
Abstract
Androgen receptor (AR) signaling pathway is required for both development of normal prostate gland and prostate cancer (PC). Patient with advanced disease are usually treated with androgen deprivation therapy. However, this treatment is only palliative, since a castration-resistant PC (CRPC) usually arises within 2–3 years of treatment. The mechanism by which CRPC develops is yet to be fully understood. However, common alteration in CRPC is the overexpression of the AR. Several studies have addressed the molecular changes occurring in AR overexpressing PC cells. The overexpression of AR enhances the binding of the receptor to chromatin in the presence of low concentrations of androgens. Furthermore, under the same conditions, AR overexpression alters also the dynamics of chromatin binding of the receptor and the binding of basic components of the transcriptional machinery. These changes translate into global epigenetic changes, which deserve more attention. Many studies have found that AR activation in CRPC cell models stimulates a different transcriptional program, which may be influenced by cooperative functions of other transcription factors. Thus, not only a single target gene but also a network of genes could be responsible for the disease progression. In fact, functional studies have shown that androgen-regulated genes, which are overexpressed in CRPC, are also likely to be important in PC progression.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
12.1 Introduction
In 1941, Huggins and Hodges reported the dependency of metastatic PC on androgens [1]. Already in the 1980s it was shown that castration reduced the levels of dihydrotestosterone (DHT) in prostate tissue of only about 50% [2]. Castration combined with ketoconazole treatment to block the production of adrenal androgen further reduced the levels of DHT [3]. Recent work by Titus et al. [4] showed that, even though the levels of DHT were reduced up to 90% in CRPCs tissues, the levels of testosterone were same as in non-malignant androgen-stimulated prostate. Moreover, the residual levels of DHT in the tissues were able to activate AR. These studies led to the idea that PC could progress to a CRPC stage as a result of androgens produced by the tumor itself [4, 5]. In fact, levels of enzymes required for intratumoral de novo androgen synthesis were recently found to be high in CRPCs [6–9]. Abiraterone was developed in order to avoid androgen synthesis in the cells, achieving an almost complete abrogation of androgens in the tissue. In a clinical trial, abiraterone extended lifespan by 4 months in patients with advanced disease and the use of this drug was approved by FDA last year [10].
Further evidence that CRPC is dependent on the androgen/AR signaling pathway was demonstrated in a recent clinical trial with next-generation antiandrogen, MDV3100. The drug improved survival of patients confirming that CRPCs are still androgen sensitive [11]. The emergence of CRPC has been associated with mutations in AR altering transactivation properties of the receptor, which also occur in about 10–20% of the CRPCs [12]. Moreover, expression of constitutively active AR splice variants and reexpression of androgen-regulated genes have also been found [13–15].
The AR gene is amplified in one-third of CRPCs [16], but not in untreated tumors suggesting that the androgen deprivation therapy selects for this genetic alteration. Furthermore, the patients with AR gene amplification respond better to a second line combined androgen blockade than patients without the amplification [17], suggesting that the tumors with the amplification are more androgen dependent than tumors without the amplification. The finding of AR gene amplification led to the hypothesis that CRPCs are androgen hypersensitive, instead of androgen independent [16].
Because AR is overexpressed in approximately 80–90% of all CRPCs, other mechanisms than gene amplification leading to AR overexpression have also been hypothesized [18–23]. It has been suggested that such mechanisms could include loss of a transcriptional repressor complex found in CRPC specimens [24]. Also the upregulation of the lymphoid enhancer-binding factor 1 (LEF1) transcription factor has been shown to induce AR gene expression in model systems [25]. More recently, the loss of RB1 signaling, which is also very frequent in CRPC [23] was shown to correlate with AR overexpression. Sharma et al. [26] demonstrated that the loss of RB1 induces E2F1-mediated AR enhanced transcription. AR expression was also shown to be inversely correlated with the androgens concentration to which cells were exposed, through an AR-mediated feedback loop involving the chromatin modifier LSD1 [27].
Understanding the molecular mechanisms driven by the AR overexpression is critical and serves as the basis for identifying new drug targets and biomarkers for this disease. Several cell line models mimicking different stages of the disease and expressing different levels of wild-type and mutated AR are available today (Table 14.1). For instance, the cell line LNCaP carrying mutated AR, as well as wild-type AR expressing VCaP cells are widely used models for CRPC. The LNCaP was derived from a human lymph node metastasis [28], whereas VCaP from a metastatic lesion in a lumbar vertebral body [29]. VCaP caries both AR gene amplification and TMPRSS2:ERG gene fusion [30, 31]. It expresses AR >tenfold higher levels than LNCaP [32]. Long-term androgen starvation of the LNCaP led to the establishment of abl-LNCaP cell line, expressing fourfold higher AR protein levels compared to parental LNCaP cell line [33]. To study the consequences of AR overexpression in PC cells in a more controlled manner, we have developed a LNCaP-based cell line model overexpressing wild type AR 4–6-fold (LNCaP-ARhi) and 2–4-fold (LNCaP-ARmo) more that the control cell line (LNCaP-pcDNA3.1) [32]. In concordance with the results by Chen et al. [20], LNCaP-ARhi cells grow faster in the presence of low levels of androgens than the control cells and adapt better to long-term androgen starvation.
In this chapter we will describe how the overexpression of AR affects androgen signaling, leads to resistance to androgen ablation therapies, and how these mechanisms could be exploited to evaluate new approaches to treat CRPC.
12.2 AR Overexpression Is the Best Known Change in CRPCs
We showed already in 2001 [18] that almost all clinical CRPC samples express more AR than hormone-naïve PC specimens. In 2004, Chen et al. [20] showed that AR is consistently upregulated in hormone-refractory xenografts. To confirm that the AR overexpression is responsible for progression, they overexpressed AR in the hormone sensitive LNCaP cells and demonstrated their acquired capability to grow in lower concentrations of androgens. Furthermore they showed that the same cells with forced expression of AR rapidly formed tumors in castrated mice, while the parental cells did not or developed tumors later. They found that the resistance was still due to activation of the AR by its ligand and that the AR overexpression was also able to induce resistance to the antiandrogen bicalutamide and to convert AR antagonist into agonists. More recently, we have demonstrated that the AR overexpression sensitizes AR signaling pathway to lower concentrations of ligand [32, 34]. Thus, the evidence that overexpression of AR is the main mechanism of castration resistance is strong and suggests that even a moderate sensitization of AR signaling may cause castration resistance. This is of fundamental importance, since it has been shown that the post-castration levels of androgens may vary significantly between individuals [5].
AR is a transcription factor that regulates the expression of a large number of genes [35–37]. Many AR target genes have been associated with the development of the disease. Therefore, the identification of genes downstream of AR signaling involved in the development and progression of PC remains an important area for future investigations. The identification of commonly altered downstream genes could potentially provide new drug targets and better biomarkers.
12.3 AR Overexpression Affects Gene Expression and AR Target Genes
The knowledge that the AR overexpression sensitizes the growth of PC cells to lower androgens concentration is not new. Already Kokontis et al. [38] showed that LNCaP cells, grown in hormone-deprived media, appeared to adapt to lowered androgen levels by increasing AR expression and transcriptional activity. The data suggested that AR is transcriptionally active in CRPC and can increase cell proliferation at low circulating levels of androgen reported in castrated men [39].
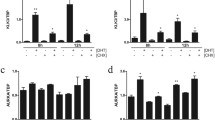
By comparing gene expression signatures of both LNCaP-ARhi and abl-LNCaP to parental LNCaP we [32] and Wang et al. [40] found independently that AR selectively upregulates M-phase cell-cycle genes in the LNCaP derivative cells, including the genes UBE2C, CDK1, and AURKA, involved in both inactivation of the M-phase checkpoint and driving cell cycle further. AR overexpression increases the number of androgen-regulated genes in the lower concentrations of androgens [32, 34]. Thus, it is conceivable that the role of the increased AR expression in CRPC cells is to sensitize the transcriptional program in order to achieve androgen-dependent cell growth in the presence of minimal androgen concentrations. The gained transcriptional program elicits expression of several cell cycle-associated genes in LNCaP-ARhi and VCaP cells, compared to control cells exposed to 10–100-fold less androgens [32]. AR regulation of CDKs and cyclins in CRPCs has previously been suggested [41]. We have demonstrated that also metabolism and mitosis-associated genes, such as ZWINT, SKP2 (S-phase kinase-associated protein 2 (p45)), and FEN1 (flap structure-specific endonuclease 1) transcripts [32, 34] are upregulated in AR overexpressing cells, as well as overexpressed in CRPC specimens [34]. The importance of the androgen regulation of metabolic pathways by AR in CRPC was pinpointed also by the finding that aerobic glycolysis, biosynthesis, and anabolism in PC cells are crucial for the disease progression [42]. The work by Massie et al. identified Calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2) as an androgen-regulated gene important in PC progression [42].
Androgen-repressed genes may also play important roles in PC cell growth and be major contributors in progression to CRPC [43]. Their reexpression during androgen deprivation is thought to contribute to disease regression, and they may become repressed once again in CRPC. Recently, the study by Zhao et al. [44] reported a systematic analysis of genomic data in order to establish the role of AR as transcriptional repressor. They presented evidences that AR directly inhibits a large number of target genes. Mechanistically, this repression is mediated by the polycomb group protein EZH2 and subsequently by repressive chromatin remodeling. These genes are developmental regulators functionally involved in cell differentiation and tumor suppression. Furthermore, forcing AR expression in LNCaP showed that increased AR binding in the AR-binding sites (ARBSs) in proximity of these genes further enhanced their repression. They also reported similar results using VCaP cells [44], thus confirming that AR overexpression alone may contribute, in CRPC to repression of this particular transcriptional program.
12.4 AR Overexpression Affects AR Coregulator’s Expression
There is a large array of coregulators required for AR-dependent transcription and so far it has been very difficult to establish the rules for their assembly into transcriptional complexes and their hierarchy of action. Studies in knock-out mice of few coregulators including KDM1A (LSD1), NCOA1 (SRC1), NCOA2 (TIF2), and FKBP4 (FKBP52) revealed only mild phenotypes [45–49], suggesting that coregulators can supplement the function of the missing ones. This suggestion leads to the hypothesis that it is the stoichiometry of the coregulators that may be mostly important for the AR physiological functions.
We performed a systematic and comprehensive study to investigate the expression profile of the AR coregulators in PC [50]. More recently, Taylor et al. [23] have done the same. We found that the levels of AR coregulators do not change dramatically in PC, excluding their involvement in disease progression. Also, the study by Taylor et al. [23] seems to support this observation, because of the coregulators, only NCOA2 was found to be upregulated in a proportion of PCs. NCOA2 expression level has been shown to correlate with early biochemical recurrence in PC patients [51] and it has been recently reported to function as an oncogene in a subset of PCs. Chromosome 8q13.3, harboring the NCOA2 gene, is the most common amplified locus in PC [23]. However, our data [50] do not support the overexpression of NCOA2 in PC. There are studies that have found coactivators to be overexpressed in PC [52–54]. For example, levels of CBP have been shown to be high in advanced PC and, in particular, in tissues from patients that failed endocrine therapy [55], although other reports do not support the finding (e.g., [23, 50]). Downregulation of AR corepressors has also been proposed to be involved in the development of CRPC. For example, the recently identified AR corepressor BTG2 [56], is frequently downregulated in PC and associated with PC aggressiveness [56–58].
We have investigated whether AR coregulators would be androgen-regulated, taking advantage of our LNCaP-AR overexpression model [32]. We studied the effect of AR overexpression on their expression and regulation. Of the over 25 coregulators studied with qRT-PCR, about half were androgen regulated. Coactivators such as AIB1, CBP, MAK, and BRCA1 showed particularly enhanced upregulation in LNCaP-ARhi cells when compared to control cells [59]. It is difficult to attribute such effect to the presence of ARBSs in the proximity of the loci of these genes, since also non-androgen-regulated AR coregulators displayed ARBSs. However, all these coactivators displayed ARBSs in a putative enhancer region. More recently, Heemers et al. [60] profiled the expression and activity of 186 AR coregulators. Similarly with our results, 30% of them resulted to be androgen regulated [60]. CBP was one of the AR coactivators upregulated in the LNCaP cell line derivative LNCaP-Rf, which was established by long-term androgen ablation of LNCaP cells [55, 60].
These data suggest the existence of a potential positive feedback loop directed to enhance AR activity in CRPC in low concentrations of androgens. The AR coregulators deserve more attention, especially since, recently, evidences showing that targeting the activity of AR coactivator such as EP300 (p300) or CBP may be therapeutically advantageous [61].
12.5 AR Overexpression Affects AR Binding to Chromatin
The first attempt to use chromatin immunoprecipitation (ChIP) coupled with genomic array (chip) to identify genomic locations bound by AR was made by Wang et al. [62]. They not only reported the first AR-binding map in chromosome 20 and 21 but also opened the way to the extensive use of this method. In the same year, Massie et al. [63] published the first ARBSs map of the AR in the LNCaP cells using a promoter array. Genome-wide AR-binding profiles of all the available cell lines models have been generated over the past few years (Table 14.2) although a few technical and experimental variations characterize each single study. Recent studies have utilized ChIP coupled with next-generation deep sequencing (ChIP-seq).
The study by Wang et al. [40] and by Yu et al. [64] reported decreased genomic binding of AR in abl-LNCaP and in VCaP cells, respectively. In contrast, two studies by Sahu et al. [65] and by Massie et al. [42] reported dramatically increased AR binding in VCaP when compared with LNCaP [42] or LNCaP-derived cells [65]. Recently, we reported that a modest overexpression of the AR gene in the LNCaP cells led to enhanced binding of AR to the chromatin when cells were treated with low concentrations of androgens [34]. However, we also observed decreased AR binding when cells were treated with a 100-times higher concentration (Table 14.2). The reasons underlying the variation in these results are still not addressed experimentally. A possible explanation may be the concentration of androgens used and the experimental settings such as type of androgen used and timing of chromatin binding assessment. Yu et al. [64] used relatively high (10 nM) concentration of synthetic, stabile androgen (R1881) and a much longer treatment time window than in the other studies. This suggests that most significant correlation between AR levels and transcriptional response affects short timepoints and initial dynamics of AR recruitment to DNA. This hypothesis needs further investigation. ChIP-chip analysis by Takayama et al. [66] reported that the number of androgen-dependent ARBSs increased in LNCaP cells treated with same concentration of R1881 (10 nM) for 6 and 24 h, suggesting that the timing of assessing chromatin binding is an important issue.
The type of androgen used may also be a source of variation. For instance, it is known that R1881 (synthetic androgen methyltrienolone) binds AR with higher affinity and is able to stimulate its activation more potently than DHT [67]. Sahu et al. [65] used 100 nM DHT while Massie et al. [42] used 1 nM R1881, which may be assumed to achieve the same range of AR activation [67]. Both found increasing amount of ARBSs in VCaP than in LNCaP, while Yu et al. [64] stimulated the cells with 10 nM R1881, which may result in downregulation of AR levels and growth inhibitory on the long run [27, 68]. Thus, one can speculate that inhibitory effect on AR binding can be observed when cells overexpressing AR are treated with high concentrations of androgens. Moreover, the non-isogenicity of the cell lines used (LNCaP vs. VCaP) does not allow a true controlled comparison between the AR binding in the different cell lines. The experiments in isogenic cell models, supports the idea that AR binding is regulated by both the amount of androgens in the media and the levels of AR protein [34, 40]. Furthermore, the binding of AR to the target genes’ regulatory regions is periodic, and it has also been demonstrated to be dependent on proteasome activity and on the activity of other cofactors [69, 70]. To address this issue, we treated our AR overexpressing cells with low and high concentrations of androgens and profiled AR binding to two well-known AR target genes: the PSA and the TMPRSS2 [71]. The binding to the regulatory regions of the genes reflected the periodicity reported previously [69, 72]. However, the overexpression of AR altered the binding to the loci studies in a manner, which seems to be gene and locus specific. We further proved that the AR binding is more rapid and potent at the enhancer and promoter region of these genes. We observed that the AR binding was affected differently in LNCaP-ARhi cells treated with higher concentrations of androgens [71], suggesting that higher concentration of ligand may mask the effect of the AR overexpression on the dynamic binding. We found also that the overexpression of AR alters also the binding dynamics of basal transcription factors, such as RNA Polymerase II as well as the chromatin structure as assessed by the enhanced histone 3 acetylation [71].
To confirm the association between the AR level and the AR-binding sites in an independent AR overexpression model, we used two CRPC tumors previously xenografted into castrated mice. One of them, LuCaP69 harbors AR gene amplification, whereas the LuCaP73 does not [18]. Consequently, the expression of AR is about tenfold higher in LuCaP69 compared to LuCaP73 according to qRT-PCR [18]. AR ChIP-seq analysis revealed approximately 19,000 and 7,000 ARBSs in LuCaP69 and LuCaP73 respectively, confirming that there is an association between the AR level and the number of ARBSs in vivo [34]. We also compared AR-binding potency in the same ARBSs map in AR overexpressing cells compared to control cells [34]. At the same loci, the AR binding in AR overexpressing cells was more potent when cells were stimulated with low concentration of androgens and tended to decrease when cells were treated with higher ligand concentrations. This finding was validated for several loci also with ChIP-qPCR [34]. For example, 100 times less ligand was needed in order to achieve the same AR recruitment to the PSA enhancer in AR overexpressing cells as compared to control cells. Thus, the AR overexpression sensitized AR binding by 100-fold. ChIP-qPCR on the PSA enhancer in the xenografts also showed that the AR binding is stronger in LuCaP69 compared to LuCaP73 confirming that the strength of the AR binding is also associated with the AR level [34].
These data indicate that both the ligand concentration and the amount of receptor affect together the chromatin binding of AR. Moreover, these data are concordant with the results of a recent work by Makkonen et al. [73] who confirmed that the binding at single gene level’s regulatory regions is enhanced in AR overexpressing cells, such as VCaP cells compared to LNCaP [73] and with the more recent report by Zhao et al. [44], associating AR binding and gene regulation.
12.6 AR Overexpression Affects the Chromatin Remodeling
It is known that the lineage-specific binding of transcription factors, such as AR, to chromatin is modulated also by other transcription factors such as FOXA1 translating epigenetic marks [65, 74, 75]. For example, the status of histone acetylation is critical for androgen receptor-mediated transcriptional activation of genes [76].
Chen et al. [20] found that a modest overexpression of AR can alter the abundance of AR coregulators recruited on the promoters of AR target genes, many of which have histone acetylation activity [77, 78]. There seems to be also a direct correlation between AR expression and chromatin modifiers. We found that many AR coactivators are targets of AR and the AR overexpression further enhances their expression [59]. Among those were CREBBP (CBP) and NCOA3 (AIB1), which are known histone acetylases [79]. Sahu et al. [65] investigated the association between AR and FOXA1 protein expression in PC, finding a direct correlation between the two [65]. The finding is confirmed also in the data by Taylor et al. [23]. Thus, the AR overexpression seems to favor expression of chromatin remodeler which facilitates AR-mediated genes transcription. A proof of principle that chromatin remodelers may be involved in the emergence of the CRPC phenotype and that the AR overexpression selects for these types of mechanisms is the recent finding that curcumin, which is able to inhibit recruitment of the complex p300/CBP to the regulatory regions of AR target genes, is able to slow growth of CRPC [61]. Indeed, to test whether the AR overexpression brings epigenetic changes to the chromatin, we investigated the chromatin structure in our AR overexpression model. LNCaP-ARhi cells showed enhanced acetylation of H3K9 and K14 [71], which are known markers of active transcription [80–82]. Furthermore we also showed preliminary results that the chromatin could be already open when cells are hormone deprived for few days [71]. These data are in agreement with the finding by Andreu-Vieyra et al. [83] showing nucleosome-depleted regions at AR enhancers in the absence of ligand. Moreover, we found that such chromatin opening was, once again, enhanced in AR overexpressing cells [71].
These data suggests that nucleosome disposal is an important mechanism to favor gene transcription and AR overexpression may affect also such mechanism. We anticipate that this mechanism is likely to favor ARBSs promiscuity for other transcription factors, which may concur to aberrantly modulate the AR transcriptional program observed in CRPC phenotype.
12.7 Summary
AR overexpression sensitizes cells to low androgen concentrations. A mechanistic explanation for such sensitization is that genome-wide chromatin binding of AR is enhanced in AR overexpressing cells. Chromatin binding of AR seems to be dependent on both the level of the receptor and the androgen concentrations to which the cells are exposed. Different concentrations are able to alter the dynamic of the AR recruitment to AR target genes regulatory regions depending on the level of the receptor. These changes translate into an enhanced AR target gene down- or upregulation, which may be different from gene to gene and due to intrinsic biological properties of such genes. The AR transcriptional program is sensitized 10–100-fold and enhances expression of AR coactivator, proliferation-associated genes, and chromatin remodelers, which may result in a positive feedback loop sustaining the AR activation in low androgen concentrations.
Altogether, these results indicate that the overexpression of AR in CRPC cells allows these cells to maintain and potentiate the AR signaling in lower androgen concentrations through several different mechanisms involving epigenetic, transcriptional, and stoichiometric changes.
12.8 Future Perspectives and Implications for Therapies
By analyzing the ARBS maps of the two xenografts, we realized that these maps overlapped poorly [34]. In order to investigate whether such variability may occur in other settings, we reanalyzed publicly available datasets and found that also in the independent study of Yu et al. [64] of a tumor specimen. Again, the ARBS maps overlapped poorly. Thus, it is difficult to attribute such variability to the different AR levels in those cells. It seems rather that, at least in the cell lines, core AR binding is conserved and additional sites are gained in consequence of the AR overexpression. This is an observation which seems to be true across all the studies (Table 14.2). If so, the expression of pioneer transcription factors such as FOXA1 or GATA2 may play a role in redistribution of transcription factor binding in different cells [65, 84].
The LNCaP model and other in vitro cell models represent great tools in order to study the mechanisms associated with the upregulation of the AR in CRPC. However, they will never mimic completely the tumor environment. Thus, in the future, it will be essential to engage in experiments of coculturing the available cell lines models with macrophages and other cell types able to nourish these cells of cytokines and other signals. This setting probably will affect the AR signaling in vivo. Moreover, recreating a tumor environment will help to explore the insight of the AR binding variability observed in the studies described above. It will help also to explain the differences between cells growing in vitro and in vivo and elucidate whether there are differences in their transcriptional program, which may be explained via a different AR binding in the genome.
It is now evident that CRPC is not androgen independent; instead, it still rely on an enhanced AR signaling in order to face the shortage of androgens in the tumor environment. Thus, the future clinical strategies for treating men with CPRC still depends on finding active drugs that inhibit the androgen signaling pathway. However, targeting AR for therapy has turned to be not an easy task. Therefore, the identification and characterization of AR target genes that are relevant in the development and progression of PC remains an important area for future investigations, as these genes could provide alternative, and perhaps more efficacious, drug targets.
Rather targeting one gene at the time, it might be advantageous to target a network of genes or few targets in combination that master different deregulated networks. Moreover, the tight stoichiometry between the abundances of AR and ligand may be exploited in order to delay tumor growth via intermittent androgen deprivation therapy in CRPC patients [85]. Some evidences suggest that patients with CRPC may benefit from these treatments and side-effects of standard therapies may be diminished [86, 87]. Moreover, as it has been established that AR acts in concert with other transcription factors establishing a network of TF, which cooperate in order to maintain the PC phenotype, a critical point is to evaluate different drugs in combinations. For instance targeting AR in combination with PI3K pathway inhibitors [88, 89] or in combination with MYC inhibitors [90].
References
Huggins C, Hodges CV (1941) Cancer Res 1:293–297
Bélanger B, Bélanger A, Labrie F et al (1989) J Steroid Biochem 32:695–698
Liu J, Albert J, Geller J (1986) Prostate 9:199–205
Titus MA, Schell MJ, Lih FB et al (2005) Clin Cancer Res 11:4653–4657
Mohler JL (2008) Adv Exp Med Biol 617:223–234
Stanbrough M, Bubley GJ, Ross K et al (2006) Cancer Res 66:2815–2825
Mostaghel EA, Page ST, Lin DW et al (2007) Cancer Res 67:5033–5041
Locke JA, Guns ES, Lubik AA et al (2008) Cancer Res 68:6407–6415
Locke JA, Guns ES, Lehman ML (2010) Prostate 70:239–251
de Bono JS, Logothetis CJ, Molina A et al (2011) N Engl J Med 364:1995–2005
Scher HI, Fizazi K, Saad F, et al (2012) N Engl J Med 367:1187–1197
Waltering KK, Urbanucci A, Visakorpi T (2012) Mol Cell Endocrinol 360:38–43
Dehm SM, Schmidt LJ, Heemers HV et al (2008) Cancer Res 68:5469–5477
Hu R, Dunn TA, Wei S et al (2009) Cancer Res 69:16–22
Guo Z, Yang X, Sun F et al (2009) Cancer Res 69:2305–2313
Visakorpi T, Hyytinen E, Koivisto P et al (1995) Nat Genet 9:401–406
Palmberg C, Koivisto P, Kakkola L et al (2000) J Urol 164:1992–1995
Linja MJ, Savinainen KJ, Saramäki OR et al (2001) Cancer Res 61:3550–3555
Edwards J, Krishna NS, Grigor KM et al (2003) Br J Cancer 89:552–556
Chen CD, Welsbie DS, Tran C et al (2004) Nat Med 10:33–39
Tamura K, Furihata M, Tsunoda T (2007) Cancer Res 67:5117–5125
Liu W, Laitinen S, Khan S et al (2009) Nat Med 15:559–565
Taylor BS, Schultz N, Hieronymus H et al (2010) Cancer Cell 18:11–22
Wang LG, Johnson EM, Kinoshita Y et al (2008) Cancer Res 68:2678–2688
Li Y, Wang L, Zhang M et al (2009) Cancer Res 69:3332–3338
Sharma A, Yeow WS, Ertel A et al (2010) J Clin Invest 120:4478–4492
Cai C, He HH, Chen S et al (2011) Cancer Cell 20:457–471
Horoszewicz JS, Leong SS, Chu TM (1980) Prog Clin Biol Res 37:115–132
Korenchuk S, Lehr JE, MClean L et al (2001) In Vivo 15:163–168
Saramäki OR, Harjula AE, Martikainen PM et al (2008) Clin Cancer Res 14:3395–3400
Tomlins SA, Rhodes DR, Perner S et al (2005) Science 310:644–648
Waltering KK, Helenius MA, Sahu B et al (2009) Cancer Res 69:8141–8149
Culig Z, Hoffmann J, Erdel M et al (1999) Br J Cancer 81:242–251
Urbanucci A, Sahu B, Seppälä J et al (2012) Oncogene 31:2153–2163
Chang C, Kokontis J, Liao S (1988) Science 240:324–326
Lubahn DB, Joseph DR, Sar M et al (1988) Mol Endocr 2:1265–1275
Perissi V, Rosenfeld MG (2005) Nat Rev Mol Cell Biol 6:542–554
Kokontis J, Takakura K, Hay N et al (1994) Cancer Res 54:1566–1573
Gregory CW, Johnson RT Jr, Mohler JL et al (2001) Cancer Res 61:2892–2988
Wang Q, Li W, Zhang Y et al (2009) Cell 138:245–256
Gregory CW, Johnson RT Jr et al (2001) J Androl 22:537–548
Massie CE, Lynch A, Ramos-Montoya A et al (2011) EMBO J 30:2719–2033
Prescott J, Jariwala U, Jia L et al (2007) Prostate 67:1371–1383
Zhao JC, Yu J, Runkle C et al (2012) Genome Res 22:322–331
Reyes JC, Barra J, Muchardt C et al (1998) EMBO J 17:6979–6991
Xu J, Qiu Y, DeMayo FJ et al (1998) Science 279:1922–1925
Gehin M, Mark M, Dennefeld C et al (2002) Mol Cell Biol 22:5923–5937
Cheung-Flynn J, Prapapanich V, Cox MB (2005) Mol Endocrinol 19:1654–1666
Yong W, Yang Z, Periyasamy S et al (2007) J Biol Chem 282:5026–5036
Linja MJ, Porkka KP, Kang Z et al (2004) Clin Cancer Res 10:1032–1040
Agoulnik IU, Vaid A, Nakka M et al (2006) Cancer Res 66:10594–10602
Li P, Yu X, Ge K et al (2002) Am J Pathol 161:1467–1474
Mestayer C, Blanchère M, Jaubert F et al (2003) Prostate 56:192–200
Culig Z, Comuzzi B, Steiner H et al (2004) J Steroid Biochem Mol Biol 92:265–271
Comuzzi B, Nemes C, Schmidt S et al (2004) J Pathol 204:159–166
Hu XD, Meng QH, Xu JY et al (2011) Biochem Biophys Res Commun 404:903–909
Ficazzola MA, Fraiman M, Gitlin J et al (2001) Carcinogenesis 22:1271–1279
Jalava SE, Urbanucci A, Latonen L et al. (2012) Oncogene. doi:10.1038/onc.2011.624
Urbanucci A, Waltering KK, Suikki HE et al (2008) BMC Cancer 8:219
Heemers HV, Regan KM, Schmidt LJ et al (2009) Mol Endocrinol 23:572–583
Shah S, Prasad S, Knudsen KE (2012) Cancer Res 72:1248–1259
Wang Q, Li W, Liu XS et al (2007) Mol Cell 27:380–392
Massie CE, Adryan B, Barbosa-Morais NL et al (2007) EMBO Rep 8:871–878
Yu J, Mani RS et al (2010) Cancer Cell 17:443–454
Sahu B, Laakso M, Ovaska K et al (2011) EMBO J 30:3962–3976
Takayama K, Tsutsumi S, Katayama S et al (2011) Oncogene 30:619–630
Pereira de Jésus-Tran K, Côté PL, Cantin L et al (2006) Protein Sci 15:987–999
Isaacs JT, D’Antonio JM, Chen S (2012) Prostate. doi:10.1002/pros.22504
Kang Z, Pirskanen A, Jänne OA et al (2002) J Biol Chem 277:48366–48371
Perissi V, Jepsen K, Glass CK et al (2010) Nat Rev Genet 11:109–123
Urbanucci A, Marttila S, Jänne OA et al (2012) Prostate 72:1223–1232
Welsbie DS, Xu J, Chen Y et al (2009) Cancer Res 69:958–966
Makkonen H, Kauhanen M, Jääskeläinen T et al (2011) Mol Cell Endocrinol 331:57–65
Lupien M, Eeckhoute J, Meyer CA et al (2008) Cell 132:958–970
Wang D, Garcia-Bassets I, Benner C et al (2011) Nature 474:390–394
Nakayama T, Watanabe M, Suzuki H et al (2000) Lab Invest 80:1789–1796
Rahman M, Miyamoto H, Chang C (2004) Clin Cancer Res 10:2208–2219
Chmelar R, Buchanan G, Need EF et al (2006) Int J Cancer 120:719–733
Heemers HV, Tindall DJ (2007) Endocr Rev 28:778–808
Strahl B, Allis C (2000) Nature 403:41–45
Verdone L, Agricola E, Caserta M et al (2006) Brief Funct Genomic Proteomic 5:209–221
Ito T (2007) J Biochem (Tokyo) 141:609–614
Andreu-Vieyra C, Lai J, Berman BP et al (2011) Mol Cell Biol 31:4648–4662
Hurtado A, Holmes KA, Ross-Innes CS et al (2011) Nat Genet 43:27–33
Chuu CP, Kokontis JM, Hiipakka RA et al (2011) J Biomed Sci 18:63
Langenhuijsen JF, Badhauser D, Schaaf B et al (2012) Urol Oncol doi:10.1016/j.urolonc
Mottet N, Van Damme J, Loulidi S et al (2012) BJU Int. doi:10.1111/j.1464-410X.2012.11120.x
Mulholland DJ, Tran LM, Li Y et al (2011) Cancer Cell 19:792–804
Carver BS, Chapinski C, Wongvipat J et al (2011) Cancer Cell 19:575–586
Delmore JE, Issa GC, Lemieux ME et al (2011) Cell 146:904–917
Klein KA, Reiter RE, Redula J et al (1997) Nat Med 3:402–408
Veldscholte J, Ris-Stalpers C, Kuiper GG et al (1990) Biochem Biophys Res Commun 173:534–540
Lee YG, Korenchuk S, Lehr J et al (2001) In Vivo 15:157–162
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Urbanucci, A., Waltering, K.K., Mills, I.G., Visakorpi, T. (2013). The Effect of AR Overexpression on Androgen Signaling in Prostate Cancer. In: Wang, Z. (eds) Androgen-Responsive Genes in Prostate Cancer. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-6182-1_12
Download citation
DOI: https://doi.org/10.1007/978-1-4614-6182-1_12
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-6181-4
Online ISBN: 978-1-4614-6182-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)