
Abstract
The clinical trial methodology of conducting experiments in humans has been, and still is, most developed in pharmacotherapy research. Although straightforward in theory, in practice the design and implementation of a conclusive clinical trial is a multifaceted exercise. Drawing valid conclusion on the effects of the experimental intervention on the investigated outcome also requires careful consideration as to how the collected data are to be analyzed. To avoid non-compliance and self-selection bias after randomization, the intention-to-treat analysis principle is widely adopted, analyzing patients according to treatment allocation, irrespective of actual exposure. After an overview of clinical trial-based therapy research, the question of the transportability of the methodology to consumer product research is addressed. Other than prescription drugs, consumer products and services are not externally allocated but selected by consumers directly, often from a variety of freely accessible alternative options. It becomes evident that this fundamental difference has profound implications on consumer product research methods, rendering intention-to-treat a meaningless concept and pointing towards the central role of considering consumer preferences, choices and actual use-based effect assessment.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Consumer Product
- Migraine Patient
- Population Attributable Risk
- Therapy Research
- Instrumental Variable Analysis
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Clinical trials are conducted in many areas, including therapy, prognosis, and prevention research, where they provide a well developed and powerful research methodology. In order to apply this methodology in consumer product research, its properties must be well understood and carefully adopted, and sometimes modified. This contribution provides an overview of the historical developments and methodological properties of clinical trials and points out aspects that require special attention in the context of consumer product research.
2 Trying Conjectures
When a consumer product or service is assumed to lead to a specific effect, it can be attempted to substantiate a claim by conducting an experimental study. The methodology of conducting experiments in humans is most developed in drug therapy research and is referred to as clinical research; clinical trial being the term to denote a specific experimental clinical study—irrespective of whether or not the participants are healthy or diseased, as the “clinical” refers to “human”, not to “disease”. Also, the “clinical” separates research conducted in humans clearly from pre-clinical research (both in-vitro and in-vivo). The element “trial” points at something being tried in some formal empirical investigation, which is already the essence of clinical trials. Trying something means, in general, raising a conjecture-based question to the world, let her speak, and then (through modus tollens) conclude whether what was conjectured is not the case or might be the case. How we come to a conjecture in the first place is a fascinating but metaphysical question, and this author agrees with intuitionistic views of the critical rationalistic philosophy of science on the matter (Popper 1935, p. 208, simply speaks of “idea” and “unjustified anticipation”), although this will not be further addressed in this contribution.
How now do we try things? By flicking a switch, we can indeed turn the lamp on; there being light. We have confirmed the conjecture empirically. This is straightforward, as the relationship between action and reaction is essentially deterministic, and in case of doubt we can simply retry. If we want to know whether switching the automatic transmission of our car from S (sporty) to E (economy) actually leads to a reduced gasoline consumption, things are already getting a bit more complicated; as the effect is not instantaneously visible (albeit possibly audible, but sound is not a direct measure of fuel consumption but merely, at best, an indicator or proxy variable) and it is also quite likely more confounded. Confounders might be our driving style, the outdoor temperature, and the route we take. Due to the more complicated, causally interwoven factors influencing our car’s fuel consumption, which we might choose to view as being at least partially, but more likely mostly, probabilistic by their nature and mode of action, and due to the gradual rather than all-or-nothing effect (as with the lamp), we are this time quite unlikely to get away with only one trial. We will need to retry, and finally, after a few weeks, to aggregate the consumption data that we obtain from test driving periods with and without the transmission set to E, using some statistics maybe. The good news is that we do not really have to worry much about a complicated study design and about sequence effects, or about the need to use a brand new car for every driving period. A car is a car after all, and it should largely respond as any machine does, in accordance to the parameters set, essentially irrespective of its mileage differing or not by a few thousand.
With respect to generalizing the findings of our trials so far, we do not have much of a problem either. Switching light switches and setting automatic transmissions to energy saving will, in the vast majority of cases, lighten up rooms and reduce fuel consumptions respectively. Things get tougher though when what we try does not relate to objects but to subjects, i.e., to human beings. As mentioned above, the methodology of clinical trials has been, and still is, most developed in therapy research, which is why this is where we will start, before moving on to research on consumer products.
Trying something in humans is much more difficult than trying a light or a transmission. Take a fictitious novel migraine pill for example. Based on the 2003 National Health Interview Survey, US migraine prevalence was 8.6% in males and twice as high in females (17.5%), with prevalence peaking in the late teens and 20s and around 50 years of age (Victor et al. 2010). A lot of research has been conducted on biological, psychological, and environmental risk factors and mechanisms. For example, there is evidence that in about one out of ten migraine patients the headache is associated with weather conditions (Hoffmann et al. 2015). Could we simply pick, for example, a female 60-years old weather-sensitive migraine patient and try the pill with her? What might happen is that the pill would relieve the headache on the first migraine day while on the subsequent episode, even four pills would have no effect. In addition consider that, had we picked another patient, one pill could have worked on both occasions—or on neither of them. Now, does the pill work or not? This is impossible to say from the data obtained by our trial so far, as obviously how humans respond to the same exposure can widely differ, both across individuals and occasions. The epidemiology of migraine already points at different subgroups and possibly different subtypes of migraine, related to sex, age, and possibly weather sensitivity. Thus, if we want to know whether in general say two of the novel pills relieve migraine headache, we can obviously not restrict ourselves to a particular patient (or two), as even for our one patient the pills might not work the same all the time. Rather, in order to be able to recommend the pill to all adult migraine patients (i.e., to generalize our findings to the whole target population of adult migraine patients), we need to investigate a whole sample of them, making sure that not all of our study sample is female and/or older than 30 years of age, as this would imply missing out on men and/or younger patients.
There are other questions we have to address when we plan our trial. How do we find the patients to participate in the trial? Sometimes there are attractive methods that allow to conveniently fill the sample. For example, one could contact the members of an online migraine support group that discusses their sensitivity to weather conditions. While those patients might be quite motivated to participate, this particular way of recruitment might select migraine patients that are not representative of the whole population of migraine patients—as their migraines are likely to be related to weather conditions whereas the majority of migraine patients’ headaches are not. Also, the particular way of recruitment can lead to other differences, both known and unknown, between the study participants and the whole population of migraine patients. Also, should we provide pills on some migraine days but not on others, and then compare the headache levels between the two types of days? We could, but what if pills per se (i.e., irrespective of their contents) would have an effect on migraine? One never knows. The problem is indeed ubiquitous and referred to as placebo effect. If in our weather-sensitive study sample the placebo effect of two white pills would be particularly strong, we might conclude that the presumably active compound that is contained in the pills would generally be efficacious, where it in fact is not. Could we mitigate this problem by sometimes using a second set of identical pills that do not contain the compound, and keep very careful track of which kind of pills were taken, when, and by whom? We could. We could also split the total study sample upfront into two halves and provide the active pills to one half and the placebo pills to the other. Of course, we then would have to take precautions that the severity of migraine would be equally distributed across the two groups, as well as other factors that could potentially influence the response to the pills. Such factors include, but are not restricted to, the duration of the disease, weather sensitivity, and age of the patients. Also, we would be better not to tell our sample what kind of pills they take, as otherwise we could introduce a differential placebo effect, most likely stronger in the active pill group. Thus, we should keep the patients blind with regard to what kind of pills they receive, and even better also the study personnel, to avoid any sort of unforeseeable influence (bias). Such a double-blind strategy can be implemented by randomly allocating the type of pill (active vs. placebo) to each patient, and to make sure that the groups are of equal size, we can deploy block randomization. Randomization also reduces the chances of having, for example, migraine severity or some unknown pill response predisposition differentially distributed across the subgroups. For these advantages, most clinical trials are actually designed as randomized clinical trials.
What this illustrates is that trialing something (i.e., some external intervention of interest, as the pill in this example) with respect to some outcome (relieve of migraine headaches) in some specified group of people (defined by inclusion- and exclusion criteria, as adult patients with migraine but not with other types of headaches in our example) is quite a bit more challenging than testing whether a light bulb can be switched on or gasoline can be saved by changing the transmission settings. Some careful thoughts are needed with respect to the target population and how the study sample can be recruited from it in an unbiased manner, and how the intervention of interest is planned and administered, so that the study results even have a chance of being conclusive with respect to the research question. Clinical trials thus require meticulous design, planning, and execution, and the devil is definitely in the details. And there are many more details to consider than those we have just lightly touched. For the taste of it: How, by the way, do we measure levels of migraine headache and its reduction in a reliable manner? Pain is a private event, and there is no direct and objective access to it, like for example to body temperature through a thermometer. This being so: could we simply switch to body temperature as our effect measure? We could. But it would not be meaningful. Temperature is not a valid surrogate endpoint for migraine pain, even if it can be measured at a high level of precision; in fact, it is a meaningless biomarker in this context, and measuring it would tell us absolutely nothing about the efficacy of the pill for relieving migraine headache. Some further thoughts must be given to even more details of the study, like study duration: We could, for each patient, only treat and record one migraine episode. However, it would probably be more relevant for assessing the value of the pill if we would extend the treatment over a few months and then look at the overall results—which of course raises the issue as to how to integrate the findings from each individual episode. But then, would it be ethical and/or scientifically smart to compare the novel pill to a placebo, or would it not be a more reasonable approach to compare it with some existing therapy? If so, should we attempt to demonstrate that the new pill is indeed better (superior) to the existing one (the active comparator, in clinical research parlance) or would we be satisfied with showing non-inferiority? We also must plan the statistical analysis of the study data and, related to this, decide on the expected size and variability of the treatment effect(s), considering of what magnitude such effects would need to be for them being of any clinical relevance, and how many patients we should consequently include in our study to make it sufficiently likely to find the expected effect when it actually exists. And so on.
As this short outline clarifies, designing good (i.e., conclusive) clinical trials is cumbersome and requires profound knowledge, specific skills, experience and diligence, let alone the huge amount of logistical planning and operative work for the execution of the study, its documentation and quality control. Also, studies conducted in humans require a lot of prerequisites, including the demonstration that the product, or pill in our example, we want to assess is produced against well-defined quality standards, evidence that the new drug is safe to take and that the dose is reasonably chosen, approval of the study protocol by an ethics committee and of course informed consent of the study participants.
3 Historical Developments
The current conceptualization, design, conduct and analysis of clinical experiments, as implemented in medicine, public health, psychology, research in education, consumer research, and many other areas, is largely based on the twentieth century works of the English geneticist and statistician Sir Ronald Aylmer Fisher and his compatriot, the epidemiologist Sir Austin Bradford Hill. Fisher conducted agricultural field research and considered rigorous experimental design as the basis for drawing valid inference on probabilistic hypotheses regarding the causal impact of the deliberate variation of experimental exposures/factors (like fertilization) on experimental units (plots of land; Fisher 1925) in terms of measured effects (crop yield). Fisher deemed randomization the cornerstone of experiments, to warrant the unbiased allocation of units to experimental groups (conditions, treatments, factor levels), so rendering all residual error in the data unsystematic noise, achieved through asymptotically balancing all background variables across the comparison groups, irrespective of whether or not these individual (baseline) covariates are even known or measured. Potential confounders so prevented from being systematically associated with the experimental manipulation renders the latter the only possible explanation of the observed effects. Aside from considering the distributional properties of the individual variables for the choice of the appropriate statistical calculations, no further prior assumption or multidimensional statistical model is needed. Rather, the “likelihood” (Fisher’s “p-value” of the statistical “test of significance”) of observing in the “dependent variable” (Tolman 1932) an effect of at least the measured magnitude under the assumption of the experimental factor (“independent variable”) having no effect (“null hypothesis”) can directly be calculated. From an epistemological point of view, Fisher had proposed a probabilistic inductive inference method for concluding on a causal effect of the experimental manipulation, by rejecting the opposing null hypothesis with a quantified likelihood of this conclusion being erroneous. Even though it is not of particular importance for the issues here addressed, it should be noted that the current practice of frequentist statistical testing largely reflects a range of variants of inconsistent amalgams of Fisher’s significance test logic and the method of hypothesis testing proposed by Neyman and Pearson (1933), and that neither party had ever intended to merge the two methods.
The institution of the experimental design and analysis method in therapy research is generally attributed to Austin Bradford Hill, who planned the first modern blinded and properly randomized controlled trial (RCT) on the effects of streptomycin in patients with pulmonary tuberculosis (launched in 1947 by the Medical Research Council in the UK; MRC 1948). Probably less well-known, it was also Hill who, seemingly in 1955, coined the expression “intention-to-treat” (Lewis and Machin 1993), which will be addressed in more detail below. As not all research questions on matters of human health relate to therapy effects and thus often cannot be addressed through experiments (which are in many circumstances impractical, irrelevant, unreliable, unethical, or a mixture thereof, coupled with the notorious issue of the questionable generalizability of experimental findings to the real world), Hill was strongly engaged in observational research and methods development. Based on a case-control study in patients from 20 hospitals in London, conducted together with Richard Doll, Hill concluded that smoking was an important risk factor for lung cancer (Doll and Hill 1950), a finding subsequently confirmed by the seminal prospective British Doctor’s cohort study which started in 1951 (Doll and Hill 1964). This etiologic endeavor, which included more than 40,000 physicians and measured chronic disease risk factors and long-term health outcomes, was indeed far beyond the scope of an experimental design. Hill was well aware of the methodological challenges of observational studies related to bias and confounding. In 1965 he proposed “viewpoints” (sometimes denoted as “Hill’s criteria for causation”) to consider in order to facilitate drawing inductive causal inferences based on observational data. While John Stuart Mill, 1843 in his System of Logic, had previously suggested methods of induction in the context of experimental data, no such an attempt had yet been made for observational data (Morabia 2013).
With the Nuremberg Code, written in 1947, and the Declaration of Helsinki, established in 1964, the framework for conducting clinical trials was defined, with a focus on protecting the rights and wellbeing of study participants by voluntary participation, and setting standards like mandatory informed consent and the ability to withdraw at any time from the study. In spite of the watershed amendments to the American Food, Drug and Cosmetics Act in 1962, which made RCTs a requirement for marketing authorization of novel drugs and providing the Food and Drug Administration with regulatory authority, acceptance of the experimental therapy research approach increased only gradually after the Second World War. Opposition towards RCTs by clinicians was driven by traditions of viewing medicine as mainly experience-based and largely grounded in clinical judgment, a widespread lack of statistical understanding, and ethical concerns against placebo arms. In his memoires, Hill (1990) pointed out that he carefully avoided using the word “randomization” in the streptomycin-trial study protocol, in order not to raise opposition from collaborating physicians. The increasing acceptance of experimental studies on treatment benefits, up to the present where the method has gained the status of a “gold standard” (cf. Cartwright 2010), occurred in the 1970s, promoted by the formation of the evidence-based medicine-movement, materially pioneered by David Sackett, who co-initiated the Cochrane-Collaboration. The collaboration was named after the Scottish epidemiologist Archibald Cochrane, whose preoccupation with closing the gap between what is known versus what is actually done in clinical medicine and his lifelong call for RCT-based substantiation of any medical intervention’s benefit outweighing its harm was thereby acknowledged. As was the case with RCTs, evidence-based medicine was initially not easily accepted by all parts of the medical establishment (e.g., Grahame-Smith 1995).
To help clinicians critically appraise the accumulating published evidence on the benefits of therapies, Sackett (1989) had developed a first design-focused hierarchy of evidence with “large randomized trials with clear-cut results (and low risk of error)” on top of the hierarchy (p. 38). This and subsequent study-design evidence hierarchies led to some confusion, as the logic originally proposed for therapy studies was not infrequently simply generalized to other domains, including diagnostic and prognostic research, even though RCTs can, for example (as briefly indicated above), contribute little or nothing to etiological research on chronic disease risks. This has been clearly pointed out early on by Sackett and others, but has not always been considered carefully. Sackett and Wennberg (1997) wrote (p. 1536): “Evidence based medicine is not restricted to randomized trials and meta-analyses. It involves tracking down the best external evidence with which to answer our clinical questions. To find out about the accuracy of a diagnostic test, we need to find proper cross sectional studies of patients clinically suspected of harboring the relevant disorder, not a randomized trial. For a question about prognosis, we need proper follow up studies of patients assembled at a uniform, early point in the clinical course of their disease.” The widely believed misconception that RCTs carry some special scientific weight in any context and would be necessary for true (“hard”) science-based conclusions (cf. Worrall 2007) has recently been addressed in a series of high-profile publications in medical journals (e.g. Ho et al. 2008), and the message seems to be gradually reaching all clinical areas. For example, DeVries and Berlet (2010), while pointing out the importance of high-quality RCTs in therapeutic research, state that prognostic studies follow different criteria, as the exposure variable being studied would not be researcher-controlled, cannot be randomly assigned, and a RCT “is inherently not possible” (p. 207).
4 Epistemological Aspects
An underlying reason for the sometimes unclear weighing of RCT-based evidence is possibly a lack of discriminating between the concepts of internal and external validity (Campbell and Stanley 1963). Internal validity depends on the tightness of built-in controls and essentially refers to the degree of certainty at which effects observed in a particular study can be causally attributed unequivocally to the experimental manipulation. This notion is reflected in Tolman’s dichotomy of dependent and independent variables, reflecting the concepts of effects of causes and of causes of effects, respectively. It is clear from the rationale underlying Fisher’s experimental method that for all non-deterministic cause-effect relationships, well-controlled randomized experiments provide the highest level of internally valid evidence—at least as long as the analysis does not deviate from the original study design, as for example in subgroup comparisons, where randomization-based protection from baseline covariate imbalance is typically lost. Obviously, high levels of experimental control are well in-line with deductivism, rigorous hypothesis testing, and concerns about internal validity.
External validity is, in contrast, a very different concept, and tends to be “at odds” with internal validity, although the latter is often considered the sine qua non of the former (Campbell and Stanley 1963, p. 5; Steckler and McLeroy 2008). External validity addresses the question as to whether research results can be generalized to other, typically real-life contexts and populations. Due to the strict and largely canonical error-prevention controls and restrictions that are applied to maximize internal validity, external validity is the notorious Achilles heel of experiments, including RCTs, in particular when research findings are to be transported to conditions of usual clinical care practices. Many typical RCT features aiming at maximizing internal validity and often referred to bluntly as “rigorous” contribute to the problem of generalizability of study results. These include highly selected patient samples free of comorbidities and concomitant medications, high compliance levels, short study durations and more or less artificial and highly restricted settings and tight procedural controls. Even the best (i.e., most “rigorous”) RCT in the world, however, does not ensure infallibility nor does it generate external validity without a strong set of assumptions regarding the generalizability of the research to the real world. Thus utmost “rigor” (in terms of maximized internal validity) and complete irrelevance (in terms of absence of external validity) can easily coexist. Unless translated into specific hypotheses for subsequent empirical testing (further research), other than with internal validity, external validity cannot be achieved by rigorous adherence to methodological standards built on deductive logic within a given experiment. As Gadenne (2013, p. 5) has clearly pointed out, “the problem of external validity is the problem of induction”. The complexity around the concepts of internal and external validity points at the challenges related to assigning weights to sets of evidence provided by different studies. It is obvious, however, that extrapolating study design-based evidence hierarchies mindlessly beyond their contexts (e.g. clinical randomized experiments to proof therapeutic concepts) and assuming their universal applicability is careless and can result in fallacious inferences and misguided policy decisions (cf. Rothman 2014).
Somewhat along the same lines as internal and external validity, the distinction of the two therapy research aspects of efficacy (i.e., whether a treatment can in principle work under ideal circumstances) and effectiveness (i.e., whether it will work under realistic circumstances) was popularized by Cochrane (1972). In line with the above considerations regarding internal validity, RCTs can, when certain assumptions hold, be the ideal approach for assessing the efficacy of drugs (Gupta 2011), and they can then be analyzed through a simple comparison of average outcomes between groups, not further adjusted for covariates, to draw causal conclusions on the efficacy of the experimental variation. As usual, the devil is in the detail or, more specifically, in the assumptions that are needed to draw valid conclusions from experimental results, in addition to more general requirements (related to the Duhem-Quine problem of required auxiliary assumptions) that need to be fulfilled (e.g. construct validity, measurement accuracy or adequate and correct data processing and analysis). From a counterfactual point of view (first introduced to biostatistics by Neyman 1923), determining the average causal effect of the novel product (a therapeutic drug, for example) would require exposing each study participant simultaneously only once to both exclusively the drug with the active substance and an indistinguishable version without that substance (placebo), which is impossible (reflecting what is sometimes referred to as the “fundamental problem of causal inference”). In any factual experiment, participants must instead be randomized to active treatment or to placebo/control. The potential outcomes model (Rubin 1974) provides a conceptual and formal framework of causal inference, grounded in counterfactual logic and accounting for the inter-individual variability of treatment responses. It provides coherent definitions to describe causal effects as they occur in empirical research. These include individual as well as average causal treatment effects and specifications of key concepts like randomization, selection bias, confounding, or compliance, and allow one to state conditions and to specify assumptions, under which factual statistics provide valid causal treatment effect estimates.
A key assumption for drawing valid conclusions from experiments (cf. West et al. 2008) is ignorability (unconfoundedness), implying that potential outcomes are independent of the assigned treatment. Even though sometimes neglected, ignorability depends on a sufficient sample size for randomization to play out. Other important assumptions are stable unit treatment value (SUTV—based on the absence of treatment variation across units and on non-interference of treatment effects across units), exclusion restriction (any effect of randomization is transmitted through the experimental exposure/treatment, which often implies the requirement of blinding of personnel and participants with regard to the allocated treatment to avoid performance bias), full compliance (post-randomization adherence to treatment regimen) and completeness (i.e., no missing data, including no post-randomization sample attrition). Fisher’s agricultural research fits, unsurprisingly enough, remarkably well with these “ideal experiment” assumptions, which his methodology in fact requires to yield valid conclusions. While indeed plots of land rarely exhibit noncompliance, this is not necessarily so with all types of experimental units, particularly not with humans, irrespective of whether they are subjects, patients, or consumers.
5 Treatment Effects
Evaluating the effects of a treatment (e.g., a drug) in a blinded manner (mainly to avoid differential ascertainment) based on an ideal RCT relies basically on comparing it statistically, with regard to an endpoint, directly (i.e., without statistical adjustment) to some control treatment (e.g., a placebo). Under the assumptions of all baseline characteristics being equally distributed across the comparison groups through randomization to the novel (R = 1) or control treatment (R = 0), no noncompliance, and no missing data, the experimental results are automatically (i.e., without the need for any mechanistic understanding, theory, or additional assumptions) turned into evidence of a causal treatment effect, i.e., an efficacy claim—the core strength of the randomized-experimental method in terms of internal validity. The mechanism of randomization renders the impact of the actual treatment (i.e., of A = 1 as compared to no treatment or to an alternative treatment, A = 0) on the potential outcomes Y(A = a) “ignorable” and participants “exchangeable” across groups (Rosenbaum and Rubin 1983), i.e., Y(a)⊥A. Ideal RCT is, however, a rather simplistic concept, as in real clinical trials compliance of study participants and completeness of data is rarely one hundred percent. This raises questions on how to deal with non-compliant participants (even treatment crossover might occur, meaning that patients randomized to the experimental treatment may have received (and actually taken) the control medication, R = 0, A = 1, or vice versa, R = 1, A = 0) and incomplete data.
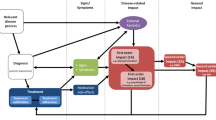
The intuitive response to broken randomization due to noncompliance (Ai ≠ Ri for some individuals i) and missing data would be to simply restrict the analysis to compliant patients with complete records. This “per-protocol” analysis strategy can provide “proof” of a therapeutic effect by answering the “can it work” (somewhere) question (cf., Cartwright 2011), i.e., for a specific outcome (Y), study and context, by demonstrating that here the outcomes were more pronounced in patients treated with the novel treatment than in those treated with the control treatment, i.e. E(Y|A = 1, R = 1)>E(Y|A = 0, R = 0), which corresponds to estimating efficacy as it might occur under ideal circumstances (Fig. 1).

Generic randomized two-arm parallel group therapy superiority study example, assuming one-sided non-compliance (patients in the control group are assumed not having access to the novel treatment). Half of the sample is randomized to the novel treatment (R = 1), the other half to the control treatment (R = 0). There is a chance that the observed treatment effect is biased by non-compliance, as 25% of the patients randomized to the novel treatment actually take the control treatment (R = 1, A = 0). There are three options to assess the effect of the novel treatment: (i) Taking into account both randomization and compliance—the per-protocol analysis; (ii) ignoring randomization—the as-treated analysis; (iii) ignoring compliance—the intention-to-treat (ITT) strategy. In an ITT analysis, patients are analyzed according to their randomized treatment, irrespective of whether they take it or not
Unfortunately, per-protocol effect estimates can be biased, as the contrasted groups are not any longer solely based on randomized treatment allocation, but also on post-randomization compliance. As factors that determine compliance can also influence the treatment effect (or can, in turn, be influenced by compliance and treatment effect), the magnitude of the association between type of treatment and effect can be confounded by such factors. The apparent benefit of the treatment can therefore be biased (typically overestimated), as the target population would be composed of a different, possibly less responsive and/or tolerant case mix than the per-protocol study population. While a per-protocol analysis does not require analyzing the details of noncompliance, it does bear the risk of introducing (self-selection) bias as the ignorability assumption cannot be maintained, and rigidly dismissing incomplete or noncompliant records always implies a loss of information and power. Also, when otherwise protocol-adherent records have missing data only in variables of minor importance or if missingness can be assumed being completely at random across participants, then excluding such records from the analysis is not a very convincing strategy.
Thus, it might be considered preferable to analyze participants according to the treatment that they have actually received, i.e., according to the “as-treated” analysis strategy, aiming at demonstrating a treatment effect on outcome Y in a specific study and context by showing that E(Y|A = 1)>E(Y|A = 0). As-treated is the only viable analysis of non-randomized (observational) cohort studies, and RCT-based safety data are usually also analyzed according to treatment received. Also, as-treated is the standard approach for analyzing preventive vaccine trials (Hudgens et al. 2004). When randomization cannot be relied on (or is absent in the first place) it is usually attempted to establish conditional exchangeability, i.e., Y(a)⊥A|C by conditioning the effect estimation on measured potential confounders (C). Conditioning can be achieved by some form (or combination of) adjustment, stratification, standardization, or matching. In order to correctly specify actual treatment (exposure) groups, an as-treated analysis necessitates the need to analyze the details of noncompliance with regard to whether treatment has simply not been taken, has been taken, but not according to the protocol, has been replaced (or supplemented) by alternative treatment(s), the correct dosing and timing of the treatment has been followed, and whether possibly physicians were noncompliant as well. The details of this pre-analysis depend to a large degree on the particular research question and circumstances, including whether or not compliance was measured in the control group and whether or not the active drug was accessible to the control group or some (active) control treatment was accessible to the treatment group. The likelihood of such complications is increased in large, long, and complex studies, in non-prescription settings, when the treatment under investigation is already on the market, under open-label treatment, and when the study is ambulatory rather than conducted in confinement.
Another classic response to protocol violations is to abstain from comparing groups according to the treatment actually received, but according to the intention-to-treat (ITT) principle. ITT analyses compare all participants according to the group to which they were randomized. Even though the approach is generally straightforward, in reality methodological problems are often encountered, as for example the need to deal with missing outcome data when participants are lost to follow up. As previously with the RCT methodology in general, the ITT approach faced considerable opposition, in particular by clinicians. This might have possibly been related to the need to statistically treat noncompliant patients as if they had taken the investigational drug, which from a clinical point of view could indeed appear being a “bizarre assumption” (Sheiner 1991, p. 4). Again like with RCTs, ITT is to date often referred to as a “gold standard”, and sometimes—less flattering—as having become gospel (Salsburg 1994). In 1990, the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), in which regulatory authorities of Europe, Japan and the United States and experts from the pharmaceutical industry participate, set out to harmonize regulation on the evaluation of medicinal products for market approval. Their 1996 E6 Good Clinical Practice guidance on clinical trials to demonstrate efficacy and safety of medicinal products acknowledges the role of statistics in trial design and analysis, which is detailed in the E9 guidance aimed at harmonizing the principles of clinical trial statistical methodology. It supports (ICH 1998, p. 28) the “intention-to-treat ideal” and states that “Preservation of the initial randomization in analysis is important in preventing bias and in providing a secure foundation for statistical tests. In many clinical trials, the use of the full analysis set provides a conservative strategy. Under many circumstances, it may also provide estimates of treatment effects that are more likely to mirror those observed in subsequent practice.” The authors of the Consolidated Standards of Reporting Trials (CONSORT; Schulz et al. 2010) recommend ITT analysis of parallel group RCTs for unbiased treatment effect estimates. Similarly, the Cochrane Collaboration (Higgins and Green 2011, Sect. 16.2.1) points out that “ITT analyses are generally preferred as they are unbiased, and also because they address a more pragmatic and clinically relevant question.” Modifications of the ITT approach, e.g. by excluding, after randomization, patients that were misdiagnosed or never had received any treatment, have been criticized for possibly introducing bias (Montedori et al. 2011).
From a causal effect estimation point of view, ITT is a form of instrumental variable analysis. In fact, the instrument (randomized treatment allocation) satisfies the key prerequisites for the validity of an instrumental variable (Greenland 2000), i.e., it is clearly linked to the actual treatment, but is unrelated to observed or unobserved prognostic factors as well as to the outcome (other than through the actual treatment; “exclusion restriction”, i.e., Y(R,A) = Y(A)). In this case, any confounding of the association between actual treatment and outcome is rendered irrelevant with respect to the association between the instrument (randomization R) and the potential outcomes, i.e., Y(a)⊥R. The reason is, based on causal-analytical considerations (Greenland and Pearl 2011), that the backdoor-path from the outcome to the instrument is blocked by the actual treatment, on which the effects of randomization and potential confounders collide; unless (incorrectly so), the ITT effect estimation would be conditioned on the actual treatment, which would open the backdoor path and (re)introduce confounding.
As pointed out above, the ITT principle to analyze the data of all participants as randomized has gained the status of the de facto standard (or even “gold standard”; Armijo-Olivo et al. 2009) for the primary analysis of randomized superiority clinical therapy trials and is broadly supported by regulatory and other authoritative bodies (Ten Have et al. 2008). There are downsides, however. The counterintuitive aspect of ITT is to some degree supported by an inherent asymmetry, which is that a treatment might be efficacious without being effective (due to a large nonadherence level). From this it can be deduced that an analysis which is exclusively based on ITT cannot provide sufficient insight into treatment effects. This is related to the fact that an ITT estimate, while avoiding confounding by self-selection through ignoring compliance, is by no means independent of compliance. In superiority settings, ITT estimates of treatment effects are being increasingly biased towards the null, i.e., diluted (compared to compliance-based estimates) as noncompliance increases. The simplicity of conducting an ITT analysis is largely restricted to parallel-group superiority designs, while deviations (e.g. crossover-designs) pose substantial conceptual and methodological problems. Moreover, for safety analyses ITT appears to be generally inappropriate (Robins and Greenland 1994). When post-randomization drop-outs occur, the ITT approach obliges some form of adjustment to avoid selection bias due to differential loss to follow-up. One of the simplest and most frequently applied forms of adjustment is by simply replacing missing outcome data points by the last known value (LOCF) of the participant. Although this is often considered to be a very conservative approach, it can introduce bias in either direction and always leads to overestimating the precision of the ITT effect estimates (Altman 2009). Under open-label conditions, the assumption that ITT provides pure estimates of effects of treatment offer/allocation does not hold anymore, as expectation effects can then introduce bias (e.g., Rosenthal, Hawthorne, and/or placebo effects). Due to the dilution of treatment effects by extending the assessment to noncompliant participants, ITT effect estimates are usually smaller than those of per-protocol and as-treated analyses, which increases the likelihood of underestimating or even failing to confirm a real effect (increased false negative/type II error rate). As a consequence, the conservativeness of ITT, compared to per-protocol and as-treated, does not extend to non-inferiority or equivalence studies, where it tends to favor equality of treatments and therefore to increase the type I (false positive) error. This becomes evident in a hypothetical study where perfect equivalence would be guaranteed under complete non-compliance of all study participants, at least as long as no additional success criterion (e.g., a minimal effect magnitude) is implemented. Even in superiority trials, to warrant external validity (generalizability, transportability) of ITT estimates, the assumption of similar levels and patterns of noncompliance under study and real-world conditions is required to hold.
Probably more importantly, however, the ITT approach addresses a different research question than non-ITT approaches. While per-protocol provides answers with respect to the effect of receiving a treatment as assigned to and in line with the protocol, and as-treated on the effect of receiving a treatment (irrespective of randomization and protocol-adherence), both are providing efficacy measures aiming at explaining effects. In contrast, ITT aims at quantifying the effect of being assigned to a treatment, regardless of whether it is received. ITT therefore does not address treatment efficacy and clinical meaning, but rather pragmatically quantifies the effectiveness of treatment allocation. This has in fact been considered an asset with regard to similarity to the real-world clinical practice and its value for informing policy decisions. However, the properties of RCT-based ITT estimates need to be handled with great care and assessed in context, in particular when comparing them to results from observational studies. An example is the controversy on the impact of hormone replacement therapy on the risk of coronary heart disease, where an observational cohort study (the Nurses Health Study) looking at more than 30,000 postmenopausal women suggested a substantial risk reduction, which was not confirmed by two subsequent RCTs (cf. Tannen et al. 2008). As Hernán et al. (2008) demonstrated, the results from the observational study estimated a different effect in a different population, and when reanalyzed by calculating an ITT-analogue effect in the sub-cohort of new hormone users and accounting for time since menopause and length of follow-up, the apparent discrepancies vanished.
While the conservativeness of ITT is often considered a major advantage, as it would protect against overestimating therapy effects, this very property might increase the risk of seriously disadvantageous public health strategies. Feinman (2009) has illustrated this point based on data from the Artery Bypass Surgery trial (Newell 1992). ITT analysis suggested a modest mortality advantage of surgery over medical treatment (5.3% vs. 7.8% mortality, respectively), while per-protocol and (more pronounced) as-treated showed a more than twofold higher mortality under medical treatment. An indifferent clinical practice regarding the therapy decision, in-line with the ITT results, might miss out on the potentially highly relevant option of embarking on an orchestrated action plan that would aim at allocating as many patients as possible to surgery. Not doing so effectively implies assuming that noncompliance rates and patterns cannot be influenced and will necessarily remain at what had been seen in the trial.
6 Consumer Products
In the medical world, treatment allocations are to a large degree made by clinicians, and patients are largely restricted to following this external allocation; they are in need of therapy and are being made an offer that they cannot easily decline. Thus, randomization appears to be an appropriate model of the external real-world. This is reflected in Fig. 2a, where the typical situation for an RCT on a therapeutic drug is summarized prior to the drug being marketed. Study participants selected from the target population in accordance with pre-specified inclusion and exclusion criteria are randomized to the novel drug (R = 1) or to some comparator (R = 0). If there is (i) no access to the novel product in those randomly allocated to the control group, i.e. a zero probability of actually taking the novel product, Pr(A = 1|R = 0) = 0, (ii) allocation to either group is equally probable through Pr(R = 1) = 0.5, and (iii) in-study exposure occurs in a double-blind and non-discriminable manner, then the effect in the study sample is essentially a function of actual treatment (exposure) and, as pointed out above, compliance.

Basic causal diagrams of (a) pre-launch effects of therapeutic drugs or consumer products in randomized parallel-group studies, of (b) clinical practice effects of prescription drugs, and of (c) in-market effects of consumer products. In the drug-therapy context, a close structural match between pre-launch clinical therapy research and post-launch clinical practice of components S, P, R, A, C, and E, to be justified on a case-by-case basis, provides support for the generalizability of the in-study findings (external validity). In consumer product contexts, the correspondence between pre-launch research and post-launch market is far more questionable, in particular with regard to the absence of external product allocation (component R) in consumer product contexts
By comparing this with Fig. 2b, denoting the situation after the therapeutic drug is on the market and can be prescribed by doctors, it becomes clear that the causal relationships are quite similar. The in-study randomization (R) corresponds to the post-market allocation of the drug by the doctor (D). All things being, while not fully equal but largely comparable, it can be expected that the study results have a good potential of predicting the real-world effectiveness of the drug once it is being marketed. Of course, in order to generalize study effects to real world effectiveness, the requirement of C1=C2 is critical: If real-world adherence to prescription differs from in-study compliance, then the study simply does not reflect the real world in that respect, and the in-study findings cannot accurately predict the post-market situation.
Transposing the above from pharmacotherapy to consumer product clinical trial settings is difficult. The first problems become evident when it comes to sampling study participants. In any research area, for generalizability, a study sample is required that represents some specific real-world population of interest. Consequently, a prerequisite of any study is that by some adequate selection mechanism S on a certain target population, a representative sample of participants P is included in the study, i.e., a sample having the same joint probability distribution over all relevant variables as the target population. Identifying and selecting participants into high-quality RCTs is in either case based on prudently defined procedures and inclusion/exclusion criteria. Drug trials typically build on the additional criterion of a confirmed medical diagnosis as well as related restrictions regarding co-morbidities and concomitant medications. Also, in particular when patients expect to benefit from the novel treatment, participation rates, i.e. Pr(P = 1|S = 1), are likely higher than when consumer products are tested for which potential study participants feel no immediate need. Target populations of drug trials are therefore likely to be more narrowly defined than those of consumer product trials. This implies that the representativeness of therapy study populations tends to be better warranted than under consumer product premises. When the rate and severity of adverse events under novel drug treatment is low and a lack of effects is not easily discernable by patients in the control group, compliance, i.e. Pr(A = R), might in general also be higher in drug as compared to consumer product trials.
Some consumer products aim at alleviating symptoms and conditions (like a cream aimed at moisturizing dry skin or a standardized diet, fitness program, or massaging device to address obesity). However, most health-related issues that are linked to consumer products are related to whether or not the use of (or exposure to) a certain consumer product is associated with improved wellbeing (rather than disease proper), or with increased or decreased risks of future adverse health-effects in currently healthy consumers. The range of consumer products and product categories that may be subjected to health-related research questions is quite broad and fuzzy, bordering on matters of lifestyle patterns, “alternative therapies”, and over-the-counter drugs. Examples are specific diets, certain fast-food items, snacks, ready-made nutrition products, sugar-enhanced soft drinks, functional food supplements, fitness programs, cosmetic products, sunglasses, alcoholic beverages, bicycle helmets, toothpaste, or tobacco products such as cigarettes. Depending on the type of consumer product tested in an RCT as outlined in Fig. 2a with regard to its health-impact, the feasibility of blinding or concealing the actual exposure is very likely generally lower (and often non-existent) than in a typical pharmacotherapy context. Also, the access to the (active) control product, which may already be on the market and then is typically freely accessible, depends very much on the study design, duration, and procedures. If the study is conducted in an ambulatory manner, all study participants typically would have access to the control product (other than in research on prescription drugs). For tobacco products, for example, this implies that all (presumably adult) noncompliant study participants randomized to the novel product (e.g., a candidate modified risk tobacco product, MRTP) would be able to obtain and consume the control product (e.g., conventional cigarettes), whereas the reverse would not be possible, as long as the novel product would not be on the market.
A key aspect of transposing research concepts from pharmacotherapy to consumer products is that prescription drugs are just that: prescribed, i.e., externally allocated. Even when consumer product RCTs follow the principles of a pharmacotherapy trial as laid out in Fig. 2a, pretty much the opposite of external allocation takes place under consumer market conditions, with largely unmediated and unrestricted product access through self-selection, i.e., consumer-internal product exposure allocation (Fig. 2c). Compared to the clinical practice world of prescription drugs, in the post-market consumer product world there are typically no diseases, no doctors, no treatments, and no patients, and products are not prescribed but freely chosen. The lack of anything only barely resembling prescription renders ITT and per-protocol based effect estimates meaningless, as there is nothing in the consumer product world that would correspond with the underlying concepts; all there is in the post-launch consumer product world is actual use (cf. Weitkunat et al. 2016, for a more detailed assessment of ITT estimates in the context of consumer product trials). In order to render an RCT-based as-treated effect estimate a valid predictor of the effect of actual use in the consumer market, it would be required that S, P, C, A, and E are identical under study and market conditions. As a comparison of Fig. 2(a) and (c) clarifies, this essentially necessitates that R1 = S3, i.e., that the self-selection to A = 1 and A = 0 under consumer market conditions is an unbiased version of what would be achieved by randomization.
7 Allocation vs. Preference
How can the problem of in-RCT randomization possibly not reflecting in-market self-selection in consumer product research be consolidated? It appears that accounting for consumer preferences is at the core of the issue. Even in the context of therapy research, concerns have been raised against randomization when external treatment allocation conflicts with patient preferences. In particular in contexts of impractical or incomplete blinding, external but also internal validity may be compromised through preference-related recruitment and compliance (King et al. 2005), and consequently preference-incorporating study designs have been proposed (e.g. Brewin and Bradley 1989; Zelen 1990; Wennberg et al. 1993). Irrespective of its relevance in therapy research, considering preference in the design of consumer studies might provide a possibility to reconcile randomization with relevance to and correspondence with the real world. By randomizing not the allocation to a certain product per se, but rather (as in the preference arm of the Wennberg et al. design) the option to choose a novel product to replace a previously used comparator product, the consumer market situation would be mirrored by the study design. Data obtained from this randomized choice option (RCO) design would lend themselves to an ITT-analogue analysis, which could be denoted as option-to-use or (for the sake of terminological similarity) intention-to-use (ITU) analysis. What ITU would estimate is actually the effect of offering a consumer product in a consumer market—something that cannot be achieved by an ITT analysis which is based on participants being externally allocated to a certain product through direct randomization. As with ITT in the therapy-research context, ITU would have the advantage of being randomization-protected against confounding by baseline variables, which of course requires analysis strictly according to randomization, irrespective of actual product choice. A critical prerequisite of ITU to provide valid effectiveness estimates is evidently the correspondence of in-study and real-world self-selection patterns and levels, which is in fact a rather strong assumption, although it can in principle be validated after the product has been launched by comparing in-study users with in-market consumers of the novel and of the comparator product. In addition to effectiveness (through ITU analysis), efficacy can be estimated from RCO data by analyzing actual use (AU)–outcome associations in the choice-option arm, necessarily by accounting for potential confounding (which corresponds to a classical observational cohort study).
From a practical point of view, the RCO design has the advantage that only those participants who are randomized to the product choice option need to be informed of the novel product, whereas the control group would reflect a market to which the novel product would never have been introduced. In addition, the RCO design provides use prevalence rate estimates based on real volitional behavior rather than solely relying on proxies of behaviors, like attitudes or intention-to-use declarations. As it may be adequate in many contexts to randomize a distinctly smaller number of consumers to the no-choice-option condition, the efficiency of an RCO design is likely comparable to a traditional RCT with direct (individual-level) randomization to a certain product. It appears worthwhile to point out that reversing the order of RCO events by first selecting participants based on their preference for the novel product (or, a weaker variant, their willingness to being randomized to it), while possibly leading to higher in-study adoption and compliance rates, will not achieve the same study-to-real-world correspondence and will lead to a very different (i.e., preference-selected) sample of participants (even though the ITT-analogue effect of product use allocation in those preferring the product can then be estimated, under the usual randomization-based protection from baseline confounding). A sensible extension of the RCO design appears to be adding a second randomization step to the scheme, allocating participants of the choice option arm who had previously expressed their preference for the novel product to actual product access versus to no access. Estimating the ITU effect of offering the product as well as estimating the AU effect would still be possible (by a slightly more complicated combination of the comparison groups), but now also a randomization-protected product effect could be estimated in those choosing the product offer and having versus not having actual access to the product.
8 Real World
Although it is often claimed that ITT would provide an effect estimate reflecting the real-world effectiveness of an intervention, this must, even under circumstances where the underlying logic applies, not be confused with population health impact estimation; ITT is restricted to quantifying effectiveness at the individual level. To quantify population-level effects, population impact measures are required, which can be based on estimates of the risk (cumulative incidence or prevalence) or rate (incidence rate) difference between the actually exposed and unexposed study groups. To estimate the population attributable risk (PAR), this risk difference (or attributable risk) is multiplied by the proportion of the total population that is actually exposed (i.e., is actually taking the drug that is investigated, or is actually using the consumer product under consideration). By multiplication with the population size, the PAR can be converted to a headcount estimate. To obtain valid PAR estimates, these calculations must be conducted in accordance with the exposure and risk strata that actually occur in the target population. If, for example, the impact of an exposure/therapy/consumer product on the outcome depends on sex, age, dose, or other factors, then stratum-specific risks as well as stratum-specific exposure prevalence estimates must be obtained in order to estimate the integrative population attributable risk (cf. Weitkunat et al. 2015).
When the generalization of therapy study results to the target population as a whole is assumed to be valid, then—in theory—a study-based ITT effect might be considered being a valid estimate of the attributable risk as it will occur in the target population, when the proportion of patients randomized to the drug in the study corresponds to the proportion of patients that the treatment will later be prescribed to and when in-study compliance corresponds to clinical practice adherence patterns and levels. Based on the considerations given to the generalizability of findings from therapy RCTs to target populations, this is, even under very favorable circumstances, a dauntingly long shot. For consumer products, it appears to be an impossible one. Here, but probably also for drug contexts, a population health impact assessment based on actual use effect estimates appears to be much more logical. What is required are stratum-specific AU estimates, based on studies where exposure-response data have been obtained for all strata (or contexts) of relevance, in particular with regard to various levels of dose, as they occur in the real world, as well as prevalence data regarding the size of all strata of relevance in the total population.
Even though this is somewhat beyond scope, contemplating the logic of how to analyze consumer product RCTs ultimately raises the question as to how useful this design is in the first place. It appears that for biomarkers of exposure or other objective short-term effects, the advantages of a randomized experimental approach apply essentially in full, even though the usually unquestioned assumption of baseline covariate balance being quasi-automatically achieved by randomization is somewhat problematic with regard to a single RCT (cf. Worrall 2007). Whenever the exposure period exceeds a few days or weeks, and whenever the outcomes are more complex (including subjective and behavioral endpoints, let alone long-term health outcomes), the question arises about what is actually being achieved through randomization. Seligman (1995, p. 974) has voiced the concern that random treatment allocation may be “less than useless” in mental disorder therapy research. In such circumstances, the likelihood of protocol deviations, allocated exposure contamination, and participants dropping out in a non-ignorable manner increases markedly, and both efficacy and effectiveness become ambiguous concepts, implying that valid analyses of outcomes cannot be conducted without accounting for post-randomization bias. Factually, the described complications render studies that have been conceived as experiments essentially observational in nature, necessitating the application of bias-correcting analysis methods, rather than a simple (sometimes denoted “naïve”) endpoint comparison across study groups. Such approaches aim at establishing conditional independence through unconfounding and include, by considering dynamic exposure as well as baseline and time-varying covariates, adjustment, inverse probability of treatment weighting, stratification for actual use patterns (irrespective of randomization), matching, propensity-score weighting (or adjustment), instrumental variable analysis, marginal structural modeling, or g-estimation (Schafer and Kang 2008, for an overview). According to Hernán and Hernández-Diaz (2012) and Hernán et al. (2013), outpatient therapy RCTs on effects of sustained interventions over long periods in real-world clinical care settings (“pragmatic trials”) conducted in large samples tend to suffer from non-differential noncompliance and sample attrition, and effectively become observational studies that require analyses beyond ITT; the authors suggest to analyze them as observational studies. It might, depending on the degree of deviation from the ideal RCT, indeed be more adequate to designate them as closed prospective cohort studies with baseline randomization.
To summarize: Clinical trials can contribute to consumer product assessment and research related to the health and wellbeing of consumers. The methodology was originally developed for and is most widely deployed in therapy research. It cannot be simply copied for consumer product research. Rather, careful consideration is required as to whether it can indeed provide sensible answers to the specific research questions at hand. Many of the critical aspects of using clinical research methods in consumer product research relate to the specific conditions of consumer’s access to freely available products. Other than patients with serious diseases, consumers usually do not have an inevitable need to use or consume a certain product and their sovereignty to choose is largely unrestricted. Such differences have far-reaching methodological implications, including the meaning of statistical data analysis strategies. To account for consumer preferences, behaviors, and contexts, study designs may more likely than not need to be adopted or even newly developed in rather unconventional ways. In general, planning and conducting research must be guided by considering whether a specific set of methods actually addresses the scientific questions at hand. Only then then collected data can have meaning, i.e., can provide evidence.
References
Altman, D. (2009). Missing outcomes in randomized trials: Addressing the dilemma. Open Medicine, 3, 2.
Armijo-Olivo, S., Warren, S., & Magee, D. (2009). Intention to treat analysis, compliance, drop-outs and how to deal with missing data in clinical research: A review. The Physical Therapy Review, 14, 36–49.
Brewin, C. R., & Bradley, C. (1989). Patient preferences and randomized clinical trials. British Medical Journal, 299, 313–315.
Campbell, D. T., & Stanley, J. C. (1963). Experimental and quasi-experimental designs for research. Chicago: Rand-McNally.
Cartwright, N. (2010). What are randomized controlled trials good for? Philosophical Studies, 147, 59–70.
Cartwright, N. (2011). A philosopher’s view of the long road from RCTs to effectiveness. Lancet, 377, 1400–1401.
Cochrane, A. L. (1972). Effectiveness and efficiency: Random reflections on health services. London: Nuffield Provincial Hospitals Trust.
DeVries, J. G., & Berlet, G. C. (2010). Understanding levels of evidence for scientific communication. Foot & Ankle Specialist, 3, 305–309.
Doll, R., & Hill, A. B. (1950). Smoking and carcinoma of the lung: Preliminary report. British Medical Journal, 2, 739–748.
Doll, R., & Hill, A. B. (1964). Mortality in relation to smoking: Ten years’ observations of British doctors. British Medical Journal, 1, 1399–1410.
Feinman, R. D. (2009). Intention-to-treat. What is the question? Nutrition and Metabolism, 6, 1.
Fisher, R. A. (1925). Statistical methods for research workers. Edinburgh: Oliver and Boyd.
Gadenne, V. (2013). External validity and the new inductivism in experimental economics. Rationality Markets and Morals, 4, 1–19.
Grahame-Smith, D. (1995). Evidence based medicine: Socratic dissent. British Medical Journal, 310, 1126–1127.
Greenland, S. (2000). Instrumental variables for epidemiologists. International Journal of Epidemiology, 29, 722–729.
Greenland, S., & Pearl, J. (2011). Causal diagrams. In M. Lovric (Ed.), International encyclopedia of statistical science (pp. 208–216). Berlin: Springer.
Gupta, S. K. (2011). Intention-to-treat concept: A review. Perspectives in Clinical Research, 2, 109–112.
Hernán, M. A., & Hernández-Diaz, S. (2012). Beyond the intention to treat in comparative effectiveness research. Clinical Trials, 9, 48–55.
Hernán, M.A., Alonso, A., Logan, R., Grodstein, F., Michels, K. B., Stampfer, M. J., Willett, W. C., Manson, J. E., & Robins, J. M. (2008). Observational studies analyzed like randomized experiments: An application to postmenopausal hormone therapy and coronary heart disease. Epidemiology, 19, 766–779.
Hernán, M. A., Hernández-Diaz, S., & Robins, J. M. (2013). Randomized trials analyzed as observational studies. Annals of Internal Medicine, 159, 560–562.
Higgins, J. P. T., & Green, S. (Eds.). (2011). Cochrane handbook for systematic reviews of interventions (V5.1.0). The Cochrane Collaboration. Accessed August 15, 2016. http://handbook.cochrane.org/
Hill, A. B. (1955). Principles of medical statistics (6th ed.). London: Lancet.
Hill, A. B. (1990). Suspended judgment: Memories of the British Streptomycin Trial in tuberculosis. The first randomized clinical trial. Controlled Clinical Trials, 11, 77–79.
Ho, P. M., Peterson, P. N., & Masoudi, F. A. (2008). Evaluating the evidence. Is there a rigid hierarchy? Circulation, 118, 1675–1684.
Hoffmann, J., Schirra, T., Lo, H., Neeb, L., Reuter, U., & Martus, P. (2015). The influence of weather on migraine—are migraine attacks predictable? Annals of Clinical and Translational Neurology, 2, 22–28.
Hudgens, G., Gilbert, P. B., & Self, S. G. (2004). Endpoints in vaccine trials. Statistical Methods in Medical Research, 13, 1–26.
ICH. (1998). Guidance for industry: E9 Statistical principles for clinical trials. Rockwell: US Department of Health and Human Services, Food and Drug Administration. Accessed August 15, 2016, from http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073137.pdf
King, M., Nazareth, I., Lampe, F., Bower, P., Chandler, M., Morou, M., Sibbald, B., & Lai, R. (2005). Impact of participant and physician intervention preferences on randomized trials. JAMA, 293, 1089–1099.
Lewis, J. A., & Machin, D. (1993). Intention to treat—who should use ITT? British Journal of Cancer, 68, 647–650.
Montedori, A., Bonacini, M. I., Casazza, G., Luchetta, M. L., Duca, F. C., & Abraha, I. (2011). Modified versus standard intention-to-treat reporting. Trials, 12, 58.
Morabia, A. (2013). Hume, Mill, Hill, and the sui generis epidemiologic approach to causal inference. American Journal of Epidemiology, 178, 1526–1532.
MRC Medical Research Council Streptomycin in Tuberculosis Trials Committee. (1948). Streptomycin treatment of pulmonary tuberculosis. British Medical Journal, 2, 769–783.
Newell, D. (1992). Intention-to-treat analysis: Implications for quantitative and qualitative research. International Journal of Epidemiology, 21, 837–884.
Neyman, J. (1923). On the application of probability theory to agricultural experiments. Essay on principles (Section 9). Statistical Science, 5, 465–472.
Neyman, J., & Pearson, E. S. (1933). On the problem of the most efficient tests of statistical hypotheses. Philosophical Transactions of the Royal Society of London Series A, 231, 289–337.
Popper, K. (1935). Logik der Forschung. Wien: Springer.
Robins, J. M., & Greenland, S. (1994). Adjusting for differential rates of PCP prophylaxis in high- versus low-dose AZT treatment arms in and AIDS randomized trial. Journal of the American Statistical Association, 89, 737–749.
Rosenbaum, P., & Rubin, D. B. (1983). The central role of the propensity score in observational studies for causal effects. Biometrika, 70, 41–55.
Rubin, D. B. (1974). Estimating causal effects of treatments in randomized and nonrandomized treatments. Journal of Education & Psychology, 66, 688–701.
Rothman, K. J. (2014). Six persistent research misconceptions. Journal of General Internal Medicine, 29, 1060–1064.
Sackett, D. L. (1989). Rules of evidence and clinical recommendations on the use of antithrombotic agents. Chest, 95(Suppl 2), 2–4.
Sackett, D. L., & Wennberg, J. E. (1997). Choosing the best research design for each question. BMJ, 315, 1636.
Salsburg, D. (1994). Intent to treat: The reduction ad absurdum that became gospel. Pharmacoepidemiology and Drug Safety, 3, 329–335.
Schafer, J. L., & Kang, J. (2008). Average causal effects from nonrandomized studies: A practical guide and simulated example. Psychological Methods, 13, 279–313.
Schulz, K. F., Altman, D. G., & Moher, D. (2010). CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. Journal of Clinical Epidemiology, 63, 834–840.
Seligman, M. E. (1995). The effectiveness of psychotherapy. The Consumer Reports study. American Psychologist, 50, 965–974.
Sheiner, L. B. (1991). The intellectual health of clinical drug evaluation. Clinical Pharmacology & Therapeutics, 50, 4–9.
Steckler, A., & McLeroy, K. R. (2008). The importance of external validity. American Journal of Public Health, 98, 9–10.
Tannen, R. L., Weiner, M. G., Xie, D., & Barnhart, K. (2008). Perspectives on hormone replacement therapy: The Women’s Health Initiative and new observational studies sampling the overall population. Fertility and Sterility, 90, 258–264.
Ten Have, T. R., Normand, S. L. T., Marcus, S. M., Brown, C. H., Lavori, P., & Duan, N. (2008). Intent-to-treat vs. non-intent-to-treat analyses under treatment non-adherence in mental health randomized trials. Psychiatric Annals, 38, 772–783.
Tolman, E. C. (1932). Purposive behavior in animals and men. New York: Appleton.
Victor, T. W., Hu, X., Campbell, J. C., Buse, D. C., & Lipton, R. B. (2010). Migraine prevalence by age and sex in the United States: A life-span study. Cephalalgia, 30, 1065–1072.
Weitkunat, R., Lee, P. N., Baker, G., Sponsiello-Wang, Z., González-Zuloeta Ladd, A. M., & Lüdicke, F. (2015). A novel approach to assess the population health impact of introducing a Modified Risk Tobacco Product. Regulatory Toxicology and Pharmacology, 72, 87–93.
Weitkunat, R., Baker, G., & Lüdicke, F. (2016). Intention-to-treat analysis but for treatment intention: How should consumer product randomized controlled trials be analyzed? International Journal Statistics Medical Research, 5, 90–98.
Wennberg, J. E., Barry, M. J., Fowler, F. J., & Mulley, A. (1993). Outcomes research, PORTs, and health care reform. Annals of the New York Academy of Sciences, 703, 52–62.
West, S. G., Duan, N., Pequegnat, W., Gaist, P., Des Jarlais, D. C., Holtgrave, D., Szapocznik, J., Fishbein, M., Rapkin, B., Clatts, M., & Mullen, P. D. (2008). Alternatives to the randomized controlled trial. American Journal of Public Health, 98, 1359–1366.
Worrall, J. (2007). Why there’s no cause to randomize. British Journal for the Philosophy of Science, 58, 451–488.
Zelen, M. (1990). Randomized consent designs for clinical trials: An update. Statistics in Medicine, 9, 645–656.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Weitkunat, R. (2017). Clinical Research-Based Product Assessment. In: Emilien, G., Weitkunat, R., Lüdicke, F. (eds) Consumer Perception of Product Risks and Benefits. Springer, Cham. https://doi.org/10.1007/978-3-319-50530-5_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-50530-5_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-50528-2
Online ISBN: 978-3-319-50530-5
eBook Packages: Economics and FinanceEconomics and Finance (R0)