
Abstract
A skeletal network of spectrin molecules provides shear stiffness to the red blood cell (RBC) membrane maintaining its shape (by providing elasticity) and thus its stability. There are two α and two β subunits of human spectrin; the α1 and β1 spectrin subunits are encoded SPTA1 and SPTB, respectively. Hereditary elliptocytosis (HE), one of the hereditary blood disorders, results in elliptical/oval, elongated RBCs due to the abnormalities that occur mainly at the atomistic level because of the mutations in SPTA1 and SPTB. In HE, the RBC membrane partly loses its elasticity and this results in a reduced overall durability of RBCs. In its severe forms, hereditary blood disorders can lead to hemolytic anemias when the abnormal RBCs start to depreciate. This study aims to observe mechanically how the abnormalities due to the mutations in SPTA1 gene affect single spectrin molecules. The stiffness of the mutated and normal/wild-type molecules are calculated using Steered Molecular Dynamics (SMD) by subjecting the spectrin α chain to displacements up to tens of nanometers and drawing force-extension maps from these computational experiments. The most common HE mutations being SPTA1 gene missense mutations in the dimer-tetramer self-association site makes it interesting to introduce mutations at the binding site and compare the change in the mechanical response of the mutated molecules to that of the wild-type. Overall, the results presented here show that the nano-mechanical tensile behaviour at the chain-level does not change under the presence of the point mutations. This suggests that the local structural disturbances the mutations cause, will affect the spectrin scaffold on the network-level rather than on the on the single chain level implying more complicated molecular interactional disorders. The work presented here is a part of a larger effort to improve understanding the functional implications of the mechanical and structural properties of proteins starting at the atomistic level.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Biomechanics
- Numerical simulation
- Molecular dynamics
- SMD
- Protein
- Nano-mechanics
- Spectrin
- Hereditary elliptocytosis
- Alpha helix
- Protein
- Deformation
1 Introduction
Many protein molecules (i.e. spectrin, titin, spider silk…) have multi-domain structures which result in a force-displacement behavior with a characteristic saw-tooth pattern when exposed to mechanical stretching due to the unfolding of their domains along the molecular chains (Fig. 1). Unfolding of molecules provide numerous advantages to proteins. Unfolding of the “hidden length” (when the contour length of a molecule is different than the end-to-end distance of the molecule) along the constituent domains of a molecule, eases the molecule into high levels of strain by keeping low force levels. This enables “softening” of the proteins. “Softening” acts as a manner to first allow for an initially stiff behavior, then to limit the overall force level and to enable large extensions at low forces and low compliance [1,2,3,4,5,6,7,8,9,10,11,12,13,14].

A schematic of the force-extension behavior yielding a “saw-tooth pattern” eventually resulting in a more compliant behavior for a single molecule containing folded domains along which the end-to-end distance (r o ) is shorter than the contour length of that particular domain
Figure 1 shows schematics for the unfolding of a single molecule with a folded-domain. When a molecule with folded domains is subjected to tensile strain, the force increases until all the domains are aligned with each other, then one of the domains unfolds revealing some hidden length dropping the force. If the tensile strain further increases, force starts increasing but this time with a more resilient response until all the domains are aligned again yet another unfolding event happens dropping the force one more time… This behavior goes on till all the hidden length is revealed as long as the molecule is subjected to higher strain levels. The force-extension map reveals the “saw-tooth” pattern which is bound to have a softer edge/lower plateau force and less peaks as the pulling speed decreases and also as the event numbers increase.
In order to understand the mechanical behaviour of structural proteins, it is useful to study their response to mechanical unfolding. These direct investigations are commonly performed by atomic force microscopy (AFM); the macromolecule is mechanically unfolded by the imposition of elongations at constant strain-rate, and the required force is measured as a function of the elongation. The unfolding patterns and force-extension maps can also be studied using computational experiments; in particular Steered Molecular Dynamics (SMD); where a harmonic potential (spring) is used to induce motion through constant velocity mimicking the AFM experiments in which a molecule is stretched by a cantilever moving at constant velocity. There are difficulties in directly comparing simulations and AFM experiments as the elongations speeds differ greatly (i.e. in the order of 10−7 for AFM experiments versus 0.5 m/s for SMD experiments). However the unfolding scenario (i.e. position of force peaks) and the order of magnitude in forces enable us to draw consistent conclusions from SMD simulations.
2 Spectrin Structure
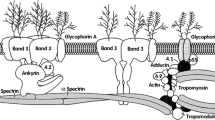
A skeletal network of spectrin molecules provides shear stiffness to the red blood cell (RBC) membrane maintaining its shape (by providing elasticity) and thus its stability. The spectrin network is composed of lateral heterodimeric association of α and β subunits which possess 16–20 tandem homologous repeats each. The principal form of the spectrin molecule constitutes of head-to-head interactions between two α/β dimers leading to the formation of a spectrin tetramer; where the tetramerization site is comprised of α-0 and β-17 partial domains (see Fig. 2) [15,16,17].

Spectrin assembly constitutes of a lateral heterodimeric association of α- and β-chains which form the spectrin tetramer by the antiparallel head-to-head interactions between two α/β dimers (Panel I). α-chain has 20 full repeats and a partial repeat (Panel III). Tetramerization site comprised of α-0 and β-17 partial domains as shown in the ribbon diagram (Panel II)
Hereditary elliptocytosis (HE) is an inherited RBC membrane disorder associated with defective spectrin due to the mutations in codons 28, 46, 48, and 49 [18]. HE disorders are characterized by visibly elliptical RBCs. HE disorders range from asymptomatic to hemolytic anemia [19]. Although many defects are known to result in HE, cases generally involve disruptions of horizontal cytoskeleton interactions; i.e. mutations in the tetramerization site which influences the function and structural integrity of the molecule by reducing the elasticity of the RBCs and thus resulting in mechanical weakness or fragility of the membrane skeleton that leads to a reduced overall durability of RBCs.
Arginine at the 28th residue (ARG28), located in the tetramerization site, is a vital residue for the conformational stability of α-spectrin [20]. There are four major types of replacements at this site: arginine replaced by cysteine, histidine, leucine or serine (ARG28 [CYS/HIS/LEU/SER]) residues [18]. The literature provides excellent structural analyses on the conformations undertaken by the mutated residues of spectrin [21]. However mechanical analysis at the atomistic level is the missing piece in studying the mechanisms that lead to RBC diseases. It has been suggested that the ARG28 mutations lead to abnormality in the folding and the bonding of the tetramerization site; however there is limited knowledge about whether these mutations cause disruptions on the dimer level in the α-chain directly or disrupt the chain-chain inter-bonding in the heterodimer/tetramerization level or even further in the network level. One of the objectives of this work is to elaborate on whether one could observe disturbances due to mutations at the local site when the site is applied external forces. Here, for that reason, the mechanical stiffness of the wild-type (WT)/healthy and the mutated (cysteine (CYS), serine (SER), histidine (HIS) and leucine (LEU) replacements) cases will be calculated using Steered Molecular Dynamics (SMD) by subjecting the spectrin tetramerization complex to displacements up to tens of nanometers drawing force-extension maps from these computational experiments.
The complete tetramer structure illustrating the interactions of the domains (mainly hydrophobic and also electrostatic) was revealed and identified in 2010 [22] providing a guide for designing further computational experiments to explore more details of the HE-related mutations and their effects. This work aims to go beyond the focus on the biochemical structural analysis and characterize tetramerization site mechanical properties and discuss the possible local effect of the ARG 28 point mutations on these properties.
3 Materials and Methods
The α-0 and α-1 domain as well as the β-16 and β-17 domain (encompassing the tetramerization site) data was obtained from the PDB with the identification (ID) 3LBX [22]. Four types of point mutations; cysteine (CYS), histidine (HIS), serine (SER) and leucine (LEU) amino acid residue, were introduced in the ARG 28 location in the α-0 residue in the spectrin tetramerization domain.
The Molecular Dynamics (MD) simulations were carried out using the all-atom CHARMM force field [23] as implemented in the NAMD program [24] and to visually observe the results in 3D, visual molecular dynamics (VMD) [25] is used. Energy minimization is performed using a conjugate gradient (CG) scheme. The equilibration is carried out at a temperature of 310 K (resembling body temperature). The stretching of the proteins is performed via the Steered Molecular Dynamics (SMD) method [26,27,28]. This method is based on the concept of pulling the center of mass of a collection of chosen atoms via a spring along the direction of the molecular axis, while keeping the center of mass of another group of atoms fixed through a stiffer spring.
Each SMD simulation is set up in three stages as follows:
-
1.
Energy minimization: Energy minimization is carried out for several thousand steps, ensuring that convergence is achieved. The convergence is determined by confirming that the total energy of the system converges to a steady value.
-
2.
Equilibration: The equilibration process ensures that the molecules start in the most likely in vivo condition at finite temperature. We find that the 20 ns equilibration, limited due to computational constraints, gives a well equilibrated structure with good geometry convergence.
-
3.
Tensile loading: SMD approach is used to pull part of the spectrin tetramerization site to model tensile loading. SMD simulations give valuable insights in the unfolding pathways of a protein. For the SMD simulations, the fixed atoms consist of the Cα (α Carbon) atoms at one end of a chain, and the pulled atom is the Cα atom at the other end of the chain. Different pulling speeds are tested (0.5, 0.05, 0.005, 0.0005 nm/ps). The SMD spring constant used is k = 400 kcal mol−1Å2.
All simulations are performed with a 2 femtosecond time step.
The simulations are carried out using an implicit solvent model [29,30,31,32,33,34] where the need for explicit water atoms is eliminated by including the effects of solvent in the inter-atomic force calculation. The advantage of implicit solvent models is that the computational cost (i.e. the necessary CPU time) of the simulation is used more efficiently to monitor longer and slower simulations as opposed to the necessity for a very large water bath to accommodate the spectrin structure. The Generalized Born model has been demonstrated to provide a good approximation of explicit solvent simulations of proteins [7, 8, 35]. In this work, employing an implicit solvent model will allow faster convergence at lower pulling speeds at a lower computational cost so that enough simulations can be done to obtain statistically meaningful results and at the same time, here, demonstrating an example of the applicability of this approach to large protein systems.
4 Results
We begin with the presentation of the SMD analysis. The Cα atom of codon 19 is fixed while the Cα atom of codon 158 is the SMD atom as shown in Fig. 3I. The figure outlines the three segments of the folded domain.

Panel (I) outlines the three segments of the folded domain in the SMD test along with the length of each segment. The Cα atom of codon 19 is fixed while the Cα atom of codon 158 is the SMD atom. Panel (II) a–d shows the force-extension plots of tensile experiments for the wild type (WT) for four different pulling speeds; 0.5 nm/ps, 0.05 nm/ps, 0.005 nm/ps, 0.0005 nm/ps; respectively
Force-extension plots of the tensile experiments for the wild type (WT) are shown in Fig. 3II for four different pulling speeds (0.5, 0.05, 0.005, 0.0005 nm/ps).
For all pulling speeds except 0.5 nm/ps, one can observe the typical three regimes;
-
1.
The elastic regime where the force increases linearly with the applied deformation.
-
2.
The plateau regime where the force remains constant with the applied deformation.
-
3.
Final regime where the force increases rapidly with the applied deformation.
It can be observed from the figure that for pulling rates 0.005 and 0.0005 nm/ps; the results are very similar. This shows the convergence of the simulations and that the pulling speed chosen is appropriate. Here, it must also be noted that in total 11 trajectories were calculated and averaged to increase the statistical precision of the results.
Figure 4I shows the force versus applied displacement behavior for the ‘wild type (WT)/healthy’ structure. The force level required to unravel the hidden length comes out to be ~200 pN. At a displacement of ~5 nm, Segment 3 (see Fig. 4IV) becomes completely aligned with the pulling direction while Segment 2 is unfolded and the force drops to zero. Figure 4II details how the length of Segment 1 remains unchanged as Segment 2 and 3 re-arrange themselves to accommodate the applied displacement. This confirms that the increase in the force for an applied displacement of ~5 nm manifests itself as stretching of Segment 3 coupled with re-orientation/conformation change of Segment 2. Further examination of Fig. 4I reveals that the force, due to the applied displacement, increases linearly until the tertiary structure changes and the triple helix starts unfolding. This process is a step-wise process and the unfolding is completed at the point “D” as shown in Fig. 4I. We can observe from Fig. 4II that as Segment 3 stretches and Sect. 2 re-orients itself, the end-to-end distance of the former section increases and that of the latter decreases until point “D” where the unfolding of the tertiary structure is completed and Segment 1 and 3 are completely aligned. Following this point, there is no further increase or decrease in the end-to-end distance of either section. Although there is some unfolding in Segment 3, the 10 nm displacement ultimately only results in tertiary re-arrangements of the helices as detailed in Fig. 4IV. Figure 4III shows the applied displacement versus the change in the contour length (here referred to as the addition of subsequent Segments 1; 2 and 3). We can observe here that the overall contour length does not change with the applied displacement of 10 nm since the increase in the end-to-end distance of Segment 3 is cancelled out by the decrease in the end-to-end distance of Segment 2 during the test as detailed in (II).

The characteristic SMD data is detailed from the tensile test on WT (wild type/healthy) case. Panel I shows the force-displacement graph with the critical points marked by A–D which are also detailed in the ribbon-diagram of α-spectrin. Segment 1 is denoted by black, segment 2 is denoted by blue and segment 3 is denoted by green. The same notation is used in panel II where each segment’s elongation is plotted with respect to the applied displacement. Panel III shows the applied displacement with respect to time together with the contour length fluctuations around -0- for the same applied displacement. Panel IV shows the ribbon diagram of the unfolding pattern: tertiary structure unraveling with the applied axial displacement
The tensile test performed here encompasses the unfolding of the protein’s tertiary structure as shown in the early experimental studies [1,2,3,4,5,6,7,8,9,10,11,12,13,14] and 10 nm displacement applied results in unraveling the ‘hidden’ 10 nm in the tertiary structure with no change in the secondary alpha-helical structure of the protein. Once the unfolding of the ‘hidden length’ is completed at point “D” as denoted in Fig. 4I, the force drops to zero revealing an initial unfolding stiffness of ~0.0125 N/m.Footnote 1
Overall, the ‘hidden length’ of protein structures gives rise to very compliant materials with high levels of ductility involved not just owing to the secondary structure of the protein (alpha helix) but mainly the tertiary structure is the reason for this very ductile behavior. Despite the gap between the pulling speeds between AFM experiments and SMD simulations where the AFM results are reproduced, the simulations here have been able to capture the peaks observed at similar force levels to those in AFM [1, 3, 6].
Following the basic SMD analysis for the WT, four types of missense mutations are introduced in the codon 28 in the α-0 region in the spectrin tetramerization site. The arginine amino acid (ARG 28) is replaced by cysteine (CYS), histidine (HIS), serine (SER) and leucine (LEU) amino acid residues. As we can see in Fig. 5, the results for the mutated structures follow a similar trend to that for the WT. The slight tendency toward the more compliant behavior for the mutated structure is possibly from the statistically varying conformations each structure assumes after the equilibration process which would also depend on factors such as the size of the structures’ side groups.

The characteristic SMD force-displacement data is shown for the WT/arginine (ARG; black) together with the cases with the point mutations; histidine (HIS; blue), cysteine (CYS; green), leucine (LEU; purple); serine (SER; red)
Figure 6 shows the force versus displacement graph for all cases for larger displacements up to ~45 nm. The contour length for the α-0 and α-1 combined alpha-helical repeat is 20 nm as shown in the latter analysis (From the fixed Cα atom of codon 19 to the SMD atom/Cα atom of codon 158; the constituent 139 codons/38 alpha-helical turns-taking into account that each turn in an alpha helix is 0.54 nm long and constitutes of 3.6 residues- are approximately 20 nm long when aligned). Further displacement applied once Segment 1–3 are all aligned will result in the unfolding of the alpha helix convolution; that is the change in the protein’s secondary structure. Figure 6 inset shows the configuration the WT structure assumes at different displacement levels. At a displacement of 25 nm (A), the alpha helix in some parts starts transforming into ‘turn’s. Once ‘coil’ secondary structure starts appearing in the structure there is a slight increase in the force levels with the applied deformation, although the change from an alpha helix to ‘turn’ secondary structure happens at a constant force level.

The characteristic SMD force-displacement data is shown for the WT/arginine (ARG; black) together with the cases with the point mutations; histidine (HIS; blue), cysteine (CYS; green), leucine (LEU; purple); serine (SER; red) for an applied larger displacement. Snapshots under the graph display the ribbon diagrams for (A), (B) and (C) for the denoted displacement for the WT (ARG 28 for the wild type/healthy case). The ribbon diagrams show the ‘alpha helix’ convolution with purple; ‘turn’s with blue and ‘coil’s with gray. ARG 28 Cα atom; the ‘hot spot’ (the mutation site) is shown with an orange sphere. After point C; the secondary structure is ‘coil’ and the backbone is completely aligned which also corresponds to the H-bonds in the backbone breaking resulting in a stiffer response shown from the force-displacement data for all cases
At (B), at a displacement of 35 nm, only the alpha helix around ARG 28 remains in-tact and at a displacement level of ~38 nm at the point (C), the whole secondary structure has turned into ‘coil’ and the 139 constituent residues have a total end-to-end distance of ~40 nm (meaning each turn almost doubles in length when stretched to completely unfold the alpha helix). After reaching an end-to-end distance of 40 nm; after which the alpha helix structure is no longer maintained, the H-bonds in the backbone start breaking giving a resistant response to the applied displacement. This stiffness is calculated to be ~8 N/m. This stiffness level also shows more clearly why the SMD spring stiffness is chosen to be 400 kcal/molȦ2 (~300 N/m) instead of the conventional choice of 10 kcal/molȦ2 (~7 N/m) which would be too low to obtain the correct stiffness. The spring stiffness has to be sufficiently large so that in a series of springs its effect is negligible (i.e. \(1/\overline{k} = \mathop \sum \limits_{i = 1}^{n} \left( {1/k_{i}} \right) + 1/k_{spring}\); where \(\overline{k}\) is the equivalent stiffness of n springs in series which is shown to be an acceptable model for the bonding of a protein structure [36]).
In order to further investigate the effect of the mutations on the particular ‘hot spot’ (codon 28), another analysis is conducted here where the stretching of the alpha helix around the ‘hot spot’ is investigated by pulling on the Cα atoms equidistant from the codon 28; in particular: Cα atoms of the codon 26 in the axial direction and the Cα atom of the residue 30 in the opposite axial direction. This tensile test is shown step-by-step (A–E) in the inset of Fig. 7.

Total force versus displacement is shown for the WT/arginine (ARG; black) together with the cases with the point mutations; histidine (HIS; blue), cysteine (CYS; green), leucine (LEU; purple); serine (SER; red) for a tensile test on the local ‘hot spot’ from the point A; folded configuration to the point E where the backbone is being stretched. Point C, in the middle, marks the complete unfolding of the ‘alpha helix’. The ribbon diagrams for each case are shown with the boundary condition: the end Cα atoms applied equal displacement in opposite directions
This segment of the α-chain structure tested here is approximately as long as a ‘turn’ in the alpha-helical convolution with an initial length of ~0.6 nm. The force versus displacement path initially follows a linear trend (with a slope of ~10 N/m) as it essentially involves breaking the alpha-helical hydrogen bonds (H-bonds) which act as linear springs. Thus in the first stage (A→B) the segment goes from one energy well (folded convolution) to another (unfolded convolution). The slope found for the four residues involved concur with the stiffness derived in the literature (~42 N/m for one residue to break the H-bonding for unfolding an alpha-helical convolution; which would then imply (42/4) N/m for residues which act like harmonic springs in series) [36]. This slope also concurs with the result shown in Fig. 6 beyond point C where the slope is measured to be ~8 N/m.
Once the ~0.6 nm length-segment is stretched another ~0.6 nm, the strain on it being 100%, the secondary structure is completely altered and alpha helix is unfolded followed by the backbone stretching. Interestingly, point (C) is the point where codon 28 secondary structure becomes a ‘coil’ where from then on the force levels increase nonlinearly in order to displace the atoms until (D) where every backbone residue is completely aligned. The constant slope observed after this point (a displacement amounting to ~1.2 nm) is the stiffness of the bonds of the backbone atoms of the polypeptide and is ~50–70 N/m. This is approximately four orders of magnitude higher than the stiffness obtained while unfolding the tertiary structure (Fig. 4).
Here, it has been presented that the unraveling of the ‘hidden length’ due to forced unfolding occurs by unfolding the tertiary structure prior to the change in the secondary structure for the healthy and mutated structures. SMD simulations help deliver a hierarchical level of stiffness values for the tetramerization site which is shown to be similar for both the healthy and the mutated structures. It is important to monitor the stability of the healthy versus mutated cases locally.
5 Discussion
The main focus of this work was to explore and assess, from a mechanical perspective, the effects of point mutations on the SPTA1 gene which cause HE (Hereditary Elliptocytosis). The crystal structure of the spectrin tetramerization site provides novel insights and enables us to do molecular testing to discuss the potential effects of mutations [22]. It was shown here that the mechanical stiffness of the tetramerization site is not directly affected by the point mutations. This suggests that the local structural disturbances the mutations cause, will affect the spectrin scaffold on the network-level rather than on the on the single chain level implying more complicated molecular interactional disorders. The local disturbances the mutations cause; due to different side-chain conformations/interactions; examined at different high-energy regions over long time periods could reveal insight to the “stability of the structure-lethality of the mutations” correlation.
Application of the SMD procedure to the tetramerization site here highlighted the unfolding pattern for a structure with folded domains where the sequence of unfolding was presented to comply: tertiary structure unraveling followed by the secondary structure change giving rise to a “softening” behaviour. From the SMD analyses, the stiffness of the backbone and the stiffness of the structure in general are discussed and compared. This provides insight into the prospective mechanical analyses that can be conducted to model different networks of poly-peptide chains [36].
Notes
- 1.
The stiffness values for protein structures from MD simulations are obtained using averaging approaches (i.e. moving average) therefore instead of an exact number, the order of magnitude of the values obtained should be considered.
References
M. Rief, M. Gautel, F. Oesterhelt, J.M. Fernandez, H.E. Gaub, Science 276, 1109–1112 (1997)
M. Rief, J.M. Fernandez, H.E. Gaub, Phys. Rev. Lett. 81, 4764–4767 (1998)
M. Rief, J. Pascual, M. Saraste, H.E. Gaub, J. Mol. Biol. 286, 553–561 (1999)
T.E. Fisher, A.F. Oberhauser, M. Carrion-Vazquez, P.E. Marszalek, J.M. Fernandez, TIBS 24, 379–384 (1999)
J. Fernandez, P. Marszalek, H. Lu, H. Li, M. Carrion-Vazquez, A. Oberhauser, K. Schulten, Nature 402, 100103 (1999)
P.-F. Lenne, A.J. Raae, S.M. Altmann, M. Saraste, J.K.H. Hörber, FEBS Lett. 476, 124–128 (2000)
E. Paci, M. Karplus, J. Mol. Biol. 288, 441–459 (1999)
S.M. Altmann, R.G. Grünberg, P.F. Lenne, J. Ylänne, A. Raae, K. Herbert, M. Saraste, M. Nilges, J.K. Hörber, Structure 10, 1085–1096 (2002)
R. Law, P. Carl, S. Harper, P. Dalhaimer, D. Speicher, D.E. Discher, Biophys. J. 84, 533–544 (2003)
H.J. Qi, M.C. Boyce, J. Mech. Phys. Solids 52, 2187–2205 (2004)
V. Ortiz, S.O. Nielsen, M.L. Klein, D.E. Discher, J. Mol. Biol. 349, 638–647 (2005)
H.J. Qi, C. Ortiz, M.C. Boyce, Trans. ASME, J. Eng. Mater. Technol. 128, 509–518 (2006)
C.P. Johnson, H. Tang, C. Carag, D.W. Speicher, D.E. Discher, Science 317, 663–666 (2007)
M. Arslan, M.C. Boyce, H.J. Qi, C. Ortiz, J. Appl. Mech. 75, 536–543 (2008)
D.W. Speicher, J.S. Morrow, W.J. Knowles, V.T. Marchesi, J. Biol. Chem. 257, 9093–9101 (1982)
D.M. Shotton, B.E. Burke, D. Branton, J. Mol. Biol. 131, 303–329 (1979)
A.M. McGough, R. Josephs, Proc. Natl. Acad. Sci. U.S.A. 87, 5208–5212 (1990)
T.L. Coetzer, K. Sahr, J. Prchal, H. Blacklock, L. Peterson, R. Koler, J. Doyle, J. Manaster, J. Palek, J. Clin. Invest. 88, 743–749 (1991)
P.G. Gallagher, Semin. Hematol. 41, 142–164 (2004)
M. Gaetani, S. Mootien, S. Harper, P.G. Gallagher, D.W. Speicher, Blood 111(12), 5712–5720 (2008)
Z. Zhang, S.A. Weed, P.G. Gallagher, J.S. Morrow, Blood 98, 1645–1653 (2001)
J.J. Ipsaro, S.L. Harper, T.E. Messick, R. Marmorstein, A. Mondragón, D.W. Speicher, Blood 115, 4843–4852 (2010)
A.D. MacKerell, D. Bashford, M. Bellott, R.L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F.T.K. Lau, C. Mattos, S. Michnick, T. Ngo, D.T. Nguyen, B. Prodhom, W.E. Reiher, B. Roux, M. Schlenkrich, J.C., Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, M. Karplus, J. Phys. Chem. B, 102, 3586–3616 (1998)
M.T. Nelson, W. Humphrey, A. Gursoy, A. Dalke, L.V. Kale´, R.D. Skeel, K. Schulten, Int. J. Supercomput. Appl. High Perform. Comput. 10, 251–268 1996
W. Humphrey, A. Dalke, K. Schulten, J. Mol. Graph. 14, 33 (1996)
H. Lu, B. Isralewitz, A. Krammer, V. Vogel, K. Schulten, Biophys. J. 75, 662–671 (1998)
B. Isralewitz, J. Baudry, J. Gullingsrud, D. Kosztin, K. Schulten, J. Mol. Graph. Model. 19, 13–25 (2001)
S. Park, F. Khalili-Araghi, E. Tajkhorshid, K. Schulten, J. Chem. Phys. 119, 3559 (2003)
M. Schaefer, C. Froemmel, J. Mol. Biol. 216, 1045–1066 (1990)
W.C. Still, A. Tempczyk, R.C. Hawley, T. Hendrickson, J. Am. Chem. Soc. 112, 6127–6129 (1990)
G.D. Hawkins, C.J. Cramer, D.G. Truhlar, J. Phys. Chem. 100, 19824–19839 (1996)
J. Srinivasan, M.W. Trevathan, P. Beroza, D.A. Case, Theor. Chem. Acc. 101, 426–434 (1999)
A. Onufriev, D. Bashford, D.A. Case, J. Phys. Chem. 104, 3712–3720 (2000)
A. Onufriev, D. Bashford, D.A. Case, Proteins: Struct., Func., Gen., 55, 383–394 (2004)
W. Cornell, R. Abseher, M. Nilges, D.A. Case, J. Mol. Graph. Model. 19, 136–145 (2001)
Z. Qin, M.J. Buehler, Phys. Rev. E 82, 061906 (2010)
Acknowledgements
This work was supported by TUBITAK (The Scientific and Technological Research Council of Turkey), grant no: 114Z733 and TED University.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter
Cite this chapter
Hunt, M. (2018). Nano-Mechanical Tensile Behavior of the SPTA1 Gene in the Presence of Hereditary Hemolytic Anemia-Related Point Mutations. In: Wriggers, P., Lenarz, T. (eds) Biomedical Technology. Lecture Notes in Applied and Computational Mechanics, vol 84. Springer, Cham. https://doi.org/10.1007/978-3-319-59548-1_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-59548-1_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-59547-4
Online ISBN: 978-3-319-59548-1
eBook Packages: EngineeringEngineering (R0)