
Abstract
The chromosome biology field at large has benefited from studies of the cell cycle components, protein cascades and genomic landscape that are required for centromere identity, assembly and stable transgenerational inheritance. Research over the past 20 years has challenged the classical descriptions of a centromere as a stable, unmutable, and transcriptionally silent chromosome component. Instead, based on studies from a broad range of eukaryotic species, including yeast, fungi, plants, and animals, the centromere has been redefined as one of the more dynamic areas of the eukaryotic genome, requiring coordination of protein complex assembly, chromatin assembly, and transcriptional activity in a cell cycle specific manner. What has emerged from more recent studies is the realization that the transcription of specific types of nucleic acids is a key process in defining centromere integrity and function. To illustrate the transcriptional landscape of centromeres across eukaryotes, we focus this review on how transcripts interact with centromere proteins, when in the cell cycle centromeric transcription occurs, and what types of sequences are being transcribed. Utilizing data from broadly different organisms, a picture emerges that places centromeric transcription as an integral component of centromere function.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
1 A Centromere Refresher
1.1 Why Centromeres?
Centromeres across eukaryotic lineages range from a relatively small, “point” within a chromosome to sprawling and complex structures that vary in size from 10’s of kilobases (KB) to 10’s of megabases (MB) (Pluta et al. 1995; Choo 1997, and reviewed in Bayes and Malik 2008; Brown and O’Neill 2014). While the location of the centromere as a single constriction on a chromosome is found broadly across the major clades of eukaryotes, some eukaryotic species do not harbor a distinct centromere; rather, there are multiple nucleating sites across chromosome arms that act as centromeres (termed holocentricity) (reviewed in Malik and Henikoff 2009). For example, chromosome segregation in Caenorhabditis elegans (and other nematodes) and some insect and plant species is mediated by sites along the entire chromosome.
The diversity in the complexity, density, and distribution of centromere forms across species lies in contrast to the uniform requisite function for the centromere: to serve as the site of kinetochore assembly and spindle attachment during meiosis and mitosis. In essence, the proper functioning of centromeres is a requirement for faithful segregation of a chromosome complement. Any failure in this function has catastrophic consequences for the cell, such as chromosome breakage, and/or loss and cellular breakdown (reviewed in Holland and Cleveland 2009); and, consequently has devastating consequences for the organism, such as infertility, loss of cell cycle control, and aberrant proliferation.
1.2 Why NOT Centromeres?
Despite the deep phylogenetic conservation of centromere function—to mediate kinetochore formation and spindle attachment—the diversity of centromere forms across species has presented a unique challenge in understanding the components that delineate centromere functionality as well as defining the minimal required elements for centromere integrity. For example, the “point centromeres” of the budding yeast, Saccharomyces cerevisiae (Fishel et al. 1988), consist of a 125-bp nucleotide sequence that supports centromere function (Meluh et al. 1998) without the requirement for any other complex repeat structures. The centromeres of the filamentous fungi Neurospora are 175–300 KB and harbor AT-rich, degenerate transposons (reviewed in Smith et al. 2012) whose sequences have been ravaged by a genome defense mechanism known as RIP (‘repeat induced point mutation’)(Smith et al. 2011). Many plants, including maize and grasses, carry satellites and transposons throughout their regional centromeres (Neumann et al. 2011; Gent and Dawe 2012). Like fungi, there does not appear to be any pattern to the repeat structure that defines the functional centromere core in most plants.
The diversity of centromere forms is not only restricted to species-specific genomic arrangements, as several species (e.g., orangutan, horse, chicken) carry a chromosome complement wherein some centromeres are characterized largely by repetitive DNA (satellites and transposable elements) while others are seemingly devoid of a highly repetitive structure (Piras et al. 2010; Shang et al. 2010; Locke et al. 2011). Thus, attempts to reconcile these diverse centromere forms with a generalizable model for centromere function across traditional eukaryotic model organisms (yeast, human, mouse, Arabidopsis, maize) have been largely unsuccessful as no simple rule appears to apply to even the majority of centromere types. Large-scale genome sequencing projects for several model species initially showed promise in capturing the DNA landscape of regional centromeres in species with diverse karyotypes [e.g., human (Schueler et al. 2001), Arabidopsis (Copenhaver et al. 1999; Kumekawa et al. 2000; Hosouchi et al. 2002), rice (Yan and Jiang 2007), wallaby (Renfree et al. 2011), and gibbon (Carbone et al. 2014)]. However, the highly repetitive nature of such centromeres, composed of expansive arrays of simple satellites [ranging in size anywhere from only 0.2 KB to more than 28 MB (Melters et al. 2013)] and other highly repeated sequences, such as transposable elements, has remained a hurdle in defining genomic maps for complex, regional centromeres. As a consequence, complex eukaryotic centromeres have, to date, remained on the “black list” (Miga et al. 2015) of regions refractive to mapping and assembly techniques (Altemose et al. 2014).
Emerging sequence techniques that afford long-range sequence information (e.g., long-read sequencers capable of sequencing >100 KB of contiguous DNA, such as PacBio and Oxford Nanopore; and, synthetic long-read sequencers such 10X Genomics) offer the potential to overcome the technical challenges of dealing with highly repeated regions of genomes. However, the overall scale of the total repeat regions that encompass the functional centromeres within model systems that are subject to genome sequencing efforts is orders of magnitude greater than the long-read sequencing capabilities and has left centromere regions in genome assemblies without the foundation of a linear genetic map in most cases, particularly human and mouse. Confounding this sequencing challenge is the sheer number of centromeres that must be tackled for assembly within any given genome—one per chromosome in a diploid cell—as each centromere contains a unique genomic sequence structure.
2 Centromeric DNA: A Descriptor or Determinant?
Studies aimed at identifying the primary sequence associated with functional centromeric chromatin have revealed a lack of conservation of centromeric sequences, even among closely related species. Thus, the genomic component of eukaryotic centromeres is relatively rapidly evolving despite its conserved role in chromosome segregation (Henikoff et al. 2001). A remarkable computational effort has led to the production of graphical models of human centromere sequences (Miga et al. 2014; Miga 2015; Rosenbloom et al. 2015), bypassing the need for strict linear assembly in the assessment of nascent genetic content. These “maps” do not delineate the order of sequences within any given centromere, yet reveal the diversity of satellites within and among centromeres, supporting earlier work demonstrating that while satellite higher order repeats (HORs) are homogenized through processes such as molecular drive and concerted evolution (Dover et al. 1982), some satellites are in fact distinct amongst different chromosomes (for an example, see Miga et al. 2014).
Defying another common misconception that each chromosome has only one location that can serve as a functional centromere, several human chromosomes have multiple HORs that act as functional centromeric epialleles (Maloney et al. 2012). Within any given chromosome, only one of these epialleles functions as the active centromere, raising the possibility of heterozygotes for different epialleles on the same chromosome pair. As the quality of sequencing and gap-filling for the human genome has increased, novel annotation workflows have also uncovered retroelements scattered throughout active centromere regions across all human chromosomes, within HORs and between epialleles (Miga et al. 2014; Rosenbloom et al. 2015). Indeed, co-option of repetitive elements, including tandem duplications, may be a general aspect of centromere ontogenesis across eukaryotes (Dawe 2003; Wong and Choo 2004; Chueh et al. 2009; O’Neill and Carone 2009; Brown and O’Neill 2010).
Most multicellular eukaryotic centromeres harbor a similar, characteristic repeat structure highly enriched in species-specific satellites (e.g., α satellites in human and minor satellites (miSAT) in mouse). The functional impact of these satellites with respect to kinetochore assembly remains less clear, however, based on multiple lines of evidence. Several studies highlight that centromeric satellites are not sufficient to form kinetochores. Placing an array of satellites in a cell is not the only requisite to form stable artificial chromosomes in all cases (Nakano et al. 2003). In fact, dicentric chromosomes often retain their satellite array but this array no longer forms functional centromeric chromatin (Warburton et al. 1997). Thus, the presence of satellite DNA alone is not the primary determinant for recruiting centromeric histones. As both ectopic centromeres in abnormal chromosomes (e.g., mini- and marker chromosomes, B chromosomes, neocentromeres) and newly formed centromeres that have only recently become fixed within a species (e.g., evolutionary new centromeres, ENC) are often devoid of satellite DNA, the absence of satellite DNA suggests such repeated DNA is also not required for centromere formation (Lo et al. 2001a; Alonso et al. 2007; Hasson et al. 2013).
While the canonical structure of species-specific satellites, and higher order arrays of groups of satellites, is neither sufficient nor required to facilitate centromere assembly, it is a pervasive feature among eukaryotic centromeres (Brown and O’Neill 2014; Plohl et al. 2014). While the fact that centromeres can form and act on genomic regions devoid of satellite DNA has lent support to the notion that centromere identity is likely under epigenetic control (Karpen and Allshire 1997; Henikoff et al. 2001). The contributions such types of genomic sequence have on defining the functional capacity of centromeric chromatin assembly and evolutionary stability of centromeres cannot be discounted. As exemplified in studies of human neocentromeres, DNA satellites alone are not required to attract centromere proteins to ectopic centromeres (e.g., Lo et al. 2001b). In such cases, another type of repeat found in most complex centromeres, retrotransposons, are found to bind the defining centromeric histone, CENP-A, and define the functional centromere (Chueh et al. 2009). These selfish entities may be the progenitors of satellite arrays (e.g., Macas et al. 2009) that experience accretion and diminution as either monomers or large homogenous arrays following centromere stabilization and fixation in a population. Just as the acquisition of repeat expansions may be linked to the ontogeny of a fixed, stable centromere within a species, the primary establishment of a new centromere may be the result of a seeding event from retroelement(s) that progressively generate novel satellites (Dawe 2003; O’Neill et al. 2004; Brown and O’Neill 2010).
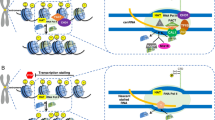
Despite the challenges in delineating a finite sequence demarcating centromere functionality across species, the protein cascade that leads to faithful centromere assembly each cell cycle is more clearly defined. The pivotal event is the loading of the centromere specific H3, CENP-A (Fig. 1a), which occurs in late telophase/early G1 in most organisms (Dunleavy et al. 2009) [n.b. in S. pombe, this occurs in S phase (Dunleavy et al. 2007)]. During replication in S phase, the levels of CENP-A are diluted to 1/2 as H3.3 is assembled into centromeric chromatin as a placeholder (Dunleavy et al. 2011). In human, HJURP (holliday junction recognition protein) associates with CENP-A in pre-nucleosomal complexes (Mellone et al. 2009; Foltz et al. 2009; Dunleavy et al. 2009) and chaperones newly synthesized CENP-A to centromeric chromatin following mitosis (telophase/early G1) (Foltz et al. 2009; Dunleavy et al. 2009) when CENP-A loading occurs (Jansen et al. 2007). After mitosis, new CENP-A loading is also facilitated by a priming mechanism involving protein complexes such as hMis18 (Fujita et al. 2007) that prepares the centromeric nucleosome for CENP-A loading (Mellone et al. 2009; Dunleavy et al. 2009). These proteins serve as the pinnacle of the DNA-chromatin interface, yet many other proteins are involved in the coordinated assembly of the kinetochore (described in Parts I and IV of this book).

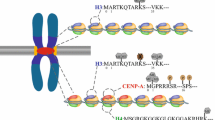
RNA binding domains of CENP-A and CENP-C. a Linear depiction of the complete CENP-A protein domain structure (Regnier et al. 2003) with amino acids that comprise each domain shown underneath. Amino acids in green were computationally predicted to have an RNA binding capability by Quenet and Dalal (2014). Most of the potential RNA interaction capability lies in the N-terminal tail region [a Alpha helix, L Loop region, CATD Centromere targeting domain (Black et al. 2004), CCBD CENP-C binding domain (Carroll et al. 2010)]. b Linear depiction of the CENP-C protein. The RNA binding domain experimentally validated by Wong et al. (2007) is located between amino acids 422 and 551, the sequence of which is shown below. Amino acids in green are most critical to RNA binding (Wong et al. 2007). The RNA binding domain of CENP-C overlaps with both the DNA binding domain (aqua) and the CATD (gray). Note that Wong et al. (2007) also found evidence for a second RNA binding domain between 552 and 943, but did not isolate the exact region. [DNA binding domain (Yang et al. 1996; Sugimoto et al. 1997; Cohen et al. 2008; Schueler et al. 2010), CATD (Yang et al. 1996), Dimerization domain (Sugimoto et al. 1997), CABD CENP-A binding domain (Trazzi et al. 2009)]
3 Active Transcription at Centromeres—Breaking Down Common Myths and Legends
Challenging another classical description of a eukaryotic centromere as a heterochromatin-rich and transcriptionally inactive region, centromeres are in fact characterized by a complex suite of different chromatin marks supporting active transcription and the production of centromeric noncoding RNAs required for proper centromere formation and function (Wong et al. 2007; Carone et al. 2009, 2013; Ting et al. 2011; Hall et al. 2012; Quenet and Dalal 2014). The chromatin encompassing the centromere core, referred to as “centrochromatin”, is distinct from that of pericentromeres and contains histone modifications associated with transcriptionally active chromatin (Sullivan and Karpen 2004; Eymery et al. 2009; Gopalakrishnan et al. 2009; Bergmann et al. 2011, 2012). CENP-A nucleosomes within centrochromatin are interspersed with modified histones, histone H3 methylation, and dimethylation of lysine 4 and di- and trimethylation of lysine 36 of histone H3 (H3K4me1, H3K4me2, H3K36me2, and H3K36me3). These modified histones are not only permissive to transcription, but differentiate centrochromatin from its neighboring pericentromere, a region that, while also characterized by a high density of repeats, is defined by histone modifications typically associated with transcriptional silencing [(Gopalakrishnan et al. 2009; Roadmap Epigenomics et al. 2015): di- and trimethylation of lysine residues 9 and 27 of histone H3 (H3K9me2, H3K9me3,H3K27me2, and H3K27me3)].
Despite the remarkably different chromatin environments that define the peri- and centromere, active transcription has been detected from both regions in many organisms (Carone et al. 2009; Ugarkovic 2005; Eymery et al. 2009; Brown et al. 2012; Gent and Dawe 2012; Hall et al. 2012; Biscotti et al. 2015; Koo et al. 2016; Rosic and Erhardt 2016). Moreover, a clear balance in transcriptional output from each region is required to maintain chromosome stability (Hall et al. 2012). The types of sequences found to produce transcripts within centromeres includes the same sequences represented in the genomic foundation of a centromere: satellites, retroelements and in some cases active genes located within the boundaries of centrochromatin (e.g., Nagaki et al. 2004).
While prevalent in complex eukaryotic centromeres, the importance of these retroelement and satellite-derived transcripts to centromere function is only recently becoming apparent; chromosome missegregation has been associated with aberrant centromere transcription in animals and satellite RNA has been implicated in the assembly of centromere components CENP-A and -C, in Drosophila, plants, mouse and human (Mejía 2002; Bergmann et al. 2011; Ting et al. 2011; Carone et al. 2013; Quenet and Dalal 2014; Leung et al. 2015).
4 Genome Engineering to Tease Apart the Transcriptional Framework of the Centromere
Advances in techniques that allow manipulation of DNA and its nascent chromatin have been used synergistically to create and modify artificial centromere constructs within living cells. For example, alpha satellite arrays from human have been isolated and, when placed in HT1080 cells, form functional human artificial chromosomes (HACs) (Harrington et al. 1997; Ikeno et al. 1998; Grimes and Monaco 2005; Lam et al. 2006; Maloney et al. 2012). Focused studies of HACs have shown that active transcription at the centromere is essential to their stable propagation (Okamoto et al. 2007; Nakano et al. 2008; Bergmann et al. 2011, 2012; Molina et al. 2016). DNA constructs that form stable HACs incorporate selectable marker genes (i.e., neo and bsr) under strong, constitutive promoters juxtaposed to the alphoid arrays. The ability of the resulting HAC to assemble a functional kinetochore and survive cell division was found to be reliant not only on the overall number of satellites but also on the transcriptional activity of these marker genes (Okamoto et al. 2007).
Human artificial chromosomes modified to carry tetO transcriptional regulatory sequences within alphoid arrays were manipulated to increase or decrease transcriptional output in attempts to define the activity for proper centromere function (Nakano et al. 2008). Switching off transcription from the tetO dramatically diminished propagation of the HACs, but upregulating transcription with tet activators had a similar effect, indicating a balanced level of transcription is a requisite for proper centromere function. Further modifications of HACs by tethering a lysine-specific demethylase (LSD1) to alphoid arrays showed that depletion of H3K4me2 from HAC centromeric chromatin results in a loss of satellite transcription and concomitant reduction in local assembly of newly synthesized CENP-A (Bergmann et al. 2011). HACs targeted to increase H3K9 acetylation, a mark permissive to transcription, showed no effect on kinetochore formation despite such a dramatic change in chromatin state. However, when this chromatin change is coupled with a dramatic increase in transcription, rapid centromere inactivation through loss of CENP-A loading results (Bergmann et al. 2012).
Recently, a study using an inducible ectopic centromere system in Drosophila showed that CENP-A assembly by the Drosophila CENP-A chaperone, CAL1, requires RNA pol II mediated transcription of nascent DNA (Chen et al. 2015). In this ectopic centromere system, transcription is mediated by CAL1’s binding partner, the chromatin remodeling complex FACT (facilitates chromatin transcription) and targets an artificial array of lacO sequences, indicating that the passage of RNA polymerase is required for CENP-A chromatin establishment rather than sequence-specific transcripts (Chen et al. 2015). A study of the primary centromere core sequence in S. pombe necessary and sufficient for CENP-A assembly was conducted wherein the core sequence was shuffled to create a de novo sequence with the same AT content and nucleosome positioning (Catania et al. 2015). This new construct was not able to effectively establish CENP-A chromatin, indicating some sequence features are required for centromere integrity. Notably, the core sequence is actively transcribed via multiple putative transcription start sites, implicating its ability to facilitate transcription (albeit stalled transcription, see below) as a defining feature of this centromere-competent sequence (Catania et al. 2015). As demonstrated by these studies, centromere integrity requires tight control of centromere transcription, suggesting that centromeric DNA sequence identity may not be an absolute requirement, but the ability to facilitate transcription and act as a fundamentally stable and immutable regulatory element(s) is needed.
5 The “How?” of Centromeric RNA: Centromere Transcripts and Protein Interactions
Centromeric RNAs have been hypothesized to perform diverse functions, including establishing and maintaining pericentromeric heterochromatin and recruiting kinetochore proteins to the centrochromatin core. Recent studies have focused on the transcription of the most prevalent centromeric sequence, satellites, with respect to centromere function, however, the identity and functional roles of satellite transcripts in diverse organisms have not been fully elucidated. Several recent studies highlighted below further support the growing evidence that transcription is an integral part of the centromere chromatin assembly cascade; how, when, and what types of transcripts impact centromere assembly are emerging areas of focus in the centromere biology field.
As the centromere is a tightly regulated network of protein and nucleic acid interactions, noncoding transcripts may only directly interact with a subset of this multi-protein network and yet, indirectly impact the function of many centromere and kinetochore proteins when these transcripts are mis-regulated. CENP-C, CENP-A, HJURP, and certain members of the chromosomal passenger complex (CPC) have been implicated as the centromere proteins that directly associate with, or bind to, noncoding RNAs (Wong et al. 2007; Ferri et al. 2009; Du et al. 2010; Carone et al. 2013; Quenet and Dalal 2014; Rosic et al. 2014; Blower 2016).
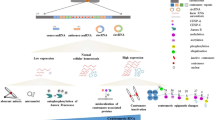
CENP-A: The first indication that CENP-A can interact with noncoding RNA was discovered in a human neocentromere; LINE-1 elements within the CENP-A binding region of a neocentromere on 10q25 are actively transcribed into a noncoding RNA that incorporates with CENP-A chromatin (Chueh et al. 2009). While less evidence exists for a direct association of centromeric RNA and CENP-A or HJURP, aberrant expression of these transcripts distinctly perturbs CENP-A localization and loading (Fig. 2). For example, overexpression of noncoding RNA from the centromeric retrotransposon KERV in tammar wallaby disrupts proper CENP-A loading into centromeres in late telophase (Carone et al. 2013). A recent study in human showed a more direct contact between CENP-A and RNA when an alpha satellite related RNA sequence was pulled down with the soluble CENP-A/HJURP complex using RNA immunoprecipitation (RIP) (Quenet and Dalal 2014). While this specific noncoding RNA sequence does not match the alpha satellite consensus, nor any other alpha satellite higher order repeat sequence, and any known repeated elements in the assembled or unassembled contigs of the human genome, DNA FISH showed it may reside in only a subset of chromosomes in the human karyotype (Quenet and Dalal 2014). Quenet and Dalal (2014) complemented their study with an in-silico prediction of potential RNA binding sites in CENP-A and HJURP, finding that 79 out of 140 residues in CENP-A and 286 out of 748 residues in HJURP had a capacity for RNA binding. Intriguingly, the entirety of the CENP-A N-terminal tail was predicted to carry RNA-binding capacity (Quenet and Dalal 2014) (Fig. 1a). CENP-A’s N-terminal tail is the most rapidly evolving portion of CENP-A (Henikoff et al. 2001; Malik and Henikoff 2001), and while it is known to be required for CENP-A stabilization at the centromere (Logsdon et al. 2015), its exact function remains elusive. Given a putative role in RNA interaction, it is possible the vast differences in amino acid sequence and overall length of the N-terminal region among species (Henikoff et al. 2001) could be to enable permissive interaction with a variety of transcripts that emanate from the rapidly evolving, underlying DNA.

Cell cycle variation of centromere transcription. Overview of when in the cell cycle centromeric transcripts have been identified in different model species in relation to critical assembly events defining centromere integrity. The cell cycle is indicated (color). Top The type of transcript for each species [from top, human (Wong et al. 2007; Chueh et al. 2009; Quenet and Dalal 2014), wallaby (Carone et al. 2013), plants (Topp et al. 2004; Koo et al. 2016), and reviewed in Gent and Dawe (2012), Drosophila (Rosic et al. 2014), frog (Blower 2016), mouse (Lu and Gilbert 2007; Ferri et al. 2009), yeast (Chen et al. 2008; Choi et al. 2011; Catania et al. 2015)] is indicated. Below the dashed line are the transcripts where the timing of transcription is known. A thinner bar represents a lower level of transcription while a thicker bar represents a higher level. Gray lines (above the dashed line) indicate that while transcripts have been identified, it is not known when in the cell cycle transcription is initiated. Above each line are the protein associations known for transcripts. Bottom The timing of protein cascade components relative to the cell cycle. Black bar indicates constitutive association with the centromere (FACT, RNA Pol II). Relevant timing of loading of the H3.3 placeholder, CPC recruitment and CENP-A loading components are indicated. Specific CENP-A assembly times are indicated for each group of species
CENP-C: CENP-C contains an experimentally validated, distinct RNA binding domain (Wong et al. 2007; Du et al. 2010) (Fig. 1b). Interestingly, the RNA binding domain of CENP-C shares homology to the RNA binding hinge domain region of the pericentromeric heterochromatin proteins HP1 alpha, beta, and gamma (Muchardt et al. 2002; Du et al. 2010). In human, CENP-C associates with single-stranded (ss) alpha satellite transcripts both in vitro and in vivo, and is lost from centromeres upon alpha satellite depletion along with the CPC proteins, INCENP and Survivin (Wong et al. 2007). DNA binding of maize CENP-C is stabilized by a ssRNA in vitro, although this stabilization appears to be independent of the ssRNA sequence (Du et al. 2010) (Fig. 2). This permissive binding in maize is in contrast to human CENP-C that showed a preferential association with alpha satellite ssRNA in competition assays with tRNA, rRNA, and mouse pericentric satellite (Wong et al. 2007). In Drosophila, the X chromosome specific satellite, SAT III, is actively transcribed into long noncoding transcripts that localize to centromeres and associate with CENP-C (Rosic et al. 2014) (Fig. 2). Upon CENP-C depletion, SAT III RNA signal is greatly reduced at centromeres, implying a similar interaction between CENP-C and RNA in Drosophila, as in human and maize. When depleting SAT III RNA levels, both newly synthesized CENP-C and CENP-A showed a reduction in centromeric signal that was also observed to cascade up through the kinetochore proteins (Rosic et al. 2014). SAT III-depleted cells also suffered errors in mitosis, including lagging chromosomes and micronuclei formation; notably, all chromosomes were susceptible to mitotic defects, indicating that SAT III RNAs, despite originating from the X chromosome, can act in trans to target the autosomes (Rosic et al. 2014). Lagging chromosomes with reduced CENP-C signal were also prevalent in human cells after RNA pol II inhibition (Chan et al. 2012).
The CPC: Ostensibly, a single centromeric noncoding transcript does not have to bind to just a single protein. When considering the fact that CENP-A assembly is facilitated by a chaperone, HJURP, and that CENP-A and CENP-C are both required for centromere integrity, it is probable that these transcripts contact multiple proteins that are associated with one another. Such RNA interactions may even serve to tether protein complexes together, or to scaffold these complexes to other components of the surrounding chromatin environment. The multi-protein interaction between Aurora-B, Dasra-a/Borealin, Survivin, and Incenp composes the CPC. The CPC aids in mitosis as a phosphorylating agent at chromosome arms, the inner centromere, and mitotic spindles (reviewed in (Carmena et al. 2012). While at the inner centromere, the CPC plays a key role in bipolar spindle attachment by acting as a “sensor” of connection between the centromere and the spindle (Lampson and Cheeseman 2011). Both cen-RNAs and spindle-enriched RNAs are known to congregate with the CPC (Ferri et al. 2009; Ideue et al. 2014; Jambhekar et al. 2014) (Fig. 2). In fact, there is direct binding of RNA to the CPC and this interaction is responsible for inner centromere localization (Jambhekar et al. 2014; Blower 2016), and is required for CPC activation (Blower 2016). Despite the fact that multiple proteins form the CPC, Ferri et al. (2009) showed that miSAT transcripts in mouse are a key partner with CPC proteins Aurora-B, INCENP and Survivin at the onset of mitosis. In Xenopus extracts, however, RNA binding was identified that is required for CPC localization, but this binding is specific only to the proteins Aurora-B and Dasra-A, and not INCENP, Survivin, and XMAP215 (Blower 2016).
In Xenopus, among the Aurora-B binding RNAs is a ~170 nt centromeric transcript (fcr1, frog centromeric repeat1) that, similar to the sat III RNA in Drosophila (Rosic et al. 2014), is only found on a subset of CENP-A defined centromeres within the karyotype (Edwards and Murray 2005). The active transcription of fcr1 is required for Aurora-B localization to the inner centromere of mitotic chromosomes and may act initially on the fcr-1-native chromosomes before diffusing to other centromeres (Blower 2016) (Fig. 2).
6 The “When?” of Centromere Transcription: It is an Around the Clock Job
The emergence of studies on RNA, transcription, and centromere function since the pivotal studies in yeast (Volpe et al. 2003), plants (Topp et al. 2004) and human neocentromeres (Wong et al. 2007) has led to accumulating evidence that transcription is a requirement for centromere function and cell stability. However, the timing of this transcription and a delineation of whether specific transcript sequences, or simply the act of transcription itself, are required for centromere integrity are not known. Studies in several model systems have begun to highlight the intricacies of transcriptional events at the centromere throughout the cell cycle, with a particular emphasis on mitotic transcription (Fig. 2).
Cell Cycle Phase G1: Late telophase/early G1 is the pivotal time in mammalian cells when CENP-A is actively loaded into centromeric chromatin. Thus, the impact of active transcription at this point in the cell cycle may have direct bearing on the ability of CENP-A to assemble functional centromeric chromatin. The 1.3 KB human centromeric transcript described above (Quenet and Dalal 2014) is transcribed by RNA pol II from late telophase into early G1, coincident with the timing of CENP-A deposition by its chaperone HJURP (Fig. 2). This RNA transcript was found to interact with these proteins, suggesting its capacity to aid in CENP-A nucleosome assembly. The transcription of one of two groups of transcripts that emanate from mouse pericentromeric gamma satellites was detected in late G1 and proceeded through mid-S phase (Lu and Gilbert 2007) (Fig. 2). This species of RNA did not show a discrete size range and transcription of this species decreased at a time coincident with the replication of pericentric heterochromatin. This RNA class is a large, heterogeneous group of gamma satellite transcripts whose transcriptional timing may simply be the result of cryptic transcription (Lu and Gilbert 2007). However, given that the appearance of these satellite transcripts is CDK (cyclin dependent kinase)-dependent, and thus is only in cells committed to proliferation, the transcripts may be required for heterochromatin reassembly at the replication fork (Lu and Gilbert 2007).
Cell Cycle Phase S and G2: Double-stranded RNAs are actively transcribed from the pericentric repeats dh and dg from within the centromeres of the yeast, Shizosaccharomyces pombe, and are subsequently processed into small interfering RNAs (siRNAs) (Volpe et al. 2002, 2003). These siRNAs are bound to a complex of proteins (the RNA-induced initiation of transcriptional gene silencing, RITS) and result in targeted H3 lysine-9 methylation through RNA interference (Volpe et al. 2002, 2003). Moreover, the disruption of RNAi components compromises heterochromatin assembly (Volpe et al. 2002) and CENP-A deposition (Folco et al. 2008), linking a small RNA component to centromere function.
The forward strand of centromeric repeats is transcribed in S phase in S. pombe, thought to be the major initiating point for siRNA production (Chen et al. 2008). siRNA levels are stably detected throughout the cell cycle but increased in S/G2, coincident with RITS complex accumulation and transcript processing (Fig. 2). Similar to that proposed for gamma satellites in mouse, these transcripts may be the result of cryptic or spurious transcription yet are required for the appropriate establishment of heterochromatin (Chen et al. 2008).
While active CENP-A loading occurs in late telophase/early G1 in mammals, H3.3 is loaded into centrochromatin during S phase, likely as a placeholder for CENP-A replenishment after mitosis (Dunleavy et al. 2011). Thus, transcription could be involved in the eviction of the placeholder (Catania et al. 2015; Chen et al. 2015; Chen and Mellone 2016). A recent study in S. pombe showed that RNA pol II stalls at centromeric DNA and that the level of stalling is directly related to the level of subsequent CENP-A nucleosome assembly. In this yeast species, CENP-A assembly occurs in S phase and G2 (Takahashi et al. 2005; Dunleavy et al. 2007; Takayama et al. 2008); an increase in RNA pol II stalling, and concomitantly permissive but “low-quality” transcription, may lead to increased CENP-A chromatin through either increased eviction rates for placeholder H3 or through demarcation of a specific environment conducive to efficient CENP-A assembly (Catania et al. 2015).
The timing of detectable increases in RNAPII stalling in S phase is coincident with DNA replication. Centromere transcription at this phase of the cell cycle [forward strands in S. pombe (Chen et al. 2008) and pericentromeric satellites in mouse (Lu and Gilbert 2007)] may position replication forks and RNA pol II to collide more often, increasing the rate of RNA pol II stalling (reviewed in Brown et al. 2012). RNA pol II stalling and collisions subsequently increase the generation of large, stable R-loop formation (Reddy et al. 2011). While the RNA–DNA hybrids present in R-loops are typically small and transient, it is notable that the transcriptional framework of the centromere may present an increase in stable R-loops in S phase since increases in R-loops are linked to phosphorylation of H3S10, a marker of subsequent entry into mitosis (M phase) (Castellano-Pozo et al. 2013; Oestergaard and Lisby 2016). Chen et al (2015) showed that FACT is required for CENP-A assembly and FACT has been previously shown to both travel in a complex with the CENP-A chaperone (Foltz et al. 2006) and localize to centromeres throughout the cell cycle (Okada et al. 2009). The presence of FACT at centromeres and in complex with key centromere assembly components supports hypotheses that FACT is an essential part of the chromatin remodeling involved in facilitating CENP-A nucleosome assembly. For example, FACT may be required to destabilize nucleosomes (Hondele and Ladurner 2013) and subsequently facilitate the transcription of centromere sequences preceding assembly of new CENP-A nucleosomes (Chen et al. 2015; Chen and Mellone 2016). Recently, FACT was found to bind the inner kinetochore proteins of the CENP-T/W complex and thus may promote the CENP-T/W deposition at centromeres (Prendergast et al. 2016). In fungi it appears that FACT is necessary to prevent spurious, ectopic incorporation of CENP-A rather than performing a function in primary CENP-A assembly at the centromere (Deyter and Biggins 2014). FACT is known to solve R-loop and replication mediated conflicts in both human and yeast (Herrera-Moyano et al. 2014). Furthermore, low levels of central core transcripts are detected in yeast cells due to an increase in RNA pol II stalling (Catania et al. 2015). We thus propose that FACT may also be present at centromeres to resolve the resulting R-loops prior to progression into mitosis. The multiple, possible roles for FACT in CENP-A assembly are not mutually exclusive, rather are an example of the dynamic state of the centromere during different phases of the cell cycle.
Cell Cycle Phase Mitosis: Whether or not centromeric transcription occurs during mitosis has been hotly debated since the majority of transcription factors and RNA polymerases are not associated with chromosomes during mitosis (Gottesfeld and Forbes 1997). However, several pieces of evidence suggest centromeric transcription occurs during M phase of the cell cycle (Liu 2016), indicating persistent transcription during mitosis may serve to further distinguish the centromere from chromosome arms. First, RNA pol II is present at kinetochores in M phase (Chan et al. 2012). Second, transcription run-on assays have shown that RNA pol II is capable of transcribing centromeres of mitotic chromosomes (Liu 2016). Third, inhibition of RNA pol II by alpha-amanitin reduces centromeric cohesion and CENP-C localization (Chan et al. 2012; Liu et al. 2015). The defective cohesion following RNA polymerase inhibition was caused by a mislocalization of Sgo1, a protein found at the inner centromere that protects cohesin during mitosis (Liu et al. 2015). Collectively, these data suggest that transcription and/or the transcripts themselves may play a functional role in a time-specific manner (i.e., restricted to specific phases of the cell cycle).
A long noncoding RNA was recently identified as actively transcribed from the centromeres of Xenopus egg extracts during mitosis; moreover, these transcripts serve a functional role via binding Aurora-B, a component of the CPC, and are required for normal kinetochore-centromere attachment (Blower 2016). Xenopus egg extracts have also led to the discovery that RNA processing assists in kinetochore and spindle assembly (Grenfell et al. 2016); inhibition of the spliceosome in egg extracts leads to an accumulation of long centromeric transcripts and a failure to efficiently recruit CENP-A, CENP-C and NDC80. What this study shows is that transcription is not only active during mitosis, further supporting the growing body of evidence indicating this occurs, but that transcripts undergo processing during this phase, contradicting the theory that RNA processing is repressed during mitosis (Shin and Manley 2002).
Centromere transcript processing is a recurring theme observed for a broad set of centromere transcripts, although the relationship of the processing machinery and/or processed RNA products to centromere integrity is less clear (with the notable exception of S. pombe, see above). Early work in mouse cells showed that a loss of DICER activity, the RNAse III enzyme that facilitates small RNA processing, results in an accumulation of larger satellite transcripts (Kanellopoulou et al. 2005). This finding implies that when DICER is available, these larger satellite transcripts are not detected as they are processed into smaller RNAs. The implication that DICER is involved in this RNA processing would also indicate these small RNAs are <40 nt based on the catalytic activity of the enzyme (MacRae et al. 2007). Other sizes of centromeric RNAs have been uncovered that are likely independent of DICER. For example, small RNAs have been detected for the maize centromere satellite CentC (Du et al. 2010) and from the wallaby centromeric retroelement KERV (Carone et al. 2009). Both of these small RNAs were also found to participate in the centromere assembly cascade: CentC associated with CENP-C directly (Du et al. 2010); a reduction in KERV small RNAs resulted in a loss of CENP-A assembly in late telophase (Carone et al. 2013) (Fig. 2). However, any connection between these types of processed, small RNA transcripts and the necessary RNA processing machinery observed in Xenopus is unexplored; likewise the timing of transcription for these processed RNAs is currently unknown.
7 Conclusion
Mounting evidence suggests that RNA species and the act of transcription itself is required for the recruitment and/or establishment of centromere and kinetochore proteins. Thus, it is clear that transcription at the centromere, and in neighboring pericentromeric heterochromatin, is functionally distinct yet critical throughout the entirety of the cell cycle. Studies are now beginning to reveal that centromeric transcripts and accompanying chromatin changes are required for different components of the centromere assembly cascade at different points in the cell cycle. Over the last decade, RNA species derived from the centromeric regions of many model species have been uncovered, as have some of their interacting partners. Closer examination of these transcripts, and indeed of the subregions of the centromere previously considered devoid of transcriptional activity, has made it clear that both the act of transcription itself and the resulting transcripts are critical to ensuring proper CENP-A assembly and faithful chromosome segregation. In the same manner that the comparative approach revealed that centromeres evolve rapidly and are established through an epigenetic framework, the use of diverse eukaryotic systems will afford the development of a model to describe key remaining questions, such as: how do specific transcripts mediate centromere function in cis and/or in trans? is splicing or RNA processing a requisite in forming functional transcripts across different cell cycles and among different species? and, how does this transcriptional landscape impact centromere evolution in both a phylogenetic and disease context? In fact, one of the reasons the myth of the centromere as “silent chromatin” prevailed for so long is that centromere transcripts have been difficult to capture and characterize. As highlighted herein, it is the very reason these transcripts are difficult to capture (e.g., RNA pol II stalling, RNA processing, protein-RNA binding) that holds the key to how centromere transcription, and their transcripts, likely function in maintaining centromere integrity.
References
Alonso A, Fritz B, Hasson D, Abrusan G, Cheung F, Yoda K, Radlwimmer B, Ladurner AG, Warburton PE (2007) Co-localization of CENP-C and CENP-H to discontinuous domains of CENP-A chromatin at human neocentromeres. Genome Biol 8(7):R148
Altemose N, Miga KH, Maggioni M, Willard HF (2014) Genomic characterization of large heterochromatic gaps in the human genome assembly. PLoS Comput Biol 10(5):e1003628. doi:10.1371/journal.pcbi.1003628
Bayes JJ, Malik HS (2008) The evolution of centromeric DNA sequences. Encycl Life Sci 1–8
Bergmann JH, Rodriguez MG, Martins NM, Kimura H, Kelly DA, Masumoto H, Larionov V, Jansen LE, Earnshaw WC (2011) Epigenetic engineering shows H3K4me2 is required for HJURP targeting and CENP-A assembly on a synthetic human kinetochore. EMBO J 30(2):328–340. doi:10.1038/emboj.2010.329 emboj2010329 [pii]
Bergmann JH, Jakubsche JN, Martins NM, Kagansky A, Nakano M, Kimura H, Kelly DA, Turner BM, Masumoto H, Larionov V, Earnshaw WC (2012) Epigenetic engineering: histone H3K9 acetylation is compatible with kinetochore structure and function. J Cell Sci 125(Pt 2):411–421. doi:10.1242/jcs.090639 jcs.090639 [pii]
Biscotti MA, Canapa A, Forconi M, Olmo E, Barucca M (2015) Transcription of tandemly repetitive DNA: functional roles. Chromosome Res 23(3):463–477. doi:10.1007/s10577-015-9494-4
Black BE, Foltz DR, Chakravarthy S, Luger K, Woods VL Jr, Cleveland DW (2004) Structural determinants for generating centromeric chromatin. Nature 430(6999):578–582
Blower MD (2016) Centromeric transcription regulates Aurora-B localization and activation. Cell Rep 15(8):1624–1633. doi:10.1016/j.celrep.2016.04.054
Brown JD, O’Neill RJ (2010) Chromosomes, conflict, and epigenetics: chromosomal speciation revisited. Annu Rev Genomics Hum Genet 11:291–316. doi:10.1146/annurev-genom-082509-141554
Brown JD, O’Neill RJ (2014) The evolution of centromeric DNA sequences. In: eLS, Wiley, USA. doi:10.1002/9780470015902.a0020827.pub2
Brown JD, Mitchell SE, O’Neill RJ (2012) Making a long story short: noncoding RNAs and chromosome change. Heredity (Edinb) 108(1):42–49. doi:10.1038/hdy.2011.10410.1038 hdy2011104 [pii]
Carbone L, Harris RA, Gnerre S, Veeramah KR, Lorente-Galdos B, Huddleston J, Meyer TJ, Herrero J, Roos C, Aken B, Anaclerio F, Archidiacono N, Baker C, Barrell D, Batzer MA, Beal K, Blancher A, Bohrson CL, Brameier M, Campbell MS, Capozzi O, Casola C, Chiatante G, Cree A, Damert A, de Jong PJ, Dumas L, Fernandez-Callejo M, Flicek P, Fuchs NV, Gut I, Gut M, Hahn MW, Hernandez-Rodriguez J, Hillier LW, Hubley R, Ianc B, Izsvak Z, Jablonski NG, Johnstone LM, Karimpour-Fard A, Konkel MK, Kostka D, Lazar NH, Lee SL, Lewis LR, Liu Y, Locke DP, Mallick S, Mendez FL, Muffato M, Nazareth LV, Nevonen KA, O'Bleness M, Ochis C, Odom DT, Pollard KS, Quilez J, Reich D, Rocchi M, Schumann GG, Searle S, Sikela JM, Skollar G, Smit A, Sonmez K, ten Hallers B, Terhune E, Thomas GW, Ullmer B, Ventura M, Walker JA, Wall JD, Walter L, Ward MC, Wheelan SJ, Whelan CW, White S, Wilhelm LJ, Woerner AE, Yandell M, Zhu B, Hammer MF, Marques-Bonet T, Eichler EE, Fulton L, Fronick C, Muzny DM, Warren WC, Worley KC, Rogers J, Wilson RK, Gibbs RA (2014) Gibbon genome and the fast karyotype evolution of small apes. Nature 513(7517):195–201. doi:10.1038/nature13679
Carmena M, Wheelock M, Funabiki H, Earnshaw WC (2012) The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol 13(12):789–803. doi:10.1038/nrm3474
Carone DM, Longo MS, Ferreri GC, Hall L, Harris M, Shook N, Bulazel KV, Carone BR, Obergfell C, O'Neill MJ, O'Neill RJ (2009) A new class of retroviral and satellite encoded small RNAs emanates from mammalian centromeres. Chromosoma 118(1):113–125. doi:10.1007/s00412-008-0181-5
Carone DM, Zhang C, Hall LE, Obergfell C, Carone BR, O’Neill MJ, O'Neill RJ (2013) Hypermorphic expression of centromeric retroelement-encoded small RNAs impairs CENP-A loading. Chromosome Res 21(1):49–62. doi:10.1007/s10577-013-9337-0
Carroll CW, Milks KJ, Straight AF (2010) Dual recognition of CENP-A nucleosomes is required for centromere assembly. J Cell Biol 189(7):1143–1155. doi:10.1083/jcb.201001013 jcb.201001013 [pii]
Castellano-Pozo M, Santos-Pereira JM, Rondon AG, Barroso S, Andujar E, Perez-Alegre M, Garcia-Muse T, Aguilera A (2013) R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol Cell 52(4):583–590. doi:10.1016/j.molcel.2013.10.006
Catania S, Pidoux AL, Allshire RC (2015) Sequence features and transcriptional stalling within centromere DNA promote establishment of CENP-A chromatin. PLoS Genet 11(3):e1004986. doi:10.1371/journal.pgen.1004986
Chan FL, Marshall OJ, Saffery R, Won Kim B, Earle E, Choo KH, Wong LH (2012) Active transcription and essential role of RNA polymerase II at the centromere during mitosis. Proc Natl Acad Sci USA 109(6):1979–1984. doi:10.1073/pnas.1108705109 1108705109 [pii]
Chen CC, Mellone BG (2016) Chromatin assembly: journey to the CENter of the chromosome. J Cell Biol 214(1):13–24. doi:10.1083/jcb.201605005
Chen ES, Zhang K, Nicolas E, Cam HP, Zofall M, Grewal SI (2008) Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature 451(7179):734–737. doi:10.1038/nature06561
Chen CC, Bowers S, Lipinszki Z, Palladino J, Trusiak S, Bettini E, Rosin L, Przewloka MR, Glover DM, O'Neill RJ, Mellone BG (2015) Establishment of centromeric chromatin by the CENP-A assembly factor CAL1 requires FACT-mediated transcription. Dev Cell 34(1):73–84. doi:10.1016/j.devcel.2015.05.012
Choi ES, Stralfors A, Castillo AG, Durand-Dubief M, Ekwall K, Allshire RC (2011) Identification of noncoding transcripts from within CENP-A chromatin at fission yeast centromeres. J Biol Chem 286(26):23600–23607
Choo K (1997) The centromere. Oxford University Press, Oxford
Chueh AC, Northrop EL, Brettingham-Moore KH, Choo KH, Wong LH (2009) LINE retrotransposon RNA is an essential structural and functional epigenetic component of a core neocentromeric chromatin. PLoS Genet 5(1):e1000354. doi:10.1371/journal.pgen.1000354
Cohen RL, Espelin CW, De Wulf P, Sorger PK, Harrison SC, Simons KT (2008) Structural and functional dissection of Mif2p, a conserved DNA-binding kinetochore protein. Mol Biol Cell 19(10):4480–4491. doi:10.1091/mbc.E08-03-0297
Copenhaver GP, Nickel K, Kuromori T, Benito MI, Kaul S, Lin X, Bevan M, Murphy G, Harris B, Parnell LD, McCombie WR, Martienssen RA, Marra M, Preuss D (1999) Genetic definition and sequence analysis of Arabidopsis centromeres. Science 286(5449):2468–2474
Dawe RK (2003) RNA interference, transposons, and the centromere. Plant Cell 15(2):297–301
Deyter GM, Biggins S (2014) The FACT complex interacts with the E3 ubiquitin ligase Psh1 to prevent ectopic localization of CENP-A. Genes Dev 28(16):1815–1826. doi:10.1101/gad.243113.114
Dover GA, Strachan T, Coen ES, Brown SD (1982) Molecular drive. Science 218(4577):1069
Du Y, Topp CN, Dawe RK (2010) DNA binding of centromere protein C (CENPC) is stabilized by single-stranded RNA. PLoS Genet 6(2):e1000835. doi:10.1371/journal.pgen.1000835
Dunleavy EM, Pidoux AL, Monet M, Bonilla C, Richardson W, Hamilton GL, Ekwall K, McLaughlin PJ, Allshire RC (2007) A NASP (N1/N2)-related protein, Sim3, binds CENP-A and is required for its deposition at fission yeast centromeres. Mol Cell 28(6):1029–1044
Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray-Gallet D, Nakamura Y, Daigo Y, Nakatani Y, Almouzni-Pettinotti G (2009) HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell 137(3):485–497. doi:10.1016/j.cell.2009.02.040 S0092-8674(09)00254-2 [pii]
Dunleavy EM, Almouzni G, Karpen GH (2011) H3.3 is deposited at centromeres in S phase as a placeholder for newly assembled CENP-A in G(1) phase. Nucleus 2(2):146–157. doi:10.4161/nucl.2.2.15211
Edwards NS, Murray AW (2005) Identification of Xenopus CENP-A and an associated centromeric DNA repeat. Mol Biol Cell 16(4):1800–1810. doi:10.1091/mbc.E04-09-0788
Eymery A, Callanan M, Vourc’h C (2009) The secret message of heterochromatin: new insights into the mechanisms and function of centromeric and pericentric repeat sequence transcription. Int J Dev Biol 53(2–3):259–268. doi:10.1387/ijdb.082673ae
Ferri F, Bouzinba-Segard H, Velasco G, Hube F, Francastel C (2009) Non-coding murine centromeric transcripts associate with and potentiate Aurora B kinase. Nucleic Acids Res 37(15):5071–5080. doi:10.1093/nar/gkp529
Fishel B, Amstutz H, Baum M, Carbon J, Clarke L (1988) Structural organization and functional analysis of centromeric DNA in the fission yeast Schizosaccharomyces pombe. Mol Cell Biol 8(2):754–763
Folco HD, Pidoux AL, Urano T, Allshire RC (2008) Heterochromatin and RNAi are required to establish CENP-A chromatin at centromeres. Science 319(5859):94–97. doi:10.1126/science.1150944 319/5859/94 [pii]
Foltz DR, Jansen LE, Black BE, Bailey AO, Yates JR 3rd, Cleveland DW (2006) The human CENP-A centromeric nucleosome-associated complex. Nat Cell Biol 8(5):458–469
Foltz DR, Jansen LE, Bailey AO, Yates JR 3rd, Bassett EA, Wood S, Black BE, Cleveland DW (2009) Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell 137(3):472–484. doi:10.1016/j.cell.2009.02.039 S0092-8674(09)00253-0 [pii]
Fujita Y, Hayashi T, Kiyomitsu T, Toyoda Y, Kokubu A, Obuse C, Yanagida M (2007) Priming of centromere for CENP-A recruitment by human hMis18alpha, hMis18beta, and M18BP1. Dev Cell 12(1):17–30. doi:10.1016/j.devcel.2006.11.002 S1534-5807(06)00507-7 [pii]
Gent JI, Dawe RK (2012) RNA as a structural and regulatory component of the centromere. Annu Rev Genet 46:443–453. doi:10.1146/annurev-genet-110711-155419
Gopalakrishnan S, Sullivan BA, Trazzi S, Della Valle G, Robertson KD (2009) DNMT3B interacts with constitutive centromere protein CENP-C to modulate DNA methylation and the histone code at centromeric regions. Hum Mol Genet 18(17):3178–3193
Gottesfeld JM, Forbes DJ (1997) Mitotic repression of the transcriptional machinery. Trends Biochem Sci 22(6):197–202
Grenfell AW, Heald R, Strzelecka M (2016) Mitotic noncoding RNA processing promotes kinetochore and spindle assembly in Xenopus. J Cell Biol 214(2):133–141. doi:10.1083/jcb.201604029
Grimes BR, Monaco ZL (2005) Artificial and engineered chromosomes: developments and prospects for gene therapy. Chromosoma 114(4):230–241
Hall LE, Mitchell SE, O’Neill RJ (2012) Pericentric and centromeric transcription: a perfect balance required. Chromosome Res 20(5):535–546. doi:10.1007/s10577-012-9297-9
Harrington JJ, Van Bokkelen G, Mays RW, Gustashaw K, Willard HF (1997) Formation of de novo centromeres and construction of first-generation human artificial microchromosomes. Nat Genet 15(4):345–355
Hasson D, Panchenko T, Salimian KJ, Salman MU, Sekulic N, Alonso A, Warburton PE, Black BE (2013) The octamer is the major form of CENP-A nucleosomes at human centromeres. Nat Struct Mol Biol 20(6):687–695. doi:10.1038/nsmb.2562
Henikoff S, Ahmad K, Malik H (2001) The centromere paradox: stable inheritance with rapidly evolving DNA. Science 293(5532):1098–1102. doi:10.1126/science.1062939
Herrera-Moyano E, Mergui X, Garcia-Rubio ML, Barroso S, Aguilera A (2014) The yeast and human FACT chromatin-reorganizing complexes solve R-loop-mediated transcription-replication conflicts. Genes Dev 28(7):735–748. doi:10.1101/gad.234070.113
Holland AJ, Cleveland DW (2009) Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol 10(7):478–487. doi:10.1038/nrm2718
Hondele M, Ladurner AG (2013) Catch me if you can: how the histone chaperone FACT capitalizes on nucleosome breathing. Nucleus 4(6):443–449. doi:10.4161/nucl.27235
Hosouchi T, Kumekawa N, Tsuruoka H, Kotani H (2002) Physical map-based sizes of the centromeric regions of Arabidopsis thaliana chromosomes 1, 2, and 3. DNA Res 9(4):117–121
Ideue T, Cho Y, Nishimura K, Tani T (2014) Involvement of satellite I noncoding RNA in regulation of chromosome segregation. Genes Cells 19(6):528–538. doi:10.1111/gtc.12149
Ikeno M, Grimes B, Okazaki T, Nakano M, Saitoh K, Hoshino H, McGill NI, Cooke H, Masumoto H (1998) Construction of YAC-based mammalian artificial chromosomes. Nat Biotechnol 16(5):431–439
Jambhekar A, Emerman AB, Schweidenback CT, Blower MD (2014) RNA stimulates Aurora B kinase activity during mitosis. PLoS ONE 9(6):e100748. doi:10.1371/journal.pone.0100748
Jansen L, Black B, Foltz D, Cleveland D (2007) Propagation of centromeric chromatin requires exit from mitosis. J Cell Biol 176(6):795–805. doi:10.1083/jcb.200701066
Kanellopoulou C, Muljo S, Kung A, Ganesan S, Drapkin R, Jenuwein T, Livingston D, Rajewsky K (2005) Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev 19(4):489–501. doi:10.1101/gad.1248505
Karpen GH, Allshire RC (1997) The case for epigenetic effects on centromere identity and function. Trends Genet 13(12):489–496
Koo DH, Zhao H, Jiang J (2016) Chromatin-associated transcripts of tandemly repetitive DNA sequences revealed by RNA-FISH. Chromosome Res. doi:10.1007/s10577-016-9537-5
Kumekawa N, Hosouchi T, Tsuruoka H, Kotani H (2000) The size and sequence organization of the centromeric region of arabidopsis thaliana chromosome 5. DNA Res 7(6):315–321
Lam AL, Boivin CD, Bonney CF, Rudd MK, Sullivan BA (2006) Human centromeric chromatin is a dynamic chromosomal domain that can spread over noncentromeric DNA. Proc Nat Acad Sci USA 103(11):4186–4191
Lampson MA, Cheeseman IM (2011) Sensing centromere tension: Aurora B and the regulation of kinetochore function. Trends Cell Biol 21(3):133–140. doi:10.1016/j.tcb.2010.10.007
Leung W, Shaffer CD, Reed LK, Smith ST, Barshop W, Dirkes W, Dothager M, Lee P, Wong J, Xiong D, Yuan H, Bedard JE, Machone JF, Patterson SD, Price AL, Turner BA, Robic S, Luippold EK, McCartha SR, Walji TA, Walker CA, Saville K, Abrams MK, Armstrong AR, Armstrong W, Bailey RJ, Barberi CR, Beck LR, Blaker AL, Blunden CE, Brand JP, Brock EJ, Brooks DW, Brown M, Butzler SC, Clark EM, Clark NB, Collins AA, Cotteleer RJ, Cullimore PR, Dawson SG, Docking CT, Dorsett SL, Dougherty GA, Downey KA, Drake AP, Earl EK, Floyd TG, Forsyth JD, Foust JD, Franchi SL, Geary JF, Hanson CK, Harding TS, Harris CB, Heckman JM, Holderness HL, Howey NA, Jacobs DA, Jewell ES, Kaisler M, Karaska EA, Kehoe JL, Koaches HC, Koehler J, Koenig D, Kujawski AJ, Kus JE, Lammers JA, Leads RR, Leatherman EC, Lippert RN, Messenger GS, Morrow AT, Newcomb V, Plasman HJ, Potocny SJ, Powers MK, Reem RM, Rennhack JP, Reynolds KR, Reynolds LA, Rhee DK, Rivard AB, Ronk AJ, Rooney MB, Rubin LS, Salbert LR, Saluja RK, Schauder T, Schneiter AR, Schulz RW, Smith KE, Spencer S, Swanson BR, Tache MA, Tewilliager AA, Tilot AK, VanEck E, Villerot MM, Vylonis MB, Watson DT, Wurzler JA, Wysocki LM, Yalamanchili M, Zaborowicz MA, Emerson JA, Ortiz C, Deuschle FJ, DiLorenzo LA, Goeller KL, Macchi CR, Muller SE, Pasierb BD, Sable JE, Tucci JM, Tynon M, Dunbar DA, Beken LH, Conturso AC, Danner BL, DeMichele GA, Gonzales JA, Hammond MS, Kelley CV, Kelly EA, Kulich D, Mageeney CM, McCabe NL, Newman AM, Spaeder LA, Tumminello RA, Revie D, Benson JM, Cristostomo MC, DaSilva PA, Harker KS, Jarrell JN, Jimenez LA, Katz BM, Kennedy WR, Kolibas KS, LeBlanc MT, Nguyen TT, Nicolas DS, Patao MD, Patao SM, Rupley BJ, Sessions BJ, Weaver JA, Goodman AL, Alvendia EL, Baldassari SM, Brown AS, Chase IO, Chen M, Chiang S, Cromwell AB, Custer AF, DiTommaso TM, El-Adaimi J, Goscinski NC, Grove RA, Gutierrez N, Harnoto RS, Hedeen H, Hong EL, Hopkins BL, Huerta VF, Khoshabian C, LaForge KM, Lee CT, Lewis BM, Lydon AM, Maniaci BJ, Mitchell RD, Morlock EV, Morris WM, Naik P, Olson NC, Osterloh JM, Perez MA, Presley JD, Randazzo MJ, Regan MK, Rossi FG, Smith MA, Soliterman EA, Sparks CJ, Tran DL, Wan T, Welker AA, Wong JN, Sreenivasan A, Youngblom J, Adams A, Alldredge J, Bryant A, Carranza D, Cifelli A, Coulson K, Debow C, Delacruz N, Emerson C, Farrar C, Foret D, Garibay E, Gooch J, Heslop M, Kaur S, Khan A, Kim V, Lamb T, Lindbeck P, Lucas G, Macias E, Martiniuc D, Mayorga L, Medina J, Membreno N, Messiah S, Neufeld L, Nguyen SF, Nichols Z, Odisho G, Peterson D, Rodela L, Rodriguez P, Rodriguez V, Ruiz J, Sherrill W, Silva V, Sparks J, Statton G, Townsend A, Valdez I, Waters M, Westphal K, Winkler S, Zumkehr J, DeJong RJ, Hoogewerf AJ, Ackerman CM, Armistead IO, Baatenburg L, Borr MJ, Brouwer LK, Burkhart BJ, Bushhouse KT, Cesko L, Choi TY, Cohen H, Damsteegt AM, Darusz JM, Dauphin CM, Davis YP, Diekema EJ, Drewry M, Eisen ME, Faber HM, Faber KJ, Feenstra E, Felzer-Kim IT, Hammond BL, Hendriksma J, Herrold MR, Hilbrands JA, Howell EJ, Jelgerhuis SA, Jelsema TR, Johnson BK, Jones KK, Kim A, Kooienga RD, Menyes EE, Nollet EA, Plescher BE, Rios L, Rose JL, Schepers AJ, Scott G, Smith JR, Sterling AM, Tenney JC, Uitvlugt C, VanDyken RE, VanderVennen M, Vue S, Kokan NP, Agbley K, Boham SK, Broomfield D, Chapman K, Dobbe A, Dobbe I, Harrington W, Ibrahem M, Kennedy A, Koplinsky CA, Kubricky C, Ladzekpo D, Pattison C, Ramirez RE Jr, Wande L, Woehlke S, Wawersik M, Kiernan E, Thompson JS, Banker R, Bartling JR, Bhatiya CI, Boudoures AL, Christiansen L, Fosselman DS, French KM, Gill IS, Havill JT, Johnson JL, Keny LJ, Kerber JM, Klett BM, Kufel CN, May FJ, Mecoli JP, Merry CR, Meyer LR, Miller EG, Mullen GJ, Palozola KC, Pfeil JJ, Thomas JG, Verbofsky EM, Spana EP, Agarwalla A, Chapman J, Chlebina B, Chong I, Falk IN, Fitzgibbons JD, Friedman H, Ighile O, Kim AJ, Knouse KA, Kung F, Mammo D, Ng CL, Nikam VS, Norton D, Pham P, Polk JW, Prasad S, Rankin H, Ratliff CD, Scala V, Schwartz NU, Shuen JA, Xu A, Xu TQ, Zhang Y, Rosenwald AG, Burg MG, Adams SJ, Baker M, Botsford B, Brinkley B, Brown C, Emiah S, Enoch E, Gier C, Greenwell A, Hoogenboom L, Matthews JE, McDonald M, Mercer A, Monsma N, Ostby K, Ramic A, Shallman D, Simon M, Spencer E, Tomkins T, Wendland P, Wylie A, Wolyniak MJ, Robertson GM, Smith SI, DiAngelo JR, Sassu ED, Bhalla SC, Sharif KA, Choeying T, Macias JS, Sanusi F, Torchon K, Bednarski AE, Alvarez CJ, Davis KC, Dunham CA, Grantham AJ, Hare AN, Schottler J, Scott ZW, Kuleck GA, Yu NS, Kaehler MM, Jipp J, Overvoorde PJ, Shoop E, Cyrankowski O, Hoover B, Kusner M, Lin D, Martinov T, Misch J, Salzman G, Schiedermayer H, Snavely M, Zarrasola S, Parrish S, Baker A, Beckett A, Belella C, Bryant J, Conrad T, Fearnow A, Gomez C, Herbstsomer RA, Hirsch S, Johnson C, Jones M, Kabaso R, Lemmon E, Vieira CM, McFarland D, McLaughlin C, Morgan A, Musokotwane S, Neutzling W, Nietmann J, Paluskievicz C, Penn J, Peoples E, Pozmanter C, Reed E, Rigby N, Schmidt L, Shelton M, Shuford R, Tirasawasdichai T, Undem B, Urick D, Vondy K, Yarrington B, Eckdahl TT, Poet JL, Allen AB, Anderson JE, Barnett JM, Baumgardner JS, Brown AD, Carney JE, Chavez RA, Christgen SL, Christie JS, Clary AN, Conn MA, Cooper KM, Crowley MJ, Crowley ST, Doty JS, Dow BA, Edwards CR, Elder DD, Fanning JP, Janssen BM, Lambright AK, Lane CE, Limle AB, Mazur T, McCracken MR, McDonough AM, Melton AD, Minnick PJ, Musick AE, Newhart WH, Noynaert JW, Ogden BJ, Sandusky MW, Schmuecker SM, Shipman AL, Smith AL, Thomsen KM, Unzicker MR, Vernon WB, Winn WW, Woyski DS, Zhu X, Du C, Ament C, Aso S, Bisogno LS, Caronna J, Fefelova N, Lopez L, Malkowitz L, Marra J, Menillo D, Obiorah I, Onsarigo EN, Primus S, Soos M, Tare A, Zidan A, Jones CJ, Aronhalt T, Bellush JM, Burke C, DeFazio S, Does BR, Johnson TD, Keysock N, Knudsen NH, Messler J, Myirski K, Rekai JL, Rempe RM, Salgado MS, Stagaard E, Starcher JR, Waggoner AW, Yemelyanova AK, Hark AT, Bertolet A, Kuschner CE, Parry K, Quach M, Shantzer L, Shaw ME, Smith MA, Glenn O, Mason P, Williams C, Key SC, Henry TC, Johnson AG, White JX, Haberman A, Asinof S, Drumm K, Freeburg T, Safa N, Schultz D, Shevin Y, Svoronos P, Vuong T, Wellinghoff J, Hoopes LL, Chau KM, Ward A, Regisford EG, Augustine L, Davis-Reyes B, Echendu V, Hales J, Ibarra S, Johnson L, Ovu S, Braverman JM, Bahr TJ, Caesar NM, Campana C, Cassidy DW, Cognetti PA, English JD, Fadus MC, Fick CN, Freda PJ, Hennessy BM, Hockenberger K, Jones JK, King JE, Knob CR, Kraftmann KJ, Li L, Lupey LN, Minniti CJ, Minton TF, Moran JV, Mudumbi K, Nordman EC, Puetz WJ, Robinson LM, Rose TJ, Sweeney EP, Timko AS, Paetkau DW, Eisler HL, Aldrup ME, Bodenberg JM, Cole MG, Deranek KM, DeShetler M, Dowd RM, Eckardt AK, Ehret SC, Fese J, Garrett AD, Kammrath A, Kappes ML, Light MR, Meier AC, O’Rouke A, Perella M, Ramsey K, Ramthun JR, Reilly MT, Robinett D, Rossi NL, Schueler MG, Shoemaker E, Starkey KM, Vetor A, Vrable A, Chandrasekaran V, Beck C, Hatfield KR, Herrick DA, Khoury CB, Lea C, Louie CA, Lowell SM, Reynolds TJ, Schibler J, Scoma AH, Smith-Gee MT, Tuberty S, Smith CD, Lopilato JE, Hauke J, Roecklein-Canfield JA, Corrielus M, Gilman H, Intriago S, Maffa A, Rauf SA, Thistle K, Trieu M, Winters J, Yang B, Hauser CR, Abusheikh T, Ashrawi Y, Benitez P, Boudreaux LR, Bourland M, Chavez M, Cruz S, Elliott G, Farek JR, Flohr S, Flores AH, Friedrichs C, Fusco Z, Goodwin Z, Helmreich E, Kiley J, Knepper JM, Langner C, Martinez M, Mendoza C, Naik M, Ochoa A, Ragland N, Raimey E, Rathore S, Reza E, Sadovsky G, Seydoux MI, Smith JE, Unruh AK, Velasquez V, Wolski MW, Gosser Y, Govind S, Clarke-Medley N, Guadron L, Lau D, Lu A, Mazzeo C, Meghdari M, Ng S, Pamnani B, Plante O, Shum YK, Song R, Johnson DE, Abdelnabi M, Archambault A, Chamma N, Gaur S, Hammett D, Kandahari A, Khayrullina G, Kumar S, Lawrence S, Madden N, Mandelbaum M, Milnthorp H, Mohini S, Patel R, Peacock SJ, Perling E, Quintana A, Rahimi M, Ramirez K, Singhal R, Weeks C, Wong T, Gillis AT, Moore ZD, Savell CD, Watson R, Mel SF, Anilkumar AA, Bilinski P, Castillo R, Closser M, Cruz NM, Dai T, Garbagnati GF, Horton LS, Kim D, Lau JH, Liu JZ, Mach SD, Phan TA, Ren Y, Stapleton KE, Strelitz JM, Sunjed R, Stamm J, Anderson MC, Bonifield BG, Coomes D, Dillman A, Durchholz EJ, Fafara-Thompson AE, Gross MJ, Gygi AM, Jackson LE, Johnson A, Kocsisova Z, Manghelli JL, McNeil K, Murillo M, Naylor KL, Neely J, Ogawa EE, Rich A, Rogers A, Spencer JD, Stemler KM, Throm AA, Van Camp M, Weihbrecht K, Wiles TA, Williams MA, Williams M, Zoll K, Bailey C, Zhou L, Balthaser DM, Bashiri A, Bower ME, Florian KA, Ghavam N, Greiner-Sosanko ES, Karim H, Mullen VW, Pelchen CE, Yenerall PM, Zhang J, Rubin MR, Arias-Mejias SM, Bermudez-Capo AG, Bernal-Vega GV, Colon-Vazquez M, Flores-Vazquez A, Gines-Rosario M, Llavona-Cartagena IG, Martinez-Rodriguez JO, Ortiz-Fuentes L, Perez-Colomba EO, Perez-Otero J, Rivera E, Rodriguez-Giron LJ, Santiago-Sanabria AJ, Senquiz-Gonzalez AM, delValle FR, Vargas-Franco D, Velazquez-Soto KI, Zambrana-Burgos JD, Martinez-Cruzado JC, Asencio-Zayas L, Babilonia-Figueroa K, Beauchamp-Perez FD, Belen-Rodriguez J, Bracero-Quinones L, Burgos-Bula AP, Collado-Mendez XA, Colon-Cruz LR, Correa-Muller AI, Crooke-Rosado JL, Cruz-Garcia JM, Defendini-Avila M, Delgado-Peraza FM, Feliciano-Cancela AJ, Gonzalez-Perez VM, Guiblet W, Heredia-Negron A, Hernandez-Muniz J, Irizarry-Gonzalez LN, Laboy-Corales AL, Llaurador-Caraballo GA, Marin-Maldonado F, Marrero-Llerena U, Martell-Martinez HA, Martinez-Traverso IM, Medina-Ortega KN, Mendez-Castellanos SG, Menendez-Serrano KC, Morales-Caraballo CI, Ortiz-DeChoudens S, Ortiz-Ortiz P, Pagan-Torres H, Perez-Afanador D, Quintana-Torres EM, Ramirez-Aponte EG, Riascos-Cuero C, Rivera-Llovet MS, Rivera-Pagan IT, Rivera-Vicens RE, Robles-Juarbe F, Rodriguez-Bonilla L, Rodriguez-Echevarria BO, Rodriguez-Garcia PM, Rodriguez-Laboy AE, Rodriguez-Santiago S, Rojas-Vargas ML, Rubio-Marrero EN, Santiago-Colon A, Santiago-Ortiz JL, Santos-Ramos CE, Serrano-Gonzalez J, Tamayo-Figueroa AM, Tascon-Penaranda EP, Torres-Castillo JL, Valentin-Feliciano NA, Valentin-Feliciano YM, Vargas-Barreto NM, Velez-Vazquez M, Vilanova-Velez LR, Zambrana-Echevarria C, MacKinnon C, Chung HM, Kay C, Pinto A, Kopp OR, Burkhardt J, Harward C, Allen R, Bhat P, Chang JH, Chen Y, Chesley C, Cohn D, DuPuis D, Fasano M, Fazzio N, Gavinski K, Gebreyesus H, Giarla T, Gostelow M, Greenstein R, Gunasinghe H, Hanson C, Hay A, He TJ, Homa K, Howe R, Howenstein J, Huang H, Khatri A, Kim YL, Knowles O, Kong S, Krock R, Kroll M, Kuhn J, Kwong M, Lee B, Lee R, Levine K, Li Y, Liu B, Liu L, Liu M, Lousararian A, Ma J, Mallya A, Manchee C, Marcus J, McDaniel S, Miller ML, Molleston JM, Diez CM, Ng P, Ngai N, Nguyen H, Nylander A, Pollack J, Rastogi S, Reddy H, Regenold N, Sarezky J, Schultz M, Shim J, Skorupa T, Smith K, Spencer SJ, Srikanth P, Stancu G, Stein AP, Strother M, Sudmeier L, Sun M, Sundaram V, Tazudeen N, Tseng A, Tzeng A, Venkat R, Venkataram S, Waldman L, Wang T, Yang H, Yu JY, Zheng Y, Preuss ML, Garcia A, Juergens M, Morris RW, Nagengast AA, Azarewicz J, Carr TJ, Chichearo N, Colgan M, Donegan M, Gardner B, Kolba N, Krumm JL, Lytle S, MacMillian L, Miller M, Montgomery A, Moretti A, Offenbacker B, Polen M, Toth J, Woytanowski J, Kadlec L, Crawford J, Spratt ML, Adams AL, Barnard BK, Cheramie MN, Eime AM, Golden KL, Hawkins AP, Hill JE, Kampmeier JA, Kern CD, Magnuson EE, Miller AR, Morrow CM, Peairs JC, Pickett GL, Popelka SA, Scott AJ, Teepe EJ, TerMeer KA, Watchinski CA, Watson LA, Weber RE, Woodard KA, Barnard DC, Appiah I, Giddens MM, McNeil GP, Adebayo A, Bagaeva K, Chinwong J, Dol C, George E, Haltaufderhyde K, Haye J, Kaur M, Semon M, Serjanov D, Toorie A, Wilson C, Riddle NC, Buhler J, Mardis ER, Elgin SC (2015) Drosophila muller f elements maintain a distinct set of genomic properties over 40 million years of evolution. G3 (Bethesda) 5 (5):719–740. doi:10.1534/g3.114.015966
Liu H (2016) Insights into centromeric transcription in mitosis. Transcription 7(1):21–25. doi:10.1080/21541264.2015.1127315
Liu H, Qu Q, Warrington R, Rice A, Cheng N, Yu H (2015) Mitotic transcription installs Sgo1 at centromeres to coordinate chromosome segregation. Mol Cell 59(3):426–436. doi:10.1016/j.molcel.2015.06.018
Lo AW, Craig JM, Saffery R, Kalitsis P, Irvine DV, Earle E, Magliano DJ, Choo KH (2001a) A 330 kb CENP-A binding domain and altered replication timing at a human neocentromere. EMBO J 20(8):2087–2096
Lo AW, Magliano DJ, Sibson MC, Kalitsis P, Craig JM, Choo KH (2001b) A novel chromatin immunoprecipitation and array (CIA) analysis identifies a 460-kb CENP-A-binding neocentromere DNA. Genome Res 11(3):448–457
Locke DP, Hillier LW, Warren WC, Worley KC, Nazareth LV, Muzny DM, Yang SP, Wang Z, Chinwalla AT, Minx P, Mitreva M, Cook L, Delehaunty KD, Fronick C, Schmidt H, Fulton LA, Fulton RS, Nelson JO, Magrini V, Pohl C, Graves TA, Markovic C, Cree A, Dinh HH, Hume J, Kovar CL, Fowler GR, Lunter G, Meader S, Heger A, Ponting CP, Marques-Bonet T, Alkan C, Chen L, Cheng Z, Kidd JM, Eichler EE, White S, Searle S, Vilella AJ, Chen Y, Flicek P, Ma J, Raney B, Suh B, Burhans R, Herrero J, Haussler D, Faria R, Fernando O, Darre F, Farre D, Gazave E, Oliva M, Navarro A, Roberto R, Capozzi O, Archidiacono N, Della Valle G, Purgato S, Rocchi M, Konkel MK, Walker JA, Ullmer B, Batzer MA, Smit AF, Hubley R, Casola C, Schrider DR, Hahn MW, Quesada V, Puente XS, Ordonez GR, Lopez-Otin C, Vinar T, Brejova B, Ratan A, Harris RS, Miller W, Kosiol C, Lawson HA, Taliwal V, Martins AL, Siepel A, Roychoudhury A, Ma X, Degenhardt J, Bustamante CD, Gutenkunst RN, Mailund T, Dutheil JY, Hobolth A, Schierup MH, Ryder OA, Yoshinaga Y, de Jong PJ, Weinstock GM, Rogers J, Mardis ER, Gibbs RA, Wilson RK (2011) Comparative and demographic analysis of orang-utan genomes. Nature 469(7331):529–533. doi:10.1038/nature09687
Logsdon GA, Barrey EJ, Bassett EA, DeNizio JE, Guo LY, Panchenko T, Dawicki-McKenna JM, Heun P, Black BE (2015) Both tails and the centromere targeting domain of CENP-A are required for centromere establishment. J Cell Biol 208(5):521–531. doi:10.1083/jcb.201412011
Lu J, Gilbert DM (2007) Proliferation-dependent and cell cycle regulated transcription of mouse pericentric heterochromatin. J Cell Biol 179(3):411–421
Macas J, Koblizkova A, Navratilova A, Neumann P (2009) Hypervariable 3′ UTR region of plant LTR-retrotransposons as a source of novel satellite repeats. Gene 448(2):198–206. doi:10.1016/j.gene.2009.06.014
MacRae IJ, Zhou K, Doudna JA (2007) Structural determinants of RNA recognition and cleavage by Dicer. Nat Struct Mol Biol 14(10):934–940. doi:10.1038/nsmb1293
Malik HS, Henikoff S (2001) Adaptive evolution of Cid, a centromere-specific histone in Drosophila. Genetics 157(3):1293–1298
Malik H, Henikoff S (2009) Major evolutionary transitions in centromere complexity. Cell 138(6):1067–1082
Maloney KA, Sullivan LL, Matheny JE, Strome ED, Merrett SL, Ferris A, Sullivan BA (2012) Functional epialleles at an endogenous human centromere. Proc Natl Acad Sci USA 109(34):13704–13709. doi:10.1073/pnas.1203126109
Mejía J (2002) Efficiency of de novo centromere formation in human artificial chromosomes. Genomics 79(3):297–304. doi:10.1006/geno.2002.6704
Mellone BG, Zhang W, Karpen GH (2009) Frodos found: behold the CENP-a “Ring” bearers. Cell 137(3):409–412. doi:10.1016/j.cell.2009.04.035 S0092-8674(09)00461-9 [pii]
Melters DP, Bradnam KR, Young HA, Telis N, May MR, Ruby JG, Sebra R, Peluso P, Eid J, Rank D, Garcia JF, DeRisi JL, Smith T, Tobias C, Ross-Ibarra J, Korf I, Chan SW (2013) Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol 14(1):R10. doi:10.1186/gb-2013-14-1-r10
Meluh PB, Yang P, Glowczewski L, Koshland D, Smith MM (1998) Cse4p is a component of the core centromere of Saccharomyces cerevisiae. Cell 94(5):607–613
Miga KH (2015) Completing the human genome: the progress and challenge of satellite DNA assembly. Chromosome Res. doi:10.1007/s10577-015-9488-2
Miga KH, Newton Y, Jain M, Altemose N, Willard HF, Kent WJ (2014) Centromere reference models for human chromosomes X and Y satellite arrays. Genome Res 24(4):697–707. doi:10.1101/gr.159624.113
Miga KH, Eisenhart C, Kent WJ (2015) Utilizing mapping targets of sequences underrepresented in the reference assembly to reduce false positive alignments. Nucleic Acids Res. doi:10.1093/nar/gkv671
Molina O, Vargiu G, Abad MA, Zhiteneva A, Jeyaprakash AA, Masumoto H, Kouprina N, Larionov V, Earnshaw WC (2016) Epigenetic engineering reveals a balance between histone modifications and transcription in kinetochore maintenance. Nat Commun 7:13334. doi:10.1038/ncomms13334
Muchardt C, Guilleme M, Seeler JS, Trouche D, Dejean A, Yaniv M (2002) Coordinated methyl and RNA binding is required for heterochromatin localization of mammalian HP1alpha. EMBO Rep 3(10):975–981. doi:10.1093/embo-reports/kvf194 kvf194 [pii]
Nagaki K, Cheng Z, Ouyang S, Talbert PB, Kim M, Jones KM, Henikoff S, Buell CR, Jiang J (2004) Sequencing of a rice centromere uncovers active genes. Nat Genet 36(2):138–145
Nakano M, Okamoto Y, Ohzeki J, Masumoto H (2003) Epigenetic assembly of centromeric chromatin at ectopic alpha-satellite sites on human chromosomes. J Cell Sci 116(Pt 19):4021–4034
Nakano M, Cardinale S, Noskov VN, Gassmann R, Vagnarelli P, Kandels-Lewis S, Larionov V, Earnshaw WC, Masumoto H (2008) Inactivation of a human kinetochore by specific targeting of chromatin modifiers. Dev Cell 14(4):507–522
Neumann P, Navratilova A, Koblizkova A, Kejnovsky E, Hribova E, Hobza R, Widmer A, Dolezel J, Macas J (2011) Plant centromeric retrotransposons: a structural and cytogenetic perspective. Mob DNA 2(1):4. doi:10.1186/1759-8753-2-4
Oestergaard VH, Lisby M (2016) Transcription-replication conflicts at chromosomal fragile sites-consequences in M phase and beyond. Chromosoma. doi:10.1007/s00412-016-0617-2
Okada M, Okawa K, Isobe T, Fukagawa T (2009) CENP-H-containing complex facilitates centromere deposition of CENP-A in cooperation with FACT and CHD1. Mol Biol Cell 20(18):3986–3995. doi:10.1091/mbc.E09-01-0065
Okamoto Y, Nakano M, Ohzeki J, Larionov V, Masumoto H (2007) A minimal CENP-A core is required for nucleation and maintenance of a functional human centromere. EMBO J 26(5):1279–1291
O’Neill RJ, Carone DM (2009) The role of ncRNA in centromeres: a lesson from marsupials. Prog Mol Subcell Biol 48:77–101. doi:10.1007/978-3-642-00182-6_4
O’Neill RJ, Eldridge MD, Metcalfe CJ (2004) Centromere dynamics and chromosome evolution in marsupials. J Hered 95(5):375–381. doi:10.1093/jhered/esh063 95/5/375 [pii]
Piras FM, Nergadze SG, Magnani E, Bertoni L, Attolini C, Khoriauli L, Raimondi E, Giulotto E (2010) Uncoupling of satellite DNA and centromeric function in the genus Equus. PLoS Genet 6(2):e1000845. doi:10.1371/journal.pgen.1000845
Plohl M, Meštrović N, Mravinac B (2014) Centromere identity from the DNA point of view. Chromosoma 123(4):313–325. doi:10.1007/s00412-014-0462-0
Pluta AF, Mackay AM, Ainsztein AM, Goldberg IG, Earnshaw WC (1995) The centromere: hub of chromosomal activities. Science 270(5242):1591–1594
Prendergast L, Muller S, Liu Y, Huang H, Dingli F, Loew D, Vassias I, Patel DJ, Sullivan KF, Almouzni G (2016) The CENP-T/-W complex is a binding partner of the histone chaperone FACT. Genes Dev 30(11):1313–1326. doi:10.1101/gad.275073.115
Quenet D, Dalal Y (2014) A long non-coding RNA is required for targeting centromeric protein A to the human centromere. eLife 3:e03254. doi:10.7554/eLife.03254
Reddy K, Tam M, Bowater RP, Barber M, Tomlinson M, Nichol Edamura K, Wang YH, Pearson CE (2011) Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Res 39(5):1749–1762. doi:10.1093/nar/gkq935
Regnier V, Novelli J, Fukagawa T, Vagnarelli P, Brown W (2003) Characterization of chicken CENP-A and comparative sequence analysis of vertebrate centromere-specific histone H3-like proteins. Gene 316:39–46
Renfree MB, Papenfuss AT, Deakin JE, Lindsay J, Heider T, Belov K, Rens W, Waters PD, Pharo EA, Shaw G, Wong ES, Lefevre CM, Nicholas KR, Kuroki Y, Wakefield MJ, Zenger KR, Wang C, Ferguson-Smith M, Nicholas FW, Hickford D, Yu H, Short KR, Siddle HV, Frankenberg SR, Chew KY, Menzies BR, Stringer JM, Suzuki S, Hore TA, Delbridge ML, Mohammadi A, Schneider NY, Hu Y, O'Hara W, Al Nadaf S, Wu C, Feng ZP, Cocks BG, Wang J, Flicek P, Searle SM, Fairley S, Beal K, Herrero J, Carone DM, Suzuki Y, Sagano S, Toyoda A, Sakaki Y, Kondo S, Nishida Y, Tatsumoto S, Mandiou I, Hsu A, McColl KA, Landsell B, Weinstock G, Kuczek E, McGrath A, Wilson P, Men A, Hazar-Rethinam M, Hall A, Davies J, Wood D, Williams S, Sundaravadanam Y, Muzny DM, Jhangiani SN, Lewis LR, Morgan MB, Okwuonu GO, Ruiz SJ, Santibanez J, Nazareth L, Cree A, Fowler G, Kovar CL, Dinh HH, Joshi V, Jing C, Lara F, Thornton R, Chen L, Deng J, Liu Y, Shen JY, Song XZ, Edson J, Troon C, Thomas D, Stephens A, Yapa L, Levchenko T, Gibbs RA, Cooper DW, Speed TP, Fujiyama A, Graves JA, O'Neill RJ, Pask AJ, Forrest SM, Worley KC (2011) Genome sequence of an Australian kangaroo, Macropus eugenii, provides insight into the evolution of mammalian reproduction and development. Genome Biol 12(8):R81. doi:10.1186/gb-2011-12-8-r81 gb-2011-12-8-r81 [pii]
Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh KH, Feizi S, Karlic R, Kim AR, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, De Jager PL, Farnham PJ, Fisher SJ, Haussler D, Jones SJ, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai LH, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M (2015) Integrative analysis of 111 reference human epigenomes. Nature 518(7539):317–330. doi:10.1038/nature14248
Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M, Harte RA, Heitner S, Hickey G, Hinrichs AS, Hubley R, Karolchik D, Learned K, Lee BT, Li CH, Miga KH, Nguyen N, Paten B, Raney BJ, Smit AF, Speir ML, Zweig AS, Haussler D, Kuhn RM, Kent WJ (2015) The UCSC genome browser database: 2015 update. Nucleic Acids Res 43 (Database issue):D670–681. doi:10.1093/nar/gku1177
Rosic S, Erhardt S (2016) No longer a nuisance: long non-coding RNAs join CENP-A in epigenetic centromere regulation. Cell Mol Life Sci 73(7):1387–1398. doi:10.1007/s00018-015-2124-7
Rosic S, Kohler F, Erhardt S (2014) Repetitive centromeric satellite RNA is essential for kinetochore formation and cell division. J Cell Biol 207(3):335–349. doi:10.1083/jcb.201404097
Schueler MG, Higgins AW, Rudd MK, Gustashaw K, Willard HF (2001) Genomic and genetic definition of a functional human centromere. Science 294(5540):109–115
Schueler MG, Swanson W, Thomas PJ, Green ED (2010) Adaptive evolution of foundation kinetochore proteins in primates. Mol Biol Evol 27(7):1585–1597. doi:10.1093/molbev/msq043 msq043 [pii]
Shang WH, Hori T, Toyoda A, Kato J, Popendorf K, Sakakibara Y, Fujiyama A, Fukagawa T (2010) Chickens possess centromeres with both extended tandem repeats and short non-tandem-repetitive sequences. Genome Res 20(9):1219–1228. doi:10.1101/gr.106245.110
Shin C, Manley JL (2002) The SR protein SRp38 represses splicing in M phase cells. Cell 111(3):407–417
Smith KM, Phatale PA, Sullivan CM, Pomraning KR, Freitag M (2011) Heterochromatin is required for normal distribution of Neurospora crassa CenH3. Mol Cell Biol 31(12):2528–2542. doi:10.1128/MCB.01285-10
Smith KM, Galazka JM, Phatale PA, Connolly LR, Freitag M (2012) Centromeres of filamentous fungi. Chromosome Res 20(5):635–656. doi:10.1007/s10577-012-9290-3
Sugimoto K, Kuriyama K, Shibata A, Himeno M (1997) Characterization of internal DNA-binding and C-terminal dimerization domains of human centromere/kinetochore autoantigen CENP-C in vitro: role of DNA-binding and self-associating activities in kinetochore organization. Chromosome Res 5(2):132–141
Sullivan BA, Karpen GH (2004) Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin. Nat Struct Mol Biol 11(11):1076–1083
Takahashi K, Takayama Y, Masuda F, Kobayashi Y, Saitoh S (2005) Two distinct pathways responsible for the loading of CENP-A to centromeres in the fission yeast cell cycle. Philos Trans R Soc Lond B Biol Sci 360 (1455):595–606; discussion 606–597. doi:10.1098/rstb.2004.1614
Takayama Y, Sato H, Saitoh S, Ogiyama Y, Masuda F, Takahashi K (2008) Biphasic incorporation of centromeric histone CENP-A in fission yeast. Mol Biol Cell 19(2):682–690. doi:10.1091/mbc.E07-05-0504
Ting DT, Lipson D, Paul S, Brannigan BW, Akhavanfard S, Coffman EJ, Contino G, Deshpande V, Iafrate AJ, Letovsky S, Rivera MN, Bardeesy N, Maheswaran S, Haber DA (2011) Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science 331(6017):593–596. doi:10.1126/science.1200801
Topp CN, Zhong CX, Dawe RK (2004) Centromere-encoded RNAs are integral components of the maize kinetochore. Proc Nat Acad Sci USA 101(45):15986–15991
Trazzi S, Perini G, Bernardoni R, Zoli M, Reese JC, Musacchio A, Della Valle G (2009) The C-terminal domain of CENP-C displays multiple and critical functions for mammalian centromere formation. PLoS ONE 4(6):e5832. doi:10.1371/journal.pone.0005832
Ugarkovic D (2005) Functional elements residing within satellite DNAs. EMBO Rep 6(11):1035–1039
Volpe TA, Kidner C, Hall IM, Teng G, Grewal SI, Martienssen RA (2002) Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 297(5588):1833–1837. doi:10.1126/science.1074973 1074973 [pii]
Volpe T, Schramke V, Hamilton GL, White SA, Teng G, Martienssen RA, Allshire RC (2003) RNA interference is required for normal centromere function in fission yeast. Chromosome Res 11(2):137–146
Warburton PE, Cooke CA, Bourassa S, Vafa O, Sullivan BA, Stetten G, Gimelli G, Warburton D, Tyler-Smith C, Sullivan KF, Poirier GG, Earnshaw WC (1997) Immunolocalization of CENP-A suggests a distinct nucleosome structure at the inner kinetochore plate of active centromeres. Curr Biol 7(11):901–904
Wong LH, Choo KH (2004) Evolutionary dynamics of transposable elements at the centromere. Trends Genet 20(12):611–616
Wong LH, Brettingham-Moore KH, Chan L, Quach JM, Anderson MA, Northrop EL, Hannan R, Saffery R, Shaw ML, Williams E, Choo KA (2007) Centromere RNA is a key component for the assembly of nucleoproteins at the nucleolus and centromere. Genome Res 17(8):1146–1160. doi:10.1101/gr.6022807
Yan H, Jiang J (2007) Rice as a model for centromere and heterochromatin research. Chromosome Res 15(1):77–84
Yang CH, Tomkiel J, Saitoh H, Johnson DH, Earnshaw WC (1996) Identification of overlapping DNA-binding and centromere-targeting domains in the human kinetochore protein CENP-C. Mol Cell Biol 16(7):3576–3586
Acknowledgements
Sincerest apologies to all of the scientists’ work that could not be covered in this review for lack of space. Thank you to J. Brown, M. O’Neill and B. Mellone for critical review of the manuscript. RJO and ZD are supported by NSF 1613803.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Duda, Z., Trusiak, S., O’Neill, R. (2017). Centromere Transcription: Means and Motive . In: Black, B. (eds) Centromeres and Kinetochores. Progress in Molecular and Subcellular Biology, vol 56. Springer, Cham. https://doi.org/10.1007/978-3-319-58592-5_11
Download citation
DOI: https://doi.org/10.1007/978-3-319-58592-5_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-58591-8
Online ISBN: 978-3-319-58592-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)