis. Front Biosci. 2002;7:d1662–75.
Baum B, Settleman J, Quinlan MP. Transitions between epithelial and mesenchymal states in development and disease. Semin Cell Dev Biol. 2008;19(3):294–308.
Beckebaum S, Zhang X, Chen X, Yu Z, Frilling A, Dworacki G, Grosse-Wilde H, Broelsch CE, Gerken G, Cicinnati VR. Increased levels of interleukin-10 in serum from patients with hepatocellular carcinoma correlate with profound numerical deficiencies and immature phenotype of circulating dendritic cell subsets. Clin Cancer Res. 2004;10(21):7260–9.
Benoit M, Barbarat B, Bernard A, Olive D, Mege JL. Coxiella burnetii, the agent of Q fever, stimulates an atypical M2 activation program in human macrophages. Eur J Immunol. 2008;38(4):1065–70.
Bode C, Bode JC. Activation of the innate immune system and alcoholic liver disease: effects of ethanol per se or enhanced intestinal translocation of bacterial toxins induced by ethanol? Alcohol Clin Exp Res. 2005;29(11 Suppl):166S–71S.
Boetticher NC, Peine CJ, Kwo P, Abrams GA, Patel T, Aqel B, Boardman L, Gores GJ, Harmsen WS, McClain CJ, Kamath PS, Shah VH. A randomized, double-blinded, placebo-controlled multicenter trial of etanercept in the treatment of alcoholic hepatitis. Gastroenterology. 2008;135(6):1953–60.
Calderaro J, Rousseau B, Amaddeo G, Mercey M, Charpy C, Costentin C, Luciani A, Zafrani ES, Laurent A, Azoulay D, Lafdil F, Pawlotsky JM. Programmed death ligand 1 expression in hepatocellular carcinoma: relationship with clinical and pathological features. Hepatology. 2016;64(6):2038–46.
Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6(11):857–66.
Callegari E, Elamin BK, Giannone F, Milazzo M, Altavilla G, Fornari F, Giacomelli L, D'Abundo L, Ferracin M, Bassi C, Zagatti B, Corra F, Miotto E, Lupini L, Bolondi L, Gramantieri L, Croce CM, Sabbioni S, Negrini M. Liver tumorigenicity promoted by microRNA-221 in a mouse transgenic model. Hepatology. 2012;56(3):1025–33.
Cao Q, Batey R, Pang G, Clancy R. Altered T-lymphocyte responsiveness to polyclonal cell activators is responsible for liver cell necrosis in alcohol-fed rats. Alcohol Clin Exp Res. 1998;22(3):723–9.
Chang HD, Radbruch A. The pro- and anti-inflammatory potential of interleukin-12. Ann N Y Acad Sci. 2007;1109:40–6.
Chen J, Katz LH, Munoz NM, Gu S, Shin JH, Jogunoori WS, Lee MH, Belkin MD, Kim SB, White JC, Andricovich J, Tzatsos A, Li S, Kim SS, Shetty K, Mishra B, Rashid A, Lee JS, Mishra L. Vitamin D deficiency promotes liver tumor growth in transforming growth factor-beta/Smad3-deficient mice through Wnt and toll-like receptor 7 pathway modulation. Sci Rep. 2016;6:30217.
Chen RH, Su YH, Chuang RL, Chang TY. Suppression of transforming growth factor-beta-induced apoptosis through a phosphatidylinositol 3-kinase/Akt-dependent pathway. Oncogene. 1998;17(15):1959–68.
Chia CS, Ban K, Ithnin H, Singh H, Krishnan R, Mokhtar S, Malihan N, Seow HF. Expression of interleukin-18, interferon-gamma and interleukin-10 in hepatocellular carcinoma. Immunol Lett. 2002;84(3):163–72.
Colnot S, Decaens T, Niwa-Kawakita M, Godard C, Hamard G, Kahn A, Giovannini M, Perret C. Liver-targeted disruption of Apc in mice activates beta-catenin signaling and leads to hepatocellular carcinomas. Proc Natl Acad Sci U S A. 2004a;101(49):17216–21.
Colnot S, Niwa-Kawakita M, Hamard G, Godard C, Le Plenier S, Houbron C, Romagnolo B, Berrebi D, Giovannini M, Perret C. Colorectal cancers in a new mouse model of familial adenomatous polyposis: influence of genetic and environmental modifiers. Lab Investig. 2004b;84(12):1619–30.
Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, Cho HI, Celis E, Quiceno DG, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207(11):2439–53.
Coulouarn C, Factor VM, Andersen JB, Durkin ME, Thorgeirsson SS. Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene. 2009;28(40):3526–36.
Cui Y, Hosui A, Sun R, Shen K, Gavrilova O, Chen W, Cam MC, Gao B, Robinson GW, Hennighausen L. Loss of signal transducer and activator of transcription 5 leads to hepatosteatosis and impaired liver regeneration. Hepatology. 2007;46(2):504–13.
Curtis BJ, Zahs A, Kovacs EJ. Epigenetic targets for reversing immune defects caused by alcohol exposure. Alcohol Res. 2013;35(1):97–113.
Cussigh A, Falleti E, Fabris C, Bitetto D, Cmet S, Fontanini E, Bignulin S, Fornasiere E, Fumolo E, Minisini R, Pirisi M, Toniutto P. Interleukin 6 promoter polymorphisms influence the outcome of chronic hepatitis C. Immunogenetics. 2011;63(1):33–41.
Davies RA, Knight B, Tian YW, Yeoh GC, Olynyk JK. Hepatic oval cell response to the choline-deficient, ethionine supplemented model of murine liver injury is attenuated by the administration of a cyclo-oxygenase 2 inhibitor. Carcinogenesis. 2006;27(8):1607–16.
de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C, Kahn A, Perret C. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95(15):8847–51.
De Larco JE, Wuertz BR, Furcht LT. The potential role of neutrophils in promoting the metastatic phenotype of tumors releasing interleukin-8. Clin Cancer Res. 2004;10(15):4895–900.
Dogru T, Ercin CN, Erdem G, Sonmez A, Tapan S, Tasci I. Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am J Gastroenterol. 2008;103(12):3217–8.
Dondeti MF, El-Maadawy EA, Talaat RM. Hepatitis-related hepatocellular carcinoma: insights into cytokine gene polymorphisms. World J Gastroenterol. 2016;22(30):6800–16.
El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, Lanyon G, Martin M, Fraumeni JF Jr, Rabkin CS. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404(6776):398–402.
Ellenrieder V, Fernandez Zapico ME, Urrutia R. TGFbeta-mediated signaling and transcriptional regulation in pancreatic development and cancer. Curr Opin Gastroenterol. 2001;17(5):434–40.
Espanol-Suner R, Carpentier R, Van Hul N, Legry V, Achouri Y, Cordi S, Jacquemin P, Lemaigre F, Leclercq IA. Liver progenitor cells yield functional hepatocytes in response to chronic liver injury in mice. Gastroenterology. 2012;143(6):1564–75. e1567
Farah IO, Mola PW, Kariuki TM, Nyindo M, Blanton RE, King CL. Repeated exposure induces periportal fibrosis in Schistosoma Mansoni-infected baboons: role of TGF-beta and IL-4. J Immunol. 2000;164(10):5337–43.
Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med. 1989;170(6):2081–95.
Flecken T, Schmidt N, Hild S, Gostick E, Drognitz O, Zeiser R, Schemmer P, Bruns H, Eiermann T, Price DA, Blum HE, Neumann-Haefelin C, Thimme R. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology. 2014;59(4):1415–26.
Fornari F, Gramantieri L, Giovannini C, Veronese A, Ferracin M, Sabbioni S, Calin GA, Grazi GL, Croce CM, Tavolari S, Chieco P, Negrini M, Bolondi L. MiR-122/cyclin G1 interaction modulates p53 activity and affects doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res. 2009;69(14):5761–7.
Forner A, Bruix J. Biomarkers for early diagnosis of hepatocellular carcinoma. Lancet Oncol. 2012;13(8):750–1.
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, Honjo T. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–34.
Friedbichler K, Themanns M, Mueller KM, Schlederer M, Kornfeld JW, Terracciano LM, Kozlov AV, Haindl S, Kenner L, Kolbe T, Mueller M, Snibson KJ, Heim MH, Moriggl R. Growth-hormone-induced signal transducer and activator of transcription 5 signaling causes gigantism, inflammation, and premature death but protects mice from aggressive liver cancer. Hepatology. 2012;55(3):941–52.
Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. 2004;4(12):941–52.
Galdiero MR, Bonavita E, Barajon I, Garlanda C, Mantovani A, Jaillon S. Tumor associated macrophages and neutrophils in cancer. Immunobiology. 2013;218(11):1402–10.
Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, Xu Y, Li YW, Tang ZY. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol. 2007;25(18):2586–93.
Garofalo M, Di Leva G, Romano G, Nuovo G, Suh SS, Ngankeu A, Taccioli C, Pichiorri F, Alder H, Secchiero P, Gasparini P, Gonelli A, Costinean S, Acunzo M, Condorelli G, Croce CM. miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell. 2009;16(6):498–509.
Gedaly R, Angulo P, Hundley J, Daily MF, Chen C, Koch A, Evers BM. PI-103 and sorafenib inhibit hepatocellular carcinoma cell proliferation by blocking Ras/Raf/MAPK and PI3K/AKT/mTOR pathways. Anticancer Res. 2010;30(12):4951–8.
Gehring AJ, Ho ZZ, Tan AT, Aung MO, Lee KH, Tan KC, Lim SG, Bertoletti A. Profile of tumor antigen-specific CD8 T cells in patients with hepatitis B virus-related hepatocellular carcinoma. Gastroenterology. 2009;137(2):682–90.
Giannelli G, Fransvea E, Marinosci F, Bergamini C, Colucci S, Schiraldi O, Antonaci S. Transforming growth factor-beta1 triggers hepatocellular carcinoma invasiveness via alpha3beta1 integrin. Am J Pathol. 2002;161(1):183–93.
Gramantieri L, Ferracin M, Fornari F, Veronese A, Sabbioni S, Liu CG, Calin GA, Giovannini C, Ferrazzi E, Grazi GL, Croce CM, Bolondi L, Negrini M. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res. 2007;67(13):6092–9.
Guillot A, Hamdaoui N, Bizy A, Zoltani K, Souktani R, Zafrani ES, Mallat A, Lotersztajn S, Lafdil F. Cannabinoid receptor 2 counteracts interleukin-17-induced immune and fibrogenic responses in mouse liver. Hepatology. 2014;59(1):296–306.
Gungor N, Knaapen AM, Munnia A, Peluso M, Haenen GR, Chiu RK, Godschalk RW, van Schooten FJ. Genotoxic effects of neutrophils and hypochlorous acid. Mutagenesis. 2010;25(2):149–54.
Hagiwara S, Kudo M, Nagai T, Inoue T, Ueshima K, Nishida N, Watanabe T, Sakurai T. Activation of JNK and high expression level of CD133 predict a poor response to sorafenib in hepatocellular carcinoma. Br J Cancer. 2012;106(12):1997–2003.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Harada N, Oshima H, Katoh M, Tamai Y, Oshima M, Taketo MM. Hepatocarcinogenesis in mice with beta-catenin and Ha-ras gene mutations. Cancer Res. 2004a;64(1):48–54.
Harada N, Shimada M, Okano S, Suehiro T, Soejima Y, Tomita Y, Maehara Y. IL-12 gene therapy is an effective therapeutic strategy for hepatocellular carcinoma in immunosuppressed mice. J Immunol. 2004b;173(11):6635–44.
Hirano S, Iwashita Y, Sasaki A, Kai S, Ohta M, Kitano S. Increased mRNA expression of chemokines in hepatocellular carcinoma with tumor-infiltrating lymphocytes. J Gastroenterol Hepatol. 2007;22(5):690–6.
Hiroishi K, Eguchi J, Baba T, Shimazaki T, Ishii S, Hiraide A, Sakaki M, Doi H, Uozumi S, Omori R, Matsumura T, Yanagawa T, Ito T, Imawari M. Strong CD8(+) T-cell responses against tumor-associated antigens prolong the recurrence-free interval after tumor treatment in patients with hepatocellular carcinoma. J Gastroenterol. 2010;45(4):451–8.
Horiguchi N, Ishac EJ, Gao B. Liver regeneration is suppressed in alcoholic cirrhosis: correlation with decreased STAT3 activation. Alcohol. 2007;41(4):271–80.
Horiike S, Kawanishi S, Kaito M, Ma N, Tanaka H, Fujita N, Iwasa M, Kobayashi Y, Hiraku Y, Oikawa S, Murata M, Wang J, Semba R, Watanabe S, Adachi Y. Accumulation of 8-nitroguanine in the liver of patients with chronic hepatitis C. J Hepatol. 2005;43(3):403–10.
Hosui A, Kimura A, Yamaji D, Zhu BM, Na R, Hennighausen L. Loss of STAT5 causes liver fibrosis and cancer development through increased TGF-{beta} and STAT3 activation. J Exp Med. 2009;206(4):819–31.
Hou J, Lin L, Zhou W, Wang Z, Ding G, Dong Q, Qin L, Wu X, Zheng Y, Yang Y, Tian W, Zhang Q, Wang C, Zhang Q, Zhuang SM, Zheng L, Liang A, Tao W, Cao X. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer Cell. 2011;19(2):232–43.
Hsu SH, Wang B, Kota J, Yu J, Costinean S, Kutay H, Yu L, Bai S, La Perle K, Chivukula RR, Mao H, Wei M, Clark KR, Mendell JR, Caligiuri MA, Jacob ST, Mendell JT, Ghoshal K. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J Clin Invest. 2012;122(8):2871–83.
Hussain SP, Schwank J, Staib F, Wang XW, Harris CC. TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene. 2007;26(15):2166–76.
Inagaki Y, Nemoto T, Kushida M, Sheng Y, Higashi K, Ikeda K, Kawada N, Shirasaki F, Takehara K, Sugiyama K, Fujii M, Yamauchi H, Nakao A, de Crombrugghe B, Watanabe T, Okazaki I. Interferon alfa down-regulates collagen gene transcription and suppresses experimental hepatic fibrosis in mice. Hepatology. 2003;38(4):890–9.
Jeong WI, Park O, Radaeva S, Gao B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology. 2006;44(6):1441–51.
Jia XQ, Cheng HQ, Qian X, Bian CX, Shi ZM, Zhang JP, Jiang BH, Feng ZQ. Lentivirus-mediated overexpression of microRNA-199a inhibits cell proliferation of human hepatocellular carcinoma. Cell Biochem Biophys. 2012;62(1):237–44.
Jin Z, Sun R, Wei H, Gao X, Chen Y, Tian Z. Accelerated liver fibrosis in hepatitis B virus transgenic mice: involvement of natural killer T cells. Hepatology. 2011;53(1):219–29.
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–8.
Kaplan MH, Whitfield JR, Boros DL, Grusby MJ. Th2 cells are required for the Schistosoma Mansoni egg-induced granulomatous response. J Immunol. 1998;160(4):1850–6.
Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124(4):823–35.
Knight B, Tirnitz-Parker JE, Olynyk JK. C-kit inhibition by imatinib mesylate attenuates progenitor cell expansion and inhibits liver tumor formation in mice. Gastroenterology. 2008;135(3):969–79. 979 e961
Kobayashi N, Hiraoka N, Yamagami W, Ojima H, Kanai Y, Kosuge T, Nakajima A, Hirohashi S. FOXP3+ regulatory T cells affect the development and progression of hepatocarcinogenesis. Clin Cancer Res. 2007;13(3):902–11.
Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J. 2000;351(Pt 2):289–305.
Komita H, Homma S, Saotome H, Zeniya M, Ohno T, Toda G. Interferon-gamma produced by interleukin-12-activated tumor infiltrating CD8+T cells directly induces apoptosis of mouse hepatocellular carcinoma. J Hepatol. 2006;45(5):662–72.
Kono H, Uesugi T, Froh M, Rusyn I, Bradford BU, Thurman RG. ICAM-1 is involved in the mechanism of alcohol-induced liver injury: studies with knockout mice. Am J Physiol Gastrointest Liver Physiol. 2001;280(6):G1289–95.
Kountouras J, Boura P, Kouklakis G. Locoregional immunochemotherapy in hepatocellular carcinoma. Hepato-Gastroenterology. 2002;49(46):1109–12.
Kovalovich K, DeAngelis RA, Li W, Furth EE, Ciliberto G, Taub R. Increased toxin-induced liver injury and fibrosis in interleukin-6-deficient mice. Hepatology. 2000;31(1):149–59.
Kroy DC, Beraza N, Tschaharganeh DF, Sander LE, Erschfeld S, Giebeler A, Liedtke C, Wasmuth HE, Trautwein C, Streetz KL. Lack of interleukin-6/glycoprotein 130/signal transducers and activators of transcription-3 signaling in hepatocytes predisposes to liver steatosis and injury in mice. Hepatology. 2010;51(2):463–73.
Kuang DM, Peng C, Zhao Q, Wu Y, Chen MS, Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma promote expansion of memory T helper 17 cells. Hepatology. 2010a;51(1):154–64.
Kuang DM, Peng C, Zhao Q, Wu Y, Zhu LY, Wang J, Yin XY, Li L, Zheng L. Tumor-activated monocytes promote expansion of IL-17-producing CD8+ T cells in hepatocellular carcinoma patients. J Immunol. 2010b;185(3):1544–9.
Kuang DM, Zhao Q, Wu Y, Peng C, Wang J, Xu Z, Yin XY, Zheng L. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J Hepatol. 2011;54(5):948–55.
Lafdil F, Miller AM, Ki SH, Gao B. Th17 cells and their associated cytokines in liver diseases. Cell Mol Immunol. 2010;7(4):250–4.
Lee HC, Tian B, Sedivy JM, Wands JR, Kim M. Loss of Raf kinase inhibitor protein promotes cell proliferation and migration of human hepatoma cells. Gastroenterology. 2006;131(4):1208–17.
Lee TK, Cheung VC, Ng IO. Liver tumor-initiating cells as a therapeutic target for hepatocellular carcinoma. Cancer Lett. 2012;338(1):101–9.
Lee TK, Cheung VC, Ng IO. Liver tumor-initiating cells as a therapeutic target for hepatocellular carcinoma. Cancer Lett. 2013;338(1):101–9.
Lemmers A, Moreno C, Gustot T, Marechal R, Degre D, Demetter P, de Nadai P, Geerts A, Quertinmont E, Vercruysse V, Le Moine O, Deviere J. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology. 2009;49(2):646–57.
Liao R, Sun J, Wu H, Yi Y, Wang JX, He HW, Cai XY, Zhou J, Cheng YF, Fan J, Qiu SJ. High expression of IL-17 and IL-17RE associate with poor prognosis of hepatocellular carcinoma. J Exp Clin Cancer Res. 2013;32:3.
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz JF, Borbath I, Haussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, Bruix J, S. I. S. Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–90.
Luedde T, Schwabe RF. NF-kappaB in the liver–linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8(2):108–18.
Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121(7):977–90.
Mantovani A. The yin-yang of tumor-associated neutrophils. Cancer Cell. 2009;16(3):173–4.
Marquardt JU, Raggi C, Andersen JB, Seo D, Avital I, Geller D, Lee YH, Kitade M, Holczbauer A, Gillen MC, Conner EA, Factor VM, Thorgeirsson SS. Human hepatic cancer stem cells are characterized by common stemness traits and diverse oncogenic pathways. Hepatology. 2011;54(3):1031–42.
McClain CJ, Song Z, Barve SS, Hill DB, Deaciuc I. Recent advances in alcoholic liver disease. IV. Dysregulated cytokine metabolism in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2004;287(3):G497–502.
Medina R, Zaidi SK, Liu CG, Stein JL, van Wijnen AJ, Croce CM, Stein GS. MicroRNAs 221 and 222 bypass quiescence and compromise cell survival. Cancer Res. 2008;68(8):2773–80.
Mentink-Kane MM, Cheever AW, Wilson MS, Madala SK, Beers LM, Ramalingam TR, Wynn TA. Accelerated and progressive and lethal liver fibrosis in mice that lack interleukin (IL)-10, IL-12p40, and IL-13Ralpha2. Gastroenterology. 2011;141(6):2200–9.
Mishra L, Banker T, Murray J, Byers S, Thenappan A, He AR, Shetty K, Johnson L, Reddy EP. Liver stem cells and hepatocellular carcinoma. Hepatology. 2009;49(1):318–29.
Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–73.
Mueller KM, Kornfeld JW, Friedbichler K, Blaas L, Egger G, Esterbauer H, Hasselblatt P, Schlederer M, Haindl S, Wagner KU, Engblom D, Haemmerle G, Kratky D, Sexl V, Kenner L, Kozlov AV, Terracciano L, Zechner R, Schuetz G, Casanova E, Pospisilik JA, Heim MH, Moriggl R. Impairment of hepatic growth hormone and glucocorticoid receptor signaling causes steatosis and hepatocellular carcinoma in mice. Hepatology. 2011;54(4):1398–409.
Murata M, Thanan R, Ma N, Kawanishi S. Role of nitrative and oxidative DNA damage in inflammation-related carcinogenesis. J Biomed Biotechnol. 2012;2012:623019.
Nagy LE. The role of innate immunity in alcoholic liver disease. Alcohol Res. 2015;37(2):237–50.
Narita Y, Wakita D, Ohkur T, Chamoto K, Nishimura T. Potential differentiation of tumor bearing mouse CD11b+gr-1+ immature myeloid cells into both suppressor macrophages and immunostimulatory dendritic cells. Biomed Res. 2009;30(1):7–15.
Naveau S, Chollet-Martin S, Dharancy S, Mathurin P, Jouet P, Piquet MA, Davion T, Oberti F, Broet P, Emilie D, F. Foie-Alcool group of the Association Francaise pour l'Etude du. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology. 2004;39(5):1390–7.
Negrini M, Gramantieri L, Sabbioni S, Croce CM. microRNA involvement in hepatocellular carcinoma. Anti Cancer Agents Med Chem. 2011;11(6):500–21.
Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140(2):197–208.
Paul WE, Seder RA. Lymphocyte responses and cytokines. Cell. 1994;76(2):241–51.
Peron JM, Couderc B, Rochaix P, Douin-Echinard V, Asnacios A, Souque A, Voigt JJ, Buscail L, Vinel JP, Favre G. Treatment of murine hepatocellular carcinoma using genetically modified cells to express interleukin-12. J Gastroenterol Hepatol. 2004;19(4):388–96.
Plum W, Tschaharganeh DF, Kroy DC, Corsten E, Erschfeld S, Dierssen U, Wasmuth H, Trautwein C, Streetz KL. Lack of glycoprotein 130/signal transducer and activator of transcription 3-mediated signaling in hepatocytes enhances chronic liver injury and fibrosis progression in a model of sclerosing cholangitis. Am J Pathol. 2010;176(5):2236–46.
Polakis P. The oncogenic activation of beta-catenin. Curr Opin Genet Dev. 1999;9(1):15–21.
Rachmilewitz D, Stamler JS, Bachwich D, Karmeli F, Ackerman Z, Podolsky DK. Enhanced colonic nitric oxide generation and nitric oxide synthase activity in ulcerative colitis and Crohn's disease. Gut. 1995;36(5):718–23.
Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130(2):435–52.
Rajoriya N, Fergusson JR, Leithead JA, Klenerman P. Gamma Delta T-lymphocytes in hepatitis C and chronic liver disease. Front Immunol. 2014;5:400.
Ramadori G, Christ B. Cytokines and the hepatic acute-phase response. Semin Liver Dis. 1999;19(2):141–55.
Rambaldi A, Gluud C. S-adenosyl-L-methionine for alcoholic liver diseases. Cochrane Database Syst Rev. 2006;2:CD002235.
Rieder G, Hofmann JA, Hatz RA, Stolte M, Enders GA. Up-regulation of inducible nitric oxide synthase in helicobacter pylori-associated gastritis may represent an increased risk factor to develop gastric carcinoma of the intestinal type. Int J Med Microbiol. 2003;293(6):403–12.
Rountree CB, Mishra L, Willenbring H. Stem cells in liver diseases and cancer: recent advances on the path to new therapies. Hepatology. 2012;55(1):298–306.
Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–62.
Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest. 2003;112(10):1486–94.
Shi F, Shi M, Zeng Z, Qi RZ, Liu ZW, Zhang JY, Yang YP, Tien P, Wang FS. PD-1 and PD-L1 upregulation promotes CD8(+) T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int J Cancer. 2011;128(4):887–96.
Sozio M, Crabb DW. Alcohol and lipid metabolism. Am J Physiol Endocrinol Metab. 2008;295(1):E10–6.
Sripa B, Mairiang E, Thinkhamrop B, Laha T, Kaewkes S, Sithithaworn P, Tessana S, Loukas A, Brindley PJ, Bethony JM. Advanced periductal fibrosis from infection with the carcinogenic human liver fluke Opisthorchis Viverrini correlates with elevated levels of interleukin-6. Hepatology. 2009;50(4):1273–81.
Stauffer JK, Scarzello AJ, Jiang Q, Wiltrout RH. Chronic inflammation, immune escape, and oncogenesis in the liver: a unique neighborhood for novel intersections. Hepatology. 2012;56(4):1567–74.
Subleski JJ, Hall VL, Back TC, Ortaldo JR, Wiltrout RH. Enhanced antitumor response by divergent modulation of natural killer and natural killer T cells in the liver. Cancer Res. 2006;66(22):11005–12.
Tang Y, Kitisin K, Jogunoori W, Li C, Deng CX, Mueller SC, Ressom HW, Rashid A, He AR, Mendelson JS, Jessup JM, Shetty K, Zasloff M, Mishra B, Reddy EP, Johnson L, Mishra L. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc Natl Acad Sci U S A. 2008;105(7):2445–50.
Tilg H, Diehl AM. Cytokines in alcoholic and nonalcoholic steatohepatitis. N Engl J Med. 2000;343(20):1467–76.
Tsai WC, Hsu PW, Lai TC, Chau GY, Lin CW, Chen CM, Lin CD, Liao YL, Wang JL, Chau YP, Hsu MT, Hsiao M, Huang HD, Tsou AP. MicroRNA-122, a tumor suppressor microRNA that regulates intrahepatic metastasis of hepatocellular carcinoma. Hepatology. 2009;49(5):1571–82.
Tsai WC, Hsu SD, Hsu CS, Lai TC, Chen SJ, Shen R, Huang Y, Chen HC, Lee CH, Tsai TF, Hsu MT, Wu JC, Huang HD, Shiao MS, Hsiao M, Tsou AP. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest. 2012;122(8):2884–97.
Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A. TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell. 2005;16(4):1987–2002.
Wan S, Kuo N, Kryczek I, Zou W, Welling TH. Myeloid cells in hepatocellular carcinoma. Hepatology. 2015;62(4):1304–12.
Wang Z, Li Y, Sarkar FH. Signaling mechanism(s) of reactive oxygen species in epithelial-mesenchymal transition reminiscent of cancer stem cells in tumor progression. Curr Stem Cell Res Ther. 2010;5(1):74–80.
Wang Z, Qiu SJ, Ye SL, Tang ZY, Xiao X. Combined IL-12 and GM-CSF gene therapy for murine hepatocellular carcinoma. Cancer Gene Ther. 2001;8(10):751–8.
Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24(6):677–88.
Wei W, Chua MS, Grepper S, So SK. Blockade of Wnt-1 signaling leads to anti-tumor effects in hepatocellular carcinoma cells. Mol Cancer. 2009;8:76.
Weng HL, Liu Y, Chen JL, Huang T, Xu LJ, Godoy P, Hu JH, Zhou C, Stickel F, Marx A, Bohle RM, Zimmer V, Lammert F, Mueller S, Gigou M, Samuel D, Mertens PR, Singer MV, Seitz HK, Dooley S. The etiology of liver damage imparts cytokines transforming growth factor beta1 or interleukin-13 as driving forces in fibrogenesis. Hepatology. 2009;50(1):230–43.
Wieckowska A, Papouchado BG, Li Z, Lopez R, Zein NN, Feldstein AE. Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am J Gastroenterol. 2008;103(6):1372–9.
Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7(10):3129–40.
Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166(5):1321–32.
Xu H, He JH, Xiao ZD, Zhang QQ, Chen YQ, Zhou H, Qu LH. Liver-enriched transcription factors regulate microRNA-122 that targets CUTL1 during liver development. Hepatology. 2010;52(4):1431–42.
Yamaguchi R, Yano H, Iemura A, Ogasawara S, Haramaki M, Kojiro M. Expression of vascular endothelial growth factor in human hepatocellular carcinoma. Hepatology. 1998;28(1):68–77.
Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, Thurman RG. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117(4):942–52.
Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity. 2007;27(4):647–59.
Zhang JP, Yan J, Xu J, Pang XH, Chen MS, Li L, Wu C, Li SP, Zheng L. Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. J Hepatol. 2009;50(5):980–9.
Zhou SL, Dai Z, Zhou ZJ, Wang XY, Yang GH, Wang Z, Huang XW, Fan J, Zhou J. Overexpression of CXCL5 mediates neutrophil infiltration and indicates poor prognosis for hepatocellular carcinoma. Hepatology. 2012;56(6):2242–54.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Ait-Ahmed, Y., Lafdil, F. (2017). Impact of Cytokines in Hepatocellular Carcinoma Initiation and Progression. In: F. Greten, T. (eds) Immunotherapy of Hepatocellular Carcinoma. Springer, Cham. https://doi.org/10.1007/978-3-319-64958-0_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-64958-0_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-64957-3
Online ISBN: 978-3-319-64958-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)