
Abstract
Sulfate-reducing bacteria (SRB) are well known for their significance in the carbon and sulfur cycles of marine sediments. Despite their general energetic restriction, some SRB are capable of anaerobic degradation of aromatic hydrocarbons in the marine environments, such as deep sea sediments characterized by recent oil formation. Proteogenomics has allowed hitherto unknown insights into the physiology of SRB from single catabolic reactions to complex metabolic networks. Best studied aromatic compound-degrading SRB at present are toluene-degrading Desulfobacula toluolica Tol2 and the naphthalene-degrading enrichment culture N47.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others

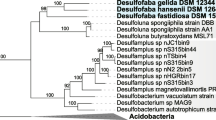
1 Introduction
Sulfate-reducing bacteria (SRB) are anaerobic microorganisms that thrive by coupling the oxidation of organic substrates to the respiratory reduction of sulfate (SO42-) to sulfide (S2-). Owing to the high concentrations (28 mM) of sulfate in sea water, SRB belong to the pivotal drivers of the biogeochemical cycles of carbon and sulfur in the marine seafloor (Jørgensen 1982). Most recently, the global rates of sulfate-reduction in marine sediments were predicted to account for 12–29% of the organic carbon mineralization in these environments (Bowles et al. 2014). Bacterial sulfate reduction has previously been reported to occur at elevated temperatures (80 to above 100 °C) in the production fluids of oil reservoirs in the North Sea (Stetter et al. 1993) and deep-sea hydrothermal vent sediments of the Guaymas Basin in the Gulf of California (Jørgensen et al. 1992). In fact SRB enriched/isolated from these two environments were shown to anaerobically grow with crude oil as sole source of organic carbon and energy by depleting n-alkanes or alkylbenzenes directly from the crude oil (Rueter et al. 1994; Harms et al. 1999; Wilkes et al. 2000). This capacity of SRB is of great interest for reservoir geochemistry, albeit an undesired process in oil industry as it deteriorates the quality of the oil by compositional alterations and by souring due to H2S generation (Head et al. 2014). Formed H2S can also contribute to the corrosion of iron in technical installations, e.g., oil rigs and pipelines, and some SRB can even use metallic iron as electron donor for sulfate reduction (Enning and Garrelfs 2014). Then again, anaerobic degradation of hydrocarbons represents a beneficial process for bioremediation efforts at hydrocarbon contaminated sites, such as aquifers (e.g., Townsend et al. 2003; Griebler et al. 2004) or marine/estuarine systems (e.g., Miralles et al. 2007; Kimes et al. 2014). For a more general overview on SRB and their role in global biogeochemical cycles, technical processes, biomedicine, and fundamental microbiology, refer to other reviews (e.g., Muyzer and Stams 2008; Rabus et al. 2015). In the following, an overview on aromatic compound-degrading, sulfate-reducing bacteria that have been studied by functional genomics is provided.
2 Desulfobacula toluolica Tol2
Desulfobacula toluolica Tol2 represents the first pure culture of a sulfate-reducing bacterium that was demonstrated to completely oxidize toluene to CO2, with this capacity found to be inducible in whole cell cultures (Rabus et al. 1993). Subsequent experiments indicated initial activation of toluene not to proceed via benzyl alcohol, but via benzylsuccinate (Rabus and Widdel 1995; Rabus and Heider 1998). The occurrence of Desulfobacula phylotypes has been reported for a tidal sand flat in the German Wadden Sea (Gittel et al. 2008), for a reduced organic-rich surface sediment in the Baltic Sea (Sinkko et al. 2013), and for a sediment core from the Caspian Sea subjected to simulated petroleum seepage in laboratory experiments (Stagars et al. 2017).
The complete 5.2 Mbp genome of D. toluolica Tol2 was the first to be determined for an aromatic compound-degrading, marine SRB, and functional predictions were refined by comprehensive differential proteomics and targeted metabolite analyses uses of substrate-adapted cells (Wöhlbrand et al. 2013). The high genome plasticity is reminiscent of the genome from the denitrifying degradation specialist “Aromatoleum aromaticum” EbN1 (Rabus et al. 2005). Anaerobic degradation of toluene and p-cresol was found to be initiated by addition to fumarate followed by modified β-oxidation of the formed succinate derivative to benzoyl-CoA and 4-hydroxybenzoyl-CoA, respectively. Notably, separated gene clusters could be assigned to these two substrates, with the p-cresol-specific initial enzyme ((4-hydroxybenzyl)succinate synthase, HbsA catalytic subunit) forming an own branch in the phylogenetic tree of alkyl-/arylalkylsuccinate synthases. Interestingly, the dehydrogenation reactions in both β-oxidation branches are apparently linked via the activity of dedicated electron transfer proteins directly to the membrane redox pool. Anaerobic degradation of phenylalanine involves nonoxidative deamination to cinnamate. Dearomatization of the central intermediate benzoyl-CoA expectedly involves an ATP-independent class II benzoyl-CoA reductase (BCR) as known from Geobacter metallireducens GS-15 (Kung et al. 2009). Complete oxidation of acetyl-CoA to CO2 proceeds via the Wood-Ljungdahl pathway as widespread in the Desulfobacteraceae family which strain Tol2 is affiliating with (Rabus et al. 2015). Detailed analysis by 1D blue native-PAGE complexome profiling and 2D blue native-/SDS-PAGE demonstrated an elaborated system of membrane protein complexes for electron transport, including, e.g., dimer and quadruple formation of the central DsrMKJOP complex and the potential capacity for Na+-based bioenergetics via the RnfABCDEG complex (Wöhlbrand et al. 2016).
3 Desulfobacula toluolica TS
A microcosm established from oil-contaminated tidal flat sediment was demonstrated to degrade toluene coupled to sulfate reduction by determining the formation of 13CO2 from 13C-labeled toluene. A metagenomics approach was applied to this microcosm yielding a draft genome of 125 scaffolds that showed highest similarity to the genome of D. toluolica Tol2. Accordingly, the degradation pathways for several aromatic compounds could be reconstructed, mirroring the network established before for strain Tol2 and underpinning its model character (Kim et al. 2014).
4 Strains NaphS2, NaphS3, and NaphS6
Strain NaphS2 originates from sulfidic marine sediment, represents the first pure culture of an anaerobic bacterium demonstrated to degrade naphthalene completely to CO2 (Galushko et al. 1999), and is closely related to sulfate-reducing strain EbS7 anaerobically degrading ethylbenzene via addition to fumarate (Kniemeyer et al. 2003). Differential proteogenomics revealed that strain NaphS2 and two further naphthalene-degrading SRBs (strains NaphS3 and NaphS6) activated 2-methylnaphthalene via addition to fumarate and that the presumptive (2-naphthylmethyl)succinate synthase was absent in naphthalene-adapted cells of all three strains. This finding indicated that the three studied marine strains did not employ a methylation as initial activation of anaerobic naphthalene degradation (Musat et al. 2009), as previously suggested for the sulfate-reducing enrichment culture N47 (Safinowski and Meckenstock 2006). Instead, carboxylation of naphthalene is discussed as a possible activation reaction (DiDonato et al. 2010; Musat et al. 2009).
5 Enrichment Culture N47
The sulfate-reducing enrichment culture N47 was obtained from soil material of a contaminated aquifer and by labeling studies (with [13C]bicarbonate) suggested to anaerobically degrade naphthalene via a possible carboxylation to 2-naphthoic acid (Meckenstock et al. 2000). Anaerobic degradation of 2-methylnaphthalene was proposed to proceed in analogy to the anaerobic degradation of toluene, i.e., via addition to fumarate followed by β-oxidation of the succinate moiety (Annweiler et al. 2000). The interim hypothesis of direct methylation of naphthalene to connect with the 2-methylnaphthalene pathway (Safinowski and Meckenstock 2006) was not sustained, as proteomic and enzymatic evidence for the possible involvement of a putative naphthalene carboxylase showing sequence similarity to phenylphosphate carboxylase emerged (Bergmann et al. 2011a; Mouttaki et al. 2012). Subsequently, a draft 4.7 Mbp genome comprised of 17 contigs was obtained for the enrichment culture N47 (Bergmann et al. 2011b), which formed the basis for a proteomics investigation. The latter allowed identification of all genes required for anaerobic degradation of 2-methylnaphthalene as well as a class I benzoyl-CoA reductase (Selesi et al. 2010). Initial biochemical studies suggested that the four-electron reduction of naphthoyl-CoA to 5,6,7,8-tetrahydro-2-naphthoyl-CoA is catalyzed by a novel type of dearomatizing reductase containing FAD, FMN, and an Fe-S cluster as cofactors (Eberlein et al. 2013). Meanwhile, it could be shown that actually two distinct enzymes are involved in the anaerobic reduction of the naphthyl ring, each catalyzing a two-electron reduction (Estelmann et al. 2015).
6 Desulfococcus multivorans
Desulfococcus multivorans strain 1be1 is a nutritionally versatile sulfate-reducing bacterium originally isolated from a sewage digester (Widdel 1980). D. multivorans is a representative of the Desulfosarcina-Desulfococcus clade within Desulfobacteraceae that has been repeatedly reported to dominate the SRB community in diverse marine sediments (e.g., Ravenschlag et al. 2000; Mußmann et al. 2005) and represent the bacterial partner in anaerobically methane-oxidizing consortia (Boetius et al. 2000). The complete 4.46 Mbp genome of D. multivorans contains a single chromosome and represents the first to be reported for a member of the Desulfosarcina-Desulfococcus clade (Dörries et al. 2016). The metabolic reconstruction was fostered by detailed differential proteome profiling across 17 different substrate conditions, revealing a catabolic network comprising 170 proteins (154 detected; ~91% coverage). Peripheral degradation of aromatic compounds feeds via the benzoyl-CoA pathway (class II benzoyl-CoA reductase) into the Wood-Ljungdahl (WL) pathway for terminal oxidation to CO2. While peripheral degradation pathways (including benzoyl-CoA pathway) displayed high substrate-specific formation, the central catabolic modules (WL and methylmalonyl-CoA pathways) as well as the membrane protein complexes involved in electron transfer were constitutively formed. This pattern appears to be a common property of to date proteogenomically studied members of the Desulfobacteraceae.
7 Desulfotomaculum gibsoniae GrollT
The mesophilic sulfate-reducing bacterium strain Groll was isolated from a black mud sample from a small freshwater ditch and shown to anaerobically grow with several aromatic compounds, including phenol, cresols, and catechol (Kuever et al. 1993). The bacterium was subsequently classified as Desulfotomaculum gibsoniae GrollT (Kuever et al. 1999). The ~4.9 Mbp genome allowed for a metabolic reconstruction (Kuever et al. 2014). The anaerobic degradation of cresols is suggested to follow the route previously suggested for D. toluolica Tol2 (see above Sect. 2), i.e., initial activation via addition to fumarate is followed by modified β-oxidation to feed via class II BCR into the central benzoyl-CoA pathway and from there to the WL-pathway for terminal oxidation to CO2. Catechol degradation is proposed to involve initial activation by phosphorylation followed by carboxylation as previously proposed for the denitrifying bacterium Thauera aromatica K172 (Ding et al. 2008).
8 Research Needs
While the biogeographical significance and biogeochemical imprint of SRB on a global scale are well established, the understanding of the catabolic properties that form the basis of an organism’s functional role in the habitat is far less advanced. Thus, more complete expert-annotated genomes of model SRBs being relevant for an intriguing degradation ability and/or abundance in the habitat are required. The integration of genomics, proteomics, and metabolite analysis has proven particularly rewarding for elucidating novel reactions and reconstructing complete catabolic networks and needs to be further developed. A broader functional genomics understanding of aromatic compound-degrading SRB promises new catabolic discoveries, insights into how these bacteria operate when degrading recalcitrant compounds at the thermodynamic limit, and to what extent the catabolic networks revealed from pure cultures of SRB can be actually found in the natural habitat (e.g., Michas et al. 2017). In this respect, metaOMICS investigations of sulfidic sites rich in aromatic compounds will be highly interesting.
References
Annweiler E, Materna A, Safinowski M, Kappler A, Richnow HH, Michaelis W, Meckenstock RU (2000) Anaerobic degradation of 2-methylnaphthalene by a sulfate-reducing enrichment culture. Appl Environ Microbiol 66:5329–5333
Bergmann FD, Selesi D, Meckenstock RU (2011a) Identification of new enzymes potentially involved in anaerobic naphthalene degradation by the sulfate-reducing enrichment culture N47. Arch Microbiol 193:241–250
Bergmann F, Selesi D, Weinmaier T, Tischler P, Rattei T, Meckenstock RU (2011b) Genomic insights into the metabolic potential of the polycyclic aromatic hydrocarbon degrading sulfate-reducing Deltaproteobacterium N47. Environ Microbiol 13:1125–1137
Boetius A, Ravenschlag K, Schubert CJ, Rickert D, Widdel F, Gieseke A, Amann R, Jørgensen BB, Witte U, Pfannkuche O (2000) A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 407:623–626
Bowles MW, Mogollón JM, Kasten S, Zable M, Hinrichs K-U (2014) Global rates of marine sulfate reduction and implications for sub-sea-floor metabolic activities. Science 344:889–891
DiDonato RJ Jr, Young ND, Butler JE, Chin K-J, Hixson KK, Mouser P, Lipton MS, DeBoy R, Methé BA (2010) Genome sequence of the deltaproteobacterial strain NaphS2 and analysis of differential gene expression during anaerobic growth on naphthalene. PLoS One 5:e14072
Ding B, Schmeling S, Fuchs G (2008) Anaerobic metabolism of catechol by the denitrifying bacterium Thauera aromatica – a result of promiscuous enzymes and regulators? J Bacteriol 190:1620–1630
Dörries M, Wöhlbrand L, Kube M, Reinhardt R, Rabus R (2016) Genome and catabolic subproteomes of the marine, nutritionally versatile, sulfate-reducing bacterium Desulfococcus multivorans DSM 2059. BMC Genomics 17:918
Eberlein C, Estelmann S, Seifert J, von Bergen M, Müller M, Meckenstock RU, Boll M (2013) Identification and characterization of 2-naphthoyl-coenzyme A reductase, the prototype of a novel class of dearomatizing reductases. Mol Microbiol 88:1032–1039
Enning D, Garrelfs J (2014) Corrosion of iron by sulfate-reducing bacteria: new views of an old problem. Appl Environ Microbiol 80:1226–1236
Estelmann S, Blank I, Feldmann A, Boll M (2015) Two distinct old yellow enzymes are involved in naphthyl ring reduction during anaerobic naphthalene degradation. Mol Microbiol 95:162–172
Galushko A, Minz D, Schink B, Widdel F (1999) Anaerobic degradation of naphthalene by a pure culture of a novel type of marine sulphate-reducing bacterium. Environ Microbiol 1:415–420
Gittel A, Mußmann M, Sass H, Cypionka H, Könneke M (2008) Identity and abundance of active sulfate-reducing bacteria in deep tidal flat sediments by direct cultivation and CARD-FISH analysis. Environ Microbiol 10:2645–2658
Griebler C, Safinowski M, Vieth A, Richnow HH, Meckenstock RU (2004) Combined application of stable carbon isotope analysis and specific metabolites determination for assessing in situ degradation of aromatic hydrocarbons in a tar oil-contaminated aquifer. Environ Sci Technol 38:617–631
Harms G, Zengler K, Rabus R, Aeckersberg F, Minz D, Rosselló-Mora R, Widdel F (1999) Anaerobic oxidation of o-xylene, m-xylene, and homologous alkylbenzenes by new types of sulfate-reducing bacteria. Appl Environ Microbiol 65:999–1004
Head IM, Gray ND, Larter SR (2014) Life in the slow lane; biogeochemistry of biodegraded petroleum containing reservoirs and implications for energy recovery and carbon management. Front Microbiol 5:566
Jørgensen BB (1982) Mineralization of organic matter in the sea bed – the role of sulphate reduction. Nature 296:643–645
Jørgensen BB, Isaksen MF, Jannasch HW (1992) Bacterial sulfate reduction above 100°C in deep-sea hydrothermal vent sediments. Science 258:1756–1757
Kim S-J, Park S-J, Jung M-Y, Kim J-G, Min U-G, Hong H-J, Rhee S-K (2014) Draft genome sequence of an aromatic compound-degrading bacterium, Desulfobacula sp. TS, belonging to the Deltaproteobacteria. FEMS Microbiol Lett 360:9–12
Kimes NE, Callaghan AV, Suflita JM, Morris PJ (2014) Microbial transformation of the Deepwater Horizon oil spill – past, present, and future perspectives. Front Microbiol 5:603
Kniemeyer O, Fischer T, Wilkes H, Glöckner FO, Widdel F (2003) Anaerobic degradation of ethylbenzene by a new type of marine sulfate-reducing bacterium. Appl Environ Microbiol 69:760–768
Kuever J, Kulmer J, Jannsen S, Fischer U, Blotevogel K-H (1993) Isolation and characterization of a new spore-forming sulfate-reducing bacterium growing by complete oxidation of catechol. Arch Microbiol 159:282–288
Kuever J, Rainey FA, Hippe H (1999) Description of Desulfotomaculum sp. Groll as Desulfotomaculum gibsoniae sp. nov. Int J Syst Bacteriol 49:1801–1808
Kuever J, Visser M, Loeffler C, Boll M, Worm P, Sousa DZ, Plugge CM, Schaap PJ, Muyzer G, Pereira IAC, Parshina SN, Goodwin LA, Kyrpides NC, Detter J, Woyke T, Chain P, Davenport KW, Rhode M, Spring S, Klenk H-P, Stams AJM (2014) Genome analysis of Desulfotomaculum gibsoniae strain GrollT a highly versatile Gram-positive sulfate-reducing bacterium. Stand Genomic Sci 9:821–839
Kung JW, Löffler C, Dörner K, Heintz D, Gallien S, Van Dorsselaer A, Friedrich T, Boll M (2009) Identification and characterization of the tungsten-containing class of benzoyl-coenzyme A reductases. PNAS 106:17687–17692
Meckenstock RU, Annweiler E, Michaelis W, Richnow HH, Schink B (2000) Anaerobic naphthalene degradation by a sulfate-reducing enrichment culture. Appl Environ Microbiol 66:2743–2747
Michas A, Vestergaard G, Trautwein K, Avramidis P, Hatzinikolaou DG, Vorgias CE, Wilkes H, Rabus R, Schloter M, Schöler A (2017) More than 2500 years of oil exposure shape sediment microbiomes with the potential for syntrophic degradation of hydrocarbons linked to methanogenesis. Microbiome 5:118
Miralles G, Grossi V, Acquaviva M, Duran R, Claude Bertrand J, Cuny P (2007) Alkane biodegradation and dynamics of phylogenetic subgroups of sulfate-reducing bacteria in an anoxic coastal marine sediment artificially contaminated with oil. Chemosphere 68:1327–1334
Mouttaki H, Johannes J, Meckenstock RU (2012) Identification of naphthalene carboxylase as a prototype for the anaerobic activation of non-substituted aromatic hydrocarbons. Environ Microbiol 14:2770–2774
Musat F, Galushko A, Jacob J, Widdel F, Kube M, Reinhardt R, Wilkes H, Schink B, Rabus R (2009) Anaerobic degradation of naphthalene and 2-methylnaphthalene by strains of marine sulfate-reducing bacteria. Environ Microbiol 11:209–219
Mußmann M, Ishii K, Rabus R, Amann R (2005) Diversity and vertical distribution of cultured and uncultured Deltaproteobacteria in an intertidal mud flat of the Wadden Sea. Environ Microbiol 7:405–418
Muyzer G, Stams AJM (2008) The ecology and biotechnology of sulphate-reducing bacteria. Nat Rev Microbiol 6:441–454
Rabus R, Heider J (1998) Initial reactions of anaerobic metabolism of alkylbenzenes in denitrifying and sulfate-reducing bacteria. Arch Microbiol 170:377–384
Rabus R, Widdel F (1995) Conversion studies with substrate analogues of toluene in a sulfate-reducing bacterium, strain Tol2. Arch Microbiol 164:448–451
Rabus R, Nordhaus R, Ludwig W, Widdel F (1993) Complete oxidation of toluene under strictly anoxic conditions by a new sulfate-reducing bacterium. Appl Environ Microbiol 59:1444–1451
Rabus R, Kube M, Heider J, Beck A, Heitmann K, Widdel F, Reinhardt R (2005) The genome sequence of an anaerobic aromatic-degrading denitrifying bacterium, strain EbN1. Arch Microbiol 183:27–36
Rabus R, Venceslau SS, Wöhlbrand L, Voordouw G, Wall JD, Pereira IAC (2015) A post-genomic view of the ecophysiology, catabolism and biotechnological relevance of sulphate-reducing prokaryotes. In: Advances in microbial physiology, vol 66. Academic Press, Oxford, pp 55–321
Ravenschlag K, Sahm K, Knoblauch C, Jørgensen BB, Amann R (2000) Community structure, cellular rRNA content, and activity of sulfate-reducing bacteria in marine arctic sediments. Appl Environ Microbiol 66:3592–3602
Rueter P, Rabus R, Wilkes H, Aeckersberg F, Rainey FA, Jannasch HW, Widdel F (1994) Anaerobic oxidation of hydrocarbons in crude oil by new types of sulphate-reducing bacteria. Nature 372:455–458
Safinowski M, Meckenstock RU (2006) Methylation is the initial reaction in anaerobic naphthalene degradation by a sulfate-reducing enrichment culture. Environ Microbiol 8:347–352
Selesi D, Jehmlich N, von Bergen M, Schmidt F, Rattei T, Tischler P, Lueders T, Meckenstock RU (2010) Combined genomic and proteomic approaches identify gene clusters involved in anaerobic 2-methylnaphthalene degradation in the sulfate-reducing enrichment culture N47. J Bacteriol 192:295–306
Sinkko H, Lukkari K, Sihvonen LM, Sivonen K, Leivuori M, Rantanen M, Paulin L, Lyra C (2013) Bacteria contribute to sediment nutrient release and reflect progressed eutrophication-driven hypoxia in an organic-rich continental sea. PLoS One 8:e67061
Stagars MH, Mishra S, Treude T, Amann R, Knittel K (2017) Microbial community response to simulated petroleum seepage in Caspian Sea sediments. Front Microbiol 8:764
Stetter KO, Huber R, Blöchl E, Kurr M, Eden RD, Fielder M, Cash H, Vance I (1993) Hyperthermophilic archaea are thriving in deep North Sea and Alaskan oil reservoirs. Nature 365:743–745
Townsend GT, Prince RC, Suflita JM (2003) Anaerobic oxidation of crude oil hydrocarbons by the resident microorganisms of a contaminated anoxic aquifer. Environ Sci Technol 37:5213–5218
Widdel (1980) Anaerober Abbau von Fettsäuren und Benzoesäure durch neu isolierte Arten Sulfat-reduzierender Bakterien. PhD thesis. Georg-August Universität zu Göttingen, Göttingen
Wilkes H, Boreham C, Harms G, Zengler K, Rabus R (2000) Anaerobic degradation and carbon isotopic fractionation of alkylbenzenes in crude oil by sulphate-reducing bacteria. Org Geochem 31:101–115
Wöhlbrand L, Jacob JH, Kube M, Mussmann M, Jarling R, Beck A, Amann R, Wilkes H, Reinhardt R, Rabus R (2013) Complete genome, catabolic sub-proteomes and key-metabolites of Desulfobacula toluolica Tol2, a marine, aromatic compound-degrading, sulfate-reducing bacterium. Environ Microbiol 15:1334–1355
Wöhlbrand L, Ruppersberg HS, Feenders C, Blasius B, Braun H-P, Rabus R (2016) Analysis of membrane-protein complexes of the marine sulfate reducer Desulfobacula toluolica Tol2 by 1 D blue native-PAGE complexome profiling and 2D blue native-/SDS-PAGE. Proteomics 16:973–988
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this entry

Cite this entry
Rabus, R., Wilkes, H. (2020). Functional Genomics of Sulfate-Reducing Bacteria Degrading Hydrocarbons. In: Boll, M. (eds) Anaerobic Utilization of Hydrocarbons, Oils, and Lipids. Handbook of Hydrocarbon and Lipid Microbiology . Springer, Cham. https://doi.org/10.1007/978-3-319-50391-2_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-50391-2_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-50390-5
Online ISBN: 978-3-319-50391-2
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences