
Abstract
The study of polymerization kinetics is of great interest in dentistry, particularly for restorative composites. Most commonly, those materials are composed of multimethacrylate monomers, photopolymerized to give a cross-linked polymer network. The rate at which that reaction takes place influences not only the final degree of conversion (extent of polymerization) but also some of the important physical and mechanical properties of the final material. Understanding how the polymerization progresses is important for researchers developing new materials and for clinicians to obtain the best performance out of the final restoration. In this chapter, the different types of polymerization will be discussed, so to contextualized the mechanisms involved in the polymerization of methacrylates and chain-transfer reactions induced by the presence of thiols. Emphasis will also be given to particular aspects of the photoinitiation reaction.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others



1 Introduction
The study of polymerization kinetics is of great interest in dentistry, particularly for restorative composites. Most commonly, those materials are composed of multimethacrylate monomers, photopolymerized to give a cross-linked polymer network. The rate at which that reaction takes place influences not only the final degree of conversion (extent of polymerization) but also some of the important physical and mechanical properties of the final material. Understanding how the polymerization progresses is important for researchers developing new materials and for clinicians to obtain the best performance out of the final restoration. In this chapter, the different types of polymerization will be discussed, so to contextualized the mechanisms involved in the polymerization of methacrylates and chain-transfer reactions induced by the presence of thiols. Emphasis will also be given to particular aspects of the photoinitiation reaction.
2 Principles of Polymer Chemistry
2.1 Types of Polymers and Polymerization Reactions
Polymers are formed by a large number of small molecules—monomers—linked with each other building up macromolecules. Polymers and polymerization reactions may be classified following the criteria presented in Table 3.1. Frequently, the terms condensation and step polymerization and addition and chain polymerization are used interchangeably because the majority of addition polymers are obtained by chain-growth polymerization reaction and, in turn, condensation polymers by step-growth polymerization. However, it is not always the case, because the terms “condensation” and “addition” are based on the polymer composition and structure, and “step” and “chain” are characterized by mechanisms of polymerization reaction. Therefore, different aspects are considered for those classifications.
Methacrylates polymerize through vinyl bonds, via a chain-growth mechanism. When combined with chain-transfer agents, such as thiols, or in thiol-ene monomer systems, a combination of chain and step-growth reactions takes place, with profound impact on reaction kinetics and the final polymer structure. Chain-transfer reactions are used as a strategy to control molecular weight in linear polymers and control gelation in cross-linked networks. These aspects will be discussed in more detail in the section on Chain-Transfer Reactions and their effect on kinetic chain length.
2.1.1 Polymer Structure
The polymer structure classification is based on compositional differences between the polymer and the monomers from which it was synthesized. According to this criterion, the polymers may be divided in condensation and addition.
2.1.1.1 Condensation Polymers
Condensation polymers are synthesized from polyfunctional monomers through condensation reactions involving organic chemistry and, in general, elimination of small molecules. Generally, the eliminated molecule is water, but hydrogen chloride (HCl), ammonia (NH3), hydrogen cyanide (HCN), or alcohol (–OH), among others, may also be eliminated. An example of condensation polymer is the polyamides:
The parentheses indicate the repeating unit, i.e., unit that repeats itself several times in the polymer chain. It is important to mention that one determined condensation polymer can be obtained from different reactions (e.g., polyamides may be synthesized from the reaction between diamides and diacids (or diacyl chlorides), but the self-condensation of amino acids is also able to produce polyamides).
It is noteworthy that some condensation reactions do not eliminate by-products, such as is the case in click reactions between alcohols or thiols and isocyanates [1,2,3]. Those are useful for the formation of several polycarbamates, such as polyurethanes and polythiourethanes, both with several uses in coating industry and, more recently, in dentistry [4,5,6].
2.1.1.2 Addition Polymers
Addition polymers are synthesized without the loss of small molecules, resulting in polymers with the same chemical composition as the monomers. The majority of the addition monomers present carbon-carbon double bonds (C=C), which convert from pi to sigma bonds during polymerization. Each pi bond that is broken originates two other sigma bonds, and those form the polymer chains. Note that the C=C bonds most commonly involved in chain-growth polymerizations are of the vinyl type, which implies a CH2=C-group linked to some constituent. One example of addition polymer is polyethylene:
2.1.1.3 Condensation Polymers Versus Addition Polymers
The classification proposed by Wallace Carothers considers only two aspects to classify a polymer as addition or condensation: (1) elemental composition of the polymer and (2) elimination or not of any small molecule. However, in some cases if only those two aspects are observed, an incorrect classification may be achieved. Therefore, the term condensation polymers is attributed to polymers constituted by repeating units linked by functional units of ester (−COO–), amide (−NHCO–), urethane (−OCONH–), sulfide (−S–), and ether linkages (−O–). It is also possible that their repeating units lack certain atoms that are present in the monomer, which could have been degraded (i.e., poly(p-xylene) produced by the dehydrogenation of p-xylene), or, as seen previously, their synthesis involved the elimination of small molecules. On the other hand, when the polymer does not fulfill any of those requirements, it is classified as an addition polymer.
2.1.2 Polymer Chain Configuration
The configuration of a polymer refers to the physical arrangement of monomers along the backbone of the chain. It strongly influences the properties of a polymer. The breaking of covalent bonds is required to change the polymer structure. Depending on the polymeric chain structure, polymers can be classified as linear, branched, or cross-linked (Fig. 3.1).

Representation of polymeric chain types. (1) Linear polymer, (2) branched polymer, and (3) cross-linked polymers
2.1.2.1 Linear Polymers
When the monomer molecules are linked together in one continuous chain of repeated units, it is classified as linear polymer. Examples are polyethylene, polyvinyl chloride, polystyrene, and nylon. In the case of addition polymers, those are formed by monofunctional monomers, such as mono-vinyls (e.g., methyl methacrylate), which are not capable of forming cross-links. They are also known as thermoplastic polymers (meaning they can be shaped by heat) and can be dissolved by solvents. Some mono-methacrylate monomers are used in the composition of dental adhesives, and their reaction kinetics is markedly different from those of cross-linking multimethacrylates. The speed of reaction and the swell ability of such materials influence their structure, particularly in the case of dentinal adhesives, which polymerize in a moist environment and are subject to phase separation during polymerization.
The size or length of the chain has significant impact on the polymer’s physical properties. For example, the longer is the chain length, the higher is the material boiling and melting temperature. The increase of chain length furthermore tends to decrease chain mobility and increase strength, toughness, viscosity, and glass transition temperature (T g ).
2.1.2.2 Branched Polymers
In some reactions, side branches of linked monomer molecules protruding from various central branch points along the main polymer chain can be observed. In this case, branched polymer is the more appropriate definition. These polymers show decreased crystallinity because the side chains sterically hinder polymer folding and packing. It is important to point out that the term branched polymer does not refer to linear polymers containing side groups that are part of the monomer structure, such as pendant groups. Only those polymers that contain side branches composed of complete monomer units are termed branched polymers.
Hyperbranched polymers are a special type of branched molecule, formed via controlled, sequential polymerizations [7]. RAFT (reversible addition fragmentation chain-transfer) mechanisms are very common to synthesize branched polymer structures. Those find applications in drug delivery systems, as well as to reinforce other polymer structures [7].
2.1.2.3 Cross-Linked Polymers
Polymer molecules linked to each other by covalent bonds at points other than their ends are defined as cross-linked. Light cross-linking is used to impart good recovery (elastic) properties to polymers to be used as rubbers. High degrees of cross-linking are used to impart high rigidity. Cross-linking tends to increase T g and increase strength and toughness. Cross-linking is necessarily a product of polymerization of multifunctional monomers, may it happen via a condensation or chain-growth mechanism. Cross-linked polymers are also known as thermosets, as they do not present a melting temperature and cannot be shaped by heat, nor dissolved by solvents. Depending on the degree of cross-linking, as already mentioned, physical properties can be tailored, including flexural properties, fracture toughness, T g , and swell ability in aqueous or organic solvents. The nature of the cross-links themselves dictates polymer properties and free volume, may they come from flexible monomers such as triethylene glycol dimethacrylates or from more rigid structures such as bisphenol A diglycidyl dimethacrylate (BisGMA), two monomers commonly used in composites. A representation of the two types of cross-links is show in Fig. 3.2.

Degrees of freedom of (a) pendant groups and (b) cross-links formed by the aliphatic TEGDMA and the aromatic BisGMA (in red). The dotted lines show the space occupied by each molecule’s motion (modified from [8])
2.1.3 Polymerization Mechanisms
A polymer chain buildup might occur by two distinct polymerization mechanisms: step- or chain-growth polymerizations. Those mechanisms differ in the types of monomers involved, as well as on the stages of polymerization at which the development of high molecular weight species is observed. Mainly, in step-growth polymerizations, the molecular weight (in the case of linear polymers) or the polymer network (in the case of cross-linked polymers) does not develop until very high degrees of conversion. This has profound effects on gelation behavior and final stress development, as will be explored further in the following sections.
In step-growth polymerization, the reaction proceeds between two different functional groups, whether they are on the same molecule or different molecules. Examples include the reactions of hydroxyl and carboxyl groups, or isocyanate and hydroxyl groups, or thiol and ene monomers. Step-growth polymerizations include esterification, amidation, aromatic substitutions, the formation of urethanes, and others. In fact, step-growth polymerizations can be divided in two subgroups:
-
(a)
Two different bifunctional and/or polyfunctional monomers, but each monomer shows only one type of functional group.
Example: nA–A + nB–B → –(−A–AB–B–)– n , where A and B are different types of functional groups.
-
(b)
Only one monomer with both types of functional groups.
Example: nA–B → –(−A–B–)– n
Importantly, in this reaction mechanism, several chains of different sizes are built, originating dimers, trimers, tetramers, pentamers, and hexamers, which then finally are linked together at the tail end of the reaction to form large molecular weight polymers (Fig. 3.3).

Representation of the stages of a step-growth polymerization. Modified from http://www.slideshare.net/guest32ca93/polymer-course
The polymer molecular weight increases gradually during the reaction, at relatively slow speed (Fig. 3.4—curve 2), and only near the end of the reaction are high molecular weight specimens found. In other words, the molecular weight does not build up until later in conversion. The main practical consequence of this fact is that, for multifunctional monomers polymerizing via step-growth (i.e., forming cross-linked networks via step growth), gelation and vitrification are delayed to very late stages in conversion and that affects the onset of modulus buildup, as well as stress development [5, 10, 11].

Graphic representation of molecular weight (g/mol) in function of the degree of conversion (%). (1) Characteristic curve for chain-growth polymerization reaction and (2) for step-growth polymerization reaction. Modified from [9]
By contrast, in chain-growth polymerizations, the final size and weight of polymer chain are reached almost immediately after the beginning of the reaction (Fig. 3.4—curve 1). The reaction occurs from the propagation of reactive species (free radical, cation, or anion) produced from an initiator, which promotes successive additions of monomer molecules in a chain reaction. In other words, very large chains or complex networks are formed at relatively low degrees of conversion and, especially for cross-linked systems, cause incomplete monomer conversion and early stress buildup [12]. Chain-growth reactions take place between monomers containing a single type of functional group, commonly a carbon-carbon double bond. The reactive species open the π-bond forming a new radical (a macroradical) and allowing the addition to a monomer molecule. In this case, monomers do not react with other monomers and different sized chains; the monomers react only with the reactive center allowing the addition of more monomer molecules in the polymer chain. The polymer chain growth is interrupted when the center is destroyed by an appropriate reaction depending on the type of reactive center and the reaction conditions. This is the mechanism through which methacrylates polymerize and will receive the most emphasis on this chapter for its importance in the polymerization of dental composites (Fig. 3.5).

Representation of mechanisms in a chain-growth polymerization. (a) Formation of high molecular weight chains immediately after polymerization starts. (b) Example of chain-growth polymerization of polyethylene
In general, chain polymerization shows three phases: initiation, propagation, and termination.
-
1.
Initiation: The initiator is activated by an influx of energy (thermal, photoactive, or redox mechanism) producing reactive centers.
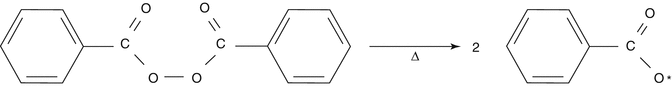
The reactive centers will react with the monomer molecule, breaking carbon double bonds leading to the formation of a macroradical:
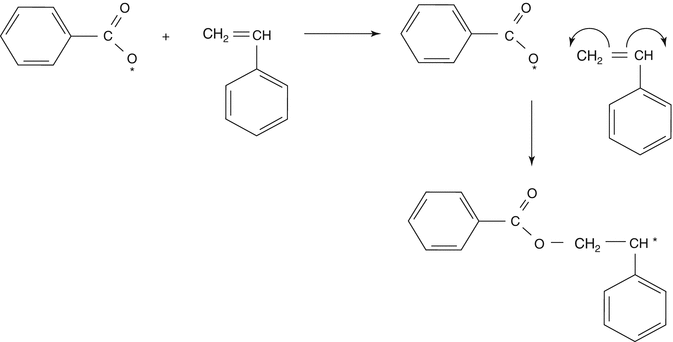
-
2.
Propagation: The reactive center is continuously transferred between the monomer molecules, and each monomer molecule addition creates a new reactive center. The polymer chain grows in high speed:
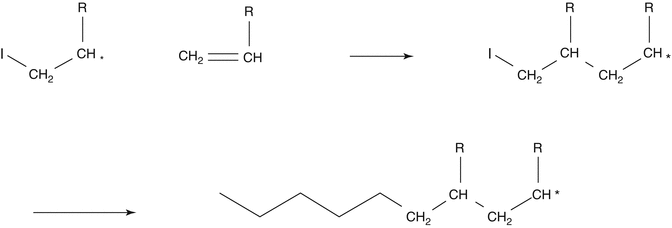
-
3.
Termination: Reactive centers are destroyed (by combination of macroradicals or disproportionation reactions), and the addition of new monomer molecules is limited.




Combination between two macroradicals

Disproportionation reaction
It is worth mentioning that the chain polymerization reaction may be classified according to the selectivity in relation to the type of reactive center. Some monomers will undergo polymerization with any reactive species, while others show high selectivity toward ionic (cationic or anionic) or radical initiator. Methacrylates are not particularly selective and can react either via radical or ionic mechanisms.
2.2 Kinetics of Radical Chain Polymerizations: Fundamental Aspects
The majority of monomers used in dental materials polymerize through a chain-growth mechanism, more specifically, a radical chain polymerization. As seen previously, the chain-growth polymerization reaction shows three different phases that are controlled by some factors which affect directly the reaction kinetics. These phases are presented in more detail in the following paragraphs.
Initiation—This phase involves two important reactions:
-
1.
Generation of free radicals—A homolytic dissociation of an initiator species (I) occurs (via a photo, thermal, or redox mechanism) to yield two radicals (R*) – initiator radical or primary radical—and there is a rate constant, k d , for this catalyst dissociation:
-
2.
Interaction between the free radical (R*) and the first monomer molecule (M) to produce the chain-initiating radical (M 1 *). There is a rate constant for the initiation step—k i —acting on this process:
Propagation—This phase consists of the growth of M 1 * by the successive additions of a large numbers of monomer molecules, creating a new radical with the same identity in each addition:
In this reaction, there is a rate constant for propagation k p .
Termination—At some point, the polymer chain stops to grow and the reaction is terminated. For this to happen, the radical centers have to be destroyed by a combination reaction (two radicals react with each other) or a disproportionation reaction (a beta hydrogen radical from one radical center is transferred to another radical center). As in previous reactions, the termination reaction occurs under a rate—termination rate k t .
In practice, these three phases happen concomitantly during polymerization, with predominance of one versus another phenomenon depending on the extent of polymerization and reaction medium mobility. Several factors contribute to the progression of the reaction, among which the radical concentration, the initial viscosity of the medium, the relative reactivity of the monomer species, the difference between cure temperature and glass transition temperature [13,14,15], etc. Those will be discussed in the following sections, with emphasis on methacrylate polymerizations.
2.2.1 Reaction Kinetics (and Its Relationship with Network Development)
For the purposes of the polymerization of dental composites and adhesives, this section will concentrate on the photoactivated reaction of methacrylates. In general terms, at the beginning of the reaction, the radicals that are generated by the excitation of the photosensitive molecule proceed to interact with the monomers and attack the first vinyl double bonds. The reaction is chemically controlled at this stage, which means the rates of propagation (k p ) and termination (k t ) are largely dependent on the supply of monomers and radicals, since there are not enough high molecular weight species that can contribute to the increase in viscosity of the medium and hinder diffusional mobility. At this stage, propagation and termination reactions occur concomitantly and at similar rates. As the reaction progresses, and more of high molecular weight species and cross-linking starts to occur, viscosity starts to increase, and the reaction becomes diffusion-controlled. This means that molecular mobility becomes more difficult, especially so for larger molecules. In turn, this leads to a decrease in termination events, because it becomes increasingly unlikely for bimolecular termination or disproportionation to take place. With the consequent decrease in the termination events, the propagation is favored, leading to autoacceleration of the reaction (Trommsdorff effect). The gelation of the network is defined as the point in conversion at which one macromolecule spans the whole specimen [9]. The propagation continues to dominate until the maximum rate of polymerization (Rpmax) is reached (Fig. 3.6). Even though it is not possible to precise the onset of gelation from this curve alone (rheology experiments are more suitable for that measurement), sometime before the Rpmax is reached, gelation of the forming network is observed. The gelation of methacrylates has been shown to occur at around 5% conversion, as determined by IR-coupled rheology experiments [16], while the Rpmax is usually observed at around 5–20% conversion, even though this second landmark is a lot more dependent on the reaction conditions [17]. The conversion at gelation and the conversion registered at the end of the autoacceleration (or at Rp max) greatly influence the limiting conversion achieved by a monomer system, as well as the final polymerization stress development.

Polymerization stress and rate of polymerization as a function of conversion for a typical multifunctional methacrylate (ethoxylated bisphenol A). Polymerization stress and degree of conversion were followed simultaneously on the same specimen. Note that the conversion at which stresses start to more markedly build up (inflection point on the stress vs. conversion curve) coincides with the end of the deceleration shown in the rate vs. conversion curve
With the progression of the polymerization reaction, the restriction to molecular mobility increases even further, to the point that even small molecule diffusion is affected. This leads to a decrease in the rate of propagation from Rp max (propagation also becomes diffusion-controlled), and the rate vs. conversion curve shows a deceleration slope (Fig. 3.6). The onset of deceleration coincides with the onset of network vitrification and that also means that opportunities for network rearrangement to compensate for shrinkage are limited, and stresses start to more significantly build up [12]. At the end of the auto-deceleration slope, the network has reached almost complete vitrification, which means any additional conversion translates into a sharp increase in stress, as has been demonstrated in simultaneous conversion vs. stress experiments [11]. One example is shown in Fig. 3.7, where for the same specimen, stress vs. conversion and polymerization rate vs. conversion plots are presented. In summary, before the network reaches vitrification, there is still opportunity for stress relaxation [11]. This is the reason why delayed gelation and vitrification strategies have gained so much attention in the development of low-stress materials [4,5,6], but it is important to point out that, for them to be effective, the delay has to occur in terms of conversion and not time.

Evidence for delayed gelation/vitrification in methacrylate networks containing up to 20 wt% thiols. Polymerization stress is significantly decreased without prejudice to mechanical properties. In this specific case, the increased conversion achieved with these materials led to increased flexural strength. Modified from [11]
The rate of polymerization ( R p ) or rate of monomer disappearance is the speed at which monomer molecules are converted to polymer. This rate is based on k p , k t , monomer concentration [M] and rate of initiation ( R i ), according to the following equation:
It can be observed that the rate of propagation is directly proportional to the square root of the rate of initiation (R i ).
Particularly for the resin composites used in dentistry, which are for the most part photoactivated, it is important to consider the intensity of absorbed light—I a (or irradiance, in moles of light quanta for liter-second)—as shown in the following equation:
This equation makes it clear that the rate of polymerization is dependent on the square root of the irradiance, with the implication that the overall rate of reaction cannot be reduced significantly unless the irradiance is reduced by at least two orders of magnitude [9]. This makes the effectiveness of so-called soft-start irradiation protocols questionable [18, 19].
2.2.1.1 Degree of Conversion
Degree of conversion is the percentage of carbon double bonds converted in single bonds or, in other words, the extent of conversion of monomer to polymer. In reactions with only one type of monomer, this percentage is obtained by the following equation:
where M 0 is the initial monomer concentration. In copolymerizations, where more than one type of monomer are reacting, the concentration of each constituent is considered. On the kinetics graph, the final degree of conversion is shown at the very end portion of the curve. A high degree of conversion is important for composite resins in order to increase general chemical and physical properties, as well as to reduce the effects of residual monomer in the organism. As already explained, the limiting conversion of methacrylates is typically around 70%. The fact that multimethacrylates do not reach complete conversion at room temperature is due to trapped radicals in the network, which in turn, stems from the fact that gelation and vitrification are reached at relatively low conversions. Also as mentioned, after deceleration, steep diffusional limitations preclude any additional conversion from taking place. This is true at least for the conditions at which methacrylates are usually polymerized in the clinical situation (room temperature at relatively high irradiances) [9, 12].
2.2.1.2 Kinetic Chain Length
Kinetic chain length (v) is the average number of monomer molecules polymerized for each free radical that initiates a polymer chain. This average is in function of the ratio of polymerization and termination/initiation rates, as per the following equation:
Kinetic chain length at constant rate of polymerization is a particular characteristic of the monomer and independent of initiation method (thermal, redox, or photochemical).
The chain length plays an important role in the kinetics process. Long chains are less mobile than short chains, and during the polymerization reaction, they reduce the termination rate. This reduction is responsible for increasing the rate of polymerization and potentiating the autoacceleration process. However, when shorter kinetic chain lengths are induced, the termination rate is increased, and then the termination-propagation rate is also increased before the vitrification point. The effect of this on the kinetics reaction is that instead of the rate of polymerization increasing too fast, it increases gradually allowing for higher degrees of conversion before the reaction reaches gelation/vitrification. In other words, shorter kinetic chain lengths are responsible for delaying the phase at which the reaction starts to become diffusion-controlled due to the mobility restriction imposed by the increased molecular weight or cross-linking. Any mechanism that is able to delay the onset of gelation and vitrification as a function of conversion has a very significant impact on polymerization stress development, since stress can be easily relaxed in the pre-gel and pre-vitrification phases. This is the mechanism through which stress reduction in methacrylates is obtained with chain-transfer reactions using thiols or in thiol-ene polymerizations [1, 11]. It is worth mentioning that kinetic chain length is inversely dependent on the radical concentration or the polymerization rate. It has a special importance because any attempt to increase the polymerization rate by increasing the radical concentration comes at the expense of producing smaller polymers.
2.2.1.3 Chain-Transfer Reactions
The effect of chain-transfer on polymerization kinetics and ultimately network development has been extensively studied [9, 12]. Chain-transfer reactions are radical displacement events, as shown below:
where XA may be a monomer, initiator, solvent, or another substance and X is the atom or species transferred.
The rate of chain-transfer reaction is given by:
where k tr is the chain-transfer constant. In other words, chain transfer is a chain breaking mechanism in which the new radical formed through the transfer is considered to be a new initiation site [9].
Some compounds, named chain-transfer agents, are known to facilitate this process, with thiols being by far the most commonly used and the most studied compounds. The chain-transfer constant depends on the molecular structure, but in general, even at low concentrations, thiols are capable of reducing the rate of polymerization and the radical chain length significantly [12]. When the radical from a propagating chain is transferred to a thiol, that molecule transfers the charge to another unreacted monomer and “caps” the growing polymer chain, which in turn becomes a “dead polymer.” The implication for this is the molecular weight and cross-linking (or network formation) are delayed in relation to the consumption of vinyl double bonds, and ultimately, the polymerization stress can be decreased. However, the potentially decreased overall cross-linking density may also impart poorer mechanical properties [3]. This is unlikely for lower thiol concentrations, and in fact, up to 10 wt% small molecule thiols have been added to methacrylate networks to delay gelation/vitrification, reducing stress without prejudice to final mechanical properties [11], as can be seen in Fig. 3.7. At higher thiol concentrations, the reaction with the methacrylate monomer becomes a more important factor in network development [10, 20], and the transfer rate constant (k tr ) is significantly higher than the propagation rate constant (k p ), accompanied by a delay in the autoacceleration [10].
2.2.1.4 Thiol-ene Reactions: Radical-Assisted Step-Growth Polymerization
For typical thiol-ene systems, the predominant polymerization mechanism is step growth [21], as long as the stoichiometric ratio is kept at 1:1 thiol to ene and that the ene monomer does not preferentially undergo homopolymerization via chain growth (ene monomers contain vinyl polymerizable functionalities, but methacrylates are not good candidates for this kind of reaction) [1, 21]. There are a couple of disadvantages with thiol-ene systems, including low T g due to the presence of flexible thioether bonds incorporated in the polymer backbone through chain-transfer reactions to the methacrylate, issues with shelf-life stability as the monomers can undergo Michael addition reactions and spontaneously polymerize in the bottle, and the malodor of low molecular weight thiol species [20]. However, when these are combined with methacrylates in ternary systems, some of these drawbacks can be overcome [11]. In this case, the reaction can be initiated via a photoactive event, which generates the first radical. In general terms, that radical can undergo transfer to another methacrylate or chain-transfer to another vinyl (from the methacrylate or the ene) by the thiol. There is, therefore, competition between chain and step-growth mechanisms as the methacrylate can undergo both homopolymerization and chain transfer with the thiol in addition to the thiol-ene reaction [21, 22]. Because the methacrylate is more likely to undergo homopolymerization, in thiol-ene/methacrylate systems, the ene consumption has been demonstrated to be delayed until the methacrylate-thiol reaction is largely complete [23], shown in Fig. 3.8. The methacrylate conversion still progresses at a slower rate, as already explained, due to the chain-transfer reactions with the thiol, which delay gelation/vitrification and consequently reduce stress (Fig. 3.8). It is important to note that the delayed gelation in this case also leads to more homogeneous networks, whose polymerization progresses further in terms of conversion because of the extended opportunity for viscous flow which, incidentally, is also capable of alleviating part of the polymerization stress [3].

(a) Functional group conversion vs. time plots of a dithiol (HDT, circle), a triene (TATATO, triangle), and a dimethacrylate (HDDMA, square) ternary mixtures. (b) Shrinkage stress of (1) BisGMA/TEGDMA 70/30, (2) thiol-ene, and (3) thiol-ene-methacrylate as a function of the overall double bond conversion. Modified from [23]
2.3 Factors Influencing the Polymerization
2.3.1 Initiators of Polymerization
Initiators can be defined as chemical substances that produce free radicals and start chemical reactions. Radicals are reactive chemical species possessing a free (unbonded or unpaired) electron. Energy is necessary to generate radicals from an initiator. Light or redox components (or a combination) are traditionally utilized as energy source for polymerization of composite dental materials. Once formed, radicals undergo two basic types of reactions: propagation reactions and termination reactions. In a propagation reaction, a radical reacts to form a covalent bond and to generate a new radical. In a termination reaction, two radicals interact in a mutually destructive reaction in which both radicals form covalent bonds and reaction ceases. The polymerization rate depends on the concentration of photosensitizer (initiator) and co-initiator [24], as well as on the interaction with specific monomer species [25].
2.3.2 Monomer Structure and Copolymerization
The monomer structure plays a crucial role during the polymerization reaction. The degree of conversion is directly dependent on the spacer group flexibility between the phenyl ring core and the methacrylate functional group. However, when ethylene glycol groups are added to BisGMA to increase the flexibility, this monomer shows an opposite behavior due to the hydrogen bonding in its network which reduces the mobility and the degree of conversion [26]. Considering that all resin composites commercially available are composed at least of two different monomers, the interaction between them determines the mixture performance. BisGMA and TEGDMA are frequently copolymerized to improve the viscosity, reactivity, and final degree of conversion of these materials. In Fig. 3.9, it is possible to see the rate of polymerization as a function of the degree of conversion for BisGMA, TEGDMA, and a mixture 50% BisGMA and 50% TEGDMA. These are the most common monomers of dental composites. The synergistic effect is clear for the mixture of monomers. It is possible to see that the polymerization rate is increased significantly and the final degree of conversion is similar to pure TEGDMA. This can be explained by the high reactivity of BisGMA when mixed to TEGDMA—a reactive diluent able to improve the mobility of the system and hence the carbon double bond conversion [9, 15].

Rate of polymerization in function of degree of conversion for (1) BisGMA, (2) TEGDMA, and (3) BisGMA + TEGDMA. Modified from [15]
2.3.3 Cross-Linking and Cyclization
Cross-linkers are monomers with functionality necessarily equal to or greater than two, capable of joining two or more molecules by a covalent bond, establishing inter- and intramolecular cross-links. Cross-links formed by BisGMA and TEGDMA are shown in Fig. 3.2b, earlier in the chapter. The concentration of cross-linkers interferes on the cyclization process. Cyclization is defined as the formation of one or more rings in a chemical compound, mainly a hydrocarbon. In free-radical cross-linking polymerizations, there are primary and secondary cyclizations. Primary cyclization happens when the free radicals attack pendant double bonds in the same chain. Secondary cyclization happens when the free radicals attack pendant double bonds on other chains already incorporated in the polymer network. Both types of cyclization processes have effects on the termination rate in copolymerization systems containing up to 4 mol% cross-linker. The effect in this case is an increase of coil size of macroradicals and hence increase of termination rate. However, during reaction development, the cross-links formed inside the macroradical coil decrease the mobility and then the termination rate. In higher concentrations, the cross-linkers show an increase in cyclization process leading to compact primary particle formation which limits the access of the radical centers and pendant vinyl groups to other polymer molecules. The result of this process is a diffusion-controlled termination rate decreased and cross-linker concentration increased. In BisGMA/TEGDMA copolymerizations, the relative ratio of each monomer has been shown to greatly interfere with the resulting polymer packing and free volume [8], as well as with the sol-gel composition of the resulting material [27].
2.3.4 Inhibitor and Catalyst
A chemical inhibitor is a reactant that slows down or delays the start of a chemical reaction. Inhibitors react with the initiating and propagating radicals, converting them to either nonradical species or radicals of reactivity too low to undergo the propagation process. In general, there are two kinds of inhibitors: reversible and irreversible inhibitors. Reversible inhibitors slow down a chemical reaction but do not stop it completely. Irreversible inhibitors prevent an unwanted reaction from occurring. The effect of the addition of inhibitors is to stop every radical, and polymerization is completely halted until they are completely consumed. In polymer chemistry, reaction inhibitors are used to modify the properties of the final polymer product as well as to prolong usefulness. The catalyst is in general a molecule used in polymers to increase the rate of a chemical reaction. Catalysts work by providing an alternative mechanism involving a different transition state and lower activation energy. Consequently, more molecular collisions have the energy needed to reach the transition state. Hence, catalysts can enable reactions that would otherwise be blocked or slowed by a kinetic barrier. The catalyst may increase reaction rate or selectivity or enable the reaction at lower temperatures.
2.3.5 Cure Temperature Versus Glass Transition Temperature
In general terms, the closer the cure and the glass transition temperature of a polymer are, the more likely it is for the material to reach full conversion [28]. This is explained by the diffusion-limited reactions in cross-linked networks described in detail in previous sections. If a material undergoes polymerization at a temperature where the translational capability of its molecular chains is high (close to its T g ), it is far more likely that all chains will be able to react, with little to no unreacted/trapped monomers. In contrast, let us consider, for example, a polymeric material presenting as glassy at room temperature (with, hypothetically, T g = 100 °C), being polymerized at room temperature. By the time the conversion reaches 20 or 30%, the network has already developed enough to increase the modulus and the T g from the monomeric state by a considerable amount. This, as already explained, undermines molecular mobility (particularly, larger molecule mobility) and impedes complete conversion. This is the reason high T g polymers generally present lower limiting conversions when polymerized at room temperature, as is the case with many dimethacrylates used in dentistry [29].
References
Hoyle CE, Bowman CN. Thiol-ene click chemistry. Angewandte Chemie – International Edition. 2010;49(9):1540–73.
Li Q, Zhou H, Wicks DA, Hoyle CE. Thiourethane-based thiol-ene high T g networks: preparation, thermal, mechanical, and physical properties. J Polym Sci A Polym Chem. 2007;45(22):5103–11.
Senyurt AF, Hoyle CE, Wei H, Piland SG, Gould TE. Thermal and mechanical properties of cross-linked photopolymers based on multifunctional thiol-urethane ene monomers. Macromolecules. 2007;40(9):3174–82.
Bacchi A, Dobson A, Ferracane JL, Consani R, Pfeifer CS. Thio-urethanes improve properties of dual-cured composite cements. J Dent Res. 2014;93(12):1320–5.
Bacchi A, Nelson M, Pfeifer CS. Characterization of methacrylate-based composites containing thio-urethane oligomers. Dent Mater. 2016;32(2):233–9.
Bacchi A, Pfeifer CS. Rheological and mechanical properties and interfacial stress development of composite cements modified with thio-urethane oligomers. Dent Mater. 2016;32(8):978–86.
Voit BI, Lederer A. Hyperbranched and highly branched polymer architectures--synthetic strategies and major characterization aspects. Chem Rev. 2009;109(11):5924–73.
Pfeifer CS, Shelton ZR, Braga RR, Windmoller D, Machado JC, Stansbury JW. Characterization of dimethacrylate polymeric networks: a study of the crosslinked structure formed by monomers used in dental composites. Eur Polym J. 2011;47(2):162–70.
Odian G. Principles of polymerization. Staten Island, NY: Wiley-Interscience; 2004. p. 198–349.
Lecamp L, Houllier F, Youssef B, Bunel C. Photoinitiated cross-linking of a thiol-methacrylate system. Polymer. 2001;42(7):2727–36.
Pfeifer CS, Wilson ND, Shelton ZR, Stansbury JW. Delayed gelation through chain-transfer reactions: mechanism for stress reduction in methacrylate networks. Polymer (Guildf). 2011;52(15):3295–303.
Berchtold KA, Lovestead TM, Bowman CN. Coupling chain length dependent and reaction diffusion controlled termination in the free radical polymerization of multivinyl (meth)acrylates. Macromolecules. 2002;35(21):7968–75.
Lovell LG, Berchtold KA, Elliott JE, Lu H, Bowman CN. Understanding the kinetics and network formation of dimethacrylate dental resins. Polym Adv Technol. 2001;12(6):335–45.
Lovell LG, Newman SM, Bowman CN. The effects of light intensity, temperature, and comonomer composition on the polymerization behavior of dimethacrylate dental resins. J Dent Res. 1999;78(8):1469–76.
Lovell LG, Stansbury JW, Syrpes DC, Bowman CN. Effects of composition and reactivity on the reaction kinetics of dimethacrylate/dimethacrylate copolymerizations. Macromolecules. 1999;32(12):3913–21.
Cook WD, Brockhurst P. The oscillating rheometer – what does it measure? J Dent Res. 1980;59(5):795–9.
Dickens SH, Stansbury JW, Choi KM, Floyd CJE. Photopolymerization kinetics of methacrylate dental resins. Macromolecules. 2003;36(16):6043–53.
Hasegawa T, Itoh K, Yukitani W, Wakumoto S, Hisamitsu H. Effects of soft-start irradiation on the depth of cure and marginal adaptation to dentin. Oper Dent. 2001;26(4):389–95.
Yap AU, Ng SC, Siow KS. Soft-start polymerization: influence on effectiveness of cure and post-gel shrinkage. Oper Dent. 2001;26(3):260–6.
Cramer NB, Couch CL, Schreck KM, Carioscia JA, Boulden JE, Stansbury JW, Bowman CN. Investigation of thiol-ene and thiol-ene-methacrylate based resins as dental restorative materials. Dent Mater. 2010;26(1):21–8.
Cramer NB, Bowman CN. Kinetics of thiol-ene and thiol-acrylate photopolymerizations with real-time Fourier transform infrared. J Polym Sci A Polym Chem. 2001;39(19):3311–9.
Reddy SK, Cramer NB, Bowman CN. Thiol-vinyl mechanisms. 2. Kinetic modeling of ternary thiol-vinyl photopolymerizations. Macromolecules. 2006;39(10):3681–7.
Lee TY, Carioscia J, Smith Z, Bowman CN. Thiol-allyl ether-methacrylate ternary systems. Evolution mechanism of polymerization-induced shrinkage stress and mechanical properties. Macromolecules. 2007;40(5):1473–9.
Musanje L, Ferracane JL, Sakaguchi RL. Determination of the optimal photoinitiator concentration in dental composites based on essential material properties. Dent Mater. 2009;25(8):994–1000.
Vaidyanathan TK, Vaidyanathan J, Lizymol PP, Ariya S, Krishnan KV. Study of visible light activated polymerization in BisGMA-TEGDMA monomers with type 1 and type 2 photoinitiators using Raman spectroscopy. Dent Mater. 2017;33(1):1–11.
Cook WD. Thermal aspects of the kinetics of dimethacrylate photopolymerization. Polymer. 1992;33(10):2152–61.
Stansbury JW, Dickens SH. Determination of double bond conversion in dental resins by near infrared spectroscopy. Dent Mater. 2001;17(1):71–9.
Wisanrakkit G, Gillham JK. The glass transition temperature (Tg) as an index of chemical conversion for a high-Tg amine/epoxy system: chemical and diffusion-controlled reaction kinetics. J Appl Polym Sci. 1990;41(11–12):2885–929.
Sideridou I, Tserki V, Papanastasiou G. Effect of chemical structure on degree of conversion in light-cured dimethacrylate-based dental resins. Biomaterials. 2002;23(8):1819–29.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter
Cite this chapter
Fugolin, A.P.P., Bacchi, A., Pfeifer, C.S. (2018). Curing Reaction and Kinetics. In: Miletic, V. (eds) Dental Composite Materials for Direct Restorations. Springer, Cham. https://doi.org/10.1007/978-3-319-60961-4_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-60961-4_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-60960-7
Online ISBN: 978-3-319-60961-4
eBook Packages: MedicineMedicine (R0)