
Abstract
Directed evolution of enzymes provides a prolific source of biocatalysts for asymmetric reactions in organic chemistry and biotechnology. Nowadays, the real challenge in this research area is the development of mutagenesis methods and strategies which ensure the formation of small and highest-quality mutant libraries requiring minimal screening effort. This chapter constitutes a critical analysis of recent developments. Saturation mutagenesis at sites lining the binding pocket using highly reduced amino acid alphabets has emerged as the superior approach for evolving stereoselectivity.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
3.1 Introduction
Asymmetric catalysis stands at the heart of modern synthetic organic chemistry. The three options are chiral transition metal catalysts, organocatalysts, and enzymes. The latter have been applied in synthetic organic chemistry for a century, but biocatalysis has not been accepted as a routine technique for several reasons. However, during the last two decades, notable progress has been made in bioprocess development, reactor design, downstream processing, immobilization, improved expression systems, and genome mining for identifying new enzymes [1]. Nevertheless, serious problems persisted due to the following often observed limitations:
-
Poor or wrong stereoselectivity
-
Limited substrate scope
-
Insufficient activity
The so-called rational design based on appropriate site-specific mutagenesis has been shown to be successful in some cases [2], but directed evolution has clearly emerged as the more general and reliable approach [3]. As in genetic optimization of thermostability [4], three basically different gene mutagenesis methods can be applied in order to enhance or invert stereoselectivity: error-prone polymerase chain reaction (epPCR), saturation mutagenesis, and/or DNA shuffling [3, 5]. The first example of directed evolution of an enantioselective enzyme concerned the hydrolytic kinetic resolution of rac-1, catalyzed by the lipase from Pseudomonas aeruginosa (PAL) (Scheme 3.1) [6]. WT PAL leads to low enantioselectivity slightly favoring the formation of (S)-2, the selectivity factor amounting to only E = 1.4.

Model reaction used in the first study of directed evolution of a stereoselective enzyme [6]
Four cycles of epPCR at low mutation rate with introduction of a single point mutation in each round enhanced enantioselectivity to E = 11 (S). Since the fifth cycle resulted in marginal improvement (E = 15), which is far from practical application, different mutagenesis strategies were developed. The combination of epPCR, saturation mutagenesis, and DNA shuffling afforded a variant characterized by six point mutations, showing a selectivity factor of E = 51 and a 250% increase in activity [7]. Only one of the mutations occurred near the active site, five being remote. A theoretical analysis based on QM/MM showed a relay mechanism to be operating. More importantly, it was predicted that only two of the six mutations are necessary for high enantioselectivity. Indeed, the respective double mutant proved to be even more effective (E = 62) [8].
These observations demonstrated that the genetic approach utilizing epPCR, saturation mutagenesis, and DNA shuffling is successful, but not efficient, a great deal of time-consuming screening being necessary (50,000 transformants). It was also possible to invert enantioselectivity in favor of (R)-2, but this also involved excessive screening [9]. At the time, several other groups joined efforts in generalizing directed evolution of stereoselectivity using the same strategies, as summarized in a 2004 review [10]. However, efficacy was not a focal point of research. Since the screening step is the bottleneck in the overall directed evolution process, methods and strategies for generating smaller and smarter libraries had to be developed.
After several years of research using PAL and other enzymes, saturation mutagenesis at sites lining the binding pocket as part of the combinatorial active-site saturation test (CAST) emerged as the optimal strategy (Scheme 3.2a) [11].

(a) Systematization of CASTing; A, B, C, etc. denote potential randomization sites, each comprising one, two, or more residues lining the binding pocket. (b) Two-, three-, and four-site ISM schemes
CAST is a convenient acronym to distinguish it from saturation mutagenesis at other (remote) sites for different purposes. When the “hits” in initial CAST libraries still display insufficient enantioselectivity, a recursive process is recommended: iterative saturation mutagenesis (ISM) [12]. Scheme 3.2b shows the case of two-, three-, and four-site ISM systems involving two, six, and 24 pathways. It is not necessary to explore all theoretically possible pathways, but some may be more productive than others. Since saturation mutagenesis at large randomization sites requires excessive screening for 95% library coverage, reduced amino acid alphabets were introduced [13]. In order to remind the reader of the relationship between the size of a randomization site, the nature of the amino acid alphabet, and the screening effort for 95% library coverage, Table 3.1 is included here which illustrates the difference between NNK and, e.g., NDT codon degeneracy, which encode 20 and 12 canonical amino acids, respectively [5]. Any other reduced amino acid alphabet that the researcher may want to use can be analyzed statistically in the same manner using the CASTER computer aid [5], which is based on the Patrick/Firth algorithm [14]. The Nov metric can also be used, in this case identifying the nth best mutant [15]. CAST/ISM should be guided by X-ray structures (or homology models) and sequence data. Initial NNK-based saturation mutagenesis at individual CAST positions, requiring the screening of only one 96-format microtiter plate, also provides information for choosing a reduced amino acid alphabet in subsequent mutagenesis experiments.
The lessons learned from these methodological developments, reported in a series of studies using different enzyme types [3f, 5], were then used as a guide in the final directed evolution study of PAL for comparison purposes [16]. It was discovered that CAST/ISM provides a notably improved triple mutant showing E = 594 (S) in the model reaction involving the kinetic resolution of rac-1, while requiring the screening of less than 10,000 transformants. This highlights the progress in methodology development. In the ISM study, three CAST sites A, B, and C, were designed, and NDT codon degeneracy was applied, leading to the highly improved triple mutant [16]. This variant has no superfluous mutations. The origin of the unusually high degree of enantioselectivity was traced on a molecular level to strong cooperative mutational effects. Such synergistic effects (more than additivity) were later found in other ISM-based studies as well [17]. In an independent study utilizing a galactosidase as the enzyme, saturation mutagenesis was likewise shown to be more efficient than DNA shuffling [18].
Since the publication of the best results concerning PAL, further progress in methodology development has been achieved [5, 19, 20]. The primary focus was placed on utilizing the smallest possible reduced amino acid alphabets, again for the purpose of minimizing screening while maximizing library quality. In doing so, two different strategies can be applied (Scheme 3.3). According to strategy 1, one and the same reduced amino acid alphabet is used for the whole CAST randomization site in a single experiment. In contrast, strategy 2 calls for a different reduced amino acid alphabet for each position of a multi-residue site, a single experiment also being involved. In both cases, ISM can be applied for further optimization.

Two strategies for applying saturation mutagenesis in order to manipulate stereoselectivity
Nowadays, automated GC or HPLC can handle typically 2000–3000 transformants within a few days. An on-plate pretest for activity is nevertheless recommended, the much smaller number of hits then being analyzed for stereoselectivity by chiral GC or HPLC. The question whether to choose a reduced amino acid alphabet such as NDT in combination with, e.g., two-residue randomization sites followed by ISM, or to opt for a much smaller reduced amino acid alphabet encoding only one, two, or three amino acids in combination with larger randomization sites, e.g., four to ten residues, has been addressed [20–22]. Triple code saturation mutagenesis (TCSM) using three- or four-residue CAST randomization sites appears to be the strategy of choice as shown by several recent studies [22]. In these cases, the initial CAST libraries often harbor stereoselective variants that fulfill all requirements for practical applications or require only one ISM step for final fine-tuning. Nevertheless, more experience is needed for final assessments. When applying CAST/ISM, several guidelines are recommended:
-
Library design by the CASTER computer aid (http://www.kofo.mpg.de/en/research/biocatalysis) or the GLUE-IT metric (http://guinevere.otago.ac.nz/cgi-bin/aef/glue-IT.pl), both available free of charge
-
Guidance by structural, mechanistic, and (consensus) sequence data [20–22]
-
Use of an on-plate pretest for activity followed by chiral GC or HPLC analysis for enantioselectivity [21]
-
Application of the quick quality control [23a] or quantitative Q-values [23b] in order to avoid screening something that does not exist
-
Application of pooling techniques for reducing the screening effort [23a, 24]
-
Techno-economical analysis which considers, inter alia, the number, quality, and cost of primers used in designing and generating mutant libraries [25]
The CAST/ISM-based approach has proven to be particularly efficient, fast, and reliable [5, 19, 20], but other gene mutagenesis methods continue to be used. Unfortunately, comparative experiments are generally not made. In some ISM-based studies, a final round of epPCR was added for further (small) improvements, as in the case of directed evolution of a glycosidase [26]. In other investigations, only epPCR and/or DNA shuffling were employed. In the sections that follow, selected recent examples of directed evolution of stereoselectivity using different gene mutagenesis techniques and strategies are critically analyzed.
3.2 Cytochrome P450 Monooxygenases
Cytochrome P450 monooxygenases (CYPs) catalyze several synthetically useful reaction types, including asymmetric oxidative hydroxylation R-H → R-OH, olefin epoxidation, and sulfoxidation. Several reviews of CYP protein engineering have appeared [27]. The first study featuring the manipulation of enantioselectivity of a cytochrome P450 monooxygenase while maintaining essentially complete regioselectivity concerned the P450-pyr catalyzed oxidative hydroxylation of N-benzyl-pyrrolidine (4) with formation of product 5 (Scheme 3.4) [28]. WT P450-pyr shows complete regioselectivity in favor of the 3-hydroxy product, but with a mere 43% ee (S). The substrate appears to be bound in two energetically similar poses within reach of the catalytically active high spin heme-Fe=O (Cpd I) in the large binding pocket. The mechanism is known to involve a radical abstraction of the H-atom with formation of a carbon-centered radical followed by rapid C-O bond formation. The ideal O-H-C angle has been calculated to be about 130ο [29].

Model reaction used in the first case of directed evolution of a P450 monooxygenase leading to high regio- and stereoselectivity [28]
Using a homology model, 17 residues were identified within 5 Å of the heme-docked substrate [28a]. With the exception of residues C366 and G256, they were subjected individually to saturation mutagenesis using NNK codon degeneracy. This would require for 95% library coverage the screening of only 94 transformants in each case. In fact, an excess of 180 transformants were screened to ensure even higher coverage [28a]. Whereas variant F403L improves (S)-selectivity to 65% ee, a single point mutation (N100S) induces the reversal of enantioselectivity. In order to enhance (R)-selectivity, a simplified version of ISM was applied by employing mutant N100S as the template in saturation mutagenesis at other CAST sites. The best double mutant proved to be N100S/T186I with an enantioselectivity of 83% ee (R) and no trade-off in regioselectivity.
In a subsequent study, (S)-selectivity was improved by exploring more of protein sequence space [28b]. First, the crystal structure of WT P450-pyr was determined and used as a guide in choosing CAST sites. Twenty residues, A77, I82, I83, L98, P99, N100, I102, A103, S182, D183, T185, T186, L251, V254, G255, D258, T259, L302, M305, and F403, were targeted using the same simplified ISM-based strategy. Only nine initial libraries had to be generated, since the remaining 11 had already been obtained in the previous study. The best variant I83H/M305Q/A77S was obtained in three rounds of iterative saturation mutagenesis (ISM), showing an ee-value of 98% (S) at fully maintained regioselectivity and little trade-off in activity [28b]. It would be interesting to test TCSM [22] in this reaction.
Particularly challenging goals arise when regio- and enantioselectivity need to be evolved. In a study dedicated to solving this problem, P450-BM3 was employed [30]. It was found that WT P450-BM3 catalyzes the hydroxylation of cyclohexene-1-carboxylic acid methyl ester (6) with insufficient regioselectivity (84%) and poor enantioselectivity (34%) ee in slight favor of (R)-7) (Scheme 3.5).

Model reaction used in the first case of directed evolution of a P450 monooxygenase leading to high regio- and stereoselectivity with optional formation of either enantiomeric product [30]
In order to achieve practical levels of regioselectivity as well as >95% (R)- and (S)-selectivity on an optional basis, ISM was applied. On the basis of the crystal structure of P450-BM3, a total of 24 CAST residues were identified (first- and second-sphere residues) (Fig. 3.1) [30].

The 24 P450-BM3 residues considered for saturation mutagenesis. They were assigned to three categories marked in green (residues closest to the heme), blue (residues further away but still lining the binding pocket), and yellow (residues at entrance to the large binding pocket) [30]
NNK-based saturation mutagenesis was initially applied at 23 of the 24 positions, leading to a limited set of improved mutants with enhanced and reversed enantioselectivity. This information formed the basis of ISM optimization. For example, the gene of the best (R)-selective variant was chosen as the template for saturation mutagenesis at other sites which had also favored (R)-selectivity, and the analogous procedure was applied for reversing (S)-selectivity. This strategy provided selective variants (97–98% ee) favoring (R)- and (S)-7, respectively, and displaying regioselectivities in the range of 93–98% [30].
ISM was also applied to P450-BM3 catalyzed oxidative hydroxylation of steroids as part of “late-stage oxidation” [31]. For example, testosterone was used as the substrate, for which 2ß- and 15ß-selective variants were evolved showing >95% regio- and diastereoselectivity.
Yet another example of late-stage oxidative hydroxylation concerns P450-BM3 catalyzed reactions of the monocyclic diterpenoid ß-cembrenediol (11), which is essentially not accepted by the WT (2% conversion) (Scheme 3.6) [32]. At the time of this project, a number of other protein engineering studies utilizing various structurally different substrates were available, the mutational data being of significant help in the new endeavor [27]. This is an important point, because it is logical to learn from past experience and not to start from “scratch” in each new project. For example, it was known that residue F87 is a sensitive position, smaller amino acids generally being necessary to enable substrate acceptance of sterically demanding compounds because the phenyl group of F87 prevents complete access to the catalytically active heme-Fe=O (Cpd I). Therefore, known variants F87A and F87G [27] were tested first, which indeed led to acceptable levels of activity. These were then used as templates for site-specific mutagenesis at selected first-sphere CAST residues A74, L75, V78, I263, A264, and L437. Iterative site-specific mutagenesis was performed in several rounds, in each case a new point mutation being introduced [32]. A total of 29 variants were produced by this technique, which resembles ISM without the need to screen libraries. Structure-guided successive site-specific mutagenesis with the creation of minimally sized libraries constitutes a fusion of rational design and directed evolution. Whereas the particular choice of the mutations limits structural diversity drastically, the results proved to be of practical interest in this project. Several variants were generated in this way, F87A/I263L catalyzing complete regioselective hydroxylation at position C-9 with an 89:11 diastereomeric ratio. The triple mutant L75A/V78A/F87G enables hydroxylation at position C-10 (97% regioselectivity) with a 74:26 diastereomeric ratio. Assignment of the absolute configuration at the new stereogenic centers was not reported [32]. This study nicely shows that very small semi-rationally designed mutant libraries may well suffice, provided sufficient previous knowledge of mutational effects is available. Indeed, after years of protein engineering of P450-BM3, the huge mutational data serves as a convenient guide when targeting new substrates using rational design or directed evolution.

P450-BM3 catalyzed oxidative hydroxylation of the diterpenoid ß-cembrenediol [32]
Another interesting example of directed evolution of a CYP concerns the late-stage regio- and stereoselective hydroxylation of the antimalaria drug artemisinin catalyzed by P450-BM3 (Scheme 3.7) [33]. Here again a semi-rational approach was implemented using saturation mutagenesis at first-sphere CAST sites, this time based on initial P450 fingerprinting followed by fingerprint-driven reactivity predictions and final ISM experiments. WT P450-BM3 does not accept the substrate. First, 125,000 transformants were screened for activity using not the actual substrate artemisinin, but five semisynthetic chromogenic probes, which gave rise to 1950 active variants that accept such sterically demanding substrates (criterion: “>10% of parent enzyme activity on at least one of the fingerprint probes”), and 522 functionally unique variants (criterion: “larger than 20% variation on at least one of the five fingerprint components compared to the parent enzyme and any other member of the library”) [33]. After correlating the P450-BM3 fingerprints with the actual artemisinin reactivity, 75 variants remained and were tested as catalysts for oxidation of the real substrate by HPLC analysis. The best hit was variant FL#62, which was found to have 16 point mutations. This work was then followed by several ISM experiments. The result of all of these efforts is summarized in Scheme 3.7 [33]. Whereas the “parent” enzyme showed 83% C7(S)-, 10% C7(R)-, and 7% C6a-selectivities, notable improvements were achieved in the final saturation mutagenesis experiments. The tendency to hydroxylate at C6 and C7 was maximized by directed evolution. If for some reason a different position were to be the target, then the challenging question of how to achieve such regioselectivity would arise.

Oxidative hydroxylation of artemisinin by P450-BM3 mutants [33]
In the directed evolution of CYPs, as in other enzyme types, a clear trend to utilize saturation mutagenesis has emerged [27, 34]. However, some researchers in recent CYP studies rely on epPCR and site-specific mutagenesis. An example is the evolution of P450-BM3 mutants that hydroxylate the contraceptive drug norethisterone at the 15ß- or 16ß-position [35a]. The combination of epPCR and DNA shuffling has also been used, as in activity optimization of the 13-hydroperoxide lyase CYP74B; the products are used for the production of C6-aldehydes [35b].
To date, the concept of P450-catalyzed late-stage oxidative hydroxylation of natural products or synthetic compounds suffers from the lack of predictability as to where hydroxylation will occur. When designing a synthetic pathway in natural product synthesis, it is generally unclear whether P450-catalyzed hydroxylation will occur at the desired position. In such a situation, the initial library must be diverse enough to harbor variants that provide many different regioisomers. If the desired product is formed to a small extent, then the respective mutant can be chosen for further mutational improvements with the aim of turning the minor into the major product.
3.3 Baeyer-Villiger Monooxygenases
The first directed evolution study of a Baeyer-Villiger monooxygenase (BVMO) concerned the optimization of cyclohexanone monooxygenase (CHMO) as the catalyst in the oxidative desymmetrization of 4-hydroxycyclohexanone [36]. One of the best variants evolved by epPCR, F345S, was then used in the desymmetrization of structurally different ketones (Table 3.2) [37]. It can be seen that in essentially all cases, high enantioselectivity as well as chemoselectivity was achieved without having to perform additional mutagenesis experiments.
These results are impressive when utilizing whole cells, but CHMO suffers from insufficient thermostability. The discovery of the unusually robust phenyl acetone monooxygenase (PAMO) aroused a great deal of interest, although its substrate scope was shown to be very narrow [38]. Such substrates as cyclohexanone or derivatives thereof are not accepted by this BVMO, apparently because the binding pocket is too small to accommodate such compounds. In order to solve this problem, a series of CAST-based directed evolution studies have appeared [39].
A bioinformatics-based study of PAMO as the catalyst in oxidative kinetic resolution of compounds 14a-b deserves special attention for three reasons (Scheme 3.8) [40]: (1) It not only utilized structural data for choosing appropriate CAST sites, but also (2) sequence alignment information in order to derive optimal reduced amino acid alphabets, and (3) a different codon degeneracy according to strategy 2 as outlined in Scheme 3.3 (Introduction). Ketone 14a was used in all screening assays, the best evolved variant then being tested in the oxidative kinetic resolution of 14b as well [40].

Oxidative kinetic resolution catalyzed by mutants of the Baeyer-Villiger monooxygenase PAMO [40]
Guided by the PAMO crystal structure [38b], four residues in loop 441–444 next to the binding pocket were chosen as CAST sites. NNK-based randomization of a four-residue CAST site would require the screening of 3.1 million transformants for 95% library coverage, while NDT codon degeneracy would still call for excessive screening( ≈ 62,000 transformants) (Table 3.1, Introduction). Therefore, eight Baeyer-Villiger monooxygenases were aligned, the loop region 441–444 being of interest (Scheme 3.9) [40]. A limited number of amino acids are conserved at the four positions: Ser and Ala (position 441); Ala, Val, Gly, and Leu (position 442); Leu, Phe, Gly, and Tyr (position 443); and Ser, Ala, Cys, and Thr (position 444). Consequently, these amino acids were used as building blocks at the respective positions of the four-residue randomization site, these reduced amino acid alphabets minimizing the screening effort drastically.

Sequence alignment of eight BVMOs (loop 441–444 in gray box) [40]
Codon degeneracies were then designed for matching the amino acids occurring at these four positions while also introducing a limited number of additional amino acids for randomization experiments in order to minimize primer costs and enhance diversity (Table 3.3) [40]. WT amino acid is maintained throughout. Interestingly, at position 441, KCA codon degeneracy correlates with the introduction of only one new amino acid (Ala). At positions 442–444, structural diversity was designed to be higher.
After screening a mere 1700 transformants (95% library coverage would require 2587), several active variants were discovered, PAMO mutant Ser441Ala/Ala442Trp/Leu443Tyr/Ser444Thr displaying the highest activity and enantioselectivity (E = 70 in favor of (R)-15a) (Scheme 3.8) [40]. This variant proved to be an even better catalyst for the oxidation of the p-chloro-derivative 14b (E >200). As in the case of 14a, WT PAMO does not accept this substrate. It was concluded that bioinformatics can be used to define a reduced amino acid alphabet and that a different designed reduced amino acid alphabet can well be effective at each position within a multi-residue randomization site in a single saturation mutagenesis experiment (strategy 2 in Scheme 3.3). This approach was later employed in the directed evolution of other enzyme types [21b, 22].
It should be mentioned that following these and other CAST-based studies of BVMOs [39], examples of epPCR as an alternative were reported. For example, BmoFI from Pseudomonas fluorescens DSM 50106 was used as the catalyst in the oxidative kinetic resolution of rac-16 with preferential formation of (S)-17 (Scheme 3.10) [41]. Enantioselectivity of the WT ranges between E = 55 (at small scale) and E = 71 (growing E. coli cells in shake flasks). Application of epPCR and screening 3500 transformants provided several improved mutants displaying E = 77–92. Upon combining the respective point mutations, an excellent variant was identified, His51Leu/Ser136Leu displaying a selectivity factor of E = 86 [41].

Oxidative kinetic resolution catalyzed by mutants of the Baeyer-Villiger monooxygenase BmoFI [41]
In a very different approach not relying on CASTing, epPCR, or DNA shuffling, a theoretically predicted remote two-residue site comprising positions 93 and 94 in PAMO was subjected to saturation mutagenesis with the expectation of an allosteric effect in the absence of an effector molecule [42]. Several four-substituted cyclohexanone derivatives (methyl, ethyl, n-butyl, tert -butyl) were chosen as substrates which are not accepted by WT PAMO. This procedure proved to be surprisingly successful, the double mutant Gln93Asn/Pro94Asp showing 97–98% (R)-selectivity for the methyl, ethyl, and n-butyl derivatives in desymmetrization reactions. The tert-butyl derivative was not accepted due to steric factors. Instead of an effector molecule inducing allostery, it is the mutational change that is causing a conformational reorganization at the binding site. Calculations of Karplus-type covariance maps [43] of the double mutant versus WT PAMO showed that the expected conformational motions were indeed occurring [42]. Deconvolution of Gln93Asn/Pro94Asp showed that the single mutants Gln93Asn and Pro94Asp are inactive, signaling pronounced cooperative mutational effects.
To date, the concept of focusing on mutationally induced remote allosteric effects has not been applied to other substrates. In further optimization, the double mutant Gln93Asn/Pro94Asp could be used as a template for CASTing. However, researchers have preferred to concentrate from the very beginning on CAST-based ISM when new projects are initiated. For example, using this strategy, PAMO was evolved as the catalyst in asymmetric sulfoxidation reactions using prochiral thioether 18 as the substrate (Scheme 3.11) [22a]. WT PAMO leads to the preferential formation of (S)-19 with notable selectivity (90% ee). The primary goal was to evolve inverted enantioselectivity in favor of (R)-19.

Asymmetric sulfoxidation catalyzed by mutants of the Baeyer-Villiger monooxygenase PAMO [22a]
Six potential randomization residues were first identified on the basis of the PAMO crystal structure [38b] and past studies which identified “hot spots”: P440, A442, and L443 (as CAST loop residues) and V54, I67, and Q152 (as traditional CAST sites). One possible strategy would be to group them into three two-residue randomization sites and to apply ISM. In this study, a different procedure was chosen [22a]. In exploratory experiments, residues V54, I67, Q152, and A442 were chosen for individual saturation mutagenesis. Instead of applying traditional NNK codon degeneracy, the “smart-intelligent” library construction according to Tang [44] was used. Four pairs of primers with degenerate codons of NDT (encoding 12 amino acids N, S, I, H, R, L, Y, C, F, D, G, and V), codon degeneracy VMA (encoding six amino acids E, A, Q, P, K, T), codon degeneracy ATG (encoding M), and codon degeneracy TGG (encoding W) were considered at the target sites [22a]. They were mixed in a ratio of 12:6:1:1, and for each library creation, only 60 transformants had to be screened for 95% coverage. Whereas saturation mutagenesis at V54 failed to provide any hits, the other libraries harbored improved hits.
Following these initial experiments, a two-site ISM scheme starting from WT PAMO was designed involving site A (P440/A442/L443) with reduced amino acid alphabets and site B (I67 as a hot spot identified earlier) with 20 amino acids. The reduced amino acid alphabet applied at P440 involved three building blocks as the components of a triple code in addition to the WT amino acid, Tyr, Leu, and Phe. This is an example of triple code saturation mutagenesis (TCSM) [22a] as part of strategy 2 (Scheme 3.3). The results of this minimal search in protein sequence space are summarized in Scheme 3.12 [22a]. It can be seen that pathway WT → A → B provided two variants showing highly reversed enantioselectivity: ZGZ-1 (I67C/P440F/A442F/L443D (92% ee) and ZGZ-2 (I67Q/P440F/A442N/L443I (95% ee) in favor of (R)-19. The opposite pathway WT → B → A was also successful, leading to variant I67A/P440Y/A442V/L443I with 94% ee (R). Overoxidation with formation of the sulfone 20 occurred to only a minimal degree.

Two ISM pathways in the directed evolution of PAMO as catalyst in the asymmetric sulfoxidation of 18, WT → A → B providing two (R)-selective variants ZGZ-1 and ZGZ-2 and WT → B → A leading to an equally (R)-selective variant ZGZ-3 [22a]
Complete reversal of enantioselectivity is impressive, because the change in energy ΔΔG# in going from WT PAMO (90% ee, S) to variant ZGZ-2 (95% ee, R) amounts to 3.9 Kcal/mol. Deconvolution of this highly (R)-selective quadruple mutant ZGZ-2 (I67Q/P440F/A442N/L443I) led to the surprising discovery that the respective single mutants are all (S)-selective: I67Q (69% ee), P440F (97% ee), A442N (69% ee), and L443I (98% ee) [22a]. If these four (S)-selective single mutants had been generated separately by other means, few researchers would combine them in order to generate the opposite (R)-selectivity! This kind of synergistic nonadditive effects continues to be observed whenever time and effort are invested in deconvolution. It is an indication of the efficacy of ISM [17].
Exhaustive deconvolution was performed, meaning the generation of variants formed by combining the four point mutations in the form of all theoretically possible double and triple mutants. It allowed a fitness pathway landscape to be constructed, comprising 4! = 24 experimental pathways which lead from (S)-selective WT PAMO to (R)-selective variant ZGZ-2 (Fig. 3.2) [22a]. Six of the 24 pathways have no local minima along the respective trajectories, meaning the absence of libraries which contain no variants with improved enantioselectivity. Eighteen pathways are characterized by at least one local minimum along the evolutionary trajectory. Analysis of the intermediate stages of all 24 pathways revealed strong cooperative mutational effects. Thus, much can be learned from deconvolution studies of this kind. In combination with MD/docking computations, they throw light on the origin of stereoselectivity while also illuminating the efficacy of ISM. It should be noted that this type of “constrained” fitness landscape [22a, 45] is different from constructing all theoretically possible pathways of an ISM scheme, as was implemented experimentally in the case of a four-site ISM system with 24 pathways [46]. The latter has been termed “unconstrained” fitness pathway landscape [17, 46], in which every trajectory leads to a different result (see Sect. 3.5).

Fitness pathway landscape featuring 24 pathways which by necessity lead from the (S)-selective WT PAMO to the best (R)-selective mutant ZGZ-2 allowing the formation of (R)- 19 [22a]. The green line denotes a typical trajectory lacking any local minima, and the red one a trajectory characterized by at least one local minimum. The upward climb is associated with 3.9 kcal/mol
3.4 Lipases and Esterases
Directed evolution has been extensively applied to lipases and esterases in order to manipulate stereoselectivity, substrate scope, and activity [3]. As delineated in the Introduction, the lipase from Pseudomonas aeruginosa (PAL) is the most systematically studied stereoselective enzyme in the field of directed evolution. In the final PAL study [16], which included deconvolution experiments, ISM was applied to the original model reaction involving the hydrolytic kinetic resolution of rac-1 with preferential formation of (S)-2 (Scheme 3.1). Based on the crystal structure of PAL, a three-site ISM scheme comprising two-residue CAST sites A, B, and C was considered. Following exploratory mutagenesis experiments, the best pathway proved to be WT → B → A which provided the triple mutant 1B2 (Leu162Asn/Met16Ala/Leu17Phe) showing a selectivity factor of E = 94 (S) and notably enhanced activity: WT PAL (k cat = 37×10−3 s−1; k cat/K m = 43.5 s−1 M−1) versus variant 1B2 (k cat = 1374×10−3 s−1; k cat/K m = 4041 s−1 M−1) [16].
This result was achieved in the following way: In the first ISM step, site B (Leu159/Leu162) was randomized using NNK codon degeneracy, leading to single mutant Leu162Asn with E = 8 (S). It was employed as the template for DNT-based saturation mutagenesis at site A (Met16/Leu17), which like NDT involves 12 amino acids as building blocks. The ISM pathway is illustrated in Scheme 3.13 [16].

ISM pathway WT → B → A in the directed evolution of PAL as the catalyst in the hydrolytic kinetic resolution of rac-1 [16]
Scheme 3.13 seems to suggest that the second mutational introduction, Met16Ala/Leu17Phe, contributes most to the overall result. However, this assumes classical mutational additivity [17]. Therefore, deconvolution was performed by generating the double mutant Met16Ala/Leu17Phe and testing it in the model reaction. Surprisingly, it proved to be a poor catalyst with E = 2.6 (S). Thus, a strong cooperative mutational effect is operating. An MD/docking analysis uncovered the molecular basis of this epistatic synergism. It involves the creation of an extended H-bond network as a consequence of the interaction of Leu162Asn in concert with Met16Ala/Leu17Phe [16].
This study required the screening of less than 10,000 transformants at a time when subsequent optimization of saturation mutagenesis strategies and techniques such as triple code saturation mutagenesis (TCSM) [22b, c] were not yet available. It is likely that TCSM as applied to PAL would require even less screening.
PAL served as a useful model system to test various mutagenesis strategies in a comparative manner. However, for several reasons, it is not likely to become a lipase of practical utility in organic chemistry. The situation is very different in the case of Candida antarctica lipase B (CALB), one of the most popular enzymes in biocatalysis [1]. It has been employed in ISM-based directed evolution in order to expand substrate scope and to invert enantioselectivity [47].
The homolog CALA has also been subjected to ISM-based directed evolution in order to accept chiral phenyl-substituted carboxylic acid esters [48], but the bulkier analogs of the ibuprofen type were not accepted. Therefore, a different strategy was tested following strategy 2 (Scheme 3.3). The plan was to use a single amino acid as building block (in addition to WT) at most of the positions of a nine-residue site in a single mutagenesis experiment [21b]. The hydrolytic kinetic resolution of rac-21 was chosen for screening, and racemic esters 22–24 were also tested following the mutagenesis experiments (Scheme 3.14). The goal was enhancing activity and enantioselectivity while minimizing screening.

Model compounds used in the directed evolution study of CALA-catalyzed hydrolytic kinetic resolution [21b]
All experiments began with the use of the triple mutant F149Y/I150N/F233G as template, obtained in the earlier ISM-based study [48]. Substrate 21 was docked into the CALA binding pocket in the oxyanion form (tetrahedral intermediate at Ser184), leading to the identification of nine residues at the acyl binding region: positions 149, 150, 215, 221, 225, 233, 234, 237, and 431 (Fig. 3.3) [21b]. Other residues near this large CAST site were not considered because they are highly conserved as shown by an alignment analysis. In view of the PAMO study [40], this would not necessarily be mandatory. Structure-based decisions were made regarding the different reduced amino acid alphabets at the nine randomization positions.

View of CALA binding pocket harboring covalently bound substrate 21 as an oxyanion [21b]. Nine CAST residues with the respective reduced amino acid alphabet(s) used in saturation mutagenesis are shown, the original WT amino acids being underlined
Substrate 21 is sterically so demanding that it is not accepted by the triple mutant with acceptable rate. Thus, small amino acids were mostly chosen for saturation mutagenesis. In the earlier CALA study [48], mutations Phe149Tyr and Ile150Asn had been shown to be important for high enantioselectivity toward similar substrates. Therefore, at these positions, Tyr and Asn were chosen as the respective building blocks (Table 3.4) [21b]. At position 233, three amino acids were employed (in addition to WT). A certain degree of intuition was involved in some of the decisions.
A single highly condensed library was created using appropriately designed primers, followed by screening about 2400 transformants in the model reaction of rac-21 (≈90% library coverage). Only a few variants proved to be active toward substrate rac-21 [21b]. The best hit was a penta-substituted variant Thr221Ser/Leu225Val/Phe233Cys/Gly237Ala/Phe431Val in which four different amino acids were introduced at five different positions. It shows high (S)-stereoselectivity (E = 100). This and other CALA variants were tested in the hydrolytic kinetic resolution of the substrates 21–24, which likewise resulted in acceptable levels of enantioselectivity [21b]. The alternative of applying conventional NNK codon degeneracy encoding all 20 canonical amino acids would have required for 95% library coverage the screening of 1014 potentially enantioselective transformants. A limited number of deconvolution experiments revealed cooperative mutational effects. This suggested that the particular variant would not be accessible by ISM. The same strategy was later successfully applied to CALA as the catalyst in acylating kinetic resolution of chiral alcohols [49].
It can be concluded that both approaches to the use of reduced amino acid alphabets are successful, strategy 1 as well as strategy 2 as delineated in Scheme 3.3. It would be interesting to test strategy 1 using triple code saturation mutagenesis. Whether such a procedure would also allow reversal of enantioselectivity is currently a matter of speculation.
Esterases have also been targeted by directed evolution for improving stereoselectivity [3], recent studies utilizing various approaches including epPCR alone [50a], epPCR in combination with saturation mutagenesis [50b], saturation mutagenesis alone [50c, d], or site-specific mutagenesis in combination with saturation mutagenesis [50e]. One study is featured here which relied solely on epPCR. The carboxyl esterase from Rhodobacter sphaeroides (RspE) was subjected to three recursive rounds of epPCR at low mutation rate, the model reaction being the hydrolytic kinetic resolution of methyl mandelate (rac-25) with preferential formation of the carboxylic acid (R)-26 (Scheme 3.15) [50a]. WT RspE shows a selectivity factor of E = 3.1(R).

Model hydrolytic kinetic resolution catalyzed by mutants of the esterase RspE [50a]
In each epPCR cycle, 4000–6000 transformants were screened for activity using an on-plate pH-dependent color test followed by conventional ee-determination. In this way, it was possible to boost (R)-selectivity to E = 30.3 (Scheme 3.16) [50a]. This study is a new example of iterative epPCR in the successful attempt to evolve enhanced stereoselectivity of an esterase. Reversal of enantioselectivity was not reported, but the best mutant in the model reaction showing E = 30.3 was used in the kinetic resolution of structurally related substrates including acetates of chiral alcohols with selectivity factors of up to E = 92. It would be interesting to see how well CASTing/ISM would perform in this enzyme system.

3.5 Epoxide Hydrolases
Several recent protein engineering studies of epoxide hydrolases have contributed to methodology development in laboratory evolution [3f, 5]. The goal of one of them was the exploration of all 24 pathways of a four-site ISM scheme [46]. To date, it is the only case of complete exploration of such an ISM system. The hydrolytic kinetic resolution of rac-27 was chosen as the model reaction, catalyzed by mutants of the epoxide hydrolase from Aspergillus niger (ANEH) (Scheme 3.17). WT is a poor catalyst in this reaction (E = 4.6 in slight favor of (S)-28).

ANEH-catalyzed hydrolytic kinetic resolution used in the construction of a complete four-site ISM system featuring 24 evolutionary pathways [46]
Four CAST randomization sites were considered on the basis of the WT ANEH crystal structure, each comprising two residues. Subsequently all saturation mutagenesis libraries were constructed using NDT codon degeneracy encoding 12 amino acids (Phe, Leu, Ile, Val, Tyr, His, Asn, Asp, Cys, Arg, Ser, and Gly), which is a balanced “cocktail” of polar/nonpolar, charged/non-charged, hydrophobic/non-hydrophobic, and aromatic/nonaromatic amino acids as building blocks. All 24 pathways provided in the final saturation mutagenesis rounds notably improved variants characterized by different sequences [46]. Some pathways proved to be more productive than others. The 12 best pathways resulting in selectivity factors in the range E = 78–159 are shown in Scheme 3.18a; the 12 pathways leading to the least improved variants (E = 28–78) are pictured in Scheme 3.18b. These results suggest that if the researcher is faced with the question of choosing an appropriate ISM pathway, an arbitrary choice has a high probability of providing improved variants. This explains why in essentially all ISM studies reported thus far arbitrarily chosen pathways led to notably improved variants [5]. Nevertheless, superior variants may have been missed. It can therefore be concluded that ISM systems should be designed in a way that involves less pathways and therefore less mutant libraries, which correlates with a lower number of decisions for the researcher to make. Indeed, methodology development since the publication of this study has focused, inter alia, on step-economy [21, 22].

Experimental exploration of a complete 24-pathway ISM system involving the ANEH-catalyzed hydrolytic kinetic resolution of rac-27 (Scheme 3.17) [46]. (a) Portion of the 24-pathway ISM scheme featuring the 12 most productive pathways leading to ANEH variants displaying E = 78–159 (S); (b) portion of the 24-pathway ISM scheme showing the 12 least productive pathways providing ANEH variants with E = 28–78 (S)
Noteworthy is another feature of the experimental results collected in Scheme 3.18. In several ISM pathways, libraries occurred which failed to harbor any improved variants. This phenomenon signals a local minimum in the fitness landscape. Such “dead ends” may occur in any directed evolution project irrespective of the mutagenesis method [3]. In the present case, ISM was not abandoned, but an inferior mutant was used as the template in the subsequent saturation mutagenesis experiment at the next randomization site. This trick led to notably improved mutants. This unique way of escaping from a local minimum is reminiscent of neutral drift [4d, 51] or the Eigen concept of quasi-species [52a]. The latter has been invoked occasionally in directed evolution studies [52b].
Maximal step-economy would involve the generation of a single saturation mutagenesis library, which should be small and ideally harbor both (R)- and (S)-selective mutants. If the degree of stereoselectivity should not be fully satisfactory, one ISM round could then be undertaken for fine-tuning. Along these lines, the directed evolution of a different epoxide hydrolase was reported recently, namely, limonene epoxide hydrolase (LEH) as the catalyst in the model hydrolytic desymmetrization of cyclohexene oxide (29) with formation of (R,R)- and (S,S)-30 (Scheme 3.19) [21a]. WT LEH shows poor enantioselectivity with minimal preference for (S,S)-30, the enantiomeric ratio (er) amounting to a mere 48:52 (4% ee).

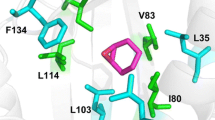
Based on the crystal structure of WT LEH, ten CAST residues were identified for saturation mutagenesis (Leu74, Phe75, Met78, Ile80, Leu103, Leu114, Ile116, Phe134, Phe139, and Leu147) [21a]. Tyr53 activates the substrate by forming an H-bond to the epoxide O-atom, Asp101 being mainly responsible for positioning water which initiates the rate-determining SN2 reaction with ring opening.
Saturation mutagenesis using NNK codon degeneracy (20-amino-acid alphabet) or NDT codon degeneracy (12-amino-acid alphabet) would require the screening of about 1015 or 1011 transformants for 95% coverage, respectively. The use the smallest amino acid alphabet, a single amino acid, would call for only ≈3000 transformants. This would reduce structural diversity dramatically, suggesting that such a strategy would fail. Nevertheless, it could be successful if the right decision is made concerning the choice of the amino acid. Such an approach constitutes single codon saturation mutagenesis (SCSM) as part of strategy 1 in Scheme 3.3 and was first tested in the directed evolution of LEH [21a]. In doing so, the choice of the single building block was crucial. The crystal structure of LEH reveals that most of the amino acids surrounding the binding pocket are hydrophobic. Therefore, valine, having a hydrophobic and sterically demanding side chain, was chosen as the smallest reduced amino acid alphabet for SCSM in a single saturation mutagenesis experiment at the ten-residue site. The results following the screening of about 3200 transformants are shown in Scheme 3.20.

Results of generating one single codon saturation mutagenesis (SCSM) library on the basis of valine as the sole building block, hydrolytic desymmetrization of cyclohexene oxide (29) serving as the model reaction [21a]
It can be seen that both (R,R)- and (S,S)-selective occur in one and the same small but high-quality library. The number of introduced valines ranges between two and five, depending upon the particular variant. In a single ISM step, fine-tuning was performed, boosting the enantiomeric ratio to 98:2 (96% ee). Crystal structures of the variants with and without product in combination with MD/docking computations uncovered the origin of stereoselectivity [21a]. In a control experiment, the use of serine failed completely, which supports the original hypothesis regarding the successful choice of valine.
Single code saturation mutagenesis cannot be expected to be general. Therefore, triple code saturation mutagenesis (TCSM) was developed [22b, c]. A reduced amino acid alphabet of three members at a ten-residue randomization site would require for 95% library coverage excessive screening. Therefore, if so many CAST residues are chosen, they need to be grouped into smaller randomization sites. As part of strategy 1 (Scheme 3.3), this approach was first tested with LEH as the catalyst in the same model reaction involving the desymmetrization of epoxide 29 (Scheme 3.19). The ten previously identified CAST residues were grouped into three randomization sites: (A) (V83/L114/I116), (B) (L74/M78/L147), and (C) (M32/L35/L103) [22b].
By considering the crystal structure of LEH, it was logical to test valine, phenylalanine, and tyrosine as the reduced amino acid alphabet in TCSM, even if the previous saturation mutagenesis experiments using these amino acids as building blocks had not been performed. The V-F-Y triple code was applied to the three randomization sites A, B, and C in separate experiments, requiring for 95% library coverage the screening of only 576, 192, and 192 transformants, respectively. The best results were obtained in library A (Scheme 3.21). As before, one and the same library harbors both (R,R)- and (S,S)-selective variants, but this time much better results were obtained. (S,S)-selectivity amounts to 96–99% ee in three different variants, while the best (R,R)-mutant results in 89% ee which was boosted to 97% ee by ISM. X-ray structures of selected mutants, MD/docking computations, and kinetic data were reported, which shed light on the origin of mutational effects [22b].

The results of triple code saturation mutagenesis (TCSM) when applied to LEH as the catalyst in the hydrolytic desymmetrization of cyclohexene oxide (29) [22b]
3.6 Transaminase
Transaminases are enzymes that catalyze the reductive amination of prochiral ketones with formation of the respective chiral primary amines [1]. An impressive industrial example of directed evolution was reported by Codexis, specifically in the asymmetric reductive amination of ketone 31 with formation of the antidiabetic drug sitagliptin (32) (Scheme 3.22) [53].

Asymmetric reductive amination with formation of the antidiabetic therapeutic drug sitagliptin (R)-32, catalyzed by mutants of transaminase ATA-117 [53]
The transaminase ATA-117 was chosen as the enzyme, which is related to the structurally well-characterized homolog from Arthrobacter sp. Both were known to be (R)-selective in the reductive amination of methyl ketones and small cyclic ketones. ATA-117 proved to be (R)-selective in the desired reaction as well, but activity was extremely low [53]. Thus, the goal was to enhance activity while maintaining stereoselectivity. At the beginning of the project, the industrial researchers did not use the “real” substrate 31, but first resorted to in vitro coevolution which means testing simpler but still structurally related compounds (substrate walking) [54]. With the help of a homology model of ATA-117, docking computations were carried out which allowed reasonable choices for randomizing sites lining the binding pocket (CAST sites). NNK-based saturation mutagenesis led to variant S223P with an 11-fold increase in activity in the reaction of a simplified model ketone. The mutant was then employed as a template for ISM experiments using the “real” substrate 31 [53]. Docking computations indicated that the trifluoromethyl group interacts with residues V69, F122, T283, and A284. Therefore, four NNK-based saturation mutagenesis libraries were generated separately at these four positions. Moreover, a combinatorial library using several residues simultaneously was created. The combinatorial library harbored an active variant characterized by four point mutations lining “small” and “large” parts of the binding pocket. Double mutants F122I/V69G, F122I/A284G, F122V/V69G, F122V/A284G, F122L/V69G, and F122L/A284G were the best hits. They all contain the parent mutation S223P. Activity was still quite low, yet without point mutation S223P, no activity whatsoever resulted, as shown by a deconvolution experiment.
The gene of the most active variant was then used as the parent for the next round of ISM, followed by combining the beneficial mutations from the small-pocket and large-pocket saturation mutagenesis libraries. This provided a variant with 12 point mutations and a 75-fold increase in activity. Subsequently, 11 additional rounds of mutagenesis/screening were performed using DNA shuffling, epPCR, rational design, and saturation mutagenesis at second-sphere sites. Process development was also performed in parallel. A total of 36,480 transformants were screened using an LC/MS/MS screen. The best variant was shown to have 27 point mutations. In 50% DMSO, this catalyst converts 200 g/L of the prositagliptin ketone 31 to sitagliptin (32) with >99.95% ee (R) [53].
The catalytic performance of the best ATA-117 variant under operating conditions underscores the success of the project. However, it is difficult to assess the efficacy of the applied mutagenesis approach. It is not clear whether the order of the mutagenesis cycles in the overall multistep process was actually planned or whether corrections in the strategy had to be undertaken. Why were the particular mutagenesis events chosen in the reported order?
3.7 Alternative Mutagenesis Approaches
As noted in the Introduction, such mutagenesis techniques such as epPCR or DNA shuffling can also be applied in order to manipulate the stereoselectivity of enzymes [3, 5, 10], but in several comparative studies, these have been shown to be less efficient [16, 18]. Nevertheless, following several cycles of saturation mutagenesis, it may be useful to add one round of epPCR for activity enhancement [26]. Other approaches such as neutral drift [4d, 51], domain swapping [55], and circular permutation [56] likewise deserve mention, but these strategies have not been applied very often to the evolution of stereoselectivity. Neutral drift can be used for identifying superior starting points for protein engineering by exploring accessible sequence space on the basis of recursive cycles of (random) mutagenesis and screening or selection. The technique identifies accumulating mutations which are neutral for the native function but which may prove to be useful for novel catalytic profiles. An example is the evolution of promiscuity by turning a ß-glucuronidase into a ß-galactosidase [51c].
Domain swapping was originally used in the study of natural evolution and for addressing mechanistic questions in protein science, but it has also been applied occasionally in directed evolution [3, 55]. For example, a glycosyltransferase was engineered for different substrate specificity [55b]. However, it is not well suited for enhancing or inverting stereoselectivity. A special form of this technique is circular permutation in which the N- and C-termini of an enzyme are relocated [56]. A seminal example concerns the engineering of enhanced activity of the lipase from Candida antarctica (CALB) [56a, b]. New locations of the N- and C-termini in WT CALB were designed to occur at positions 282 and 283 in hope of influencing local backbone flexibility and perhaps active site accessibility. Indeed, this led to higher activity [56b], but enantioselectivity was not addressed at the time nor in a subsequent review [56a].
Conclusions and Perspectives
This chapter provides a summary of the most important recent methodological developments in the directed evolution of stereoselective enzymes as catalysts in synthetic organic chemistry and biotechnology. Rather than being comprehensive, five different types of enzymes were chosen which illustrate important recent advances in strategies and methods.
It can safely be concluded that structure-guided saturation mutagenesis at sites in vicinity of the active site (CASTing) is a distinctly logical approach to reshape the bonding pockets of enzymes in the quest to manipulate stereoselectivity, substrate scope, and activity. The use of reduced amino acid alphabets at relatively large CAST randomization sites has emerged as the preferred strategy, allowing for the creation of small yet high-quality mutant libraries requiring less screening than in the past. In this respect, triple code saturation mutagenesis (TCSM) appears to be an optimal compromise between limited structural diversity and screening effort (bottleneck of directed evolution). TCSM allows for step-economy, since initial mutant libraries already contain notably improved (R)- as well as (S)-variants. If needed, further tuning is possible by iterative saturation mutagenesis (ISM). It has been demonstrated that it is better to strive for higher library coverage at reduced structural diversity regarding the chosen amino acid alphabet rather to maintain maximum structural diversity using NNK codon degeneracy at the same screening effort correlating with considerably lower library coverage [13a].
One of the remaining fundamental challenges in directed evolution is the development of a general guide for optimizing more than one or two enzyme parameters, e.g., stereo-/regioselectivity, substrate scope, rate, and thermostability. Efforts are underway in several laboratories.
References
(a) Drauz K, Gröger H, May O (eds) (2012) Enzyme catalysis in organic synthesis, 3rd edn. Wiley-VCH, Weinheim; (b) Faber K (2011) Biotransformations in organic chemistry, 6th edn. Springer, Heidelberg; (c) Liese A, Seelbach K, Wandrey C (eds) (2006) Industrial biotransformations, Wiley-VCH, Weinheim.
(a) Pleiss J (2012) Rational design of enzymes. In: Drauz K, Gröger H, May O (eds) Enzyme catalysis in organic synthesis, 3rd edn. Wiley-VCH, Weinheim, p 89–117; (b) Ema T, Nakano Y, Yoshida D, Kamata S, Sakai T (2012) Redesign of enzyme for improving catalytic activity and enantioselectivity toward poor substrates: manipulation of the transition state. Org Biomol Chem 10:6299–6308; (c) Steiner K, Schwab H (2012) Recent advances in rational approaches for enzyme engineering. Comput Struct Biotechnol J 2:e201209010.
Reviews of directed evolution: (a) Bommarius AS (2015) Biocatalysis, a status report. Annu Rev Chem Biomol Eng 6:319–345; (b) Denard CA, Ren H, Zhao H (2015) Improving and repurposing biocatalysts via directed evolution. Curr Opin Chem Biol 25:55–64; (c) Currin A, Swainston N, Day PJ, Kell DB (2015) Synthetic biology for the directed evolution of protein biocatalysts: navigating sequence space intelligently. Chem Soc Rev 44:1172–1239; (d) Gillam EMJ, Copp JN, Ackerley DF (eds) (2014) Directed evolution library creation. In: Methods in molecular biology, Humana Press, Totowa; (e) Widersten M (2014) Protein engineering for development of new hydrolytic biocatalysts. Curr Opin Chem Biol 21:42–47; (f) Reetz MT (2012) Directed evolution of enzymes. In Drauz K, Gröger H, May O (eds) Enzyme catalysis in organic synthesis, 3rd edn. Wiley-VCH, Weinheim, p 119–190
Review of directed evolution of protein thermostability: (a) Arnold FH (1998) Design by directed evolution. Acc Chem Res 31:125–131; (b) Petrounia IP, Arnold FH (2000) Designed evolution of enzymatic properties. Curr Opin Biotechnol 11: 325–330; (c) Polizzi KM, Bommarius AS, Broering JM, Chaparro-Riggers JF (2007) Stability of biocatalysts. Curr Opin Chem Biol 11:220–225; (d) Tokuriki N, Tawfik DS (2009) Stability effects of mutations and protein evolvability. Curr Opin Struct Biol 19:596–604
Reviews of directed evolution of stereoselectivity[3f]: (a) Reetz MT (2011) Laboratory evolution of stereoselective enzymes: a prolific source of catalysts for asymmetric reactions. Angew Chem Int Ed 50:138–174; (b) Reetz MT (2013) Biocatalysis in organic chemistry and biotechnology: past, present, and future. J Am Chem Soc 135:12480–12496
Reetz MT, Zonta A, Schimossek K, Liebeton K, Jaeger KE (1997) Creation of enantioselective biocatalysts for organic chemistry by in vitro evolution. Angew Chem Int Ed Eng 36:2830–2832
Reetz MT, Wilensek S, Zha D, Jaeger KE (2001) Directed evolution of an enantioselective enzyme through combinatorial multiple-cassette mutagenesis. Angew Chem Int Ed 40:3589–3591
Reetz MT, Puls M, Carballeira JD, Vogel A, Jaeger KE, Eggert T, Thiel W, Bocola M, Otte N (2007) Learning from directed evolution: further lessons from theoretical investigations into cooperative mutations in lipase enantioselectivity. Chembiochem 8:106–112
Zha DX, Wilensek S, Hermes M, Jaeger KE, Reetz MT (2001) Complete reversal of enantioselectivity of an enzyme-catalyzed reaction by directed evolution. Chem Commun 24:2664–2665
Reetz MT (2004) Controlling the enantioselectivity of enzymes by directed evolution: practical and theoretical ramifications. Proc Natl Acad Sci U S A 101:5716–5722
Reetz MT, Bocola M, Carballeira JD, Zha DX, Vogel A (2005) Expanding the range of substrate acceptance of enzymes: combinatorial active-site saturation test. Angew Chem Int Ed 44:4192–4196
(a) Reetz MT, Wang LW, Bocola M (2006) Directed evolution of enantioselective enzymes: iterative cycles of CASTing for probing protein-sequence space. Angew Chem Int Ed 45:1236–1241; (b) Reetz MT (2005) Evolution im Reagenzglas: Neue Perspektiven für die Weiße Biotechnologie. Tätigkeitsberichte der Max-Planck-Gesellschaft 327–331
(a) Reetz MT, Kahakeaw D, Lohmer R (2008) Addressing the numbers problem in directed evolution. Chembiochem 9:1797–1804; (b) Clouthier CM, Kayser MM, Reetz MT (2006) Designing new Baeyer-Villiger monooxygenases using restricted CASTing. J Org Chem 71:8431–8437
(a) Firth AE, Patrick WM (2008) GLUE-IT and PEDEL-AA: new programmes for analyzing protein diversity in randomized libraries. Nucleic Acids Res 36 (Web Server issue):W281–285; (b) Denault M, Pelletier JN (2007) Protein library design and screening: working out the probabilities. In: Arndt KM, Müller KM (eds) Protein engineering protocols, Humana Press, Totowa, p 127–154
Nov Y (2012) When second best is good enough: another probabilistic look at saturation mutagenesis. Appl Environ Microbiol 78:258–262
Reetz MT, Prasad S, Carballeira JD, Gumulya Y, Bocola M (2010) Iterative saturation mutagenesis accelerates laboratory evolution of enzyme stereoselectivity: rigorous comparison with traditional methods. J Am Chem Soc 132:9144–9152
Reetz MT (2013) The importance of additive and non-additive mutational effects in protein engineering. Angew Chem Int Ed 52:2658–2666
Parikh MR, Matsumura I (2005) Site-saturation mutagenesis is more efficient than DNA shuffling for the directed evolution of beta-fucosidase from beta-galactosidase. J Mol Biol 352:621–628
Acevedo-Rocha CG, Hoebenreich S, Reetz MT (2014) Iterative saturation mutagenesis: a powerful approach to engineer proteins by systematically simulating Darwinian evolution. Methods Mol Biol 1179:103–128
Sun Z, Wikmark Y, Bäckvall J-E, Reetz MT (2016) New concepts for increasing the efficiency in directed evolution of stereoselective enzymes. Chem Eur J 22:5046–5054
(a) Sun Z, Lonsdale R, Kong XD, Xu JH, Zhou J, Reetz MT (2015) Reshaping an enzyme binding pocket for enhanced and inverted stereoselectivity: use of smallest amino acid alphabets in directed evolution. Angew Chem Int Ed 54:12410–12415; (b) Sandström AG, Wikmark Y, Engström K, Nyhlen J, Bäckvall JE (2012) Combinatorial reshaping of the Candida antarctica lipase A substrate pocket for enantioselectivity using an extremely condensed library. Proc Natl Acad Sci U S A 109:78–83
(a) Zhang ZG, Lonsdale R, Sanchis J, Reetz MT (2014) Extreme synergistic mutational effects in the directed evolution of a baeyer-villiger monooxygenase as catalyst for asymmetric sulfoxidation. J Am Chem Soc 136:17262–17272; (b) Sun Z, Lonsdale R, Wu L, Li G, Li A, Wang J, Zhou J, Reetz MT (2016) Structure-guided triple code saturation mutagenesis: efficient tuning of the stereoselectivity of an epoxide hydrolase. ACS Catal 6:1590–1597; (c) Sun Z, Lonsdale R, Ilie A, Li G, Zhou J, Reetz MT (2016) Catalytic asymmetric reduction of difficult-to-reduce ketones: triple code saturation mutagenesis of an alcohol dehydrogenase. ACS Catal 6:1598–1605
(a) Bougioukou DJ, Kille S, Taglieber A, Reetz MT (2009) Directed Evolution of an enantioselective enoate-reductase: testing the utility of iterative saturation mutagenesis. Adv Synth Catal 351:3287–3305; (b) Sullivan B, Walton AZ, Stewart JD (2013) Library construction and evaluation for site saturation mutagenesis. Enzym Microb Technol 53:70–77
Polizzi KM, Parikh M, Spencer CU, Matsumura I, Lee JH, Realff MJ, Bommarius AS (2006) Pooling for improved screening of combinatorial libraries for directed evolution. Biotechnol Prog 22:961–967
Acevedo-Rocha CG, Reetz MT, Nov Y (2015) Economical analysis of saturation mutagenesis experiments. Sci Rep 5:10654
Kwan DH, Constantinescu I, Chapanian R, Higgins MA, Kotzler MP, Samain E, Boraston AB, Kizhakkedathu JN, Withers SG (2015) Toward efficient enzymes for the generation of universal blood through structure-guided directed evolution. J Am Chem Soc 137:5695–5705
(a) Whitehouse CJ, Bell SG, Wong LL (2012) P450(BM3) (CYP102A1): connecting the dots. Chem Soc Rev 41:1218–1260; (b) Fasan R (2012) Tuning P450 enzymes as oxidation catalysts. ACS Catal 2:647–666; (c) Bernhardt R, Urlacher VB (2014) Cytochromes P450 as promising catalysts for biotechnological application: chances and limitations. Appl. Microbiol Biotechnol 98:6185–6203; (d) Holtmann D, Fraaije MW, Arends IW, Opperman DJ, Hollmann F (2014) The taming of oxygen: biocatalytic oxyfunctionalisations. Chem Commun 50:13180–13200; (e) Roiban GD, Reetz MT (2015) Expanding the toolbox of organic chemists: directed evolution of P450 monooxygenases as catalysts in regio- and stereoselective oxidative hydroxylation. Chem Commun 51:2208–2224; (f) Girvan HM, Munro AW (2016) Applications of microbiol cytochrome P450 enzymes in biotechnology and synthetic biology. Curr Opin Chem Biol 31:136–145
(a) Tang WL, Li Z, Zhao H (2010) Inverting the enantioselectivity of P450pyr monooxygenase by directed evolution. Chem Commun 46:5461–5463; (b) Pham SQ, Pompidor G, Liu J, Li XD, Li Z (2012) Evolving P450pyr hydroxylase for highly enantioselective hydroxylation at non-activated carbon atom. Chem Commun 48:4618–4620
Lonsdale R, Harvey JN, Mulholland AJ (2010) Inclusion of dispersion effects significantly improves accuracy of calculated reaction barriers for cytochrome P450 catalyzed reactions. J Phys Chem Lett 1:3232–3237
Agudo R, Roiban GD, Reetz MT (2012) Achieving regio- and enantioselectivity of P450-catalyzed oxidative CH activation of small functionalized molecules by structure-guided directed evolution. Chembiochem 13:1465–1473
Kille S, Zilly FE, Acevedo JP, Reetz MT (2011) Regio- and stereoselectivity of P450-catalysed hydroxylation of steroids controlled by laboratory evolution. Nat Chem 3:738–743
Le-Huu P, Heidt T, Claasen B, Laschat S, Urlacher VB (2015) Chemo-, regio-, and stereoselective oxidation of the monocyclic diterpenoid beta-cembrenediol by P450 BM3. ACS Catal 5:1772–1780
Zhang K, Shafer BM, Demars MD 2nd, Stern HA, Fasan R (2012) Controlled oxidation of remote sp3 C-H bonds in artemisinin via P450 catalysts with fine-tuned regio- and stereoselectivity. J Am Chem Soc 134:18695–18704
(a) Chen MM, Snow CD, Vizcarra CL, Mayo SL, Arnold FH (2012) Comparison of random mutagenesis and semi-rational designed libraries for improved cytochrome P450 BM3-catalyzed hydroxylation of small alkanes. Prot Eng Des Sel 25:171–178; (b) Ritter C, Nett N, Acevedo-Rocha CG, Lonsdale R, Kraling K, Dempwolff F, Hoebenreich S, Graumann PL, Reetz MT, Meggers E (2015) Bioorthogonal enzymatic activation of caged compounds. Angew Chem Int Ed 54:13440–13443; (c) Dennig A, Lulsdorf N, Liu H, Schwaneberg U (2013) Regioselective o-hydroxylation of monosubstituted benzenes by P450 BM3. Angew Chem Int Ed 52:8459–8462; (d) Agudo R, Roiban GD, Lonsdale R, Ilie A, Reetz MT (2015) Biocatalytic route to chiral acyloins: P450-catalyzed regio- and enantioselective alpha-hydroxylation of ketones. J Org Chem 80:950–956; (e) Hoebenreich S, Zilly FE, Acevedo-Rocha CG, Zilly M, Reetz MT (2015) Speeding up directed evolution: combining the advantages of solid-phase combinatorial gene synthesis with statistically guided reduction of screening effort. ACS Synth Biol 4:317–331; (f) Roiban GD, Agudo R, Reetz MT (2014) Cytochrome P450 catalyzed oxidative hydroxylation of achiral organic compounds with simultaneous creation of two chirality centers in a single C-H activation step. Angew Chem Int Ed 53:8659–8663; (g) Hu S, Huang J, Mei LH, Yu Q, Yao SJ, Jin ZH (2010) Altering the regioselectivity of cytochrome P450 BM-3 by saturation mutagenesis for the biosynthesis of indirubin. J Mol Catal B-Enzym 67:29–35; (h) Nguyen KT, Virus C, Gunnewich N, Hannemann F, Bernhardt R (2012) Changing the regioselectivity of a P450 from C15 to C11 hydroxylation of progesterone. Chembiochem 13:1161–1166; (i) Yang Y, Liu J, Li Z (2014) Engineering of P450pyr hydroxylase for the highly regio- and enantioselective subterminal hydroxylation of alkanes. Angew Chem Int Ed 53:3120–3124
(a) Reinen J, Vredenburg G, Klaering K, Vermeulen NPE, Commandeur JNM, Honing M, Vos JC (2015) Selective whole-cell biosynthesis of the designer drug metabolites 15-or 16-ß-hydroxynorethisterone by engineered Cytochrome P450 BM3 mutants. J Mol Catal B-Enzym 121:64–74; (b) Bruhlmann F, Bosijokovic B, Ullmann C, Auffray P, Fourage L, Wahler D (2013) Directed evolution of a 13-hydroperoxide lyase (CYP74B) for improved process performance. J Biotechnol 163:339–345
Reetz MT, Brunner B, Schneider T, Schulz F, Clouthier CM, Kayser MM (2004) Directed evolution as a method to create enantioselective cyclohexanone monooxygenases for catalysis in Baeyer-Villiger reactions. Angew Chem Int Ed 43:4075–4078
Mihovilovic MD, Rudroff F, Winninger A, Schneider T, Schulz F, Reetz MT (2006) Microbial Baeyer-Villiger oxidation: stereopreference and substrate acceptance of cyclohexanone monooxygenase mutants prepared by directed evolution. Org Lett 8:1221–1224
(a) Fraaije MW, Wu J, Heuts DP, van Hellemond EW, Spelberg JH, Janssen DB (2005) Discovery of a thermostable Baeyer-Villiger monooxygenase by genome mining. Appl Microbiol Biotechnol 66:393–400; (b) Malito E, Alfieri A, Fraaije MW, Mattevi A (2004) Crystal structure of a Baeyer-Villiger monooxygenase. Prod Natl Acad Sci 101:13157–13162
Reviews of directed evolution of Baeyer-Villiger monooxygenases: (a) Zhang ZG, Parra LP, Reetz MT (2012) Protein engineering of stereoselective Baeyer-Villiger monooxygenases. Chem Eur J 18:10160–10172; (b) Balke K, Kadow M, Mallin H, Sass S, Bornscheuer UT (2012) Discovery, application and protein engineering of Baeyer-Villiger monooxygenases for organic synthesis. Org Biomol Chem 10:6249–6265
Reetz MT, Wu S (2008) Greatly reduced amino acid alphabets in directed evolution: making the right choice for saturation mutagenesis at homologous enzyme positions. Chem Commun 43:5499–5501
Kirschner A, Bornscheuer UT (2006) Kinetic resolution of 4-hydroxy-2-ketones catalyzed by a Baeyer-Villiger monooxygenase. Angew Chem 45:7004–7006
Wu S, Acevedo JP, Reetz MT (2010) Induced allostery in the directed evolution of an enantioselective Baeyer-Villiger monooxygenase. Prod Natl Acad Sci USA 107:2775–2780
Ichiye T, Karplus M (1991) Collective motions in proteins – a covariance analysis of atomic fluctuations in molecular-dynamics and normal mode simulations. Proteins 11:205–217
Tang L, Gao H, Zhu X, Wang X, Zhou M, Jiang R (2012) Construction of “small-intelligent” focused mutagenesis libraries using well-designed combinatorial degenerate primers. BioTechniques 52:149–158
Reetz MT, Sanchis J (2008) Constructing and analyzing the fitness landscape of an experimental evolutionary process. Chembiochem 9:2260–2267
Gumulya Y, Sanchis J, Reetz MT (2012) Many pathways in laboratory evolution can lead to improved enzymes: how to escape from local minima. Chembiochem 13:1060–1066
Wu Q, Soni P, Reetz MT (2013) Laboratory evolution of enantiocomplementary Candida antarctica lipase B mutants with broad substrate scope. J Am Chem Soc 135:1872–1881
Engström K, Nyhlen J, Sandström AG, Bäckvall JE (2010) Directed evolution of an enantioselective lipase with broad substrate scope for hydrolysis of alpha-substituted esters. J Am Chem Soc 132:7038–7042
Wikmark Y, Svedendahl Humble M, Bäckvall JE (2015) Combinatorial library based engineering of Candida antarctica lipase A for enantioselective transacylation of sec-alcohols in organic solvent. Angew Chem Int Ed 54:4284–4288
(a) Ma J, Wu L, Guo F, Gu J, Tang X, Jiang L, Liu J, Zhou J, Yu H (2013) Enhanced enantioselectivity of a carboxyl esterase from Rhodobacter sphaeroides by directed evolution. Appl Microbiol Biotechnol 97:4897–4906; (b) Luan ZJ, Li FL, Dou S, Chen Q, Kong XD, Zhou JH, Yu HL, Xu JH (2015) Substrate channel evolution of an esterase for the synthesis of cilastatin. Catal Sci Technol 5:2622–2629; (c) Godinho LF, Reis CR, van Merkerk R, Poelarends GJ, Quax WJ (2012) An esterase with superior activity and enantioselectivity towards 1,2-o-isopropylideneglycerol esters obtained by protein design. Adv Synth Catal 354:3009–3015; (d) Nobili A, Tao Y, Pavlidis IV, van den Bergh T, Joosten HJ, Tan T, Bornscheuer UT (2015) Simultaneous use of in silico design and a correlated mutation network as a tool to efficiently guide enzyme engineering. Chembiochem 16:805–810
(a) Gupta RD, Tawfik DS (2008) Directed enzyme evolution via small and effective neutral drift libraries. Nat Methods 5:939–942; (b) Kaltenbach M, Tokuriki, N (2014) Generation of effective libraries by neutral drift. Methods Molec Biol 1179:69–81; (c) Smith WS, Hale JR, Neylon C (2011) Applying neutral drift to the directed molecular evolution of a ß-glucuronidase into a ß-galactosidase: two different evolutionary pathways lead to the same variant. BMC Res Notes 4:138; (d) Bernath-Levin K, Shainsky J, Sigawi L, Fishman A (2014) Directed evolution of nitrobenzene dioxygenase for the synthesis of the antioxidant hydroxytyrosol. Appl Microbiol Biotechnol 98:4975–4985
(a) Eigen M, McCaskill J, Schuster P (1988) Molecular quasi-species. J Phys Chem 92:6881–6891; (b) Kurtovic S, Mannervik B (2009) Identification of emerging quasi-species in directed enzyme evolution. Biochemistry 48:9330–9339
Savile CK, Janey JM, Mundorff EC, Moore JC, Tam S, Jarvis WR, Colbeck JC, Krebber A, Fleitz FJ, Brands J, Devine PN, Huisman GW, Hughes GJ (2010) Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science 329:305–309
Chen Z, Zhao H (2005) Rapid creation of a novel protein function by in vitro coevolution. J Mol Biol 348:1273–1282
(a) Ostermeier M, Benkovic SJ (2000) Evolution of protein function by domain swapping. Adv Protein Chem 55:29–77; (b) Park AH, Park HY, Sohng JK, Lee HC, Liou K, Yoon YJ, Km BG (2009) Expanding substrate specificity of GT-B fold glycosyltransferase via domain swapping and high-throughput screening. Biotechnol Bioeng 102:988–994
(a) Yu Y, Lutz S (2011) Circular permutation: a different way to engineer enzyme structure and function. Trends Biotechnol 29:18–25; (b) Qian Z, Lutz S (2005) Improving the catalytic activity of Candida antarctica lipase B by circular permutation. J Am Chem Soc 127:13466–13467; (c) Yu Y, Lutz S (2010) Improved triglyceride transesterification by circular permutated Candida antarctica lipase B. Biotechnol Bioeng 105:44–50.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Reetz, M.T. (2017). Recent Advances in Directed Evolution of Stereoselective Enzymes. In: Alcalde, M. (eds) Directed Enzyme Evolution: Advances and Applications. Springer, Cham. https://doi.org/10.1007/978-3-319-50413-1_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-50413-1_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-50411-7
Online ISBN: 978-3-319-50413-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)