
Abstract
Prenatal development constitutes a critical time for shaping adult behaviour and may set the stage for vulnerability to disease later in life. A wealth of information from humans as well as from animal research has revealed that exposure to hostile conditions during gestation may result in a series of coordinated biological responses aimed at enhancing the probability of survival, but could also increase the susceptibility to mental illness. Prenatal stress has been linked to abnormal cognitive, behavioural and psychosocial outcomes both in animals and in humans, but the underlying molecular and physiological mechanisms remain largely unknown. In this chapter, we shall review experimental data from studies reported for rats, since more information is available for them than for other species. The major focus of the present chapter is to update and discuss data on behavioural, functional and morphological effects of prenatal stress in rats that may have counterparts in prospective and/or retrospective studies of gestational stress in humans. This work contributes to understanding the role of neuronal plasticity in the long-term effects of developmental adversity on brain function and its implications for vulnerability to disease.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Gestational stress
- Developmental programming
- Glucocorticoids
- Neurotrophins
- Hypothalamic-pituitary-adrenal (HPA) axis
Introduction
As discussed in Chap. 6, preclinical and clinical studies have shown that adverse events early in life induce profound and persistent effects on brain structure and function, which may lead to cognitive deficits and increased incidence of psychopathology later in life (Fumagalli et al. 2007). The concept of ‘ developmental programming ’ was put forward in an attempt to explain the association between environmental challenges during pregnancy, altered foetal growth and development, and later pathophysiology. During programming, non-genetic factors such as stress or exposure to excess glucocorticoids , the main hormonal mediator of stress, are transmitted to the foetus and act on specific tissues during sensitive windows of development to change developmental trajectories and thus their organisation and function (Maccari et al. 2003; Harris and Seckl 2011). Since different cells and tissues are sensitive to various factors at different times, the effects of adversity on an animal’s biology will be tissue, time and challenge specific (Sandman et al. 2011; Harris and Seckl 2011; Connors et al. 2008). It can be speculated that prenatal plasticity of physiological systems is biologically adaptive, allowing environmental factors acting on the mother or the foetus or both to alter the functions of an organ or tissue system to prepare the unborn animal optimally for the environmental conditions ex-utero (Del Giudice 2012; Maccari et al. 2003). Nevertheless, in extreme conditions like gestational stress , offspring display short and long-term physiological and behavioural abnormalities that increase the risk of disease. In humans, current information available from retrospective and prospective studies has reported that foetuses exposed to maternal stress at various times during gestation are at greater subsequent risk for cardiovascular and metabolic disorders than species with shorter lifespans. Such stress can result from a wide range of natural or man-made disasters or pressures, including earthquakes, floods, freezing storms, war, terrorist acts, chronic interpersonal tensions, or adverse conditions in the home or workplace (Weinstock 2008). Prenatal stress during pregnancy is associated with intrauterine growth restrictions and an increased risk of premature birth (Rondo et al. 2003), as well as with emotional and cognitive deficits in early life. Higher incidence of developing autism, hyperactivity and attention-deficit hyperactivity disorder were found in prenatally stressed children (Weinstock 2008; Cottrell and Seckl 2009). In adults, prenatal stress is linked to depression, anxiety and schizophrenia (Charil et al. 2010; Fumagalli et al. 2007; Weinstock 2008). Studies performed on mothers that self-reported elevated levels of anxiety during pregnancy indicated that their infants were more irritable and showed a higher incidence of sleeping and feeding problems than those of non-anxious mothers (Huizink et al. 2002).
By studying the effects of prenatal stress in experimental animals, one can control the timing, intensity and duration of stress exposure and evaluate the interaction of the mother with her offspring in a controlled environment (Fumagalli et al. 2007; Weinstock 2008). Most animal studies have been performed in rodents, for which the most comprehensive behavioural, morphological and histological information is available. The studies show the deleterious effects of prenatal stress on the offspring’s neuronal development and brain morphology, as well as changes in cerebral asymmetry that persist into adulthood (Fride and Weinstock 1988; Barros et al. 2006a; Adrover et al. 2007; Baier et al. 2012; Pallares et al. 2013b; Sandman et al. 2011; Zuena et al. 2008).
Foetal development can also be compromised by factors other than prenatal stress, including maternal malnutrition, mental illness, drugs, alcohol, viral infection, or environmental toxins (Valenzuela et al. 2012; Sandman et al. 2011; Connors et al. 2008; Barker 1998). Nevertheless, in this chapter, we will concentrate only on some effects of gestational stress in rats on behaviour, particularly in relation to the possible underlying morphological and neurochemical changes in brain regions that are involved in human disease vulnerability later in life.
Prenatal Stress Effects on Offspring Brain Plasticity and Related Behavioural Dysfunctions in Rats
Experimental Paradigms
Animal models of prenatal stress were developed to mimic etiological features of human diseases. Most studies with those models have exposed pregnant dams to a stressful situation and later explored short- and long-term behavioural and molecular changes in the newborn. Numerous protocols are described in the literature, which vary in the type of stressor applied, daily frequency, length of application, and week of gestation chosen. The types of stressors used in rodents have been summarized by Baier et al. (2012) and range from suspension, crowding, repeated tail shocks, restraint, immobilization, and saline injections to unpredictable stress with noise and flashing lights. According to Archer and Blackman (1971), the intensity of the response of the mother is more important than the intensity of the stimulus.
In our laboratory, we employ a prenatal restraint stress model in which pregnant rats are randomly assigned either to a control group that stays undisturbed throughout the pregnancy or to the stress group. “Stressed dams” are placed individually in a transparent plastic restrainer fitted closely to body size for 45 min three times a day (9.00, 12.00 and 16.00 h) during the last week of gestation (i.e. from gestational day 14 until delivery), which approximately coincides with the second trimester of gestation in humans and is considered critical for vulnerability to psychiatric disorders (Bayer et al. 1993). The restraint stress protocol is a preferred means of stressing animals because it is painless, straightforward, does not involve bodily harm and is inexpensive. Moreover, the physiological changes associated with restraint result from the distress and aversive nature of having to remain immobile, and the changes mainly manifest as increases in adrenocorticotropic hormone and corticosterone (Buynitsky and Mostofsky 2009; Ward and Weisz 1984). In our hands, the intensity and duration of the stress applied are sufficient to induce alterations in a variety of assessed parameters, including behavioural and physiological dysfunction related to glutamatergic and dopaminergic system development in the offspring (Berger et al. 2002; Barros et al. 2006a, b; Adrover et al. 2007; Katunar et al. 2010; Pallares et al. 2013b). We have also observed that prenatal stress induces long-term effects on the male offspring reproductive system and spermatogenesis development, particularly by inducing a long term imbalance of circulating sexual hormone levels (Pallares et al. 2013a).
Prenatal Stress and Behavioural Dysfunctions in Rats
The effect of prenatal stress on baseline motor activity in rodents has been regarded as a main parameter of altered behaviour in order to evaluate coping and responsiveness of specific neurotransmitter pathways related to locomotion. However, the results that have been reported are controversial, since increased, decreased and unchanged basal motor activity have all been reported in prenatal stress animals (Pallares et al. 2007; Koenig et al. 2005; Fumagalli et al. 2007). On the other hand, amphetamine injection increases locomotion in prenatally stressed rats (Lehmann et al. 2000; Koenig et al. 2005). Schizophrenic patients respond abnormally to psychostimulants much as do prenatally stressed rats, suggesting that prenatal exposure to stress might cause an enhanced responsiveness of the dopaminergic system , which represents a core feature of schizophrenia (Fumagalli et al. 2007). In support of this observation, our group demonstrated that basal and amphetamine-stimulated dopamine output in the Nucleus Accumbens of adolescent and adult prenatal stress rats is higher than in controls (Silvagni et al. 2008). In addition, schizophrenic patients have deficits in filtering or discriminating relevant from irrelevant information (i.e. signal to noise ratio) and the pathology is characterized by deficits in operative measures of filter ability, such as prepulse inhibition. It has been demonstrated that exposure to chronic stress during gestation disrupts prepulse inhibition (Koenig et al. 2005).
Despite the similarities between schizophrenia and long-lasting behavioural effects resulting from prenatal stress, this paradigm has also been proposed as a model of major depression. The symptoms of clinical depression in humans involve changes in mood that cannot be assessed in animal models. Nevertheless, behavioural patterns related to human depression, such as loss of active coping, social withdrawal and anhedonia (inability to feel pleasure), can be measured under appropriate conditions. Prenatal stress rats and mice show increased duration of immobility in the forced swim Porsolt test when compared to rats with normal gestation (Morley-Fletcher et al. 2003; Alonso et al. 1991). Furthermore, prenatal stress reduces exploratory behaviour in adult animals, presumably due to decreased motivation (Vallee et al. 1997), and the animals are more likely to develop anhedonia, as indicated by a decrease in sucrose solution consumed when performing the sucrose preference test (Sun et al. 2013).
Evidence from experimental research also demonstrates that prenatal stress affects emotions of the offspring by increasing anxiety-like behaviour. In 1957, Thompson was the first to demonstrate that trauma in females during pregnancy increased emotionality in their offspring (Thompson 1957). Anxiety state in experimental animals can be measured in tests that evaluate fear-like reactions, which can be elicited in unfamiliar open spaces like the “open field” or in the “elevated plus maze.” Increased anxiety in both sexes of offspring of mothers subjected to unpredictable noise throughout gestation were reported by Fride and Weinstock (1988) and Vallee et al. (1997). In our hands, prenatally stressed adult rats spend more time in the closed arms of the elevated plus maze than control arms, suggesting anxiety (Barros et al. 2006b).
Cognitive impairment is also related to gestational stress . Chronic maternal restraint stress slows the acquisition of spatial learning (Lemaire et al. 2000; Son et al. 2006; Yang et al. 2006) and reduces long-term potentiation, a primary physiological model of memory (Son et al. 2006) in offspring of rats and mice (see Chap. 5). The principal region for the regulation of cognition is the hippocampus, which also has the highest density of glucocorticoid receptors. Hippocampal integrity is essential for the development of physiological cognitive processes, including learning and memory. The cytoarchitecture of the rat hippocampus is altered by maternal gestational stress; prenatal stress decreases the hippocampal volume of the progeny by reducing the number of granule cells within the hippocampal dentate gyrus (Lemaire et al. 2000) and neurogenesis in this region (Lemaire et al. 2006). By mimicking the effects of prenatal stress through intramuscular injection of synthetic glucocorticoids during pregnancy, Uno et al. (1994) have demonstrated a dose-dependent degenerative change and reduction of the offspring hippocampal neurons. Moreover, hippocampus is smaller in schizophrenic and depressed subjects, so gestational stress might be etiologically relevant for the morphological changes that contribute to cognitive deterioration in psychiatric patients (Fumagalli et al. 2007). Furthermore, prenatal stress reduces the number of hippocampal synapses in the CA3 area (Hayashi et al. 1998) and, as we reported in our studies, prenatal stress induces a long term reduction in dendritic arborisation of the hippocampal CA1 region (Barros et al. 2006a; Pallares et al. 2013b).
Other brain areas that play a key role in controlling cognitive processes, such as the amygdala and the cerebral cortex, are also reported to be affected by prenatal stress. The amygdala is involved in mood regulation and in the mediation of emotional processes of fear and anxiety. Prenatal stress diminishes the size of most of the amygdalar nuclei , with exception of the lateral amygdalar nuclei, for which the effects of prenatal stress on size are age-specific: during early postnatal development a decreased volume and reduced number of neurons and glial cells are observed in prenatally stressed rats, whereas postnatally prenatal stress increases the number of neurons and glial cells in the same nuclei (Salm et al. 2004; Kraszpulski et al. 2006; Kawamura et al. 2006). The cerebral cortex is responsible for a variety of higher order brain processes such as memory, planning, problem solving and voluntary muscle movement. We found that prenatal stress alters subtypes of dopamine and glutamate receptors in different cortical regions in males offspring (Berger et al. 2002) and that prenatal stress induces a long term reduction of dendritic arborisations (Barros et al. 2006a; Pallares et al. 2013a). Furthermore, dendritic spine densities on both the apical and basal dendrites of pyramidal neurons are reduced by approximately 20% in the dorsal anterior cingulate cortex and orbitofrontal cortex for both male and female prenatally stressed rats (Murmu et al. 2006).
Possible Mechanisms of Prenatal Stress-Related Brain Programming
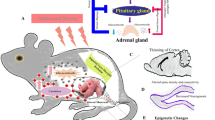
The mechanism by which the pregnant mother translates stress to the developing brain is still a matter of debate. Nevertheless, in this chapter we present several hypotheses that have been proposed (Fig. 7.1).

Possible mechanism linking prenatal stress with plastic changes in the developing brain. The scheme depicts three of the possible links between prenatal stress and changes in the developing brain of the offspring, As explained in the text one of the mechanism proposed suggests that the increase of glucocorticoids and catecholamines in the stressed pregnant dam binds to their specific receptors in the placenta, reducing foetal blood flow, decreasing the supply of essential nutrients and oxygen to the foetus concomitantly with oxidative damage. A second mechanism directly relates the increase of circulating hormones in the mother to induce a dysfunctional HPA axis in the offspring, triggering alterations in the capability to cope with a stressful event. A third mechanism is related to changes in maternal behaviour that produces a long-term deregulation of the functional activity of the offspring HPA axis and alterations of DNA methylation
Maternal Stress Affects by Altering Blood Flow to the Placenta
Maternal stress has also been shown to constrict the placental arteries. The placenta has a high number of adrenergic receptors that in presence of hormones reduce foetal blood flow and the supply of essential nutrients and oxygen from the uterus. Thereby, the morphology and development of the placenta is affected as well as the foetus’ oxygenation and nutrition, inducing, for example, low birth weight among other effects (Weinstock 2005; Charil et al. 2010). Moreover, fluctuations in the oxygen availability induce oxidative stress in the placenta, increasing the number of oxygen free radicals and the release of cytokines that could also contribute to the development of neural disorders by oxidative damage (Charil et al. 2010).
Deregulation of Neurotrophins
Neurotrophins are small polypeptides that mediate the enduring effects of perinatal adversities on brain function by exerting a complex array of actions on various cellular phenotypes. In the central nervous system, they regulate survival and maturation of developing neurons including axonal growth and synaptic plasticity. Moreover, they also modulate neurotransmitter function (Fumagalli et al. 2007). Therefore, neurotrophins may be a route through which developmental manipulation, such as stress, can alter cellular resilience and modify brain structure. In addition, interfering with neurotrophin receptor function during development determines behavioural abnormalities such as dopaminergic hyper-responsivity and disrupted prepulse inhibition of acoustic startle, both of which are characteristic of schizophrenia (Rajakumar et al. 2004). Brain derived neurotrophic factor (BDNF) regulates synaptic transmission across a broad temporal spectrum ranging from short-term modulation, which occurs in the order of seconds to minutes, to prolonged effects that persist for many hours, such as long-term potentiation (Fumagalli et al. 2007). Several studies have demonstrated that prenatal stress may reduce the biosynthesis of the BDNF in the infant (Van den Hove et al. 2006) and adult (Zuena et al. 2008; Monteleone et al. 2014) hippocampus of prenatally stressed male rats. Moreover, the expression of the fibroblast growth factor (FGF-2) , which plays a relevant role as a neuroprotective molecule during development and adulthood, but is also involved in several psychiatric disorders, was found to be reduced in prefrontal cortex of prenatal stress animals (Fumagalli et al. 2007). The reduced expression of both trophic factors could occur as a result of perinatal stressors and may account for the vulnerability of specific neuronal systems.
Excess Glucocorticoids and Dysfunctional Stress Response in the Offspring
One potential mechanism whereby prenatal stress influences foetal development is by modifying the programming of the hypothalamic-pituitary-adrenal (HPA) axis, a major system controlling the organism’s response to stress and regulating certain circadian activity. Behavioural alterations induced by a dysfunctional HPA axis (i.e. alterations in the individual capability to cope with a stressful event) show similarities with psychiatric disorders including major depression and schizophrenia (Fumagalli et al. 2007). Maternal stress can affect the offspring’s stress response later in life (Levine 1967). Moreover, prolonged restraint stress exposure in pregnant rats during the last week of gestation reprograms their foetal HPA axis (Weinstock 2005, 2008): prenatally stressed rats secrete higher amounts of total- and free basal corticosterone at the end of the light period (Koehl et al. 1997) and exhibit prolonged elevation in plasma glucocorticoid levels following acute exposure to restraint stress (Vallee et al. 1997). That both prenatal stress and prenatal synthetic glucocorticoid exposure during the last week of gestation in rats permanently diminish corticosterone receptors in the hippocampus and hypothalamus could explain the reported imbalance in stress hormone levels during resting or even after a stressful episode of exposure. Reduction of central glucocorticoid receptors leads to an attenuation of the HPA axis feedback loop sensitivity (Henry et al. 1994; Maccari et al. 1995; Welberg et al. 2000).
During pregnancy, glucocorticoids (GCs) are naturally elevated. GCs are essential for foetal growth and induction of certain substances, such as pulmonary surfactant. GCs are also involved in normal brain development, where they exert a wide spectrum of effects in most regions of the developing brain, ranging from sub-cellular re-organization to neuron-neuron and neuron-glial interactions . Since sustained elevation of these hormones or their removal from the foetal brain is detrimental to normal processes, it is not surprising that most studies agree that glucocorticoids are the main agent conveying the effects of maternal stress to developing foetuses (Mastorci et al. 2009; Charil et al. 2010; Matthews 2001). In rats, maturation of the HPA axis starts early in development and extends to the early postnatal period. The glucocorticoid type II receptor (GR) mRNA can be detected in the hippocampus, hypothalamus and pituitary from day 13 of gestation and increases rapidly after birth (Cintra et al. 1993). The long-term effects of prenatal stress might be the consequence of excessive exposure of the foetus to maternal corticosterone. Such effects were shown to be prevented if the adrenal glands were surgical removed from the dams (Barbazanges et al. 1996). Additionally, inhibition of the enzyme 11beta-hydroxysteroid dehydrogenase type 2 (11beta-HSD2), which rapidly inactivates glucocorticoids when passing through placenta and other foetal tissues, induces permanent alterations of the HPA axis and increases anxiety-like behaviour (Welberg et al. 2000) suggesting that foetal overexposure to endogenous glucocorticoids may represent a common link between the prenatal environment and disorders linked to adult HPA axis dysfunction.
On the other hand, persistent effects of prenatal stress on HPA axis activity can be simulated by experimental models of maternal deprivation from postnatal day 2 to 14 (Liu et al. 2000), or it can be reversed by early adoption (Maccari et al. 1995) and neonatal handling (Meaney 2001), indicating that maternal behaviour may be crucial for the long-term regulation of functional activity of their offspring’s HPA axis. Indeed, as discussed in Chap. 6, in recent years it has been demonstrated that the mother’s behaviour produces stable alterations of DNA methylation and chromatin structure in the offspring (Meaney and Szyf 2005). Offspring in high mother-pup interactions have a reduced number of methylation of CpG dinucleotides in the GR promoter sequence than do offspring from lower maternal care mothers. That hypomethylation might be responsible for increased transcription of the GR gene, providing an epigenetic mechanism by which maternal care affects the gene expression of the offspring (Weaver et al. 2004).
Overall, adverse life conditions during prenatal or early postnatal life may be highly detrimental to the function and responsiveness of the HPA axis to stress. This could ultimately lead to a more persistent exposure of the brain to elevated levels of glucocorticoids that may reduce cellular resiliency and lead to damage of function in certain brain regions (McEwen 2000).
Concluding Remarks
Prenatal stress has been linked to abnormal outcomes in rodents, non-human primates, and humans (Charil et al. 2010; Huizink et al. 2004; Weinstock 2001). The data described in this chapter provide evidence that maternal stress at critical periods of development may alter the programming of foetal brain areas controlling and processing important behaviours, thereby increasing susceptibility to psychopathology. The increase in stress hormones during critical windows of brain development is detrimental for normal neuronal differentiation and function. Depending on intensity and the time of gestation the stress takes place, the behavioural effects on the offspring range from learning and attention deficits, anxiety- and depressive-like behaviour to abnormal stress response, among others. It is clear from animal studies that prenatal stress affects the morphology of the offspring brains, but the mechanisms that lead to these effects remain poorly understood. Nevertheless, the maternal and foetal HPA axis and the placenta seem to represent the main candidates for these mechanisms.
As early as 1941, Sontag (1941) drew attention to the long-lasting consequences of gestational stress on infant development and behaviour. Extrapolation of experimental research results from prenatal stress studies in animals to gestational disturbances in humans has been attempted. Although we are aware that direct comparison between experimental animals and humans is a complex issue, understanding of brain mechanism underlying the link between prenatal stress and adult psychopathologies still relies on animal studies. In this context, it can be speculated that the impairments observed in limbic areas in animal studies after prenatal stress might induce behavioural effects in animals that correspond to similar effects in human subjects. Therefore, animal studies could provide a neurochemical and morphological basis to the observed human psychopathologies associated with gestational stress.
Abbreviations
- 11-beta-HSD2:
-
11-beta-hydroxysteroid dehydrogenase type II enzyme
- BDNF:
-
Brain derived neurotrophic factor
- FGF-2:
-
Fibroblast grow factor type II
- GCs:
-
Glucocorticoids
- GR:
-
Glucocorticoid receptor type II
- HPA:
-
Hypothalamic-pituitary-adrenal axis
References
Adrover E, Berger MA, Perez AA, Tarazi FI, Antonelli MC (2007) Effects of prenatal stress on dopamine D2 receptor asymmetry in rat brain. Synapse 61(6):459–462
Alonso SJ, Arevalo R, Afonso D, Rodriguez M (1991) Effects of maternal stress during pregnancy on forced swimming test behavior of the offspring. Physiol Behav 50:511–517
Archer JE, Blackman DE (1971) Prenatal psychological stress and offspring behavior in rats and mice. Dev Psychobiol 4(3):193–248. doi:10.1002/dev.420040302
Baier CJ, Katunar MR, Adrover E, Pallares ME, Antonelli MC (2012) Gestational restraint stress and the developing dopaminergic system: an overview. Neurotox Res. doi:10.1007/s12640-011-9305-4
Barbazanges A, Piazza PV, Le Moal M, Maccari S (1996) Maternal glucocorticoid secretion mediates long-term effects of prenatal stress. J Neurosci 16(12):3943–3949
Barker DJ (1998) In utero programming of chronic disease. Clin Sci (Lond) 95(2):115–128
Barros VG, Duhalde-Vega M, Caltana L, Brusco A, Antonelli MC (2006a) Astrocyte-neuron vulnerability to prenatal stress in the adult rat brain. J Neurosci Res 83(5):787–800
Barros VG, Rodriguez P, Martijena ID, Perez A, Molina VA, Antonelli MC (2006b) Prenatal stress and early adoption effects on benzodiazepine receptors and anxiogenic behavior in the adult rat brain. Synapse 60(8):609–618
Bayer SA, Altman J, Russo RJ, Zhang X (1993) Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology 14(1):83–144
Berger MA, Barros VG, Sarchi MI, Tarazi FI, Antonelli MC (2002) Long-term effects of prenatal stress on dopamine and glutamate receptors in adult rat brain. Neurochem Res 27(11):1525–1533
Buynitsky T, Mostofsky DI (2009) Restraint stress in biobehavioral research: recent developments. Neurosci Biobehav Rev 33(7):1089–1098. doi:10.1016/j.neubiorev.2009.05.004. S0149-7634(09)00074-8 [pii]
Charil A, Laplante DP, Vaillancourt C, King S (2010) Prenatal stress and brain development. Brain Res Rev 65(1):56–79. doi:10.1016/j.brainresrev.2010.06.002. S0165-0173(10)00072-X [pii]
Cintra A, Solfrini V, Bunnemann B, Okret S, Bortolotti F, Gustafsson JA, Fuxe K (1993) Prenatal development of glucocorticoid receptor gene expression and immunoreactivity in the rat brain and pituitary gland: a combined in situ hybridization and immunocytochemical analysis. Neuroendocrinology 57(6):1133–1147
Connors SL, Levitt P, Matthews SG, Slotkin TA, Johnston MV, Kinney HC, Johnson WG, Dailey RM, Zimmerman AW (2008) Fetal mechanisms in neurodevelopmental disorders. Pediatr Neurol 38(3):163–176. doi:10.1016/j.pediatrneurol.2007.10.009. S0887-8994(07)00542-5 [pii]
Cottrell EC, Seckl JR (2009) Prenatal stress, glucocorticoids and the programming of adult disease. Front Behav Neurosci 3:19. doi:10.3389/neuro.08.019.2009
Del Giudice M (2012) Fetal programming by maternal stress: insights from a conflict perspective. Psychoneuroendocrinology 37(10):1614–1629. S0306-4530(12)00191-6 [pii]. doi:10.1016/j.psyneuen.2012.05.014
Fride E, Weinstock M (1988) Prenatal stress increases anxiety related behavior and alters cerebral lateralization of dopamine activity. Life Sci 42(10):1059–1065
Fumagalli F, Molteni R, Racagni G, Riva MA (2007) Stress during development: impact on neuroplasticity and relevance to psychopathology. Prog Neurobiol 81(4):197–217. S0301-0082(07)00014-7 [pii]. doi:10.1016/j.pneurobio.2007.01.002
Harris A, Seckl J (2011) Glucocorticoids, prenatal stress and the programming of disease. Horm Behav 59(3):279–289. S0018-506X(10)00167-4 [pii]. doi:10.1016/j.yhbeh.2010.06.007
Hayashi A, Nagaoka M, Yamada K, Ichitani Y, Miake Y, Okado N (1998) Maternal stress induces synaptic loss and developmental disabilities of offspring. Int J Dev Neurosci 16(3–4):209–216
Henry C, Kabbaj M, Simon H, Le Moal M, Maccari S (1994) Prenatal stress increases the hypothalamo-pituitary-adrenal axis response in young and adult rats. J Neuroendocrinol 6(3):341–345
Huizink AC, de Medina PG, Mulder EJ, Visser GH, Buitelaar JK (2002) Psychological measures of prenatal stress as predictors of infant temperament. J Am Acad Child Adolesc Psychiatry 41(9):1078–1085
Huizink AC, Mulder EJ, Buitelaar JK (2004) Prenatal stress and risk for psychopathology: specific effects or induction of general susceptibility? Psychol Bull 130(1):115–142
Katunar M, Saez T, Brusco A, Antonelli M (2010) Ontogenetic expression of dopamine-related transcription factors and tyrosine hydroxylase in prenatally stressed rats. Neurotox Res 18(1):69–81
Kawamura T, Chen J, Takahashi T, Ichitani Y, Nakahara D (2006) Prenatal stress suppresses cell proliferation in the early developing brain. Neuroreport 17(14):1515–1518. doi:10.1097/01.wnr.0000236849.53682.6d. 00001756-200610020-00012[pii]
Koehl M, Barbazanges A, Le Moal M, Maccari S (1997) Prenatal stress induces a phase advance of circadian corticosterone rhythm in adult rats which is prevented by postnatal stress. Brain Res 759(2):317–320
Koenig JI, Elmer GI, Shepard PD, Lee PR, Mayo C, Joy B, Hercher E, Brady DL (2005) Prenatal exposure to a repeated variable stress paradigm elicits behavioral and neuroendocrinological changes in the adult offspring: potential relevance to schizophrenia. Behav Brain Res 156(2):251–261. S0166-4328(04)00216-5 [pii]. doi:10.1016/j.bbr.2004.05.030
Kraszpulski M, Dickerson PA, Salm AK (2006) Prenatal stress affects the developmental trajectory of the rat amygdala. Stress 9(2):85–95. J740525J80R55214 [pii]. doi:10.1080/10253890600798109
Lehmann J, Stohr T, Feldon J (2000) Long-term effects of prenatal stress experiences and postnatal maternal separation on emotionality and attentional processes. Behav Brain Res 107(1–2):133–144
Lemaire V, Koehl M, Le Moal M, Abrous DN (2000) Prenatal stress produces learning deficits associated with an inhibition of neurogenesis in the hippocampus. Proc Natl Acad Sci USA 97(20):11032–11037. 97/20/11032 [pii]
Lemaire V, Lamarque S, Le Moal M, Piazza PV, Abrous DN (2006) Postnatal stimulation of the pups counteracts prenatal stress-induced deficits in hippocampal neurogenesis. Biol Psychiatry 59(9):786–792. S0006-3223(05)01400-9 [pii]. doi:10.1016/j.Biopsych.2005.11.009
Levine S (1967) Maternal and environmental influences on the adrenocortical response to stress in weanling rats. Science 156(3772):258–260
Liu D, Diorio J, Day JC, Francis DD, Meaney MJ (2000) Maternal care, hippocampal synaptogenesis and cognitive development in rats. Nat Neurosci 3(8):799–806. doi:10.1038/77702
Maccari S, Piazza PV, Kabbaj M, Barbazanges A, Simon H, Le Moal M (1995) Adoption reverses the long-term impairment in glucocorticoid feedback induced by prenatal stress. J Neurosci 15(1 Pt 1):110–116
Maccari S, Darnaudery M, Morley-Fletcher S, Zuena AR, Cinque C, Van Reeth O (2003) Prenatal stress and long-term consequences: implications of glucocorticoid hormones. Neurosci Biobehav Rev 27(1–2):119–127. S0149763403000149 [pii]
Mastorci F, Vicentini M, Viltart O, Manghi M, Graiani G, Quaini F, Meerlo P, Nalivaiko E, Maccari S, Sgoifo A (2009) Long-term effects of prenatal stress: changes in adult cardiovascular regulation and sensitivity to stress. Neurosci Biobehav Rev 33(2):191–203. S0149-7634(08)00122-X [pii]. doi:10.1016/j.neubiorev.2008.08.001
Matthews SG (2001) Antenatal glucocorticoids and the developing brain: mechanisms of action. Semin Neonatol 6(4):309–317. doi:10.1053/siny.2001.0066. S1084-2756(01)90066-1[pii]
McEwen BS (2000) Effects of adverse experiences for brain structure and function. Biol Psychiatry 48(8):721–731. S0006-3223(00)00964-1 [pii]
Meaney MJ (2001) Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu Rev Neurosci 24:1161–1192. doi:10.1146/annurev.neuro.24.1.1161. 24/1/1161[pii]
Meaney MJ, Szyf M (2005) Maternal care as a model for experience-dependent chromatin plasticity? Trends Neurosci 28(9):456–463. S0166-2236(05)00189-X [pii]. doi:10.1016/j.tins.2005.07.006
Monteleone MC, Adrover E, Pallares ME, Antonelli MC, Frasch AC, Brocco MA (2014) Prenatal stress changes the glycoprotein GPM6A gene expression and induces epigenetic changes in rat offspring brain. Epigenetics 9(1):152–160. 25925 [pii]. doi:10.4161/epi.25925
Morley-Fletcher S, Darnaudery M, Koehl M, Casolini P, Van Reeth O, Maccari S (2003) Prenatal stress in rats predicts immobility behavior in the forced swim test. Effects of a chronic treatment with tianeptine. Brain Res 989(2):246–251. S0006899303032931 [pii]
Murmu MS, Salomon S, Biala Y, Weinstock M, Braun K, Bock J (2006) Changes of spine density and dendritic complexity in the prefrontal cortex in offspring of mothers exposed to stress during pregnancy. Eur J Neurosci 24(5):1477–1487. EJN5024 [pii]. doi:10.1111/j.1460-9568.2006.05024.x
Pallares ME, Scacchi Bernasconi PA, Feleder C, Cutrera RA (2007) Effects of prenatal stress on motor performance and anxiety behavior in Swiss mice. Physiol Behav 92(5):951–956. S0031-9384(07)00267-3 [pii]. doi:10.1016/j.physbeh.2007.06.021
Pallares ME, Adrover E, Baier CJ, Bourguignon NS, Monteleone MC, Brocco MA, Gonzalez-Calvar SI, Antonelli MC (2013a) Prenatal maternal restraint stress exposure alters the reproductive hormone profile and testis development of the rat male offspring. Stress 16(4):429–440. doi:10.3109/10253890.2012.761195
Pallares ME, Baier CJ, Adrover E, Monteleone MC, Brocco MA, Antonelli MC (2013b) Age-dependent effects of prenatal stress on the corticolimbic dopaminergic system development in the rat male offspring. Neurochem Res 38(11):2323–2335. doi:10.1007/s11064-013-1143-8
Rajakumar N, Leung LS, Ma J, Rajakumar B, Rushlow W (2004) Altered neurotrophin receptor function in the developing prefrontal cortex leads to adult-onset dopaminergic hyperresponsivity and impaired prepulse inhibition of acoustic startle. Biol Psychiatry 55(8):797–803. doi:10.1016/j.biopsych.2003.12.015. S0006322304000551[pii]
Rondo PH, Ferreira RF, Nogueira F, Ribeiro MC, Lobert H, Artes R (2003) Maternal psychological stress and distress as predictors of low birth weight, prematurity and intrauterine growth retardation. Eur J Clin Nutr 57(2):266–272. doi:10.1038/sj.ejcn.1601526. 1601526[pii]
Salm AK, Pavelko M, Krouse EM, Webster W, Kraszpulski M, Birkle DL (2004) Lateral amygdaloid nucleus expansion in adult rats is associated with exposure to prenatal stress. Brain Res Dev Brain Res 148(2):159–167. doi:10.1016/j.devbrainres.2003.11.005. S0165380603003468[pii]
Sandman CA, Davis EP, Buss C, Glynn LM (2011) Prenatal programming of human neurological function. Int J Pept 2011:837596. doi:10.1155/2011/837596
Silvagni A, Barros VG, Mura C, Antonelli MC, Carboni E (2008) Prenatal restraint stress differentially modifies basal and stimulated dopamine and noradrenaline release in the nucleus accumbens shell: an ‘in vivo’ microdialysis study in adolescent and young adult rats. Eur J Neurosci 28(4):744–758. EJN6364 [pii]. doi:10.1111/j.1460-9568.2008.06364.x
Son GH, Geum D, Chung S, Kim EJ, Jo JH, Kim CM, Lee KH, Kim H, Choi S, Kim HT, Lee CJ, Kim K (2006) Maternal stress produces learning deficits associated with impairment of NMDA receptor-mediated synaptic plasticity. J Neurosci 26(12):3309–3318. 26/12/3309 [pii]. doi:10.1523/JNEUROSCI.3850-05.2006
Sontag LD (1941) The significance of fetal environmental differences. Am J Obstet Gynecol 42(6):996–1003
Sun H, Guan L, Zhu Z, Li H (2013) Reduced levels of NR1 and NR2A with depression-like behavior in different brain regions in prenatally stressed juvenile offspring. PLoS One 8(11):e81775. doi:10.1371/journal.pone.0081775. PONE-D-13-37163[pii]
Thompson WR (1957) Influence of prenatal maternal anxiety on emotionality in young rats. Science 125(3250):698–699
Uno H, Eisele S, Sakai A, Shelton S, Baker E, DeJesus O, Holden J (1994) Neurotoxicity of glucocorticoids in the primate brain. Horm Behav 28(4):336–348. S0018-506X(84)71030-0 [pii]. doi:10.1006/hbeh.1994.1030
Valenzuela CF, Morton RA, Diaz MR, Topper L (2012) Does moderate drinking harm the fetal brain? Insights from animal models. Trends Neurosci 35(5):284–292. S0166-2236(12)00018-5 [pii]. doi:10.1016/j.tins.2012.01.006
Vallee M, Mayo W, Dellu F, Le Moal M, Simon H, Maccari S (1997) Prenatal stress induces high anxiety and postnatal handling induces low anxiety in adult offspring: correlation with stress-induced corticosterone secretion. J Neurosci 17(7):2626–2636
Van den Hove DL, Steinbusch HW, Scheepens A, Van de Berg WD, Kooiman LA, Boosten BJ, Prickaerts J, Blanco CE (2006) Prenatal stress and neonatal rat brain development. Neuroscience 137(1):145–155. S0306-4522(05)00985-1[pii]. doi:10.1016/j.neuroscience.2005.08.060
Ward IL, Weisz J (1984) Differential effects of maternal stress on circulating levels of corticosterone, progesterone, and testosterone in male and female rat fetuses and their mothers. Endocrinology 114(5):1635–1644. doi:10.1210/endo-114-5-1635
Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ (2004) Epigenetic programming by maternal behavior. Nat Neurosci 7(8):847–854. doi:10.1038/nn1276. nn1276[pii]
Weinstock M (2001) Alterations induced by gestational stress in brain morphology and behaviour of the offspring. Prog Neurobiol 65(5):427–451. S0301-0082(01)00018-1 [pii]
Weinstock M (2005) The potential influence of maternal stress hormones on development and mental health of the offspring. Brain Behav Immun 19(4):296–308. S0889-1591(04)00132-1 [pii]. doi:10.1016/j.bbi.2004.09.006
Weinstock M (2008) The long-term behavioural consequences of prenatal stress. Neurosci Biobehav Rev 32(6):1073–1086
Welberg LA, Seckl JR, Holmes MC (2000) Inhibition of 11beta-hydroxysteroid dehydrogenase, the foeto-placental barrier to maternal glucocorticoids, permanently programs amygdala GR mRNA expression and anxiety-like behaviour in the offspring. Eur J Neurosci 12(3):1047–1054. ejn958 [pii]
Yang J, Han H, Cao J, Li L, Xu L (2006) Prenatal stress modifies hippocampal synaptic plasticity and spatial learning in young rat offspring. Hippocampus 16(5):431–436. doi:10.1002/hipo.20181
Zuena AR, Mairesse J, Casolini P, Cinque C, Alema GS, Morley-Fletcher S, Chiodi V, Spagnoli LG, Gradini R, Catalani A, Nicoletti F, Maccari S (2008) Prenatal restraint stress generates two distinct behavioral and neurochemical profiles in male and female rats. PLoS One 3(5):e2170. doi:10.1371/journal.pone.0002170
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Pallarés, M.E., Antonelli, M.C. (2017). Prenatal Stress and Neurodevelopmental Plasticity: Relevance to Psychopathology. In: von Bernhardi, R., Eugenín, J., Muller, K. (eds) The Plastic Brain. Advances in Experimental Medicine and Biology, vol 1015. Springer, Cham. https://doi.org/10.1007/978-3-319-62817-2_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-62817-2_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-62815-8
Online ISBN: 978-3-319-62817-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)