
Abstract
Fifty years ago, little was known of the role of the prevailing light:dark environment in terms of its impact on the circadian pathophysiology of organisms. In the intervening years the field of photoperiodic regulation of the master circadian oscillator, i.e., the suprachiasmatic nucleus (SCN), has advanced at a rapid pace. The importance of the regulatory actions of the light:dark cycle, and particularly of perturbed light:dark cycles, not only on the SCN but also on the circadian production of pineal melatonin as well as the cyclic metabolism of cells throughout the body are by no means trivial. When the regular cyclic information generated and dispensed by the SCN is dysregulated, the negative consequences in terms of cellular and organismal physiology can be dire to the extent that the rate of aging and the onset and progression of a variety of age-related diseases have now been at least provisionally linked to circadian disruption and/or melatonin suppression. While the findings are not definitive, there is certainly credible data to warrant the conclusion that regular circadian rhythms at multiple levels, including a stable day:night melatonin cycle, enhance life quality and potentially delay senescence and forestall diseases normally associated with advanced age. As a result, the prolonged health span may also predispose to a longer life span . In view of the critical role of an abnormal or unusual light environment in terms of perturbing essential circadian physiological events, serious consideration should be given to rational thought about the misuse of artificial light and the consequences thereof.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1.1 Introduction
Properly-timed circadian rhythms are a sine qua non for optimal cellular and organismal health and for the prolongation of health span , which may also translate into an enhanced life span (Agorastos and Lindhorst 2016). In the absence of well-regulated circadian biology, the molecular physiology of cells is in a state of relative chaos, the degree of which presumably is determined in part by the severity of the dysregulation (chronodisruption) and the frequency and duration of the circadian insults. There is compelling evidence that subcellular turmoil due to any influence is associated with altered metabolic pathways that jeopardize the health of cells (Hardeland et al. 2012; Reiter et al. 2012a, b). Unhealthy cells promote less vigorous organs which surely compromise the welfare and survival of organisms. The “bottom line” is that regular circadian rhythms improve cellular physiology at the molecular level.
An estimated 15% of the genes possessed by cells relate to circadian rhythms (Scott 2015). The rhythms resulting from the expression of these genes at the cellular level have been referred to as slave oscillations given that many of them are under the influence of a more superior time-giver (Lamia et al. 2008; Golombek and Rosenstein 2010; Hardeland et al. 2012; Coelho et al. 2015; Buijs et al. 2016). While there are several factors that influence the expression of rhythms within cells, a major one is the cyclic information received from the master circadian oscillator or pacemaker, the suprachiasmatic nuclei (SCN) (Pauls et al. 2016). In mammals including the human, the activity of the SCN in turn is governed in large part by the light:dark environment as perceived by the eyes (Hughes et al. 2016). The unique and intricate molecular mechanisms whereby specific, especially blue, wavelengths of light impact distinctive intrinsically photosensitive retinal ganglion cells (ipRGC) in the outer layer of the mammalian retinas along with the retinohypothalamic tract which transfers this information to the SCN are well described elsewhere and are not reviewed here (Lucas et al. 2014).
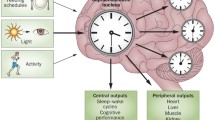
Once the photoperiodic information is received by the SCN, it must convey the associated message to the slave oscillators at the cellular level, whether or not this information is accurate relative to regularly-changing daily and seasonal changes in the light:dark environment as determined by the rotation of the Earth on its axis and its journey around the sun. The SCN has two known routes by which to inform the peripheral elements about the photoperiodic state. Thus, the central oscillator apprises the more distant cells via either a neural or humoral pathway (Fig. 1.1).

Regulation of the master circadian oscillator (the suprachiasmatic nuclei or SCN) and its impact on peripheral cellular rhythms. The major regulatory input to the SCN comes from the light:dark cycle as perceived by melanopsin in the intrinsically photoreceptic ganglion cells of the eyes. Other factors that impact the circadian system, but to a lesser degree than the light:dark environment, include physical activity and eating. The SCN communicates its circadian information to peripheral cellular clocks via two routes, the autonomic nervous system and by means of a humoral signal, most conspicuously the melatonin cycle. Desynchronization of circadian rhythms leads to pathological disorders, represented here by the “physiological variants.” Chronic disruption of the circadian system likely contributes to aging and age-related diseases
Since the SCN are located near the base of the telencephalic/diencephalic interface, they are in close proximity to the “head ganglion” of the autonomic nervous system, i.e., the hypothalamus. The central pacemaker has solicited the autonomic sympathetic/parasympathetic pathways as the neural route for contacting peripheral cells. One limitation of using the autonomic nervous system is that its postganglionic fibers are restricted in terms of their peripheral distribution. To solve this problem, the SCN also selected a humoral route for communication. To achieve this, the SCN utilizing the central and peripheral sympathetic nervous system, communicates with the pineal gland and dictates its metabolism (Stehle et al. 2011). In the absence of the light, the message is stimulatory at the level of the pinealocytes such that they are induced to synthesize and secrete, both into the blood (Vaughan et al. 1976) and into the cerebrospinal fluid (CSF) (Skinner and Malpaux 1999), a humoral mediator, i.e., melatonin, which contacts every cell in the organism. Any cell capable of “reading” this message, therefore, knows the status of the external light:dark environment. By utilizing both neural and humoral routes, information from the SCN is imposed on the genome of every cell in vertebrates.
With the advent of the introduction of artificial light in 1879 when the light bulb was invented and with the development of rapid transmeridian travel via airplanes, the photoperiodic information received by the SCN from the ipRGC is often not representative of the true daily/seasonal changes in the light:dark environment. Since the SCN is not allowed a choice in passing the altered information forward, every cell sometimes receives misinformation which disrupts their cycles and leads to circadian disturbances, i.e., chronodisruption (Erren and Reiter 2009). This contributes to malfunction of intrinsic molecular processes which create the chaos mentioned above; chaos always translates into accelerated cellular deterioration, i.e., aging. There are factors in addition to the light:dark cycle, specifically the rest/activity cycle and the feeding regimen, that also influence the function of the SCN (Fig. 1.1). This review, however, is primarily concerned with the instructions the SCN receives from the retinas and how perturbations of this information impacts cellular and organism senescence .
A major point of this report is that the aging-related consequences of chronodisruption cannot be easily distinguished from those caused by a perturbed melatonin cycle. Altered SCN rhythms are always accompanied by either a changed melatonin rhythm or a total suppression of melatonin synthesis and secretion. Given that this humoral message reaches every cell, circadian disorder becomes widespread. The problem is confounded by the fact that the plasma and CSF melatonin cycles are also designed to strengthen the central circadian clock message (Cassone 1990; Reiter et al. 2014b). In the absence of this regularly-repeating humoral message, the function of the SCN is weakened causing additional disorder which, via feedback and feedforward processes, may become a vicious cycle of gradually deteriorating molecular physiology. This leads to an acceleration of age-related processes.
The most common example of chronodisruption is that caused by light at inappropriate times, i.e., light at night. Throughout the world, well-developed societies, due to electrification, are experiencing something that organisms have never experienced during a very long period of evolution; they are experiencing what is referred to as the “end of night.” It was the dependence of organisms on the regularly-repeating periods of light and darkness during evolution that was surely consequential in the evolution and the development of the SCN; after all, it was the most reliable environmental variable on which to evolve a “clock.”
Light pollution is unquestionably a major factor that negatively impacts the function of the central circadian oscillator and, by necessity, the slave oscillators as well. Many studies have documented that light-at-night (LAN) changes the circadian output of the SCN, typically measured as perturbations in the blood melatonin cycle (Lewy et al. 1980) or in the urinary melatonin metabolite (Dumont et al. 2012) rhythms in humans and animals. These disturbances in the melatonin cycle have been linked to a variety of diseases/disorders that contribute to aging per se and indirectly to age-related diseases (Reiter 1995, 1997; Hardeland 2013; Hardeland et al. 2015; Opie and Lecour 2016), i.e., the promotion of pathologies. Of special note is the accelerated cancer growth, changes in glucose metabolism, skin deterioration, etc. (Kleszczynski and Fischer 2012; Haus and Smolensky 2013; Cipolla-Neto et al. 2014) Again, the reader is reminded that any alteration in the melatonin rhythm does not occur without a corresponding change in the neural message conveyed to the periphery by the SCN. Hence, it is never possible to distinguish whether an age-related change or pathology is a specific consequence of a malfunctional melatonin rhythm or abnormal circadian neural information; most likely, they are due to a combination of these factors (Reiter et al. 2012a; Hardeland 2013, 2015).
1.2 Hallmarks of Aging: Effects of Melatonin
Aging, the rate of which varies widely among animal and plant species, is usually characterized by a time-related functional deterioration of living creatures (Jones et al. 2014). More formally, aging is defined “as a progressive loss of physiological integrity leading to impaired function and increased vulnerability to death.” Historically, aging was judged on the basis of longevity in addition to the propensity of an individual to develop overt diseases. In the current era, however, aging is being investigated at the molecular level with the goal of identifying the processes that actually contribute to the aging phenotype. When these molecular processes are defined, it is the assumption that they will be modifiable and an increased healthy life span can be realized.
In a recent review, Lopez-Otin et al. (2013) suggested nine markers of aging which they feel characterize the degenerative processes at the subcellular level. While these markers are listed as being distinctly different entities, in fact, they have significant functional overlap consistent with the undoubtedly great complexity of the aging process. This entanglement will likely make identifying the specific mechanisms that underlie aging very difficult and equally problematic to treat.
A common denominator for many of the processes that are characteristics of aging is the oxidative microenvironment within cells (Forman 2016). Cells utilize oxygen as a basis of metabolism in the production of energy. Because of the relative inefficiency of the mitochondrial electron transport chain, the generation of oxygen free radicals and related non-radical oxygen-based derivatives is unavoidable (Brand 2016). This occurs when electrons are fumbled during their transfer between respiratory complexes and chemically reduce molecular oxygen to the superoxide anion radical (O ∙−2 ). While there is a formidable antioxidant defense system, some toxic species escape detoxification and harm neighboring molecules. As a consequence, over the course of a life time, oxidatively-damaged molecules gradually accumulate which impedes efficient molecular functions. These less than proficient systems further contribute to physiological disability and the greater deterioration typical of older individuals (Sohal and Allen 1990).
Oxidative stress is certainly not the only factor that accounts for the aging phenotype; but it surely subsidizes the amount of damage molecules sustain. If these injured molecules are not repaired or removed, they become a burden to cellular physiology. Accumulated oxidative stress is mentioned as a feature of many theories that have attempted to explain aging (Ames 1989). Melatonin, the endogenous levels of which diminish with age in most individuals, normally functions as a potent antioxidant to reduce the accumulation of oxidized molecules which accelerate senescence and aging.
Cells typically have a finite number of cell divisions they can undergo before they functionally collapse. This phenomenon was described by Hayflick several decades ago (Hayflick 1979). It is our prediction that properly-timed melatonin exposure would extend the Hayflick limit, i.e., defer cellular aging. This assumption is based on melatonin’s ability to both organize the circadian biology of cells and to its actions as a multifaceted direct free radical scavenger and its indirect actions in the promotion of enzymatic antioxidative defense processes. Moreover, melatonin’s high concentration in mitochondria (Venegas et al. 2012), a site where free radical formation is abundant, is consistent with the option. In this regard, melatonin has recently been designated as a mitochondria-targeted antioxidant (Reiter et al. 2016) which was proven equivalent to or more effective than synthetic antioxidants, Mito E and Mito Q, in resisting oxidative damage and inflammation. Mito E and Mito Q are industry-produced antioxidants that, because of their increased lipid solubility, concentrate in the mitochondria up to 100-500-fold (Oyewole and Birch-Machin 2015). Despite this, they are no more effective than endogenous-produced melatonin in preventing free radical-mediated molecular damage resulting from the simultaneous exposure of animals to two highly toxic bacterial molecules, lipopolysaccharide and peptidoglycan (Galley 2010; Lowes et al. 2013). Ramis et al. (2015) have recently reviewed the literature related to the relative effectiveness of synthetic antioxidants and melatonin in determining the degree of oxidative stress. Given that melatonin is so effective in curtailing the actions to toxic free radicals and the fact that its levels in both animals and many humans wane with advancing age (Scholtens et al. 2016), it seems safe to assume that the loss of melatonin including the suppression of its rhythm which contributes to circadian disruption in late life contributes to the aging phenotype which is a consequence of accumulated oxidatively-damaged molecules that occur throughout life.
More than 15 years ago, we reported that early-life pinealectomy in rats, which deprived the animals of a circadian melatonin message and dropped their circulating melatonin concentrations to barely-measurable values, caused augmented oxidative damage to all tissues in which it was measured when the rats reached 24 months of age; rats of this age are generally considered old (Reiter et al. 1999). These results likely related to the high efficiency of melatonin as a multifaceted antioxidant which, due to its low levels, allowed many mitochondrial-generated free radicals to go uncontested. This would certainly contribute to accelerated aging. These animals, however, likely had weakened circadian rhythms at the peripheral cellular level which also presumably supported the generation of an increased numbers of partially-reduced toxic oxygen derivatives. This illustrates the difficulty in distinguishing which factor, melatonin deprivation or circadian disorder, contributes most significantly to the more abundant oxidative stress in the old animals.
Some resolution to this conundrum could perhaps be gained by comparing the degree of oxidative damage in different tissues of old animals (that have been pinealectomized at an early age). In the study of Reiter et al. (1999), for example, all tissues in which lipid peroxidation and protein carbonyls were measured (lung, liver, skin, pancreas, kidney, etc.), normally rely on the blood melatonin cycle for the majority of their circadian direction. Other tissues (e.g., salivary glands) which also received a significant amount of their circadian information via the autonomic nervous system, may exhibit less oxidative damage than cells which are not directly innervated by the sympathetic/parasympathetic fibers. These glands, since the activity of the SCN is still synchronized by the prevailing light: dark environment as perceived by the ipRGC, may receive regular circadian information which may better synchronize their inherent cellular rhythms thereby reducing free radical generation.
The melatonin rhythm, as a reflection of information provided by the SCN, as suggested above, likely has a significant role in directing the circadian biology of peripheral cells. The melatonin cycle, unlike the SCN-mediated circadian information conveyed via the autonomic nervous system, contacts every cell in the organism. That melatonin is in fact capable of synchronizing cellular rhythms has been documented (Jung-Hynes et al. 2010). This group showed that the intrinsic circadian rhythms of cultured cells were brought into synchrony when melatonin was added to the culture medium. This action would likely aid in slowing molecular degeneration that contributes to aging. This circadian regulatory action of melatonin may also be an explanation for the recent findings which document that melatonin sensitizes previously chemotherapeutic-resistant cancer cells to these treatments (Martin et al. 2010; Xiang et al. 2015). Given the findings of Jung-Hynes et al. (2010) relative to melatonin’s ability to properly direct circadian gene expression at the peripheral level, the necessity of maintaining a regular alternating light:dark environment which propels the melatonin rhythm seems essential if aging is to be slowed. These findings are also consistent with the observations that excessive light pollution or frequent long-haul transmeridian travel, both of which cause chronodisruption and melatonin rhythm alterations/suppression, are associated with an increased risk of developing cancer and other age-related diseases. Melatonin has been commonly used by frequent long-haul travelers to combat the sleep deprivation and fatigue related to travel across multiple time zones (Cardinali et al. 2002). Besides helping to correct these disturbances, melatonin’s use for this reason may also reduce the likelihood of developing age-related cancer predictably linked to frequent long-haul travel. Whether these beneficial effects of melatonin derive from is intrinsic oncostatic activity or as a result of its rhythm-synchronizing activity is difficult to determine.
1.3 Consequences of Disruption of Circadian Rhythms
During aging, there is a clear diminution in the strength of circadian rhythms which compromises organismal homeostasis in a negative way; the resulting changes contribute to morphological and physiological aging. The molecular aging phenotype is manifested at many levels. In old individuals of any species, molecular damage comes in a variety of forms and has been studied for many years. This damage includes elevated levels of lipid peroxidation products (Grinna 1977; Zs-Nagy 1978), increased damaged protein concentrations (Steinberg and Witztum 1990; Clarke et al. 1991) and enhanced amounts of oxidized DNA (Alexander 1967; Imlag and Linn 1988). These changes also are a result of a number of factors including disease processes and perturbations of circadian rhythms, among others.
A more recently defined disorder that contributes to aging and especially to age-related diseases is the gradually-developing instability of the genome. This instability becomes greater as organisms age and is best known for its association with conditions such as progeroid-like syndromes where aging is highly accelerated and in cancer (Campisi 2005; Vijg and Suh 2013). A relationship of the deteriorating stability of the genome and cancer incidence is not unexpected since specific mutations are a requirement for a cancer to develop. While humans possess genes, e.g., SIRT1, that foster longevity (Jazwinski et al. 2010; Kim et al. 2012), the longest lived individuals often do not develop cancer; this possibly aids in their long survival (Kovacic and Somanthan 2014). SIRT1 also influences PER2 in the master circadian clock which aids in stabilizing circadian rhythms which helps to delay/prevent disease of aging (Asher et al. 2008).
The stability of the genome declines during aging due to a variety of factors; many of these relate to an unhealthy life style (cigarette smoking, poor diet, etc.) and those that predispose to cancer (exposure to volatile carcinogens, ingestion of heavy metals, ionizing and ultraviolet radiation). A more recently defined, previously thought to be innocuous factor that aggravates genomic stability is light-at-night (LAN), i.e., circadian rhythm disturbance and/or melatonin suppression (Belancio et al. 2015). These occur especially during night shift work, late night recreational activities (so called, social jet lag), light exposure at night due to light pollution and intentional light after darkness onset when awakening. Each of these disturbs the function of the master biological clock and suppresses the melatonin synthesis/secretion cycle thereby contributing to circadian dysregulation and genomic instability (Fig. 1.2). Equally as disruptive to the circadian system are rapid changes in time zones, especially when moving in an eastwardly direction; the resulting jet lag has been linked to an increased likelihood of developing cancer (Ptacek et al. 2007), probably due to suppression of melatonin (Reiter et al. 2007; Hill et al. 2015). Genomic instability due to any reason leads to accelerated DNA damage and the consequences thereof; this includes accelerated aging.

Light exposure at night disrupts the function of the master circadian oscillator, the central clock (CC; also known as the suprachiasmatic nuclei), which results in an associated deterioration in the normally synchronized oscillations in peripheral cells. Under regularly alternating light:dark cycle, these DNA damage response (DDR) are well synchronized. The perturbed rhythms due to light-at-night negatively impacts the melatonin cycle, DDR and metabolism all of which are believed to contribute to aging and/or age-related diseases . From Belancio et al. (2015) with permission
Recently, Belancio et al. (2010a) have summarized the data that defines the role of transposable elements in genomic instability. These elements are capable of rearranging the genetic material and support for their involvement in cancer has accumulated over the last decade (Gasior et al. 2006; Belancio et al. 2010b; Lee et al. 2012; Scott et al. 2016). Genomic instability contributes to many different lesions in DNA including single base-pair substitutions or deletions and large genomic rearrangements including deletions, insertions, inversions and translocations (Belancio et al. 2010a). The large genomic changes likely contribute to the aging phenotype (Vijg and Dolle 2002; Hsieh et al. 2013).
Research has shown there are clear connections between genomic stability transposable elements (retrotransposons), circadian organization, metabolism and aging (Belancio et al. 2010a). Retro elements are mobile genetic entities that consist of two related groups, i.e., long terminal repeat (LTR) and non-LTR retrotransposons; these are represented by long (LINE) and short interspersed elements (SINE) (Belancio et al. 2008). There is now credible evidence showing that L1 activity and the circadian system are inextricably linked (deHaro et al. 2014). Hence, in an in vivo cancer model, the interaction of melatonin with its MT1 membrane receptor suppressed L1 expression; melatonin receptor had a similar effect in cultured cells. Given that melatonin is an important component of the endogenous circadian networks, it is plausible that L1 expression is under the influence of the circadian system.
As noted above, LAN is a major factor capable of disturbing the function of the master circadian oscillator, the SCN (Ikeno and Yan 2016). Since the prevailing light:dark environment drives the circadian network including the intrinsic rhythms of the slave oscillators in perhaps all mammalian cells, when darkness is interrupted by light genomic stability is also impacted as suggested by the findings of deHaro et al. (2014). The manifestations of the genomic damage that likely occurs as a result of interruption of darkness probably accounts, at least in part, for the reported elevated cancer risk in night shift workers (Lewy et al. 1980; Reiter et al. 2007; Erren and Reiter 2008). Beyond this, LAN also accelerates the growth of already established cancer because of the suppression of melatonin (Blask et al. 2005; Hill et al. 2015). Finally, there are other metabolic consequences resulting from circadian disruption due to LAN; some of these include obesity (Cipolla-Neto et al. 2014; Coomans et al. 2015; Scott 2015) and diabetes (Ingenwerth et al. 2016) and at the cellular level depressed SIRT1 activity (Asher et al. 2008; Jung-Hynes et al. 2010) and clock gene expression (Granados-Fuentes et al. 2015). These perturbations contribute to the functional decline normally known as aging and certainly in part relate to chronodisruption- and melatonin suppression-mediated genomic instability.
1.4 Cellular Aging: The Case of Stem Cell Senescence
The ability of melatonin to forestall senescence of stem cells grown in vitro and subjected to multiple passages was recently documented. Judging from the number of approved clinical trials, it seems likely that MSC-based therapies will continue to be a common treatment paradigm in the field of regenerative medicine (Caplan and Correa 2011). The recovery of stem cells for therapy is often a limiting factor because of their numbers; this is particularly the case with certain stem cells (Jones and Wagers 2008). For example, bone marrow donation remains rather infrequent and even when marrow samples are collected there is a very low ratio of stem cells to the total number of blood forming cells. Hence, in vitro expansion of the relatively small number of stem cells that are recovered is critical to obtain the necessary number for implantation therapy. Repeated in vitro passaging of stem cells, however, introduces the likelihood of a number of disorders which compromise their usefulness, i.e., they lose their stemness and exhibit signs of senescence (Baker et al. 2015).
In an attempt to preserve bone marrow MSC (BMMSC) in a more youthful state during multiple passaging, Shuai et al. (2016) compared four low molecular weight molecules (rapamycin, resveratrol, quercetin and melatonin) in terms of preserving rat and human BMMSC in a more original state. These molecules were selected as candidates because of their previously-reported anti-aging and antioxidant actions and for their ability to enhance stem cell protection.
Shuai et al. (2016) initially showed that BMMSC lost their self-renewal potential and their osteogenic differentiation capacity during growth through 15 passages. When the four molecules (each at 10 nM) were compared for their efficacy in maintaining stemness, melatonin proved to be far superior to the other three molecules (rapamycin, resveratrol and quercetin). For example, melatonin proved highly effective in maintaining the ectopic osteogenic activity of BMMSC during long-term passaging and when the cells were transplanted into nude mice (for 8 weeks). In other models in which melatonin was tested (calvarial defect repair, bone loss after ovariectomy, reduction of immune competence), it also had a very positive effect on the outcome measured and clearly delayed senescence.
To establish the mechanism by which melatonin functioned in their study, Shuai et al. (2016) explored the possibility that the potent antioxidant functions of the indoleamine (Manchester et al. 2015; Reiter et al. 2016) accounted for its protective actions given that cellular senescence during in vitro passaging of stem cells is known to be related to ROS generation (Busuttil et al. 2003; Parrinello et al. 2003). The incubation of BMMSC with melatonin reduced ROS formation by more than 50%; this drop was associated with a 2-fold elevation in the expression of superoxide dismutase 2 (SOD2). Likewise, the p53 pathway is stimulated by both ROS and as a result of telomere shortening limits stem cell proliferation and renewal (Bonizzi et al. 2002). When Shuai et al. (2016) examined p53 and the downstream regulator, p16, melatonin was found to significantly prevent the rise in p53 which normally would have suppressed stem cell renewal. Finally, clusters of master genes, one of which is designated NONAG, control the multipotency of stem cells by preserving them in an undifferentiated state (Tsai et al. 2012). NONAG expression is normally lost during senescence of BMMSC. Melatonin treatment preserved NONAG expression at a level equivalent to that in just-isolated stem cells. Thus, the collective results show that melatonin preserves stemness of BMMSC by reducing oxidative stress, inhibiting the p53 pathway and maintaining NONAG expression. These beneficial actions seem not to involve MT1/MT2 melatonin membrane receptors (Luchetti et al. 2014) but rather melatonin’s direct scavenging actions on ROS (Tan et al. 1993; Reiter et al. 2014a) and its ability to promote antioxidant enzymes (Rodriguez et al. 2004). In the study reported by Shuai et al. (2016), treating BMMSC with luzindole (an MT1/MT2 receptor antagonist) did not interfere with melatonin’s ability to safeguard these cells; thus, these receptors are not involved in anti-senescent action of melatonin.
These findings are directly applicable to aging and age-related diseases. Elevated free radical generation is commonly associated with advanced age as metabolic pathways become less efficient (Hocman 1979; Allen 1990). Simultaneously, blood melatonin levels wane as does the antioxidant capacity of this fluid (Benot et al. 1999) (Fig. 1.3). Also, early-life pinealectomy, which deprives animals of their daily melatonin rhythm and contributes to circadian dysregulation exaggerates the amount of oxidatively-damaged molecules these animals accumulate when they are 24 months of age (Reiter et al. 1999). Interestingly, the premier means of delaying aging, i.e., caloric-restriction (Meites 1990; Rae 2004), preserves molecular and cellular function and, likewise, prevents the normal reduction in pineal melatonin synthesis associated with aging (Stokkan et al. 1991). Mechanistically, the preserved melatonin production is accompanied by the retention of β-adrenergic receptors on the pinealocyte membranes; these receptors mediate the sympathetic stimulation of melatonin synthesis (Henden et al. 1992). Whether the conserved melatonin rhythm contributed to the preserved physiology of the caloric-restricted old animals has yet to be proven.

The nocturnal levels of circulating melatonin usually drop as humans age (bottom panel). Associated with the nighttime reduction in melatonin is a loss of the total antioxidant status (TAS) (upper panel) of the blood. Thus, as humans age the reduction in the level of the antioxidant, melatonin, likely contributes to the accumulation of free radical damage to cells which hastens aging. From Benot et al. (1999) with permission
The ability of melatonin to forestall the functional capacity of stem cells is not exclusive to long-term passaged senescing BMMSC. Similarly, aging of the umbilical cord-derived (Lee et al. 2014) and adipose-derived stem cells (Yip et al. 2013) is also reduced when they are grown in a melatonin-containing medium. By retarding the aging of these cells, melatonin treatment also preserves their growth and physiology when they are used for transplantation therapy (Chen et al. 2014a, b; Yip et al. 2013). Finally, Zhou et al. (2015) reported that melatonin prevented MSC premature senescence when the cells were treated with the oxidizing agent, H2O2. This reversal was associated with an upregulation of the SIRT1 pathway, an event known to be associated with anti-aging processes (Favero et al. 2015; Ghosh and Zhou 2015). In addition to prolonging the functional half life of MSC, we had earlier predicted that culturing more differentiated cells in a melatonin-containing medium would allow them to undergo more mitoses than prescribed by the Hayflick limit (Hayflick 1979; Reiter et al. 2016).
Beyond upregulating the expression of the longevity-promoting gene, SIRT1, in MSC by melatonin, little is known about other molecular events that were changed as a consequence of melatonin treatment. Thus, while melatonin’s antioxidant functions are usually used as an explanation for the enhanced functional state and prolonged survival of MSC, the circadian biology of these cells under the influence of melatonin is yet to be examined. Under in vivo situations, melatonin presumably provides stem cells with timing information as it does for terminally-differentiated elements.
Collectively, the ability of melatonin to maintain a more optimal physiological state of stem cells and reduce the rate at which they become non-functional and undergo apoptosis and aging, has applications to a number of human conditions beyond aging per se. The more rapid stabilization of dental implants, wound healing and bone fracture repair are examples where melatonin’s capability to improve the functions of stem cells may come into play (Cutando et al. 2011; Clafshenkel et al. 2012; Lee et al. 2014). Even more important may be the use of melatonin to prolong the survival of stem cells when they are injected in vivo to restore tissues such as the cardiomyocytes and neurons which have no capability of regeneration. Typically, the vast majority of stem cells die when injected before they have any restorative benefit. Since the pathological conditions that require stem cell treatment are most common in the elderly, the improved function due to the use of melatonin may enhance the quality of life in older individuals. Again, in these conditions the circadian-organizing actions of melatonin should not be overlooked. Perhaps older individuals with stem cell implants should be given melatonin at a specific time of the day (night) to aid in the synchronization of the circadian rhythms of not only stem cells but other non-diseased cells as well. This may be of importance since the endogenous melatonin signal is often severely weakened in older patients because of the diminished melatonin cycle.
In vivo, stem cells are maintained in their optimal functional state by a complex signaling network (Hawkins et al. 2014). Under culture conditions due to a change in their microenvironment, these essential signals are lost; as a result researchers have sought means to defer senescence of these cells when undergoing multiple passages (Coutu et al. 2011; Eom et al. 2014). These procedures, however, have disadvantages (Guitart et al. 2010). Here, we propose that melatonin is one signaling molecule that should be tested for its ability to prolong the undifferentiated state of the cells. Other signaling molecules have already been used for this purpose (Danet et al. 2003; Lin et al. 2012) and have been shown to have only slight efficacy in this regard. Based on the comparative investigation of Shaui et al. (2016), however, melatonin should be seriously considered as an agent to delay aging of stem cells in vitro as well as after their injection. Finally, melatonin should be considered for use to prolong the survival of differentiated cells in vivo which are limited in the number of mitoses they can experience (Hayflick 1979).
1.5 Circadian Disruption and Age-Related Diseases
Epidemiological data have long suggested that circadian dysregulation due to light exposure at night (LAN) (or rapid transmeridian travel) is associated with an increased cancer risk (Hansen 2001; Erren et al. 2010; Rao et al. 2015). These findings also have strong support from experimental laboratory studies. Blask et al. (2005) and Hill et al. (2015) have repeatedly shown that LAN stimulates xenografted human tumors transplanted into immune-compromised rodents grow more aggressively; this exaggerated growth is likewise inhibited by properly-timed melatonin administration. As noted in this survey, LAN always simultaneously disrupts the circadian melatonin cycle which also perturbs circadian rhythms more generally.
That disruption of the master clock promotes cancer growth was also recently documented in a circadian-based study reported by Papagiannakopoulos et al. (2016). Using a genetically-engineered mouse model that can be induced to develop lung adenocarcinoma, this group reported that circadian disruption due to imposed jet lag or genetic manipulation of central clock components promoted lung cancer growth and reduced the survival of the animals. Loss of the central clock components, Per2 and Bmal were associated with elevated c-Myc expression, enhanced tumor cell proliferation and metabolic disruption. Melatonin levels were not measured in these animals nor was it given as a supplement to determine whether it would reverse the observed effects on exaggerated tumor growth.
The studies summarized above are only a few of the many that document that circadian disintegration and melatonin suppression may be consequential in the increased cancer risk experienced by aging individuals (Erren et al. 2016). The majority of cancers are age-related and both accurate circadian regulation and a well-preserved melatonin rhythm decrease with age. The implication is that disturbances in these two related systems contribute to cancer development and, more generally, to the degenerative changes of aging (Reiter et al. 2007).
In addition to cancer, a number of degenerative nervous system disorders are presumed to be mediated by the gradual loss of a robust melatonin cycle in the elderly; moreover, during aging the intrinsic central circadian clock is weakened and the 24-hour cycles are progressively disrupted. As an example of this, the sleep/wake cycle is often significantly advanced and sleep itself is fragmented in the elderly. The loss of adequate and restful sleep may itself be a contributing factor to aging.
Neural conditions that may be aggravated by the deteriorating melatonin cycle and a more general disturbance of the biological clock include Alzheimer disease (AD) (Pappolla et al. 1997; Hardeland 2012; Rosales-Corral et al. 2012; Ali and Kim 2015; Miller et al. 2015; Zhang et al. 2016), Parkinson disease (PD) (Mayo et al. 2005; Santos, 2012), Huntington disease (HD) (Escribano et al. 2014; van Wamelen et al. 2015), amyotrophic lateral sclerosis (ALS) (Anderson and Rodriguez 2011; Farez et al. 2016), and multiple sclerosis (MS) (Kashani et al. 2014; Wen et al. 2016). These conditions often are associated with altered circadian rhythms; whether these disturbances are a cause or an effect of these states has not been unequivocally determined. The ability of melatonin to defer the progressive development of these conditions in limited clinical trials, however, has been documented (Weishaupt et al. 2006; Medeiros et al. 2007; Cardinali et al. 2012; Lopez-Gonzalez et al. 2015). Moreover, preliminary studies with the use of melatonin to treat AD patients also suggest that this treatment may retard the progression of this devastating condition in humans (Hardeland 2016). While it is usually surmised that the antioxidant actions of melatonin account for its ability to slow the causes of AD, as noted this indole also has the capability of synchronizing the circadian system generally as well as promoting more successful sleep (Erren et al. 2016) which may aid in forestalling CNS degeneration. Finally, melatonin may also reduce aging in the central nervous system by stimulating hippocampal progenitor cell proliferation which leads to the restoration of lost neurons in the dentate gyrus (Ethuwapranee et al. 2015).
That the rhythmic machinery underlying the sleep/wake cycle is altered becomes apparent in the older population where the timing of sleep onset shifts to an earlier time. Additionally, sleep architecture is changed since there is an increased frequency of awakenings during the nighttime sleep period along with a reduction in the total amount of non-rapid eye movement (REM) sleep (Lo et al. 2016; Mattis and Sehgal 2016). Concurrent with these changes there is often a marked attenuation of the nocturnal peak of circulating melatonin. On the other hand, there are reports of some elderly individuals who do not experience a substantial drop in maximal night melatonin values. To the knowledge of the current authors, there are no reports which examined whether the retained robust melatonin rhythm was associated with sleep architecture of younger individuals with fewer nocturnal awakenings, etc. Also, whether the well preserved melatonin cycle in a small percentage of the aged are beneficial in maintaining sleep quality seems not to have been investigated.
The differential rate of deterioration of the melatonin cycle and sleep efficiency is of special interest in light of data published by Dauchy et al. (2013) using animals. This group reported that the quality of daytime light has a major impact on the amplitude of the nighttime melatonin peak; when rats were exposed to blue wavelength-enriched light during the day, the maximal nighttime melatonin levels were increased up to ten-fold. This remarkably augmented nighttime melatonin peak enhanced the ability of the animals to resist the growth of transplanted cancer cells. Given the other actions of melatonin, the animals would be expected to have a much higher total antioxidant capacity and perhaps greater longevity. The findings also have potential implications for older individuals who may be able to improve nighttime sleep by changing their daytime light exposure to one rich in blue wavelengths.
Cardiovascular physiology also exhibits changes that have been classified as negative during aging. It is common that mean arterial blood pressure (MAP) gradually increases as individuals advance in age (Shen et al. 2015). This hypertensive response may be related to the concurrent drop in the nocturnal circulating melatonin peak and the perturbed circadian system in the elderly. In young individuals, the nighttime rise in melatonin is functionally correlated with a reduction in nocturnal systolic blood pressure (Pechanova et al. 2014). The loss of this drop in systolic pressure at night contributes to a rise in the MAP throughout life. This is consistent with what occurs in the aged. As the amplitude of the melatonin rhythm declines, the MAP rises. Hypertension is an independent risk factor for cardiovascular death and, therefore, it reduces longevity. Melatonin’s benefits, besides mediating the nocturnal dip in systolic blood pressure, via its antioxidant actions likely contribute to its protection of the myocardium, as seen during cardiac ischemic/reperfusion injury (Dominguez-Rodriguez et al. 2012; Liu et al. 2015). Additionally, melatonin has been shown to exert cardiovascular protection due to its capacity to improve the bioavailability of nitric oxide which leads to vasodilation and a drop in blood pressure (Simko et al. 2016). Thus, the loss of a stout nocturnal melatonin rise in the aged may well contribute to the elevation of MAP during late life. Elevated blood pressure invariably compromises longevity if it goes untreated.
In addition to melatonin making nitric oxide more available to provide relief from elevated MAP, its loss during aging also impacts the autonomic nervous system which may contribute to mortality. Melatonin influences sympathetic drive by enhancing GABA-ergic signaling involved in inhibiting the paraventricular nucleus by the SCN (Wang et al. 2003). Additionally, neurons in the area postrema, which are central to blood pressure regulation, are epigenetically influenced by melatonin (Irmak and Sizlan 2006). Finally, intermittent ventricular dyssynchrony has been shown to be treatable with melatonin (Dominguez-Rodriguez et al. 2016). Clearly, the melatonin rhythm has direct effects on blood pressure as well as indirect actions via its ability to modulate circadian rhythms of the autonomic nervous system (Simko et al. 2016).
1.6 Concluding Remarks and Perspectives
Even though no one has ever avoided aging and death and they are unquestionably inevitable, there is still unbounding interest in the possibility of deferring these conditions when means of delaying aging are discussed. The major consideration usually relates to prolonging life. Increasing longevity without maintaining optimal health, i.e., without extending health span , may, however, be counterproductive. On the other hand, prolonging good health into advanced age may concurrently contribute to enhanced longevity. In experimental rodents, the most reproducible means of extending life and limiting pathologies is to rather severely limit caloric intake; but the success of this procedure when applied in primates has been equivocal (Roth et al. 1999; McKiernan et al. 2011; Mattison et al. 2012). Thus, the potential benefits of reducing food intake for the purpose of stretching the life span of human remains obscure. This, however, has not dissuaded scientists from trying to identify molecules that can be consumed and that mimic the cellular molecular actions observed in underfed rodents (Ingram and Roth 2015). The amount of resources dedicated to identifying elixirs that may slow down time is currently massive and even well-established, renowned scientists are self-testing molecules that may defer age-related diseases and enhance longevity.
In the current report, we examined the data that suggests a relationship between circadian biology and the processes of aging. Circadian rhythm disturbances cannot be considered in terms of their effects without taking into account the melatonin cycle. This is a requirement since the central Zeitgeber, i.e., the SCN, and the melatonin rhythm are mutually interactive. Any disturbance of the message sent out from the SCN is always accompanied by a perturbation and often a suppression of the pineal melatonin production and release. Moreover, any alteration in melatonin synthesis and secretion changes the function of the master clock given that melatonin in the blood, or more likely in the CSF (Reiter et al. 2014b), feedbacks on the SCN to modulate circadian rhythmicity, i.e., it serves as a chronobiotic.
The data summarized herein shows that the SCN-pineal axis may well impact the progress of senescence development and especially the likelihood of delaying the onset or severity of age-related diseases. A major difficulty with assessing the specific role of an imposed circadian disorder on cellular function stems from the fact that there is no procedure nor drug that uniquely and precisely destroys circadian biology without also seriously impacting the melatonin rhythm. While exogenously-administered melatonin has been used to potentially define the functional relevance of this molecule in altering the rate of aging and disease prevention, the evidence remains sketchy whether this relates to the chronobiotic actions of this indole or is exclusively a result of the diverse antioxidant functions. Certainly, melatonin has been successfully used experimentally to delay the onset and progression of age-related disorders; however, whether it directly defers aging per se remains unclear.
Relative to the circadian system and aging, it has been difficult to link the functional deterioration of this system specifically to the rate of aging or of age-related diseases . Rather than contributing to aging, the weakening circadian rhythms may be a consequence of aging rather than being causative. Also, while there is no doubt that the circadian system suffers dysfunction in the aged, whether this relates to specific disease phenotypes remains to be defined.
As summarized by Lopez-Otin et al. (2013), there are many facets that likely contribute to the aging phenotype and to mortality. These include genomic damage that goes unrepaired and therefore accumulates with increasing age. This issue is particularly obvious in humans suffering with progeria-related diseases where DNA damage occurs at an unprecedented rate. What are defined as epigenetic shifts also may advance age-related functional deterioration (Wu et al. 1993). These authors found that when DNA methylation in human cells is mapped over time, some areas of the genome become hypermethylated while some do not. Likewise, histone modification changes as a function of age in some tissues. It seems likely that the accumulated damage and epigenetic shifts may relate to circadian disturbances and changes in the melatonin cycle although this has yet to be determined. While the accumulated DNA damage that occurs is often a result of oxidative stress, it is probable that melatonin, because of its antioxidant properties, slows DNA damage and thereby contributes to slowing cellular aging.
Telomeres, the repetitive sequences on the ends of linear chromosomes shorten with age and contribute to the instability of the genome. DNA polymerases are incapable of replicating these chromosomal segments. As telomeres shrink, the cell undergoes senescence and implodes due to apoptosis. Agents that stimulate telomerase, which renews telomeres, delay cellular aging (Hewitt et al. 2012). Experimentally, melatonin does enhance telomerase activity in normal cells (Hardeland 2013) suggesting that this indole may contribute to preserving good health and deferring aging; however, there are no reports that definitively prove either blunting of the melatonin cycle or disturbed circadian rhythms in the aged as being related to changes in telomerase activity.
There is a great diversity in the trajectories of the rate of aging across the tree of life and rather few species have been investigated as to why these species age differently (Promislow and Harvey 1990; Jones et al. 2008). Some rodents are relatively short lived, e.g., the rat, while others have an uncommonly long life, e.g., naked mole rat (Austad 2007). Bird species also show a similar wide diversity in their duration of survival (Barja 1998; Herrero and Barja 1998). A satisfactory explanation as to why these species differ so markedly in terms of their survival has yet to be determined.
The functional connections between either altered circadian rhythms or perturbations of the melatonin cycle and aging remain nebulous. The reader is reminded that these rhythms developed over a very long period of evolution during which the light:dark cycle was precisely reproduced from year to year as dictated by the rising and setting of the sun. Hence, severe disturbances of these precise rhythms that result from the introduction of inappropriately-timed light would be expected to have health and age-related consequences, some of which may be dire. This may also have to be taken into consideration when humans finally populate planets where the light:dark environment differs significantly from that on Earth.
References
Agorastos A, Linthorst ACE (2016) Potential pleiotropic beneficial effects of adjuvant melatonergic treatment in posttraumatic stress disorder. J Pineal Res 61:3–26
Alexander P (1967) The role of DNA lesion in processes leading to aging in mice. Symp Soc Exp Biol 21:29
Ali T, Kim MO (2015) Melatonin ameliorates amyloid beta-induced memory deficits, tau hyperphosphorylation and neurodegeneration via P13/Akt/GSk3β pathway in the mouse hippocampus. J Pineal Res 59:47–59
Allen RG (1990) Role of free radicals in senescence. Annu Rev Gerontal Geriatr 10:198–213
Ames BN (1989) Endogenous DNA damage as related to cancer and aging. Mutat Res 214:41–46
Anderson G, Rodriguez M (2011) Multiple sclerosis, seizures, and antiepileptics: role of IL-18, IDO, and melatonin. Eur J Neurol 18:680–685
Asher G, Gatfield D, Stratmann LM, Reinke H, Dibner C, Kreppel E Mostoslavsky R, Alt FW, Schibler U (2008) SIRT1 regulates the circadian clock gene expression through PER2 deacetylation. Cell 134:317–328
Austad SN (2007) Vertebrate aging research 2006. Aging Cell 6:135–138
Baker N, Boyette LB, Tuan RS (2015) Characterization of bone marrow-derived mesenchymal stem cells in aging. Bone 70:37–47
Barja G (1998) Mitochondrial free radical production and aging in mammals and birds. Ann NY Acad Sci 854:224–238
Belancio VP, Hedges DJ, Deininger P (2008) Mammalian non-LTR retrotransposons for better or worse, in sickness and health. Genome Res 18:343–358
Belancio VP, Roy-Engel AM, Deininger PL (2010a) All y’all need to know ‘bout retroelements in cancer’. Semin Cancer Biol 20:200–210
Belancio VP, Roy-Engel AM, Pochampally RR, Deininger P (2010b) Somatic expression of LINE-1 elements in human tissues. Nuclei Acids Res 38:3909–3922
Belancio VP, Blask DE, Deininger P, Hill SM, Jazwinski SM (2015) The aging clock and circadian control of metabolism and genomic stability. Front Genet 5:455
Benot S, Gobema R, Reiter RJ, Garcia-Maurino S, Osuna C, Guerrero JM (1999) Physiological levels of melatonin contribute to the antioxidant capacity of human serum. J Pineal Res 27:59–64
Blask DE, Brainard GC, Dauchy RT, Hanifin JP, Davidson LK, Krause JA, Sauer LA, Rivera-Bermudez MA, Dubocovich ML, Jasser SA, Lynch DT, Rollag MD, Zalatan F (2005) Melatonin-depleted blood from premenopausal woman exposed to light at night stimulates growth of human cancer xenografts in nude rats. Cancer Res 65:11174–11184
Bonizzi G, Cicalese A, Insigna A, Pelicci PG (2002) The emerging role of p53 in stem cells. Trenda Mol Med 18:6–12
Brand MD (2016) Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med 100:14–31
Buijs FN, Leon-Mercado L, Guzman-Ruiz R, Guerrero-Vargas NN, Romo-Nava F, Buijs RM (2016) The circadian system: a regulatory feedback network of periphery and brain. Physiology (Bethesda) 31:170–181
Busuttil RA, Rubio M, Dolle ME, Campisi J, Vijg J (2003) Oxygen accelerates the accumulation of melatonin during the senescence and immortalization of murine cells in culture. Aging Cell 2:287–294
Campisi J (2005) Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120:513–522
Caplan AL, Correa D (2011) The MSC: an injury drugstore. Cell Stem Cell 9:11–15
Cardinali DP, Bortman GP, Liotta G, Perez-Lloret S, Albornoz LE, Cutrera RA, Batista J, Ortega Gallo P (2002) A multifactorial approach employing melatonin to accelerate resynchronization of sleep-wake cycle after a 12 time-zone westerly transmeridian flight in elite soccer athletes. J Pineal Res 32:41–46
Cardinali DP, Vigo DE, Olivar N, Vidal MF, Furio AM, Brusco LI (2012) Therapeutic application of melatonin in mild cognitive impairment. Am J Neurodegener Dis 1:280–291
Cassone VM (1990) Effects of melatonin on vertebrate circadian systems. Trends Neurosci 13:457–464
Chen HH, Lin KC, Wallace CG, Chen YT, Yang Y, Leu S, Chen YC, Sun CK, Tsai TH, Chen YL, Chung SY, Chang CL, Yip HK (2014a) Additional benefit of combined therapy with melatonin and apoptotic adipose derived mesenchymal stem cells against sepsis-induced kidney injury. J Pineal Res 57:16–32
Chen YT, Chiang HJ, Chen CH, Sung PH, Lee FY, Tsai TH, Chang CL, Chen HH, Sun CK, Leu S, Chang HW, Yang CC, Yip HK (2014b) Melatonin treatment further improves adipose-derived mesenchymal stem cell therapy for acute interstitial cystitis in rat. J Pineal Res 57:248–261
Cipolla-Neto J, Amaral FG, Afeche SC, Tan DX, Reiter RJ (2014) Melatonin, energy metabolism, and obesity: a review. J Pineal Res 56:371–381
Clafshenkel WP, Rutkowski JL, Palchesko RN, Romeo JD, McGowan KA, Gawalt ES, Witt-Enderby PA (2012) A novel calcium aluminate-melatonin scaffold enhances bone regeneration within a calvarial defect. J Pineal Res 53:206–218
Clarke RC, Daly L, Robinson K, Naughten E (1991) Hyperhomocysteinemia: an independent risk factor for vascular disease. N Engl J Med 324:1149–1155
Coelho LA, Peres R, Amaral FG, Reiter RJ, Cipolla-Neto J (2015) Daily differential expression of melatonin-related genes and clock genes in rat cumulus-oocyte complex: changes after pinealectomy. J Pineal Res 58:490–499
Coomans CP, Lucassen EA, Kooljman S, Fifel K, Deboer T, Rensen PC, Michel S, Meijer JH (2015) Plasticity of circadian clocks and consequences for metabolism. Diabetes Obes Metab 17(Supp1):65–75
Coutu DL, Francois M, Galipeau J (2011) Inhibition of cellular senescence by developmentally regulated FGF receptors in mesenchymal stem cells. Blood 117:6112–6801
Cutando A, Aneiros-Fernandez J, Lopez-Valverde A, Arias-Santiago S, Aneiros-Cachaza J, Reiter RJ (2011) A new perspective in oral health: potential importance and actions of melatonin receptors MT1, MT2, MT3 and RZR/ROR in the oral cavity. Arch Oral Biol 56:944–950
Danet GH, Pan Y, Luongo JL, Bonnet DA, Simon MC (2003) Expansion of human SCID-repopulating cells under hypoxic conditions. J Clin Invest 112:126–135
Dauchy RT, Dauchy EM, Hanifin JP, Gauthreaux SL, Mao L, Belancio VP, Ooms TG, Dupepe LM, Jablonski MR, Warfield B, Wren MA, Brainard GC, Hill SM, Blask DE (2013) Effects of spectral transmissance through standard laboratory cages on circadian metabolism and physiology in nude rats. J Am Assoc Lab Anim Sci 52:146–156
deHaro D, Kines KJ, Sokolowski M, Dauchy RT, Va Streva, Hill SM (2014) Regulation of L1 expression and retro transposition by melatonin and its receptor: implications for cancer risk associated with light exposure at night. Nucleic Acids Res 42:7694–7707
Dominguez-Rodriguez A, Abreu-Gonzalez P, Avanzas P (2012) The role of melatonin in acute myocardial infarction. Front Biosci 17:2433–2441
Dominguez-Rodriguez A, Abreu-Gonzalez P, Picollo R, Galasso G, Reiter RJ (2016) Melatonin is associated with reverse remodeling after cardiac resynchronization therapy in patients with heart failure and ventricular dyssynchrony. Int J Cardiol 221:359–363
Dumont M, Lanctot V, Cadieux-Viau R, Paquet J (2012) Melatonin production and light exposure of rotating night workers. Chronobiol Int 29:203–210
Eom YW, Oh JE, Lee JI, Baik SK, Rhee KJ, Shin HC, Kim YM, Ahn CM, Kong JH, Kim HS, Shim KY (2014) The role of growth factors in maintenance of stemness in bone marrow-derived mesenchymal stem cells. Biochem Biophys Res Commun 445:16–22
Erren TC, Reiter RJ (2008) A generalized theory of carcinogenesis due to chronodisruption. Neuroendocrinol Lett 29:815–821
Erren TC, Reiter RJ (2009) Defining chronodisruption. J Pineal Res 46:245–247
Erren TC, Falaturi P, Reiter RJ (2010) Research into the chronodisruption-cancer theory: the imperative for causal clarification and the danger of causal reductionism. Neuroendocr Lett 31:1–3
Erren TC, Morfeld P, Foster RG, Reiter RJ, Gross JV, Westermann IK (2016) Sleep and cancer: synthesis of experimental data and meta-analyses of cancer incidence among some 1,500,000 study individuals in 13 countries. Chronobiol Int 22:1–26
Escribano BM, Colin-Gonzalez AL, Santamaria A, Tunez I (2014) The role of melatonin in multiple sclerosis, Huntington’s disease and cerebral ischemia. CNS Neurol Disord: Drug Targets 13:096–1119
Ethuwapranee K, Sotthibundhu A, Govitropong P (2015) Melatonin attenuates methamphetamine-induced inhibition of proliferation of adult rat hippocampal progenitor cells in vitro. J Pineal Res 58:418–428
Farez MF, Calandri IL, Correale J, Quintana FJ (2016) Anti-inflammatory effects of melatonin multiple sclerosis. Bioassays
Favero G, Franceschetti L, Rodella LF, Rezzani R (2015) Sirtuins, aging and cardiovascular risk. Age 37:9804
Forman HJ (2016) Redox signaling: an evolution from free radicals to aging. Free Radic Biol Med 97:398–704
Galley HF (2010) Bench-to beside review: targeting antioxidants to mitochondria in sepsis. Crit Care 14:230–239
Gasior SL, Wakeman TP, Xu B, Deininger PL (2006) The human LINE-1 retrotransposon creates DNA double-strand breaks. J Mol Biol 357:1383–1395
Ghosh S, Zhou Z (2015) SIRTain regulators of premature senescence and accelerated aging. Protein Cell 6:322–333
Golombek DA, Rosenstein RE (2010) Physiology of circadian entrainment. Physiol Rev 90:1063–1072
Granados-Fuentes D, Hermanstyne TO, Carrasquillo Y, Nerbonne JM, Herzog ED (2015) IA channels encoded by kv1.4 and Kv4.2 regulate circadian period of PER2 expression in the suprachiasmatic nucleus. J Biol Rhythms 30:396–407
Grinna LS (1977) Changes in cell membranes during aging. Gerontology 23:452–464
Guitart AV, Hammoud M, Dello Sbarba P, Ivanovic Z, Praloran V (2010) Slow-cycling/quiescence balance of hematopoietic stem cells is related to physiological gradient of oxygen. Exp Hematol 38:847–851
Hansen J (2001) Increased breast cancer risk among women who work predominantly at night. Epidemiology 12:74–77
Hardeland R (2012) Melatonin in aging and disease—multiple consequences of reduced secretion, options and limits of treatment. Aging Dis 3:194–225
Hardeland R (2013) Melatonin and the theories of aging: a critical appraisal of melatonin’s role in antiaging mechanisms. J Pineal Res 55:325–356
Hardeland R (2015) Melatonin and circadian oscillators in aging—a dynamic approach to the multiple connected players. Interdiscip Top Gerontol 40:128–140
Hardeland R (2016) Melatonin and synthetic melatoninergic agonists in psychiatric and age-associated disorders: successful and unsuccessful approaches. Curr Pharm Res 22:1086–1101
Hardeland R, Madrid JA, Tan DX, Reiter RJ (2012) Melatonin, the circadian multioscillator system and health: the need for detailed analyses of peripheral melatonin signaling. J Pineal Res 52:139–166
Hardeland R, Cardinali DP Brown GM, Pandi-Perumal SR (2015) Melatonin and brain inflammaging. Prog Neurobiol 127–128:46–63
Haus EL, Smolensky MH (2013) Shift work and cancer risk: potential mechanistic roles of circadian disruption, light at night, and sleep deprivation. Sleep Med Rev 17:273–284
Hawkins K, Joy S, McKay T (2014) Cell signalling pathways underlying induced pluripotent stem cell reprogramming. World J Stem Cells 6:620–628
Hayflick L (1979) The cell biology of aging. J Invest Dermatol 73:8–14
Henden T, Stokkan KA, Reiter RJ, Nonaka K, Lerchl A, Jones DJ (1992) Age-associated reduction in pineal β-adrenergic receptor density is prevented by life-long food restriction in rats. Biol Signals 1:34–39
Herrero A, Barja G (1998) H2O2 production of heart mitochondria and aging rats are slower in canaries and parakeets than in mice: sites of free radical generation and mechanisms involved. Mech Aging Dev 103:133–146
Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, Anderson R, Paschok M, Mann J, Passos JF (2012) Telomeres are favored targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun 3:1–9
Hill SM, Belancio VP, Dauchy RT, Xiang S, Brimer S, Mao L, Hauch A, Lundberg DW, Summers W, Yuan L, Frash T, Blask DE (2015) Melatonin: an inhibitor of breast cancer. Endocr Relat Cancer 22:R183–R201
Hocman G (1979) Biochemistry of aging. Int J Biochem 10:867–876
Hsieh JC, Van Den BD, Kang H, Hsieh CL, Lieber MR (2013) Large chromosome deletions, duplications, and gene conversion events accumulate with age in normal human colon crypts. Aging Cell 12:269–279
Hughes S, Jagannath A, Rodgers J, Hankins MW, Peirson SN, Foster RG (2016) Signaling by melanopsin (OPN4) expressing photosensitive retinal ganglion cells. Eye 30:247–254
Imlay JA, Linn S (1988) DNA damage and oxygen radical toxicity. Science 240:1302–1309
Ingenwerth M, Reinbeck AL, Stahr A, Partke HJ, Roden M, Burkart V, von Gall C (2016) Perturbation of the molecular clockwork in the SCN of non-obese diabetic mice prior to diabetes onset. Chronobiol Int 2:1–7
Ingram DK, Roth GS (2015) Caloric restriction mimetrics: can you have your cake and eat it too? Aging Res Rev 20:46–62
Ikeno T, Yan L (2016) Chronic light exposure in the middle of the night disturbs the circadian system and emotional regulation. J Biol Rhythms 31:352–364
Irmak MK, Sizlan A (2006) Essential hypertension seems to result from melatonin-induced epigenetic modifications in area postrema. Med Hypotheses 66:1000–1007
Jazwinski SM, Kim S, Dai J, Li L, Bi X, Jiang JC, Arnold J, Batzer MA, Walker JA, Welsh DA, Lefante CM (2010) HRAS1 and LASS1 with APOE are associated with human longevity and healthy aging. Aging Cell 9:698–708
Jones DL, Wagers AJ (2008) No place like home: anatomy and function of stem cells. Mol Cell Biol 9:11–21
Jones OR, Gaillard JM, Tuljapunkar S (2008) Senescence rates are determined by ranking on the fast-slow life-history continuum. Ecol Lett 11:664–673
Jones OR, Scheuerlein A, Salguero-Gomez R, Camarda CG, Schaible R, Casper BB (2014) Diversity of ageing across the tree of life. Nature 505:169–173
Jung-Hynes B, Reiter RJ, Ahmad N (2010) Sirtuins, melatonin and circadian rhythms: building a bridge between aging and cancer. J Pineal Res 48:9–19
Kashani IR, Rajabi Z, Akbari M, Hassanzadeh G, Mohseni A, Eramsadati MK, Rafiee K, Bever C, Kipp M, Zendedel A (2014) Protective effects of melatonin against mitochondrial injury in a mouse model of multiple sclerosis. Exp Brain Res 232:2835–2846
Kim S, Bi X, Czarny-Ratajczak M, Dai J, Welsh DA, Myers L, Welsch MA, Cherry KE, Arnold J, Poor LW, Jazwinski SM (2012) Telomere maintenance genes SIRT1 and XRCC6 impact age-related decline in telomere length but only SIRT1 is associated with human longevity. Biogerontology 13:119–131
Kleszczynski K, Fischer TW (2012) Melatonin and skin aging. Dermatoendocrinology 4:245–252
Kovacic P, Somanathan R (2014) Melatonin and circadian rhythm: aging, cancer and mechanism. J Prevent Med 4:545–560
Lamia KA, Storch KF, Wectz CJ (2008) Physiological significance of a peripheral tissue circadian clock. Proc Nat Acad Sci USA 105:15172–15177
Lee E, Iskow R, Yang L, Gokcumen O, Haseley P, Luquette LJ 3rd, Lohr JG, Harris CC, Ding L, Wilson RK, Wheeler DA, Gibbs RA, Kucherlapati R, Lee C, Kharchenko PV, Park RJ (2012) Cancer genome atlas research network. Science 337:967–971
Lee SJ, Jung YH, Oh SY, Yun SP, Han HJ (2014) Melatonin enhances the human mesenchymal stem cells motility via melatonin receptor 2 coupling with Gαq in skin wound healing. J Pineal Res 57:393–407
Lewy AJ, Wehr TA, Goodwin FK, Newsome DA, Markey SP (1980) Light suppresses melatonin secretion in humans. Science 210:1267–1269
Lin H, Yang G, Tan J, Tuan RS (2012) Influence of decellularized matrix derived from human mesenchymal stem cells on their proliferation, migration and multi-lineage differentiation potential. Biomaterials 33:4480–4489
Liu LF, Qian ZH, Qin Q, Shi M, Zhang H, Tao XM, Zhu WP (2015) Effect of melatonin on oncosis of myocardial cells in the myocardial ischemia/reperfusion injury rat and the role of the mitochondrial permeability transition pore. Genet Mol Res 14:7481–7489
Lo JL, Groeger JA, Cheung CH, Dijk DJ, Chee MW (2016) Self reported sleep duration and cognitive performance in older adults: a systematic review and metric-analysis. Sleep Med 17:87–98
Lopez-Gonzalez A, Alvarez-Sanchez N, Lardone PJ, Cruz-Chamorro I, Martinez-Lopez A, Guerrero JM, Reiter RJ, Carrillo-Vico A (2015) Melatonin treatment improves primary progressive multiple sclerosis: a case report. J Pineal Res 58:173–177
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153:1194–1217
Lowes DA, Webster NR, Murphy MP, Galley HF (2013) Antioxidants that protect mitochondria reduce interleukin-6 and oxidative stress, improve mitochondria function, and reduce biochemical markers of organ dysfunction in a rat model of acute sepsis. Brit J Anaesth 110:472–480
Lucas RJ, Peirson SN, Berson DM, Brown TM, Cooper HM, Czeisler CA et al (2014) Measuring and using light in the melanopsin age. Trends Neurosci 37:1–9
Luchetti F, Canonico R, Bartolini D, Arcangeletti M, Ciffolilli S, Murdolo G, Piroddi M, Papa S, Reiter RJ, Galli F (2014) Melatonin regulates mesenchymal stem cell differentiation: a review. J Pineal Res 56:382–397
Manchester LC, Coto-Montes A, Boga JA, Andersen LP, Zhou Z, Galano A, Vriend J, Tan DX, Reiter RJ (2015) Melatonin: an ancient molecule that makes oxygen metabolically tolerable. J Pineal Res 59:403–419
Martin V, Garcia-Santos G, Rodriguez-Blanco J, Casado-Zapico S, Sanchez-Sanchez A, Antolin I, Rodriguez C (2010) Melatonin sensitizes human malignant glioma cells against TRAIL-induced cell death. Cancer Lett 287:216–233
Mattis J, Sehgal A (2016) Circadian rhythms, sleep and disorders of aging. Trends Endocrinol Metab 27:192–203
Mattison JA, Roth GS, Beasley TM, Tilmont EM, Handy AM, Herbert RL, Longo DL, Allison DB, Young JE, Bryant W, Barnard D, Ward WF, Qi W, Ingram DK (2012) Impact for caloric restriction on health and survival in rhesus monkeys from the NIA study. Nuture 489:318–321
Mayo JC, Sainz RM, Tan DX, Antolin I, Rodriguez C, Reiter RJ (2005) Melatonin and Parkinson’s disease. Endocrine 27:169–178
McKiernan SH, Colman RJ, Lopez M, Beasley TM, Aiken JM, Anderson RM, Weindruch R (2011) Caloric restriction delays aging-induced cellular phenotypes in rhesus monkey skeletal muscle. Exp Gerontol 46:23–29
Medeiros CA, Carvalhedo de Bruin PF, Lopes LA, Magalhaes ML, de Lourdes Seabra M, de Bruin VM (2007) Effect of exogenous melatonin on sleep and motor dysfunction in Parkinson’s disease. A randomized, double blind, placebo-controlled study. J Neurol 254:459–464
Meites J (1990) Aging: hypothalamic catecholamines, neuroendocrine-immune interactions, and dietary restriction. Proc Soc Exp Biol Med 195:304–311
Miller E, Morel A, Saso L, Saluk J (2015) Melatonin redox activity. Its potential clinical applications in neurodegenerative disorders. Curr Top Med Chem 15:163–169
Opie LH, Lecour S (2016) Melatonin has multiorgan effects. Eur Heart J Cardiovasc Pharmaocother 2:258–264
Oyewole AO, Birch-Machin MA (2015) Mitochondria-targeted antioxidants. FASEB J 29:4766–4771
Papagiannakopoulos T, Bauer MR, Davidson SM, Heimann M, Subbaraj L, Bhutar Bartlebaugh J, Vander Heiden MG, Jacks T (2016) Circadian rhythm disruption promotes lung tumorigenesis. Cell Metab 24:324–331
Pappolla MA, Sos M, Omar RA, Bick RJ, Hickson-Bick DLM, Reiter RJ, Efthimiopoulos S, Robakis NK (1997) Melatonin prevents death of neuroblastoma cells exposed to the Alzheimer amyloid peptide. J Neurosci 17:1683–1690
Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J (2003) Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 5:741–747
Pauls SD, Honma K, Honma S, Silver R (2016) Deconstructing circadian rhythmicity with models and manipulations. Trends Neurosci 39:405–419
Pechanova O, Paulis L, Simko F (2014) Peripheral and central effects of melatonin on blood pressure regulation. Int J Mol Sci 15:1937–17920
Promislow DEL, Harvey PH (1990) Living fast and dying young: a comparative analysis of life-history variation among mammals. J Zool 220:417–437
Ptacek LJ, Jones CR, Fu YH (2007) Novel insights from genetic and molecular characterization of the human clock. Cold Spring Harbor Symp Quat Biol 72:273–277
Rae M (2004) It’s never too late: calorie restriction is effective in older mammals. Rejuvenation Res 7:3–8
Ramis MR, Esteban S, Miralles A, Tan DX, Reiter RJ (2015) Calorie restriction, resveratrol and melatonin: role of SIRT1 and implications for aging and related diseases. Mech Ageing Dev 146:28–41
Rao D, Yu H, Bai Y, Zheng X, Xie L (2015) Does night-shift work increase the risk of prostate cancer? A systematic review and meta-analysis. Onco Targets Ther 8:2817–2826
Reiter RJ (1995) The pineal gland and melatonin in relation to aging: a summary of the theories and the data. Exp Gerontol 30:199–212
Reiter RJ (1997) Aging and oxygen toxicity: relation to changes in melatonin. Age 20:201–213
Reiter RJ, Tan DX, Kim SJ, Manchester LC, Qi W, Garcia JJ, Cabrera J, El-Sokkary G, Rouvier-Garay V (1999) Augmentation of indices of oxidative damage in life-long melatonin-deficient rats. Mech Aging Dev 110:157–173
Reiter RJ, Tan DX, Korkmaz A, Erren TC, Piekarski C, Tamura H, Manchester LC (2007) Light-at-night, chronodisruption, melatonin suppression and cancer risk: a review. Crit Rev Oncol 13:303–328
Reiter RJ, Tan DX, Korkmaz A, Ma S (2012a) Obesity and metabolic syndrome: association with chronodisruption, sleep deprivation, and melatonin suppression. Ann Med 44:564–577
Reiter RJ, Tan DX, Madrid JA, Erren TC (2012b) When the circadian clock becomes a ticking time bomb. Chronobiol Int 29:1286–1287
Reiter RJ, Tan DX, Galano A (2014a) Melatonin: exceeding expectations. Physiology (Bethesda) 29:325–333
Reiter RJ, Tan DX, Kim SJ, Cruz MH (2014b) Delivery of pineal melatonin to the brain and SCN: role of canaliculi, cerebrospinal fluid, tanycytes and Virchow-Robin perivascular spaces. Brain Str Funct 219:1873–1887
Reiter RJ, Mayo JC, Tan DX, Sainz RM, Alatorre-Jimenez M, Qin L (2016) Melatonin as an antioxidant: under promises but over delivers. J Pineal Res 61:253–278
Rodriguez C, Mayo JC, Sainz RM, Antolin I, Herrera F, Martin V, Reiter RJ (2004) Regulation of antioxidant enzymes: a significant role for melatonin. J Pineal Res 36:1–9
Rosales-Corral SA, Acuna-Castroviejo D, Coto-Montes A, Boga JA, Manchester LC, Fuentes-Broto L, Korkmaz A, Ma S, Tan DX, Reiter RJ (2012) Alzheimer’s disease: pathological mechanisms and the role of melatonin. J Pineal Res 52:167–202
Roth SS, Ingram DK, Lane MA (1999) Caloric restriction in primates: will it work and how will we know. J Am Geriatr Sci 47:896–903
Santos CM (2012) New agents promote neuroprotection in Parkinson’s disease models. CNS Neurol Disorder Drug Targets 11:410–418
Scholtens R, van Munster BC, van Kempen MF, de Rooij SE (2016) Physiological melatonin levels in healthy old people: a systematic review. J Psychosom Res 86:20–27
Scott EC, Gardner EJ, Masood A, Chuang NT, Vertino PM, Devine SE (2016) A hot L1 retrotransposon evades somatic regression and initiates human colorectal cancer. Genome Res 26:745–755
Scott EM (2015) Circadian clocks, obesity and cardiometabolic function. Diabetes Obes Metab 17(Suppl 1):84–89
Shen S, He T, Chu J, He J, Chen X (2015) Uncontrolled hypertension and orthostatic hypotension in relation to standing balance in elderly hypertensive subjects. Clin Interv Aging 10:897–906
Shuai Y, Liao L, Su X, Yu Y, Shao B, Jing H, Zhang X, Dang Z, Jin Y (2016) Melatonin treatment improves stem cell therapy by preserving stemness during long-term in vitro expression. Theranostics 6:1899–1917
Simko F, Baka T, Paulis L, Reiter RJ (2016) Elevated heart rate and nondipping heart rate as potential targets for melatonin: a review. J Pineal Res 61:127–137
Skinner DC, Malpaux B (1999) High melatonin concentrations in third ventricular cerebrospinal fluid are not due to Galen vein blood recirculating through the choroid plexus. Endocrinology 140:4399–4405
Sohal RS, Allen RG (1990) Oxidative stress as a causal factor in differentiation and aging: a unifying hypothesis. Exp Gerontol 25:499–522
Stehle JH, Saade A, Rawasdeh O, Ackermann K, Jilg A, Sebesteny T, Maronde E (2011) A survey of molecular details in the human pineal gland in the light of phylogeny, structure, function and chronobiological diseases. J Pineal Res 51:17–43
Steinberg D, Witztum JL (1990) Lipoproteins and atherogenesis. J Am Med Assoc 264:3047–3052
Stokkan KA, Reiter RJ, Nonaka KO, Lerchl A, Yu BP, Vaughan MK (1991) Food restriction retards aging of the pineal gland. Brain Res 545:66–72
Tan DX, Chen LD, Poeggeler B, Manchester LC, Reiter RJ (1993) Melatonin: a potent endogenous hydroxyl radical scavenger. Endocr J 1:57–60
Tsai CC, Su PF, Huang YF, Yew TL, Hung SC (2012) Oct4 and Nanog directly regulate Dnmt1 to maintain self-renewal and undifferentiated state in mesenchymal stem cells. Mol Cell 47:162–182
van Wamelen DJ, Roos RA, Aziz NA (2015) Therapeutic strategies for circadian rhythm and sleep disturbances in Huntington disease. Neurodegener Dis Manag 5:549–559
Vaughan GM, Pelham RW, Pang SF, Loughlin LL, Wilson KM, Sandock KL, Vaughan MK, Koslow SH, Reiter RJ (1976) Nocturnal elevation of plasma melatonin and urinary 5-hydroxyindoleacetic acid in young men: attempts at modification by brief changes in environmental lighting and sleep and by autonomic drugs. J Clin Endocrinol Metab 42:752–764
Venegas C, Garcia JA, Escames G, Ortiz F, Lopez A, Doerrier C, Garcia-Corzo L, Lopez LC, Reiter RJ (2012) Extrapineal melatonin: analysis of its subcellular distribution and daily fluctuations. J Pineal Res 52:217–227
Vijg J, Dolle ME (2002) Large genome rearrangements as a primary cause of aging. Mech Aging Dev 123:907–915
Vijg J, Suh Y (2013) Genome instability and aging. Annu Rev Physiol 75:645–668
Wang F, Li J, Wu C, Yang J, Xu F, Zhao Q (2003) The GABA(A) receptor mediates the hypnotic activity of melatonin in rats. Pharmacol Biochem Behav 74:573–578
Weishaupt JH, Bartels C, Polking E, Dietrich J, Rohde G, Poeggeler B, Mertens N, Sperling S, Bohn M, Huther G, Ehrenreich H (2006) Reduced oxidative damage in ALS by high-dose enteral melatonin treatment. J Pineal Res 41:313–323
Wen J, Ariyannur PS, Ribeiro R, Tanaka M, Moffett JR, Kirmani BF, Namboodiri AM, Zhang Y (2016) Efficacy of N-acetylserotonin and melatonin in the EAE model of multiple sclerosis. J Neuroimmune Pharmacol 11:763–773
Wu J, Issa JP, Herman J, Bassett DE Jr, Nelkin BD, Baylin SB (1993) Expression of an exogenous eukaryotic DNA methyltransferase gene induces transformation of NIH 3T3 cells. Proc Natl Acad Sci USA 90:8891–8895
Xiang S, Dauchy RT, Hauch A, Mao L, Yuan L, Wren MA, Belancio V, Mondal D, Frash T, Blask DE, Hill SM (2015) Doxorubicin resistance in breast cancer is driven by light at night-induced disruption of the circadian melatonin signal. J Pineal Res 59:60–69
Yip HK, Chang YC, Wallace CG, Chang LT, Tsai TH, Chen YL, Chang HW, Leu S, Zhen YY, Tsai CY, Yeh SH, Sun CK, Yen CH (2013) Melatonin treatment improves adipose-derived mesenchymal stem therapy for acute lung ischemia-reperfusion injury. J Pineal Res 54:207–221
Zhang S, Wang P, Ren L, Hu C, Bi J (2016) Protective effect of melatonin soluble Aβ1-42-induced memory impairment, astrogliosis, and synaptic dysfunction via the Musashi1/Notch1/Hes1 signaling pathway in the rat hippocampus. Alzheimer’s Res Ther 8
Zhou L, Chen X, Liu T, Gong Y, Chen S, Pan G, Cui W, Luo ZP, Pei M, Yang H, He F (2015) Melatonin reverses H2O2-induced premature senescence in mesenchymal stem cells via the SIRT1-dependent pathway. J Pineal Res 59:190–205
Zs-Nagy I (1978) A membrane hypothesis of aging. J Theoret Biol 75:189–195
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Reiter, R.J., Rosales-Corral, S.A., Tan, D.X., Alatorre-Jimenez, M., Lopez, C. (2017). Circadian Dysregulation and Melatonin Rhythm Suppression in the Context of Aging. In: Jazwinski, S., Belancio, V., Hill, S. (eds) Circadian Rhythms and Their Impact on Aging. Healthy Ageing and Longevity, vol 7. Springer, Cham. https://doi.org/10.1007/978-3-319-64543-8_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-64543-8_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-64542-1
Online ISBN: 978-3-319-64543-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)