
Abstract
Colorectal cancer is one of the most intensively studied cancers with well-documented precursor lesions. The acquisition of genomic instability plays a central role in its development. In the majority of cases, tumor growth results from different combinations of sporadic genetic events and epigenetic alterations, resulting in increased cell proliferation and decreased cell death. Three main pathways have been identified: chromosomal instability (CIN) pathway, microsatellite instability (MSI) pathway, and CpG island hypermethylation phenotype (CIMP) pathway. Within these pathways, somatic BRAF and/or KRAS mutations have been identified as major players. Up to 5% of colorectal cancers develop in the setting of inherited syndromes, such as Lynch syndrome, familial adenomatous polyposis, MUTYH-associated polyposis, and certain hamartomatous polyposis conditions, including Peutz-Jeghers syndrome and juvenile polyposis syndrome. In this chapter, we describe the above-mentioned pathways and syndromes in detail, referring to different molecular events and different precursor lesions. In the last part, we address possible future perspectives in colorectal carcinogenesis.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Familial Adenomatous Polyposis
- Adenomatous Polyposis Coli
- Lynch Syndrome
- KRAS Mutation
- Adenomatous Polyposis Coli Gene
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
4.1 Introduction
Colorectal cancer (CRC) ranks the third most frequent cancer in men (after lung and prostate cancer) and second in women (after breast cancer), representing approximately 9.7% of all new cancer cases diagnosed worldwide [1, 2]. In 2012, an estimated 746.000 men and 614.000 women were diagnosed with CRC, and 694.0000 died of the disease [1, 2]. In the last decade (2001–2010), the global incidence rate decreased by approximately 3% per year [3].
On the molecular level, CRC is one of the most intensively studied cancers. The existence of well-documented precursor lesions indicates multistep cancer development. In fact, this type of cancer represents a very heterogeneous disease regarding the clinical presentation, likelihood of cure, pattern of extension, and response to treatment [4]. The acquisition of genomic instability plays a central role in its development. In the majority of cases, CRC results from different combinations of sporadic genetic events and epigenetic alterations, resulting in increased cell proliferation and decreased cell death [5]. Kindred and twin studies, also studies based on family clusters, estimated that approximately 30% of all CRC cases are inherited [6, 7].
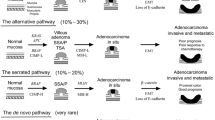
In the last decade, a growing body of scientific evidence demonstrated that different CRC subtypes can be separated based upon combinations of different genetic markers. Three major signaling pathways have been recognized, all characterized by specific precursor lesions, mechanisms of carcinogenesis, and natural history: the chromosomal instability (CIN) pathway, the microsatellite instability (MSI) pathway, and the CpG island hypermethylation phenotype (CIMP) pathway [5, 8] (Fig. 4.1). Within these pathways, somatic BRAF and/or KRAS mutations have been identified as major players [5]. Up to 5% of CRCs develop in the setting of inherited syndromes like Lynch syndrome (LS), familial adenomatous polyposis (FAP), MUTYH-associated polyposis (MAP), and certain hamartomatous polyposis conditions [9].

Three major carcinogenic pathways have been identified in colorectal cancer (CRC): chromosomal instability (CIN), microsatellite instability (MSI), and CpG island methylator phenotype (CIMP)
4.2 Molecular Classification of Colorectal Cancer
In this chapter, we will describe the three major pathways responsible for CRC: chromosomal instability (CIN), microsatellite instability (MSI), and CpG island hypermethylation phenotype (CIMP). We will also refer to the MAP and to hamartomatous polyposis syndrome, such as Peutz-Jeghers syndrome (PJS) and juvenile polyposis syndrome (JPS), and will finally address possible future perspectives.
4.2.1 The Chromosomal Instability (CIN) Pathway
The CIN pathway, also called the “traditional pathway,” is characterized by imbalance in chromosomal number (aneuploidy), subsequent loss of heterozygosity (LOH) of genes, and subchromosomal genetic amplifications [10]. The time of tumor development via this pathway is approximately 10–15 years. The initial lesion in this pathway is the dysplastic aberrant crypt focus (ACF) [11]. It is a microscopic mucosal lesion that develops into conventional adenomas, i.e., tubular adenomas, tubulovillous, and villous adenomas (Fig. 4.2), which are the macroscopically discernable precursor lesions of sporadic CRCs arising via this pathway [12]. It is of note that traditional adenomas are also considered to be the precursor lesions in the hereditary cancers, namely, LS and FAP [12, 13].

Tubular colorectal adenoma with low-grade dysplasia, characterized by well-formed glands and pseudostratified, polarized, hyperchromatic nuclei (a). High-grade dysplasia characterized by increased architectural complexity and more severe atypia with loss of nuclear polarity (b)
Already in 1990, Fearon and Vogelstein [14] proposed a multistep model of sequential genetic alterations, responsible for adenoma and ultimately carcinoma formation within the colorectum (Fig. 4.3). Mutation in the adenomatous polyposis coli (APC) tumor-suppressor gene located on chromosome 5q21 has been identified as the first step of this model [15]. Both copies of the APC gene must be functionally inactivated for adenomas to develop. Specifically, APC mutation interferes with phosphorylation of β-catenin, a component of the Wnt signaling pathway that regulates apoptosis, growth, and differentiation. Consequently, β-catenin is not ubiquitinated and destroyed by the proteasome. It accumulates in the cytosol and is translocated to the nucleus, where it interacts with T-cell factor (TCF)/lymphoid enhancer factor (LEF), converting these effectors into transcriptional activators [16]. Activation of the Wnt pathway is present in up to 80% of adenomas [11].

Multistep genetic model of colorectal carcinogenesis (adenoma-carcinoma sequence): chromosomal instability is observed in benign adenomas and increases in conjunction with tumor progression (from [12], S. Karger AG, with permission)
The second molecular event is an activating mutation of Kirsten-rat sarcoma 2 viral oncogene (KRAS), which is, however, not unique for this pathway. This mutation is found in approximately 45% of CRCs and constitutively activates the MAPK signaling pathway [17]. The genomic change in adenoma-carcinoma sequence also includes LOH of chromosome 18q, which is present in up to 60% of tumors [18]. Many important tumor-suppressor genes are located at 18q21.1—DCC, SMAD2, and SMAD4, the latter being involved in TGF-β signaling, responsible for regulation of growth and apoptosis. Mutational inactivation of the tumor-suppressor TP53 (17p13) occurs as a late event (at the transition from high-grade adenoma to invasive cancer) in up to 80% of CRC [17]. Mutational activation of phosphatidylinositol-4, 5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) occurs also in the late phase, but in a small number of cases [10].
Recognition of the central role of APC mutations in tumorigenesis has improved our understanding of FAP, the second most common inherited CRC syndrome. APC-associated polyposis conditions also include attenuated FAP, Gardner syndrome (FAP with epidermoid cysts, osteomas, dental anomalies, and/or desmoid tumors), and Turcot syndrome (colonic polyps with central nervous system tumors) [9, 19]. FAP is characterized by the development of hundreds to thousands of conventional adenomas beginning in early adolescence (Fig. 4.4). The disease inevitably leads to CRC, thereby prompting prophylactic colectomy. This syndrome accounts for only <1% of all CRCs. The neoplastic polyps are distributed among the colorectum and can also be observed in the stomach and small bowel, in particular the duodenum. Attenuated FAP is a less severe form, characterized by <100 colonic adenomatous polyps with tendency for proximal location. In individuals with attenuated FAP, adenoma and CRC development is delayed by 15 years when compared to classic FAP [20].

Gross presentation of familial adenomatous polyposis (FAP): the colectomy specimen shows numerous adenomatous polyps
4.2.2 Microsatellite Instability (MSI) Pathway and Lynch Syndrome (LS)
Errors that occur during DNA replication are corrected by the mismatch repair (MMR) system, which includes the following proteins: MLH1, PMS2, MSH2, and MSH6. This system is necessary for maintaining genomic stability. During mismatch repair, the MMR proteins form heterodimers, that is, MLH1 builds a complex with PMS2, and MSH2 builds another with MSH6 [21, 22]. It is well known that the MLH1 and MSH2 proteins are the dominant components of their heterodimers. In consequence, mutations in MLH1 or MSH2 gene lead to proteolytic degradation of the corresponding dimer and subsequent loss of both, the main and the auxiliary partner proteins (Fig. 4.5) [23]. If a mutation occurs in one of the auxiliary genes, i.e., PMS2 or MSH6, this results in a loss of the respective PMS2 or MSH6 protein, but does not cause secondary loss of the dominant protein, that is, MLH1 or MSH2 [3].

Mismatch repair (MMR) protein expression in a cancer with high-level microsatellite instability (MSI-H): MLH1 (a) and PMS2 (b) staining is lost in the tumor cell nuclei, while the expression of MSH2 (c) and MSH6 (d) is retained. Nonneoplastic stromal and inflammatory cells serve as internal positive control (serial sections)
When the MMR system does not function properly, the cells accumulate genetic errors. These may happen also in so-called microsatellites, that is, repetitive segments of DNA (two to five nucleotides in length), which are found scattered throughout the genome in the noncoding regions between genes or within genes [24]. MSI is defined as a change of any length of these repeating units, due to deletion or insertion [25].
For MSI testing, different panels of microsatellite markers have been used. The first consensus of the National Cancer Institute (NCI) recommended the use of a panel of five markers for MSI testing [26]. This included two mononucleotide repeats (BAT-25 and BAT-26) and three dinucleotide repeats (D5S346, D2S123, and D17S250) [27]. Other panels are solely based upon mononucleotide repeat markers, which can be amplified and analyzed in a single assay [28, 29]. Tumors with instability in two or more of the five markers qualify for high-level MSI (MSI-H; Fig. 4.6), whereas those with instability at one repeat qualify for low-level MSI (MSI-L). When all markers are stable, the lesion is called microsatellite stable (MSS) [4, 28].

Example of cancer with high-level microsatellite instability (MSI-H): The MSI profile is assessed by a panel of five monomorphic mononucleotide repeats. Instability for all markers is observed, as shown by additional alleles (allelic shifts). Two polymorphic pentanucleotide repeats (Penta C and Penta D) are included for sample identification
Approximately 15% of CRCs are genetically instable due to MSI [30]. The majority of these tumors (80%) are sporadic and arise due to hypermethylation of the MLH1 gene promoter [31]. Other 20% of tumors are inherited, that is, caused by germ line mutation in one of the MMR genes and associated with LS [32]. This syndrome follows an autosomal dominant trait of inheritance and accounts for 2–4% of all CRCs [33, 34]. Specifically, mutations in MLH1 and MSH2 account for most cases (approximately 40% each), while mutations in MSH6 and PMS2 account for only 10% and 5%, respectively [33, 35].
CRCs in LS usually occur at early age (approximately 45 years) and are right-sided (approximately 70% proximal to the splenic flexure) [33]. In addition, they may be multifocal with syn- and/or metachronous tumor development, and there is also a higher risk for extracolonic tumors [36]. These mainly include endometrial, ovary, and gastric tumors [9].
The lifetime risk of cancer in LS is depending on sex and the mutated MMR gene [37,38,39,40,41,42,43,44]. In patients with MLH1 or MSH2 mutation, the risk of CRC has been calculated 27–74% for males and 22–53% for females, respectively, with mean age at diagnosis varying from 27 to 46 years (69 years for sporadic cancers). The risk of endometrium cancer is 14–54% [45]. When MSH6 is mutated, the CRC risk appears to be lower (18%), while the endometrium cancer risk is not changed. Smaller studies reported a lower PMS2 mutation penetrance for CRC and endometrium cancer as compared with MLH1 and MSH2 mutation carriers and similar or even lower risks as compared with MSH6 mutation carriers [46]. A large European cohort recently reported a cumulative risk of CRC of 19% for males and 11% for females, while the risk of endometrium cancer was 12%. In this cohort, the mean age at diagnosis for both CRC and endometrium cancer was higher as compared with MLH1 or MSH2 mutation carriers. When compared with MSH6, the mean age at diagnosis of CRC was lower, and the mean age at diagnosis of endometrium cancer was similar [46].
Several tools are available to assist the clinical diagnosis of LS, including analyses of family history, tumor testing, mutation prediction models, and genetic testing. The Amsterdam criteria were created first in 1990 and then reestablished in 1999 as Amsterdam criteria II defining clinical criteria needed for the diagnosis of HNPCC [45, 47,48,49,50,51]. These criteria include individual patient and family history of colonic and extracolonic tumors. They are listed in Table 4.1.
The revised Bethesda guidelines are a third set of clinicopathologic criteria developed to identify individuals that should be investigated for LS by evaluation of MSI and/or immunohistochemical (IHC) testing (Table 4.2) [52].
Adenomas and CRCs in LS arise earlier and at more proximal location when compared to sporadic neoplasm. The rate of adenoma development is similar to the rate of adenoma development in the sporadic setting, but progression to cancer occurs at increased rate. This is in contrast to FAP, which has an increased rate of adenoma formation, while progression to cancer is believed to occur at a similar rate to that of sporadic adenomas. In LS, the germ line inactivation of one of the mismatch repair genes, coupled with somatic inactivation of the remaining allele, increases the mutation rate and, subsequently, the rate of progression from adenoma to cancer (Fig. 4.7) [12, 53].

Relative effects of germ line mutations on the rate of tumor initiation and progression: In sporadic tumors, adenoma formation and cancer development are rate-limiting steps. In familial adenomatous polyposis, adenoma formation occurs at an increased rate, while adenomas progress to cancer at a rate similar to the sporadic setting. The mutation rate within adenomatous polyps and, subsequently, the rate of progression from adenoma to cancer are increased in Lynch syndrome (from [12], S. Karger AG, with permission)
4.2.3 CpG Island Methylator Phenotype (CIMP) and Serrated Pathway
CpG dinucleotides (cytosine nucleotide followed by a guanine nucleotide) are uncommon in the human genome. However, in the promoter region of about half of all genes, clusters of these nucleotides, called CpG islands, are found [54]. Aberrant (hyper)methylation of CpG-rich promoters leads to epigenetic silencing of tumor-suppressor genes and ultimately cancer. The methylation status of the tumor can be assessed according to the degree of methylation as CIMP high, CIMP low, or CIMP negative [55]. However, molecular analysis of CIMP and classification of methylation level appear to be poorly standardized. Hence, up to date, no precise definition of CIMP and no consensus recommendation are available [3].
Sporadic MSI-H CRCs occur in patients without germ line mutation in a MMR gene. These tumors occur preferably in the right colon. They are diagnosed more commonly in women, often at advanced age [56, 57]. These cancers develop from serrated precursor lesions [31] through the CIMP or “serrated pathway” (Fig. 4.8), characterized by BRAF mutation (characteristically V600E) and hypermethylation in CpG-rich gene promoters, which leads to silencing of distinct tumor-suppressor genes, including the MMR gene MLH1, as well as p16, MGMT, and IGFB7 [58,59,60,61,62].

Colorectal carcinogenesis according to the “serrated (CIMP) pathway”: sporadic colorectal adenocarcinomas with high-level microsatellite instability (MSI-H) develop from serrated precursor lesions due to promoter hypermethylation of the MLH1 gene (from [12], S. Karger AG, with permission)
Sessile serrated adenomas/polyps (SSA/P) are considered to be the main precursor lesions of the serrated pathway. They account for approximately 5–25% of all serrated lesions occurring in the colorectum [13, 63, 64]. They may arise from large microvesicular hyperplastic polyps or develop de novo from normal colonic mucosa. Uncomplicated SSA/Ps do not show dysplasia. Dysplasia may, however, occur during neoplastic progression (Fig. 4.9). There appears to be a histological continuum from non-dysplastic ACF to microvesicular hyperplastic polyps to SSA/P to SSA/P with cytological dysplasia and ultimately to invasive (“serrated”) adenocarcinoma [12].

Sessile serrated adenoma/polyp (SSA/P) with increased serration of non-dysplastic crypts, with T-shaped (“anchor”) crypts and mature goblet cells at the crypt bases (a). Cytological dysplasia is not present in uncomplicated SSA/P, but develops with progression toward carcinoma (b), often in conjunction with promoter hypermethylation of the MLH1 gene, as illustrated by loss of nuclear MLH1 expression (c). The proliferation rate (MIB-1) is markedly increased in the dysplastic glands (d)
Serrated lesions can also occur in familial setting. Serrated polyposis syndrome is a rare condition characterized by multiple and/or large serrated polyps of the colon. Guarinos et al. [65] identified BRAF mutations in 63% and KRAS mutations in 10% of lesions occurring in this syndrome; 43% of the lesions were CIMP high. A single per patient analysis showed that all patients had BRAF or KRAS mutation in more than 25% of the polyps, and 84.8% of patients had a mutation in BRAF or KRAS in more than 50% of their polyps [65]. Germ line loss-of-function mutations in oncogene-induced senescence pathways may play an additional role in the disease [66].
Traditional serrated adenomas (TSAs) are much less common than the other serrated lesions, accounting for approximately 1% of colorectal polyps (Fig. 4.10). The majority of lesions are detected in the distal colon [12]. TSAs may originate from preexisting non-dysplastic serrated polyps, including hyperplastic polyps and SSA/Ps. On the molecular level, these lesions are characterized by BRAF mutations, giving rise to BRAF-mutated MSS CRCs [67, 68]. TSAs may alternatively develop de novo. These lesions mainly show mutations in the KRAS gene. Malignant progression occurs via TP53 mutation and Wnt pathway activation regardless of mutation status [67,68,69].

Traditional serrated adenoma (TSA) with slit-like serration, cytoplasmic eosinophilia, and proliferative “ectopic crypts” (a). In high-grade dysplasia, marked architectural complexity and nuclear atypia with increased nuclear/cytoplasmic ratio are observed (b)
4.2.4 MUTYH-Associated Polyposis (MAP)
MAP is a hereditary condition caused by biallelic germ line mutations in MUTYH gene and has an autosomal recessive pattern of inheritance [70]. It is characterized by the development of multiple neoplastic polyps in the colorectum and increased risk of CRC [9]. The colonic phenotype of MAP mimics FAP—however, in addition to multiple adenomatous polyps, hyperplastic polyps and SSA/Ps can also be found [71].
The MUTYH gene product is involved in the base-excision repair pathway and protects against oxidative DNA damage. Individuals with >10 colorectal adenomas who do not have mutation in APC should undergo genetic testing for MAP [9].
4.2.5 Hamartomatous Polyposis Conditions
Hamartomatous polyposis conditions include PJS, JPS, hereditary mixed polyposis syndrome, Cowden syndrome, Bannayan-Riley-Ruvalcaba syndrome, and Cronkhite-Canada syndrome [72]. This group of disorders is characterized by the development of multiple benign-appearing polyps in the gastrointestinal tract. Affected individuals bear an increased risk of cancer, not only in the gastrointestinal tract but also in other organs [9]. Carcinogenesis, that is, progression of the hamartomatous polyps to cancer or cancer development de novo, is still largely unclear, suggesting different pathways from adenomatous polyposis. In this chapter, we will concentrate on the two most common conditions, that is, PJS and JPS. They can both be sporadic or familial, in the hereditary setting having an autosomal dominant pattern of inheritance.
In PJS the key clinical features are hyperpigmentation (melanosis) of the lips, mouth, and oral mucosa and polyposis of the small intestine. Affected individuals harbor a mutation in the STK11 gene [73]. Lifetime cancer risk is as high as 81–93%, with 50% risk for breast cancer and 39% risk for colon cancer [74].
JPS is caused by germ line mutations in either MADH4 (SMAD4, DPC4) or BMPR1A, which can be found in 18.2% or 20.8% of affected individuals, respectively [75]. The condition is characterized by multiple hamartomatous polyps, most commonly arising in the colon but rarely also in the stomach, duodenum, and small bowel. For both sporadic and familial JPS, mean age of CRC diagnosis is 37 years [76]. Lifetime cancer risk has been estimated 38% for colonic and 21% for upper GI cancers, including the stomach, pancreas, and small bowel [77].
4.3 Future Perspectives
Recent data indicate an even greater complexity of cancer development in the colorectum. Thus, germ line exonuclease domain mutations (EDMs) of POLE and POLD1 have been shown to confer high risk of multiple colorectal adenomas and carcinoma, a condition named polymerase proofreading-associated polyposis (PPAP). Somatic POLE EDMs have also been found in sporadic CRCs and endometrial cancers. It is believed that both the germ line and the somatic mutations cause impair polymerase proofreading resulting in “ultramutated,” yet microsatellite stable (MSS), tumors [78].
In addition, Guinney et al. [79] reported four “consensus molecular subtypes” (CMS) of colorectal cancer: CMS1 (MSI immune, 14%), hypermutated, microsatellite unstable, and strong immune activation; CMS2 (canonical, 37%), epithelial, marked WNT, and MYC signaling activation; CMS3 (metabolic, 13%), epithelial and evident metabolic dysregulation; and CMS4 (mesenchymal, 23%), prominent transforming growth factor-β activation, stromal invasion, and angiogenesis. It is of note that 13% of samples tested showed mixed features, which could be explained by intratumoral heterogeneity or by a transition phenotype. The significance of this “consensus” publication is, however, currently unclear.
Conclusion
Different molecular and cellular mechanisms of carcinogenesis have been identified in the large bowel. These mainly include CIN, MSI, and CIMP pathways. Familial cancers may arise within FAP, LS, and MAP syndromes. Hamartomatous polyposis syndromes likewise harbor increased cancer risk. Four consensus molecular subtypes (CMS1-CMS4) have been described recently, awaiting validation by other groups.
References
Cancer fact sheet n°297. WHO Media Center. 2015. http://www.who.int/mediacentre/factsheets/fs297/en/. Accessed 2 Dec 2015.
GLOBOCAN. Estimated cancer incidence, mortality and prevalence in 2012 (2012) International Agency for Research on Cancer–WHO. 2012. http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx. Accessed 2 Dec 2015.
Setaffy L, Langner C. Microsatellite instability in colorectal cancer: clinicopathological significance. Pol J Pathol. 2015;66:203–21.
Bosman FT, Yua P. Molecular pathology of colorectal cancer. Pol J Pathol. 2014;65:257–66.
Samadder NJ, Vierkant RA, Tillmans LS, et al. Associations between colorectal cancer molecular markers and pathways with clinicopathologic features in older women. Gastroenterology. 2013;145:348–56. doi:10.1053/j.gastro.2013.05.001.
Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer—analyses of co-horts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78–85.
Grady WM. Genetic testing for high-risk colon cancer patients. Gastroenterology. 2003;124:1574–94.
Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology. 2010;138:2088–100. doi:10.1053/j.gastro.2009.12.066.
Jasperson KW, Tuohy TM, Neklason DW. Hereditary and familial colon cancer. Gastroenterology. 2010;138:2044–58. doi:10.1053/j.gastro.2010.01.054.
Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. 2009;138:2059–72. doi:10.1053/j.gastro.2009.12.065.
Takayama T, Ohi M, Hayashi T, et al. Analysis of K-ras, APC, and beta-catenin in aberrant crypt foci in sporadic adenoma, cancer, and familial adenomatous polyposis. Gastroenterology. 2001;121:599–611.
Langner C. Serrated and non-serrated precursor lesions of colorectal cancer. Dig Dis. 2015;33:28–37. doi:10.1159/000366032.
Bettington M, Walker N, Clouston A, et al. The serrated pathway to colorectal carcinoma: current concepts and challenges. Histopathology. 2013;62:367–86. doi:10.1111/his.12055.
Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67.
Bodmer WF, Bailey CJ, Bodmer J, et al. Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature. 1987;328:614–6.
White BD, Chien AJ, Dawson DW. Dysregulation of Wnt/β-catenin signaling in gastrointestinal cancers. Gastroenterology. 2012;142:219–32. doi:10.1053/j.gastro.2011.12.001.
Leslie A, Carey FA, Pratt NR, et al. The colorectal adenoma-carcinoma sequence. Br J Surg. 2002;89:845–60.
Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–32.
Claes K, Dahan K, Tejpar S, et al. The genetics of familial adenomatous polyposis (FAP) and MutYH-associated polyposis (MAP). Acta Gastroenterol Belg. 2011;74:421–6.
Burt RW, Leppert MF, Slattery ML, et al. Genetic testing and phenotype in a large kindred with attenuated familial adenomatous polyposis. Gastroenterology. 2004;127:444–51.
Acharya S, Wilson T, Gradia S, et al. hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc Natl Acad Sci U S A. 1996;93:13629–34.
Kadyrov FA, Dzantiev L, Constantin N, et al. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006;126:297–308.
Young J, Simms LA, Biden KG, et al. Features of colorectal cancers with high-level microsatellite instability occurring in familial and sporadic settings: parallel pathways of tumorigenesis. Am J Pathol. 2001;159:2107–16.
Sideris M, Papagrigoriadis S. Molecular biomarkers and classification models in the evaluation of the prognosis of colorectal cancer. Anticancer Res. 2014;34:2061–8.
Boland CR, Koi M, Chang DK, et al. The biochemical basis of microsatellite instability and abnormal immunohistochemistry and clinical behavior in Lynch syndrome: from bench to bedside. Familial Cancer. 2008;7:41–52.
Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–57.
Mead LJ, Jenkins MA, Young J, et al. Microsatellite instability markers for identifying early-onset colorectal cancers caused by germ-line mutations in DNA mismatch repair genes. Clin Cancer Res. 2007;13:2865–9.
Suraweera N, Duval A, Reperant M, et al. Evaluation of tumor microsatellite instability using five quasimonomorphic mononucleotide repeats and pentaplex PCR. Gastroenterology. 2002;123:1804–11.
Patil DT, Bronner MP, Portier BP, et al. A five-marker panel in a multiplex PCR accurately detects microsatellite instability-high colorectal tumors without control DNA. Diagn Mol Pathol. 2012;21:127–33. doi:10.1097/PDM.0b013e3182461cc3.
Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007;50:113–30.
Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138:2073–87. doi:10.1053/j.gastro.2009.12.064.
Lynch HT, Smyrk T, Lynch JF. Molecular genetics and clinical-pathology features of hereditary nonpolyposis colorectal carcinoma (Lynch syndrome): historical journey from pedigree anecdote to molecular genetic confirmation. Oncology. 1998;55:103–8.
Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919–32.
Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26:5783–8. doi:10.1200/JCO.2008.17.5950.
Cicek MS, Lindor NM, Gallinger S, et al. Quality assessment and correlation of microsatellite instability and immunohistochemical markers among population- and clinic-based colorectal tumors results from the Colon Cancer Family Registry. J Mol Diagn. 2011;13:271–81. doi:10.1016/j.jmoldx.2010.12.004.
Bellizzi AM, Frankel WL. Colorectal cancer due to deficiency in DNA mismatch repair function: a review. Adv Anat Pathol. 2009;16:405–17. doi:10.1097/PAP.0b013e3181bb6bdc.
Dunlop MG, Farrington SM, Carothers AD, et al. Cancer risk associated with germline DNA mismatch repair gene mutations. Hum Mol Genet. 1997;6:105–10.
Quehenberger F, Vasen HF, van Houwelingen HC. Risk of colorectal and endometrial cancer for carriers of mutations of the hMLH1 and hMSH2 gene: correction for ascertainment. J Med Genet. 2005;42:491–6.
Jenkins MA, Baglietto L, Dowty JG, et al. Cancer risks for mismatch repair gene mutation carriers: a population-based early onset case-family study. Clin Gastroenterol Hepatol. 2006;4:489–98.
Alarcon F, Lasset C, Carayol J, et al. Estimating cancer risk in HNPCC by the GRL method. Eur J Hum Genet. 2007;15:831–6.
Senter L, Clendenning M, Sotamaa K, et al. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology. 2008;135:419–28. doi:10.1053/j.gastro.2008.04.026.
Choi YH, Cotterchio M, McKeown-Eyssen G, et al. Penetrance of colorectal cancer among MLH1/MSH2 carriers participating in the colorectal cancer familial registry in Ontario. Hered Cancer Clin Pract. 2009;7:14. doi:10.1186/1897-4287-7-14.
Baglietto L, Lindor NM, Dowty JG, et al. Risks of Lynch syndrome cancers for MSH6 mutation carriers. J Natl Cancer Inst. 2010;102:193–201. doi:10.1093/jnci/djp473.
Bonadona V, Bonaïti B, Olschwang S, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA. 2011;305:2304–10. doi:10.1001/jama.2011.743.
Giardiello FM, Allen JI, Axilbund JE, et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-Society Task Force on colorectal cancer. Gastroenterology. 2014;147:502–26. doi:10.1053/j.gastro.2014.04.001.
ten Broeke SW, Brohet RM, Tops CM, et al. Lynch syndrome caused by germline PMS2 mutations: delineating the cancer risk. J Clin Oncol. 2015;33:319–25. doi:10.1200/JCO.2014.57.8088.
Vasen HFA, Mecklin J-P, Meera Khan P, et al. The International Collaborative Group on hereditary non-polyposis colorectal cancer (ICG-HNPCC). Dis Colon Rectum. 1991;34:424–5.
Vasen HF, Watson P, Mecklin JP, et al. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453–6.
Giardiello FM, Allen JI, Axilbund JE, et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the U.S. Multi-Society Task Force on Colorectal Cancer. Gastrointest Endosc. 2014;80:197–220. doi:10.1016/j.gie.2014.06.006.
Giardiello FM, Allen JI, Axilbund JE, et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-Society Task Force on Colorectal Cancer. Dis Colon Rectum. 2014;57:1025–48.
Giardiello FM, Allen JI, Axilbund JE, et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-society Task Force on colorectal cancer. Am J Gastroenterol. 2014;109:1159–79. doi:10.1038/ajg.2014.186.
Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–8.
Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi:10.1146/annurev-pathol-011110-130235.
Bird AP. CpG-rich islands and the function of DNA methylation. Nature (London). 1986;321:209–13.
Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4:988–93.
Lee S, Cho NY, Choi M, et al. Clinicopathological features of CpG island methylator phenotype-positive colorectal cancer and its adverse prognosis in relation to KRAS/BRAF mutation. Pathol Int. 2008;58:104–13. doi:10.1111/j.1440-1827.2007.02197.x.
Yamamoto E, Suzuki H, Yamano HO, et al. Molecular dissection of premalignant colorectal lesions reveals early onset of the CpG island methylator phenotype. Am J Pathol. 2012;181:1847–61. doi:10.1016/j.ajpath.2012.08.007.
Chirieac LR, Shen L, Catalano PJ, et al. Phenotype of microsatellite-stable colorectal carcinomas with CpG island methylation. Am J Surg Pathol. 2005;29:429–36.
Juo YY, Johnston FM, Zhang DY, et al. Prognostic value of CpG island methylator phenotype among colorectal cancer patients: a systematic review and meta-analysis. Ann Oncol. 2014;25:2314–27. doi:10.1093/annonc/mdu149.
Kriegl L, Neumann J, Vieth M. Up and downregulation of p16(Ink4a) expression in BRAF-mutated polyps/adenomas indicates a senescence barrier in the serrated route to colon cancer. Mod Pathol. 2011;24:1015–22. doi:10.1038/modpathol.2011.43.
Wajapeyee N, Serra RW, Zhu X, et al. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363–74. doi:10.1016/j.cell.2007.12.032.
Guarinos C, Sánchez-Fortún C, Rodríguez-Soler M, et al. Serrated polyposis syndrome: molecular, pathological and clinical aspects. World J Gastroenterol. 2012;18:2452–61. doi:10.3748/wjg.v18.i20.2452.
Rex DK, Ahnen DJ, Baron JA, et al. Serrated lesions of the colorectum: review and recommendations from an expert panel. Am J Gastroenterol. 2012;107:1315–29. doi:10.1038/ajg.2012.161.
Fernando WC, Miranda MS, Worthley DL, et al. The CIMP phenotype in BRAF mutant serrated polyps from a prospective colonoscopy patient cohort. Gastroenterol Res Pract. 2014;2014:374926. doi:10.1155/2014/374926.
Guarinos C, Sánchez-Fortún C, Rodríguez-Soler M, et al. Clinical subtypes and molecular characteristics of serrated polyposis syndrome. Clin Gastroenterol Hepatol. 2013;11:705–11. doi:10.1016/j.cgh.2012.12.045.
Gala MK, Mizukami Y, Le LP, et al. Germline mutations in oncogene-induced senescence pathways are associated with multiple sessile serrated adenomas. Gastroenterology. 2014;146:520–19.
Bettington ML, Chetty R. Traditional serrated adenoma: an update. Hum Pathol. 2015;46:933–8. doi:10.1016/j.humpath.2015.04.002.
Bettington ML, Walker NI, Rosty C, et al. A clinicopathological and molecular analysis of 200 traditional serrated adenomas. Mod Pathol. 2015;28:414–27. doi:10.1038/modpathol.2014.122.
Bettington M, Walker N, Rosty C, et al. Clinicopathological and molecular features of sessile serrated adenomas with dysplasia or carcinoma. Gut. 2017;66(1):97–106. doi:10.1136/gutjnl-2015-310456.
Al-Tassan N, Chmiel NH, Maynard J, et al. Inherited variants of MYH associated with somatic G:C → T:A mutations in colorectal tumors. Nat Genet. 2002;30:227–32.
Boparai KS, Dekker E, Van Eeden S, et al. Hyperplastic polyps and sessile serrated adenomas as a phenotypic expression of MYH-associated polyposis. Gastroenterology. 2008;135:2014–8. doi:10.1053/j.gastro.2008.09.020.
Calva D, Howe JR. Hamartomatous polyposis syndromes. Surg Clin North Am. 2008;88:779–817. doi:10.1016/j.suc.2008.05.002.
Jenne DE, Reimann H, Nezu J-I, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18:38.
McGarrity TJ, Amos C. Peutz-Jeghers syndrome: clinicopathology and molecular alterations. Cell Mol Life Sci. 2006;63:2135–44.
Howe JR, Sayed MG, Ahmed AF, et al. The prevalence of MADH4 and BMPR1A mutations in juvenile polyposis and absence of BMPR2, BMPR1B, and ACVR1 mutations. J Med Genet. 2004;41:484.
Giardiello FM, Hamilton SR, Kern SE, et al. Colorectal neoplasia in juvenile polyposis or juvenile polyps. Arch Dis Child. 1991;66:971.
Howe JR, Mitros FA, Summers RW. The risk of gastrointestinal carcinoma in familial juvenile polyposis. Ann Surg Oncol. 1998;5:751.
Briggs S, Tomlinson I. Germline and somatic polymerase ε and δ mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol. 2013;230:148–53. doi:10.1002/path.4185.
Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–6. doi:10.1038/nm.3967.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Brčić, I., Callé, C., Langner, C. (2017). Molecular and Cellular Mechanisms of Carcinogenesis in the Large Bowel. In: Haybaeck, J. (eds) Mechanisms of Molecular Carcinogenesis – Volume 2. Springer, Cham. https://doi.org/10.1007/978-3-319-53661-3_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-53661-3_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-53660-6
Online ISBN: 978-3-319-53661-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)