
Abstract
Androgen receptor (AR) is a hormone-dependent transcription factor, involved in male dimorphism from embryogenesis to adulthood and highly expressed in the prostate. Upon activation by androgen binding, AR translocates to the nucleus, homodimerizes, and activates the transcription of specific androgen-responsive genes. This signaling pathway is subject to regulation by numerous factors such as AR transcriptional cofactors and co-regulators, posttranslational AR modifications, and crosstalk with other signaling pathways. Androgen signaling is required for the development and maintenance of normal prostate but also plays a key role in the initiation and progression of prostate cancer. Therefore, targeting the AR pathway is the first line of intervention for treating this disease. Unfortunately, most treated patients develop resistance through a variety of mechanisms that, for the most part, reactivate AR signaling. These mechanisms involve alterations of AR itself (amplification, mutation, alternative splicing, and posttranslational modifications), alterations of co-regulators, and/or activation through crosstalk with other signaling pathways. Although newer AR-directed strategies are being developed and tested in the clinical setting, the most effective approach in treating lethal prostate cancer remains an active area of research.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Androgen receptor
- Androgen signaling
- Prostate transcription factor
- Castration-resistant prostate cancer
- Treatment resistance
- Pathway crosstalk
Androgen Receptor (AR): Description and Function
AR Gene and Protein Description
The androgen receptor (AR) is encoded by a 186,588 base-pair, 8-exon gene located on chromosome X (Xq11–12) and belongs to the steroid hormone nuclear receptor superfamily. AR expression is cell-dependent and regulated by androgen [1]. The AR gene encodes for a 919 amino acid nuclear receptor (~110 kDa) that acts as a hormone-dependent DNA-binding transcription factor. This modular protein is composed of four domains [2]: the N-terminal domain (NTD) , the DNA-binding domain (DBD) , the hinge region , and the C-terminal domain (CTD) . The NTD contains residues involved in the recruitment of transcriptional co-regulators and is required for the activation of transcriptional activity. The autonomous transcriptional activation function (AF1) of the NTD is ligand-independent and required for maximal activity of AR [3]. The DBD is composed of two zinc-finger motifs involved in the recognition of specific DNA sequences, located in AR target genes [4, 5], known as androgen response elements (AREs : canonical consensus sequence = 5′-AGAACA-3′ in direct repeats or inverted repeats separated by a spacer of three base pairs for classical and selective AREs, respectively [6]). Noncanonical elements have also been described but are bound by AR with a lower affinity [7]. The DBD is separated from the CTD by the hinge region , a flexible region that includes the nuclear localization signal sequence (NLS). The CTD contains the ligand-binding domain (LBD) , which is formed by 12 α[alpha]-helix arranged in a globular structure surrounding the hydrophobic ligand-binding cavity, and a second transcriptional activation function (AF2). The AF2 is ligand-dependent and has a synergistic effect with AF1 that results in full AR transactivation [8]. Despite a high similarity of sequence between members of the hormone nuclear receptor superfamily, some key residues in the AR AF2 core domain are essential for AR functionality and make it distinct from the other hormone nuclear receptors [9]. The CTD also contains the nuclear export signal (NES), which is used to export AR to the cytoplasm upon ligand withdrawal.
The Role of AR in Normal Development
AR is weakly expressed in numerous tissues and cells yet highly expressed in the adipocytes, liver, and prostate (http://biogps.org/#goto=genereport&id=367), directly linking its function to male dimorphism. Following androgen activation, AR participates widely in the male reproductive tract including gonadal development and maintenance [10]. During embryogenesis, AR signaling regulates the reproductive tract patterning by determining the Wolffian duct differentiation and inducing the development of the male reproductive tissues [11, 12]. In adults, AR remains essential for seminiferous tubule and prostate function in terms of epithelial and stromal compartment maintenance [13] and maintenance of paracrine factor-mediated secretory functions of the epithelial cells [14]. AR signaling is also involved in other physiological processes linked to sexual dimorphism, such as muscle development, lipid accumulation, and bone homeostasis [13].
AR Transcriptional Regulation
The activation of the AR signaling pathway depends on ligand binding to AR. In the absence of ligand, AR is localized in the cytoplasm and forms a complex with HSP90/HSP70-based multiprotein chaperone machinery, protecting it from degradation [15]. Barring 10% that is produced in the adrenal cortex, the vast majority of circulating androgen is testosterone produced by the testes. Circulating testosterone enters prostate epithelial cells and is rapidly converted to the potent AR ligand 5α[alpha]-dihydrotestosterone (5α[alpha]-DHT) by the enzyme 5α[alpha]-reductase to DHT (dihydrotestosterone or androstanolone) [16]. DHT binds the LBD of AR with a twofold higher affinity and a fivefold decreased dissociation rate compared to its precursor [17]. Binding of DHT to the LBD of AR induces conformational changes resulting in AR dissociation from the HSP90/HSP70 complex and the exposition of the AF2 domain and the NLS. AR is then shuttled to the nucleus via the microtubule network [18, 19]. The AR NLS interaction with importin-α[alpha] and importin-β[beta] mediates the translocation of AR across the nuclear membrane [20]. Upon entry into the nucleus, AR homodimerizes and binds to AREs associated with AR target genes. These sequences are found in both proximal promoters and distal enhancers, up to several thousand base pairs upstream or downstream of the transcription start site [21]. The AR cistrome has been defined by multiple research groups in cell lines, such as LNCaP [22,23,24] and VCaP cells [25], in transgenic mouse models [26], and in human clinical samples of prostate cancer tissues [27] including castrate-resistant prostate cancer [28]. These studies clearly demonstrated that the AR cistrome is reprogrammed in human prostate tumorigenesis [27, 28]. AR binding to AREs leads to recruitment of co-regulators (replacement of transcriptional corepressors with coactivators), general transcription factors, and RNA polymerase II to induce transcription activation. Two well-described AR target genes and indicators of androgen signaling pathway activation are prostate-specific antigen (PSA) and transmembrane protease, serine 2 (TMPRSS2). A noncanonical pathway of androgen signaling, independent of androgen binding and involving cofactors or crosstalk with other intracellular signaling pathways, has also been characterized.
AR Signaling Regulation
AR Co-regulators and Cofactors
AR activity is modulated through an interaction with specific cofactors. Depriest et al. recently generated a database referencing 274 AR-associated co-regulator genes [29]. According to Heemers and Tindall [30], AR-interacting proteins can be classified into three main groups: (1) general transcription factors, comprising the classical transcriptional machinery, (2) co-regulators that shift the balance toward expression or repression of the transcriptional activity, and (3) specific transcription factors.
The first class includes TFIID/B/F proteins , required for the recruitment of RNA polymerase II, and TFIIE/H. Of these, AR directly interacts with TFIIF, TFIIH, and polymerase II through its RPB2 subunit.
The second class of cofactors is composed of more than 160 proteins [30] including components of the chromatin remodeling complex (e.g., ARIP4 [31], BAF57 [32], the SWI3-related gene product SRG3 [33], and SRCAP [34]) and histone modifiers (e.g., members of the p160 SRC gene family [35, 36]). P300 [37] and CREB-binding protein (CBP) also directly acetylate AR through their acetyltransferase activity. This acetylation allows for the recruitment of other coactivators to serve as molecular bridges between AR and the transcriptional machinery [37]. Histone modifiers either promote (e.g., demethylases such as LSD1 and JMJD2C [38, 39]) or repress (e.g., deacetylases such as SIRT1 [40]) AR-mediated transcriptional activity. AR also interacts with ubiquitination/proteasome and SUMOylation pathway components, proteins involved in splicing and RNA metabolism, DNA repair proteins, chaperones, cell cycle proteins, signal integrators, and apoptosis regulators [30, 41].
The third category of AR-interacting proteins [30] regulates AR signaling by defining the temporal, spatial, and functional binding pattern of AR [42]. This class of cofactors acts by modifying the interaction of AR with DNA [43], titrating other co-regulators [44], or recruiting AR on non- or partial AREs [42]. For example, DNA motifs recognized by the three transcription factors FoxA1, GATA2, and Oct1 are enriched at AR half-site motifs [42]. FoxA1 is a known pioneering factor that is essential for maximal prostatic gene activation [45] by facilitating AR binding on FoxA1-dependent AR binding sites. More recently, FoxA1 has been shown to regulate AR function by masking AR binding sites, which become functional upon FoxA1 depletion [23]. While FoxA1-AR interaction is not affected by ligand binding, AR interacts with GATA2 and Oct1 in a hormone-dependent manner. These collaborating partners have distinct functional roles in androgen-dependent gene transcription and cell proliferation [42].
These observations highlight the balance between coactivators and corepressors of AR and their importance on its activity. AR cofactors are differentially expressed in prostate cancer and have the potential to drive disease progression.
AR Protein Posttranslational Modifications and Signaling Crosstalk
Although androgen binding is the primary means for AR activation, protein posttranslational modifications also influence AR activity. AR, at the protein level, can be altered by up to five well-described modifications (phosphorylation, acetylation (discussed above), SUMOylation, methylation , and ubiquitination) on a subset of 23 different amino acids [46].
AR phosphorylation occurs at serine 16, 81, 256, 308, and 424 in the presence of androgen, but the impact of phosphorylation at any one of these sites in terms of AR activity remains unclear. For example, stress-induced JNK1 phosphorylation on serine 650 regulates nuclear export of AR, antagonizing AR-mediated transcription [47]. AR phosphorylation at specific tyrosines results from specific growth factor signaling. Growth factors such as IGF-I (insulin-like growth factor-I) , KGF (keratinocyte growth factor ), and EGF (epidermal growth factor) are able to activate androgen signaling through AR phosphorylation, leading to an increase of PSA level. This activation is inhibited by the AR antagonist Casodex, highlighting the specificity of the mechanism for AR [48]. EGF, for example, induces the activity of Src and Ack1 kinases, which, in turn, phosphorylate AR at tyrosines 534 and 267, respectively. Phosphorylation at these sites increases AR transcriptional activity by enhancing its nuclear translocation and DNA binding. EGF can also modify AR activity by inducing IL-6 upregulation in prostate cancer cells.
AR SUMOylation , occurring at regulatory amino acid SUMO acceptor motifs at lysines 386 and 520, results in an inhibition of both the ligand-activated and the basal ligand-independent activity of AR [49]. AR is also regulated by mono- and polyubiquitination , which impact the stability and turnover of the protein or its activity, depending on the topology of the polyubiquitin chains [28, 50, 51]. The position of the lysine residue used for ubiquitin chain branching dictates the fate of the substrate. For example, the polyubiquitination of AR mediated by MDM2 induces AR degradation by the 26S proteasome [51], while the one driven by RNF6 promotes AR transcriptional activity [50].
AR Involvement in Primary Prostate Cancer
After skin cancer, prostate cancer is the most common and the third most lethal form of cancer in men in the United States, with 161,360 estimated new cases in 2017 [52]. Over 50 years ago, Huggins’ work [53, 54] demonstrated a direct relation between androgen and prostate cancer, reporting a regression of prostate cancer after orchiectomy. High levels of AR in prostate cancer luminal epithelial cells are associated with a high tumor grade, deregulation of cell-cycle genes [55,56,57], inhibition of apoptosis [58], increased angiogenesis [59], and crosstalk with PI3K-AKT-PTEN, RAF, Wnt, and DNA repair signaling pathways [60]. AR has also been implicated in the development of chromosomal rearrangements , such as the TMPRSS2-ERG gene fusion, detected in around 50% of prostate cancer patients [61,62,63]. Clinically, androgen signaling is monitored using the prostate-specific antigen (PSA) level, encoded by the AR target gene KLK3 [64]. The level of circulating PSA is measured to track prostate cancer progression and disease recurrence in the context of androgen deprivation therapy (ADT) [64,65,66], which is the first line of treatment for advanced prostate cancers. Current ADT approaches are aimed at chemically lowering circulating testosterone levels by the administration of luteinizing hormone-releasing hormone (LHRH ) analogs . Since these approaches target only 90% of androgen production (not from the adrenal glands), they are often used in combination with the classic antiandrogen compounds (e.g., flutamide, bicalutamide, and nilutamide). Despite encouraging initial response following ADT, relapse occurs for almost all cases within several months and leads to a more aggressive form of prostate cancer defined as castration-resistant prostate cancer (CRPC).
Mechanisms of AR Reactivation Associated with CRPC
There are several mechanisms of resistance associated with the onset of metastatic CRPC (mCRPC) tumors, among which include AR-related alterations (e.g., AR gene amplification or mutations [60, 67, 68], alternative splicing of AR mRNA [69,70,71,72], and posttranslational modifications of AR protein (Fig. 20.1)), crosstalk with other cancer-promoting signaling pathways, genomic alterations involving cofactors/co-regulators and other AR signaling proteins, and intraprostatic generation of androgen [73, 74]. These findings have led to the development of second-generation antiandrogens, which are improved AR antagonists (e.g., enzalutamide) or inhibitors targeting the biosynthesis of AR (e.g., abiraterone acetate). Enzalutamide (MDV3100) is a targeted AR inhibitor that competitively binds to the LBD of the androgen receptor and inhibits androgen-receptor translocation to the cell nucleus, recruitment of AR cofactors, and AR binding to DNA [75, 76]. The 17α[alpha]-hydroxylase/C17,20-lyase (CYP17) inhibitor abiraterone acetate acts as an antagonist to AR and inhibits 3β[beta]-hydroxysteroid dehydrogenase blocking androgen synthesis in the adrenal glands, testes, and within the prostate tumor [77, 78]. Despite improved response rates and overall survival with these molecules [79], almost all metastatic CRPC patients develop resistance to these agents as well. Recent genomic sequencing studies of large cohorts with resistance to these molecules have recently been reported and show further AR signaling reactivation alterations [60]. Below, we discuss the mechanisms, linked to AR or AR signaling, known to be involved in the resistance of mCRPC to second-generation ADT.

AR modifications associated with castrate-resistant prostate cancer development. (a) Wild-type AR gene, RNA, and protein. (b–d) Alterations to the AR gene DNA, mRNA, or protein-associated androgen deprivation therapy resistance. These alterations result in constitutive activation of AR, due to mutation in the LBD (b) or splicing variant (c). (d) The most common posttranslational modifications of AR that enhance its transcriptional activity and that are driven by one of the AR cofactors (e.g., p300) or crosstalk with other signaling pathways (e.g., AKT, MAPK, Ack, and Src). Black arrows represent phosphorylation, while the red arrow represents acetylation. CE = cryptic exon, NTD = N-terminal domain, DBD = DNA-binding domain, hinge = hinge domain, LBD = ligand-binding domain
AR Gene Amplification
Early studies using both targeted or genome-wide approaches of hormone-naïve versus hormone-refractory primary prostate cancers led to the finding of an acquired increased copy number (up to 60 copies per cell) at chromosome Xq11–13 including the genomic loci of AR in roughly 30% of recurrent tumors [80,81,82]. Other studies have also reported an AR amplification in more than 50% of circulating tumor cells (CTC) from metastasized CRPC [83, 84]. This observation is consistent with the frequency of AR amplification found in recent genome sequencing studies [60, 85]. AR amplification drives its overexpression and increases the likelihood of androgen-AR interaction, thus reactivating the AR signaling pathway.
AR and AR-Associated Gene Mutations
AR mutations in the context of CRPC were first described roughly 20 years ago [67, 68] and have since been characterized in around 20% of CRPC [86]. Numerous AR mutations have been described in prostate cancer, approximately 45% of which are somatic single-base substitution occurring in the LBD [87]. Several mutations in this region affect the ligand specificity of AR, allowing its activation by non-androgenic steroids or antiandrogens [88]. Recent genomic sequencing analyses of metastatic prostate cancers have shed a considerable amount of light regarding mutations to AR and AR signaling genes such as NCOR1, NCOR2, FOXA1, and NKX3.1 [85, 86, 89,90,91]. The most recent study, based on a large sequenced CRPC patient cohort treated with the most up-to-date standard-of-care antiandrogen therapy (abiraterone or enzalutamide) or through a cohort of prospective clinical trials (n = 150), found that upward of 70% of cases harbored AR pathway aberrations [60]. The majority (63%) of alterations impacted AR directly, through amplifications and mutations including hotspot mutations that confer agonism to AR antagonists such as flutamide (T878A) and bicalutamide (W742C) [92]. This agonism to enzalutamide has also been described with the F876L mutation [93] as well as to glucocorticoids in case of L702H mutation. In addition to AR mutations itself, Robinson et al. also observed alterations in AR pathway members such as NCOR1, NCOR2, and FOXA1 [51] and AR-associated genes such as deletion of ZBTB16 [94, 95] and SPOP mutations [89, 96].
AR Splice Variants
More than 20 different AR variants have been described in preclinical or clinical CRPC samples ([60, 69,70,71,72], reviewed by Lu C. and Luo J. [97] and more recently by Wadosky and Koochekpour [98]). The AR variants are generated from multiple alternative splicing events (e.g., aberrant splicing, inclusion of an alternative exon, or insertion of cryptic exons) of the AR mRNA. Structural alterations in the AR gene resulting in AR variant expression have also been described [99, 100]. Insertions of cryptic exons downstream of the sequences encoding the DBD or deletions of the exons encoding the LBD result in a truncated AR protein devoid of the functional LBD. AR splice variants that lack the LBD (encoded by exons 5–6-7–8) generate constitutively active forms of AR [70, 71]. The activity of these AR variants is no longer regulated by androgens. They are thus resistant to antiandrogen therapies and constitutively activate the AR signaling pathway [71, 72]. Moreover, while AR is translocated to the nucleus via microtubule transport, AR-V7 (the most characterized AR variant lacking the LBD) exploits another way to its translocation that is still under investigation [101]. AR-V7 and AR-v567es are the most commonly detected AR variants in prostate cancer and thus, the most studied to date (Fig. 20.1). A genome-wide occupancy study using ChIP-seq found that AR variants bind DNA as dimers and display a binding preference for the same canonical high-affinity AREs that are engaged by AR-FL, albeit with lower affinity [102]. While initially described as heterodimers with AR full length (AR-FL), the variants have since been implicated in homodimerization and driving AR signaling independently of AR-FL [102,103,104,105]. Based on nuclear AR expression using N- and C-terminal-specific AR antibodies, Zhang et al. found an increase in the prevalence of AR variants in CRPC clinical samples compared to primary prostate cancer [106]. Another study of 13 CRPC bone metastasis samples found that the level of AR variant protein constituted 32% (range 0–95%) of the AR full length. Meanwhile, the RNA level was relatively weak compared to the full length, suggesting that AR variants could be posttranscriptionally stabilized in CRPC [107]. AR-FL, AR-V1, and AR-V7 transcripts were detected in most of the nonmalignant primary tumors and metastatic samples examined, while the AR-V567es transcript was detected in only 7 (23%) CRPC bone metastases. The expression of these variants is also associated with a poor prognosis of patients, most likely due to their constitutive activation [107]. AR variants drive androgen-independent cell proliferation in a manner that is resistant to antiandrogens, including enzalutamide [108], and are widely expressed in the context of metastatic CRPC (SU2C cohort) and to a lower extent in pre-abiraterone/enzalutamide primary prostate cancer (TCGA cohort [60]). While controversial [109], AR-V7 expression has been associated with abiraterone and enzalutamide resistance [110,111,112,113]. More recently AR-V9 has also been associated with abiraterone resistance [114]. AR variants are hypothesized to induce epithelial-to-mesenchymal transition and stem cell phenotypes [115], but a further validation of this notion is needed.
AR Signaling and Crosstalk with Other Signaling Pathways Associated with CRPC
Many signaling pathways interacting with AR have been observed as altered or dysregulated in prostate cancer cells. For example, (1) the loss of PTEN and subsequent activation of PI3K/AKT are critical event in human prostate cancer [116, 117], (2) increased expression of EGFR correlates with the evolution of prostate cancer [118], (3) elevated circulating IL-6 and IL-8 levels have been observed and associated with advanced prostate cancer cases [119,120,121,122,123], and (4) members of SRC family have been described as increased in prostate cancer, even at higher levels in CRPC [124, 125].
Some of them have been shown to enhance AR signaling in the context of CRPC that arises either as a feedback following androgen withdrawal and/or compensation from growth factors and other signaling ligands. The expressions of several peptide growth factors, such as EGF/TGFα[alpha] and IGF-1 , have been shown to be increased during progression to CRPC [118, 123, 126] and either induce AR transcriptional activity irrespective of androgen stimulation or sensitize AR to low concentrations of androgens (Fig. 20.2) [48, 127, 128]. More recently, another growth factor, CXCL12 , has been characterized as androgen-independent AR activator in prostate cells [129]. Interleukins are also able to induce androgen-independent AR activity. IL-6 (interleukin-6), a multifunctional cytokine produced by prostate cells, binds to its specific receptor and induces a signaling cascade including JAK, STAT3, and p300. The N-terminal domain of AR directly interacts with STAT3, after IL-6 induction through phosphorylation of mitogen-activated protein kinase (MAPK) pathway [130]. This interaction leads to the activation of the AR NTD. IL-8 is also able to increase AR expression and promote its activity in an androgen-independent manner [131]. In addition, several protein kinases (e.g., MAPK, Akt/PKB, PKA, and PKC) and non-receptor tyrosine kinases (ERBB2/HER-2/neu, Src, FAK, and Etk/BMX) modulate AR activity by direct phosphorylation of serine/threonine or tyrosine residues, respectively, on AR or one of its cofactors (e.g., TIF2 and SRC1) [127, 128, 132]. The ERBB2/HER-2/neu tyrosine kinase modulates AR signaling [133], through MAP kinase pathway [134] or AKT pathway [135] or when associated with ERBB3 through a mechanism that remains to be elucidated [136]. As mentioned above, some specific AR polyubiquitinations serve as negative regulators of AR by enhancing its degradation [51]. This degradation is inhibited by HER2/ERBB3/PI3-kinase pathway in the context of hormone-refractory prostate cancer, providing a mechanism of enhanced AR stability as an additional mechanism of resistance [136]. PKA (protein kinase A), whose activity is dependent on the cellular level of cAMP, activates AR in the absence of androgen (Fig. 20.2) [137, 138]. There is a well-described and dynamic interplay between PI3K/AKT/mTOR and AR signaling axes during prostate cancer progression as well as a mechanism of ADT resistance. In the presence of androgen, AKT phosphorylates AR on Ser 213 and Ser 791, inducing a modification in AR signaling [139,140,141]. However, activation of the PI3K/AKT/mTOR pathway resulting from PTEN loss is associated with androgen insensitivity and the development of CRPC [142]. To address the mechanism underlying this finding, two independent groups found that the loss of PTEN in prostates results in a decrease in transcription of AR target genes through derepression of negative regulators of AR activity, EGR1, and c-Jun [143, 144]. In addition, loss of AR signaling either through genetic or pharmacological manipulation with enzalutamide leads to a reduction of FKBP5, an AR target gene. Low FKBP5 levels lower the AKT phosphatase and negative regulator PHLPP protein levels. In addition, mTOR inhibition in the background of PTEN loss leads to an increase in AR levels through upregulation of HER3, which increases AR stability. Altogether, these data show how PI3K/AKT/mTOR pathway activity in the context of CRPC alters the need of the restricted levels of circulating androgens.

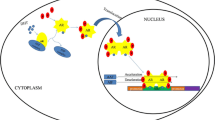
Androgen-dependent and independent AR activation . AR can induce androgen-signaling pathway upon androgen binding (on the left) or activation through interaction with other signaling pathways (on the right)
Moreover, an increase of AR acetylation enhances the binding of p300 on AR, reducing N-CoR/HDAC/Smad3 corepressor binding. This effect leads to a modulation of the transcriptional activation on AR-responsive genes, resulting in an aberrant cell growth in prostate cancer stable cells [37].
The activity of the glucocorticoid receptor (GR) has also been described as a potential mechanism of resistance to enzalutamide and ARN-509 in a preclinical model and has been confirmed in patient samples [145]. Glucocorticoids administrated at a low dose inhibit adrenocorticotropic hormone (ACTH) production by the pituitary and initially result in reduced androgen levels. However, a high expression of GR by the prostate cancer will result in GR activation in tumor cells. In this context, a more efficient strategy could be to combine AR and GR inhibition. This is currently being explored in an early phase clinical trial combining enzalutamide and mifepristone (NCT02012296).
Next Generation of AR-Targeted Therapies
Even in the face of potent second-line AR antagonists (enzalutamide) or CYP17 inhibitors (abiraterone), metastatic CRPC tumors continue to evolve different mechanisms to reactivate AR signaling, which has spurned the development of further agents targeting the AR signaling axis. New inhibitors have emerged that efficiently block both AR full length and variant action by targeting the N-terminal domain of AR [146, 147] or inducing degradation of AR mRNA [148] or protein [149,150,151]. None of them have been fully tested on patients and FDA-approved. One of the two most promising compounds is EPI-506 (binds the NTD of AR), which is currently in phase I/II clinical trial (NCT02606123). While the patients will not be selected based on AR-V7 status, responses to treatment will be stratified based on AR-V7 expression in CTCs. The second promising compound is the FDA-approved niclosamide, which promotes AR-V7 degradation and potentially restores the sensitivity of the tumors to second-generation ADT [152,153,154]. Two clinical trials are currently ongoing to assess the efficiency of niclosamide in combination with enzalutamide (NCT02532114) or abiraterone (NCT02807805).
The concept of bipolar androgen therapy is also emerging. This is based on the observation that the growth of AR-positive human CRPC cell lines is inhibited by supraphysiologic levels of androgens [155,156,157]. This has recently been the subject of a pilot clinical trial [158]. These newer strategies may also prove beneficial in combination with inhibitors targeting key crosstalk pathways (e.g., PI3K/AKT/mTOR or ERBB2/Her2/neu) or microtubule-targeting agents (e.g., taxanes). One consequence of taxane treatment is the inhibition of AR nuclear trafficking [18, 19, 159, 160]. This mechanism predicts synergy between effective AR-targeted therapy and taxanes, clinically validated by the unprecedented survival results for men with advanced, hormone-naïve prostate cancer in CHAARTED and STAMPEDE clinical trials (Sweeney ASCO 2014; James ASCO 2015 [161,162,163]).
Conclusion
Androgen signaling is a cellular pathway activated upon androgen binding to its specific receptor AR, leading to the transcriptional activation of androgen-responsive genes. Regulation of this pathway occurs through the action of numerous co-regulators of AR and is influenced by crosstalk from other signaling pathways in the cell and the microenvironment. Activation of AR is essential for the male dimorphism and also determinant for the development and maintenance of the prostate. Androgen signaling is also essential for the maintenance and progression of prostate cancer, making chemical castration, in the case of non-organ confined disease, the first line of intervention. While the involvement of androgen receptor in prostate cancer progression is established, the therapeutic strategy targeting androgen signaling-driven prostate cancer still needs to be improved for the incurable forms of the disease.
Despite continued hormonal therapy, including the most robust and potent second-line antiandrogens (e.g., enzalutamide) or CYP17 inhibitors (e.g., abiraterone), metastatic CRPC tumors evolve complex and ever-adapting mechanisms to reactivate AR signaling or other mechanisms that render the tumor cells indifferent to AR signaling. Recent evidence shows that neuroendocrine prostate cancer can arise in later stages of prostate cancer progression from a pre-existing adenocarcinoma during the course of treatment resistance to AR-directed therapies [164]. This is as an adaptive resistance mechanism.
The complexity and variability of mechanisms of resistance that have been described to date emphasize the growing need for better model systems that recapitulate clinically relevant mechanisms and a precision strategy adapted to each patient. The synergistic effect observed in recent combinatory treatments also highlights the important question of the timing, order, and/or combination of drugs in future strategies.
References
Blok LJ, Themmen AP, Peters AH, Trapman J, Baarends WM, Hoogerbrugge JW, et al. Transcriptional regulation of androgen receptor gene expression in Sertoli cells and other cell types. Mol Cell Endocrinol. 1992;88(1-3):153–64.
Brinkmann AO, Faber PW, van Rooij HC, Kuiper GG, Ris C, Klaassen P, et al. The human androgen receptor: domain structure, genomic organization and regulation of expression. J Steroid Biochem. 1989;34(1-6):307–10.
Callewaert L, Van Tilborgh N, Claessens F. Interplay between two hormone-independent activation domains in the androgen receptor. Cancer Res. 2006;66(1):543–53.
Nelson PS, Clegg N, Arnold H, Ferguson C, Bonham M, White J, et al. The program of androgen-responsive genes in neoplastic prostate epithelium. Proc Natl Acad Sci U S A. 2002;99(18):11890–5.
Shaffer PL, Jivan A, Dollins DE, Claessens F, Gewirth DT. Structural basis of androgen receptor binding to selective androgen response elements. Proc Natl Acad Sci U S A. 2004;101(14):4758–63.
Claessens F, Denayer S, Van Tilborgh N, Kerkhofs S, Helsen C, Haelens A. Diverse roles of androgen receptor (AR) domains in AR-mediated signaling. Nucl Recept Signal. 2008;6:e008.
Wang Q, Carroll JS, Brown M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol Cell. 2005;19(5):631–42.
Matsumoto T, Sakari M, Okada M, Yokoyama A, Takahashi S, Kouzmenko A, et al. The androgen receptor in health and disease. Annu Rev Physiol. 2013;75:201–24.
Slagsvold T, Kraus I, Bentzen T, Palvimo J, Saatcioglu F. Mutational analysis of the androgen receptor AF-2 (activation function 2) core domain reveals functional and mechanistic differences of conserved residues compared with other nuclear receptors. Mol Endocrinol. 2000;14(10):1603–17.
Wilson JD. The role of androgens in male gender role behavior. Endocr Rev. 1999;20(5):726–37.
Murashima A, Kishigami S, Thomson A, Yamada G. Androgens and mammalian male reproductive tract development. Biochim Biophys Acta. 2015;1849(2):163–70.
Renfree MB, Fenelon J, Wijiyanti G, Wilson JD, Shaw G. Wolffian duct differentiation by physiological concentrations of androgen delivered systemically. Dev Biol. 2009;334(2):429–36.
De Gendt K, Verhoeven G. Tissue- and cell-specific functions of the androgen receptor revealed through conditional knockout models in mice. Mol Cell Endocrinol. 2012;352(1-2):13–25.
Kurita T, Wang YZ, Donjacour AA, Zhao C, Lydon JP, O'Malley BW, et al. Paracrine regulation of apoptosis by steroid hormones in the male and female reproductive system. Cell Death Differ. 2001;8(2):192–200.
Pratt WB, Galigniana MD, Morishima Y, Murphy PJ. Role of molecular chaperones in steroid receptor action. Essays Biochem. 2004;40:41–58.
Penning TM, Jin Y, Rizner TL, Bauman DR. Pre-receptor regulation of the androgen receptor. Mol Cell Endocrinol. 2008;281(1-2):1–8.
Grino PB, Griffin JE, Wilson JD. Testosterone at high concentrations interacts with the human androgen receptor similarly to dihydrotestosterone. Endocrinology. 1990;126(2):1165–72.
Darshan MS, Loftus MS, Thadani-Mulero M, Levy BP, Escuin D, Zhou XK, et al. Taxane-induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer Res. 2011;71(18):6019–29.
Thadani-Mulero M, Nanus DM, Giannakakou P. Androgen receptor on the move: boarding the microtubule expressway to the nucleus. Cancer Res. 2012;72(18):4611–5.
Black BE, Paschal BM. Intranuclear organization and function of the androgen receptor. Trends Endocrinol Metab. 2004;15(9):411–7.
Nickols NG, Dervan PB. Suppression of androgen receptor-mediated gene expression by a sequence-specific DNA-binding polyamide. Proc Natl Acad Sci U S A. 2007;104(25):10418–23.
Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138(2):245–56.
Sahu B, Laakso M, Ovaska K, Mirtti T, Lundin J, Rannikko A, et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J. 2011;30(19):3962–76.
Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474(7351):390–4.
Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17(5):443–54.
Chen Y, Chi P, Rockowitz S, Iaquinta PJ, Shamu T, Shukla S, et al. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat Med. 2013;19(8):1023–9.
Pomerantz MM, Li F, Takeda DY, Lenci R, Chonkar A, Chabot M, et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat Genet. 2015;47(11):1346–51.
Sharma NL, Massie CE, Ramos-Montoya A, Zecchini V, Scott HE, Lamb AD, et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell. 2013;23(1):35–47.
DePriest AD, Fiandalo MV, Schlanger S, Heemers F, Mohler JL, Liu S, et al. Regulators of androgen action resource: a one-stop shop for the comprehensive study of androgen receptor action. Database (Oxford). 2016;2016:pii: bav125.
Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007;28(7):778–808.
Rouleau N, Domans'kyi A, Reeben M, Moilanen AM, Havas K, Kang Z, et al. Novel ATPase of SNF2-like protein family interacts with androgen receptor and modulates androgen-dependent transcription. Mol Biol Cell. 2002;13(6):2106–19.
Link KA, Burd CJ, Williams E, Marshall T, Rosson G, Henry E, et al. BAF57 governs androgen receptor action and androgen-dependent proliferation through SWI/SNF. Mol Cell Biol. 2005;25(6):2200–15.
Hong CY, Suh JH, Kim K, Gong EY, Jeon SH, Ko M, et al. Modulation of androgen receptor transactivation by the SWI3-related gene product (SRG3) in multiple ways. Mol Cell Biol. 2005;25(12):4841–52.
Monroy MA, Schott NM, Cox L, Chen JD, Ruh M, Chrivia JC. SNF2-related CBP activator protein (SRCAP) functions as a coactivator of steroid receptor-mediated transcription through synergistic interactions with CARM-1 and GRIP-1. Mol Endocrinol. 2003;17(12):2519–28.
Bevan CL, Hoare S, Claessens F, Heery DM, Parker MG. The AF1 and AF2 domains of the androgen receptor interact with distinct regions of SRC1. Mol Cell Biol. 1999;19(12):8383–92.
Axlund SD, Lambert JR, Nordeen SK. HOXC8 inhibits androgen receptor signaling in human prostate cancer cells by inhibiting SRC-3 recruitment to direct androgen target genes. Mol Cancer Res. 2010;8(12):1643–55.
Fu M, Rao M, Wang C, Sakamaki T, Wang J, Di Vizio D, et al. Acetylation of androgen receptor enhances coactivator binding and promotes prostate cancer cell growth. Mol Cell Biol. 2003;23(23):8563–75.
Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437(7057):436–9.
Wissmann M, Yin N, Muller JM, Greschik H, Fodor BD, Jenuwein T, et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol. 2007;9(3):347–53.
Fu M, Liu M, Sauve AA, Jiao X, Zhang X, Wu X, et al. Hormonal control of androgen receptor function through SIRT1. Mol Cell Biol. 2006;26(21):8122–35.
Heinlein CA, Chang C. Androgen receptor (AR) coregulators: an overview. Endocr Rev. 2002;23(2):175–200.
Wang Q, Li W, Liu XS, Carroll JS, Janne OA, Keeton EK, et al. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell. 2007;27(3):380–92.
Holter E, Kotaja N, Makela S, Strauss L, Kietz S, Janne OA, et al. Inhibition of androgen receptor (AR) function by the reproductive orphan nuclear receptor DAX-1. Mol Endocrinol. 2002;16(3):515–28.
Aarnisalo P, Palvimo JJ, Janne OA. CREB-binding protein in androgen receptor-mediated signaling. Proc Natl Acad Sci U S A. 1998;95(5):2122–7.
Gao N, Zhang J, Rao MA, Case TC, Mirosevich J, Wang Y, et al. The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol Endocrinol. 2003;17(8):1484–507.
Gioeli D, Paschal BM. Post-translational modification of the androgen receptor. Mol Cell Endocrinol. 2012;352(1-2):70–8.
Gioeli D, Black BE, Gordon V, Spencer A, Kesler CT, Eblen ST, et al. Stress kinase signaling regulates androgen receptor phosphorylation, transcription, and localization. Mol Endocrinol. 2006;20(3):503–15.
Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, et al. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994;54(20):5474–8.
Mukherjee S, Cruz-Rodriguez O, Bolton E, Iniguez-Lluhi JA. The in vivo role of androgen receptor SUMOylation as revealed by androgen insensitivity syndrome and prostate cancer mutations targeting the proline/glycine residues of synergy control motifs. J Biol Chem. 2012;287(37):31195–206.
Xu K, Shimelis H, Linn DE, Jiang R, Yang X, Sun F, et al. Regulation of androgen receptor transcriptional activity and specificity by RNF6-induced ubiquitination. Cancer Cell. 2009;15(4):270–82.
Lin HK, Wang L, Hu YC, Altuwaijri S, Chang C. Phosphorylation-dependent ubiquitylation and degradation of androgen receptor by Akt require Mdm2 E3 ligase. EMBO J. 2002;21(15):4037–48.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30.
Huggins C. Endocrine-induced regression of cancers. Cancer Res. 1967;27(11):1925–30.
Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J Clin. 1972;22(4):232–40.
Balk SP, Knudsen KE. AR, the cell cycle, and prostate cancer. Nucl Recept Signal. 2008;6:e001.
Knudsen KE, Arden KC, Cavenee WK. Multiple G1 regulatory elements control the androgen-dependent proliferation of prostatic carcinoma cells. J Biol Chem. 1998;273(32):20213–22.
Xu Y, Chen SY, Ross KN, Balk SP. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. 2006;66(15):7783–92.
Raclaw KA, Heemers HV, Kidd EM, Dehm SM, Tindall DJ. Induction of FLIP expression by androgens protects prostate cancer cells from TRAIL-mediated apoptosis. Prostate. 2008;68(15):1696–706.
Stewart RJ, Panigrahy D, Flynn E, Folkman J. Vascular endothelial growth factor expression and tumor angiogenesis are regulated by androgens in hormone responsive human prostate carcinoma: evidence for androgen dependent destabilization of vascular endothelial growth factor transcripts. J Urol. 2001;165(2):688–93.
Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–28.
Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310(5748):644–8.
Mosquera JM, Mehra R, Regan MM, Perner S, Genega EM, Bueti G, et al. Prevalence of TMPRSS2-ERG fusion prostate cancer among men undergoing prostate biopsy in the United States. Clin Cancer Res. 2009;15(14):4706–11.
Cancer Genome Atlas Research N. The molecular taxonomy of primary prostate cancer. Cell. 2015;163(4):1011–25.
Riegman PH, Vlietstra RJ, van der Korput JA, Brinkmann AO, Trapman J. The promoter of the prostate-specific antigen gene contains a functional androgen responsive element. Mol Endocrinol. 1991;5(12):1921–30.
Nash AF, Melezinek I. The role of prostate specific antigen measurement in the detection and management of prostate cancer. Endocr Relat Cancer. 2000;7(1):37–51.
Ryan CJ, Smith A, Lal P, Satagopan J, Reuter V, Scardino P, et al. Persistent prostate-specific antigen expression after neoadjuvant androgen depletion: an early predictor of relapse or incomplete androgen suppression. Urology. 2006;68(4):834–9.
Culig Z, Hobisch A, Cronauer MV, Cato AC, Hittmair A, Radmayr C, et al. Mutant androgen receptor detected in an advanced-stage prostatic carcinoma is activated by adrenal androgens and progesterone. Mol Endocrinol. 1993;7(12):1541–50.
Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332(21):1393–8.
Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68(13):5469–77.
Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69(6):2305–13.
Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69(1):16–22.
Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120(8):2715–30.
Dai C, Heemers H, Sharifi N. Androgen signaling in prostate cancer. Cold Spring Harb Perspect Med. 2017;7(9):pii: a030452.
Sharifi N, Auchus RJ. Steroid biosynthesis and prostate cancer. Steroids. 2012;77(7):719–26.
Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371(5):424–33.
Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324(5928):787–90.
de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364(21):1995–2005.
Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368(2):138–48.
Kang M, Jeong CW, Kwak C, Ku JH, Kim HH. Comparing the clinical efficacy of abiraterone acetate, enzalutamide, and orteronel in patients with metastatic castration-resistant prostate cancer by performing a network meta-analysis of eight randomized controlled trials. Oncotarget. 2017;8(35):59690–97.
Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, et al. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997;57(2):314–9.
Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9(4):401–6.
Palmberg C, Koivisto P, Kakkola L, Tammela TL, Kallioniemi OP, Visakorpi T. Androgen receptor gene amplification at primary progression predicts response to combined androgen blockade as second line therapy for advanced prostate cancer. J Urol. 2000;164(6):1992–5.
Leversha MA, Han J, Asgari Z, Danila DC, Lin O, Gonzalez-Espinoza R, et al. Fluorescence in situ hybridization analysis of circulating tumor cells in metastatic prostate cancer. Clin Cancer Res. 2009;15(6):2091–7.
Attard G, Swennenhuis JF, Olmos D, Reid AH, Vickers E, A'Hern R, et al. Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res. 2009;69(7):2912–8.
Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–43.
Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. 2013;63(5):920–6.
Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, Trifiro M. The androgen receptor gene mutations database: 2012 update. Hum Mutat. 2012;33(5):887–94.
Steketee K, Timmerman L, Ziel-van der Made AC, Doesburg P, Brinkmann AO, Trapman J. Broadened ligand responsiveness of androgen receptor mutants obtained by random amino acid substitution of H874 and mutation hot spot T877 in prostate cancer. Int J Cancer. 2002;100(3):309–17.
Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44(6):685–9.
Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JM, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520(7547):353–7.
Hong MK, Macintyre G, Wedge DC, Van Loo P, Patel K, Lunke S, et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat Commun. 2015;6:6605.
Hara T, Miyazaki J, Araki H, Yamaoka M, Kanzaki N, Kusaka M, et al. Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003;63(1):149–53.
Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 2013;3(9):1030–43.
Kikugawa T, Kinugasa Y, Shiraishi K, Nanba D, Nakashiro K, Tanji N, et al. PLZF regulates Pbx1 transcription and Pbx1-HoxC8 complex leads to androgen-independent prostate cancer proliferation. Prostate. 2006;66(10):1092–9.
Cao J, Zhu S, Zhou W, Li J, Liu C, Xuan H, et al. PLZF mediates the PTEN/AKT/FOXO3a signaling in suppression of prostate tumorigenesis. PLoS One. 2013;8(12):e77922.
Geng C, He B, Xu L, Barbieri CE, Eedunuri VK, Chew SA, et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc Natl Acad Sci U S A. 2013;110(17):6997–7002.
Lu C, Luo J. Decoding the androgen receptor splice variants. Transl Androl Urol. 2013;2(3):178–86.
Wadosky KM, Koochekpour S. Androgen receptor splice variants and prostate cancer: from bench to bedside. Oncotarget. 2017;8(11):18550–76.
Li Y, Hwang TH, Oseth LA, Hauge A, Vessella RL, Schmechel SC, et al. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene. 2012;31(45):4759–67.
Nyquist MD, Li Y, Hwang TH, Manlove LS, Vessella RL, Silverstein KA, et al. TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer. Proc Natl Acad Sci U S A. 2013;110(43):17492–7.
Thadani-Mulero M, Portella L, Sun S, Sung M, Matov A, Vessella RL, et al. Androgen receptor splice variants determine taxane sensitivity in prostate cancer. Cancer Res. 2014;74(8):2270–82.
Chan SC, Selth LA, Li Y, Nyquist MD, Miao L, Bradner JE, et al. Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine-based therapies. Nucleic Acids Res. 2015;43(12):5880–97.
Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010;107(39):16759–65.
Cao B, Qi Y, Zhang G, Xu D, Zhan Y, Alvarez X, et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget. 2014;5(6):1646–56.
Xu D, Zhan Y, Qi Y, Cao B, Bai S, Xu W, et al. Androgen receptor splice variants dimerize to transactivate target genes. Cancer Res. 2015;75(17):3663–71.
Zhang X, Morrissey C, Sun S, Ketchandji M, Nelson PS, True LD, et al. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS One. 2011;6(11):e27970.
Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, et al. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One. 2011;6(4):e19059.
Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013;73(2):483–9.
Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15(12):701–11.
Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371(11):1028–38.
Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17(18):5913–25.
Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Zhu Y, et al. Clinical significance of androgen receptor splice variant-7 mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first- and second-line abiraterone and enzalutamide. J Clin Oncol. 2017;35:2149.
Qu F, Xie W, Nakabayashi M, Zhang H, Jeong SH, Wang X, et al. Association of AR-V7 and prostate-specific antigen RNA levels in blood with efficacy of abiraterone acetate and enzalutamide treatment in men with prostate cancer. Clin Cancer Res. 2017;23(3):726–34.
Kohli M, Ho Y, Hillman DW, Van Etten JL, Henzler C, Yang R, et al. Androgen receptor variant AR-V9 is co-expressed with AR-V7 in prostate cancer metastases and predicts abiraterone resistance. Clin Cancer Res. 2017;23:4704.
Kong D, Sethi S, Li Y, Chen W, Sakr WA, Heath E, et al. Androgen receptor splice variants contribute to prostate cancer aggressiveness through induction of EMT and expression of stem cell marker genes. Prostate. 2015;75(2):161–74.
Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1(3):E59.
Majumder PK, Sellers WR. Akt-regulated pathways in prostate cancer. Oncogene. 2005;24(50):7465–74.
Di Lorenzo G, Tortora G, D'Armiento FP, De Rosa G, Staibano S, Autorino R, et al. Expression of epidermal growth factor receptor correlates with disease relapse and progression to androgen-independence in human prostate cancer. Clin Cancer Res. 2002;8(11):3438–44.
Twillie DA, Eisenberger MA, Carducci MA, Hseih WS, Kim WY, Simons JW. Interleukin-6: a candidate mediator of human prostate cancer morbidity. Urology. 1995;45(3):542–9.
Nakashima J, Tachibana M, Horiguchi Y, Oya M, Ohigashi T, Asakura H, et al. Serum interleukin 6 as a prognostic factor in patients with prostate cancer. Clin Cancer Res. 2000;6(7):2702–6.
Shariat SF, Andrews B, Kattan MW, Kim J, Wheeler TM, Slawin KM. Plasma levels of interleukin-6 and its soluble receptor are associated with prostate cancer progression and metastasis. Urology. 2001;58(6):1008–15.
Veltri RW, Miller MC, Zhao G, Ng A, Marley GM, Wright GL, Jr., et al. Interleukin-8 serum levels in patients with benign prostatic hyperplasia and prostate cancer. Urology 1999;53(1):139-147.
George DJ, Halabi S, Shepard TF, Sanford B, Vogelzang NJ, Small EJ, et al. The prognostic significance of plasma interleukin-6 levels in patients with metastatic hormone-refractory prostate cancer: results from cancer and leukemia group B 9480. Clin Cancer Res. 2005;11(5):1815–20.
Tien JC, Liu Z, Liao L, Wang F, Xu Y, Wu YL, et al. The steroid receptor coactivator-3 is required for the development of castration-resistant prostate cancer. Cancer Res. 2013;73(13):3997–4008.
Tatarov O, Mitchell TJ, Seywright M, Leung HY, Brunton VG, Edwards J. SRC family kinase activity is up-regulated in hormone-refractory prostate cancer. Clin Cancer Res. 2009;15(10):3540–9.
Krueckl SL, Sikes RA, Edlund NM, Bell RH, Hurtado-Coll A, Fazli L, et al. Increased insulin-like growth factor I receptor expression and signaling are components of androgen-independent progression in a lineage-derived prostate cancer progression model. Cancer Res. 2004;64(23):8620–9.
Gregory CW, Fei X, Ponguta LA, He B, Bill HM, French FS, et al. Epidermal growth factor increases coactivation of the androgen receptor in recurrent prostate cancer. J Biol Chem. 2004;279(8):7119–30.
Ueda T, Mawji NR, Bruchovsky N, Sadar MD. Ligand-independent activation of the androgen receptor by interleukin-6 and the role of steroid receptor coactivator-1 in prostate cancer cells. J Biol Chem. 2002;277(41):38087–94.
Kasina S, Macoska JA. The CXCL12/CXCR4 axis promotes ligand-independent activation of the androgen receptor. Mol Cell Endocrinol. 2012;351(2):249–63.
Hobisch A, Eder IE, Putz T, Horninger W, Bartsch G, Klocker H, et al. Interleukin-6 regulates prostate-specific protein expression in prostate carcinoma cells by activation of the androgen receptor. Cancer Res. 1998;58(20):4640–5.
Seaton A, Scullin P, Maxwell PJ, Wilson C, Pettigrew J, Gallagher R, et al. Interleukin-8 signaling promotes androgen-independent proliferation of prostate cancer cells via induction of androgen receptor expression and activation. Carcinogenesis. 2008;29(6):1148–56.
Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006;10(4):309–19.
Craft N, Shostak Y, Carey M, Sawyers CL. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med. 1999;5(3):280–5.
Yeh S, Lin HK, Kang HY, Thin TH, Lin MF, Chang C. From HER2/Neu signal cascade to androgen receptor and its coactivators: a novel pathway by induction of androgen target genes through MAP kinase in prostate cancer cells. Proc Natl Acad Sci U S A. 1999;96(10):5458–63.
Wen Y, Hu MC, Makino K, Spohn B, Bartholomeusz G, Yan DH, et al. HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Res. 2000;60(24):6841–5.
Mellinghoff IK, Vivanco I, Kwon A, Tran C, Wongvipat J, Sawyers CL. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell. 2004;6(5):517–27.
Nazareth LV, Weigel NL. Activation of the human androgen receptor through a protein kinase A signaling pathway. J Biol Chem. 1996;271(33):19900–7.
Sadar MD. Androgen-independent induction of prostate-specific antigen gene expression via cross-talk between the androgen receptor and protein kinase A signal transduction pathways. J Biol Chem. 1999;274(12):7777–83.
Taneja SS, Ha S, Swenson NK, Huang HY, Lee P, Melamed J, et al. Cell-specific regulation of androgen receptor phosphorylation in vivo. J Biol Chem. 2005;280(49):40916–24.
Lin HK, Hu YC, Yang L, Altuwaijri S, Chen YT, Kang HY, et al. Suppression versus induction of androgen receptor functions by the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer LNCaP cells with different passage numbers. J Biol Chem. 2003;278(51):50902–7.
Lin HK, Hu YC, Lee DK, Chang C. Regulation of androgen receptor signaling by PTEN (phosphatase and tensin homolog deleted on chromosome 10) tumor suppressor through distinct mechanisms in prostate cancer cells. Mol Endocrinol. 2004;18(10):2409–23.
Jiao J, Wang S, Qiao R, Vivanco I, Watson PA, Sawyers CL, et al. Murine cell lines derived from Pten null prostate cancer show the critical role of PTEN in hormone refractory prostate cancer development. Cancer Res. 2007;67(13):6083–91.
Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19(5):575–86.
Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19(6):792–804.
Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155(6):1309–22.
Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17(6):535–46.
Antonarakis ES, Chandhasin C, Osbourne E, Luo J, Sadar MD, Perabo F. Targeting the N-terminal domain of the androgen receptor: a new approach for the treatment of advanced prostate cancer. Oncologist. 2016;21(12):1427–35.
Mashima T, Okabe S, Seimiya H. Pharmacological targeting of constitutively active truncated androgen receptor by nigericin and suppression of hormone-refractory prostate cancer cell growth. Mol Pharmacol. 2010;78(5):846–54.
Cao B, Liu X, Li J, Liu S, Qi Y, Xiong Z, et al. 20(S)-protopanaxadiol-aglycone downregulation of the full-length and splice variants of androgen receptor. Int J Cancer. 2013;132(6):1277–87.
Li J, Cao B, Liu X, Fu X, Xiong Z, Chen L, et al. Berberine suppresses androgen receptor signaling in prostate cancer. Mol Cancer Ther. 2011;10(8):1346–56.
Li X, Liu Z, Xu X, Blair CA, Sun Z, Xie J, et al. Kava components down-regulate expression of AR and AR splice variants and reduce growth in patient-derived prostate cancer xenografts in mice. PLoS One. 2012;7(2):e31213.
Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP, et al. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin Cancer Res. 2014;20(12):3198–210.
Liu C, Armstrong CM, Lou W, Lombard AP, Cucchiara V, Gu X, et al. Niclosamide and bicalutamide combination treatment overcomes enzalutamide and bicalutamide resistant prostate cancer. Mol Cancer Ther. 2017;16:1521.
Liu C, Armstrong C, Zhu Y, Lou W, Gao AC. Niclosamide enhances abiraterone treatment via inhibition of androgen receptor variants in castration resistant prostate cancer. Oncotarget. 2016;7(22):32210–20.
Denmeade SR, Isaacs JT. Bipolar androgen therapy: the rationale for rapid cycling of supraphysiologic androgen/ablation in men with castration resistant prostate cancer. Prostate. 2010;70(14):1600–7.
Isaacs JT, D'Antonio JM, Chen S, Antony L, Dalrymple SP, Ndikuyeze GH, et al. Adaptive auto-regulation of androgen receptor provides a paradigm shifting rationale for bipolar androgen therapy (BAT) for castrate resistant human prostate cancer. Prostate. 2012;72(14):1491–505.
Umekita Y, Hiipakka RA, Kokontis JM, Liao S. Human prostate tumor growth in athymic mice: inhibition by androgens and stimulation by finasteride. Proc Natl Acad Sci U S A. 1996;93(21):11802–7.
Schweizer MT, Antonarakis ES, Wang H, Ajiboye AS, Spitz A, Cao H, et al. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer: results from a pilot clinical study. Sci Transl Med. 2015;7(269):269ra2.
Gan L, Chen S, Wang Y, Watahiki A, Bohrer L, Sun Z, et al. Inhibition of the androgen receptor as a novel mechanism of taxol chemotherapy in prostate cancer. Cancer Res. 2009;69(21):8386–94.
Zhu ML, Horbinski CM, Garzotto M, Qian DZ, Beer TM, Kyprianou N. Tubulin-targeting chemotherapy impairs androgen receptor activity in prostate cancer. Cancer Res. 2010;70(20):7992–8002.
Vale CL, Burdett S, Rydzewska LH, Albiges L, Clarke NW, Fisher D, et al. Addition of docetaxel or bisphosphonates to standard of care in men with localised or metastatic, hormone-sensitive prostate cancer: a systematic review and meta-analyses of aggregate data. Lancet Oncol. 2016;17(2):243–56.
Miller RE, Sweeney CJ. Chemotherapy for metastatic castrate-sensitive prostate cancer. Prostate Cancer Prostatic Dis. 2016;19(2):139–44.
James ND, Sydes MR, Clarke NW, Mason MD, Dearnaley DP, Spears MR, et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet. 2016;387(10024):1163–77.
Rickman DS, Beltran H, Demichelis F, Rubin MA. Biology and evolution of poorly differentiated neuroendocrine tumors. Nat Med. 2017;23(6):1–10.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter
Cite this chapter
Berger, A., Rickman, D.S. (2018). The Role of Androgen Receptor in Prostate Cancer. In: Robinson, B., Mosquera, J., Ro, J., Divatia, M. (eds) Precision Molecular Pathology of Prostate Cancer. Molecular Pathology Library. Springer, Cham. https://doi.org/10.1007/978-3-319-64096-9_20
Download citation
DOI: https://doi.org/10.1007/978-3-319-64096-9_20
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-64094-5
Online ISBN: 978-3-319-64096-9
eBook Packages: MedicineMedicine (R0)