
Abstract
The sulfate-reducing highly enriched culture N47 is capable to anaerobically degrade naphthalene, 2-methylnaphthalene, and 2-naphthoic acid. A proteogenomic investigation was performed to elucidate the initial activation reaction of anaerobic naphthalene degradation. This lead to the identification of an alpha-subunit of a carboxylase protein that was two-fold up-regulated in naphthalene-grown cells compared to 2-methylnaphthalene-grown cells. The putative naphthalene carboxylase subunit showed 48% similarity to the anaerobic benzene carboxylase from an iron-reducing, benzene-degrading culture and 45% to alpha-subunit of phenylphosphate carboxylase of Aromatoleum aromaticum EbN1. A gene for the beta-subunit of putative naphthalene carboxylase was located nearby on the genome and was expressed with naphthalene. Similar to anaerobic benzene carboxylase, there were no genes for gamma- and delta-subunits of a putative carboxylase protein located on the genome which excludes participation in degradation of phenolic compounds. The genes identified for putative naphthalene carboxylase subunits showed only weak similarity to 4-hydroxybenzoate decarboxylase excluding ATP-independent carboxylation. Several ORFs were identified that possibly encode a 2-naphthoate-CoA ligase, which is obligate for activation before the subsequent ring reduction by naphthoyl-CoA reductase. One of these ligases was exclusively expressed on naphthalene and 2-naphthoic acid and might be the responsible naphthoate-CoA-ligase.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Naphthalene is the smallest polycyclic aromatic hydrocarbon and it exhibits a low chemical reactivity due to its aromatic 10π electron system. Since several decades, aerobic naphthalene degradation has been extensively studied and the initial reactions by oxygenase enzymes have been described in detail (Habe and Omori 2003). In contrast, the biochemical mechanism of anaerobic naphthalene degradation with alternative electron acceptors like sulfate or nitrate has not yet been adequately elucidated although several nitrate- and sulfate-reducing pure and highly enriched microbial cultures are available (Zhang and Young 1997; Galushko et al. 1999; Meckenstock et al. 2000; Rockne et al. 2000; Musat et al. 2009). However, in the absence of molecular oxygen, totally different and new biochemical reactions must be used by the microorganisms to activate the non-substituted aromatic ring structure of naphthalene.
Two different biochemical activation reactions have been proposed for naphthalene degradation under anoxic conditions namely methylation of naphthalene to 2-methylnaphthalene (Annweiler et al. 2000; Safinowski and Meckenstock 2006) and direct carboxylation to 2-naphthoic acid (Zhang and Young 1997; Musat et al. 2009). A hydroxylation to 1-naphthol or 2-naphthol as initial attack is unlikely, as culture N47 is not growing with these compounds as substrate, nor could hydroxylated metabolites be detected by GC–MS during growth on naphthalene (Meckenstock et al. 2000). The proposal of the methylation reaction was based on metabolite analyses of culture supernatants of the sulfate-reducing culture N47 during growth on deuterated D8-naphthalene. Here, deuterated D7-naphthyl-2-methyl-succinate and D7-naphthyl-2-methylene-succinate where identified which are specific metabolites of the anaerobic 2-methylnaphthalene degradation pathway (Safinowski and Meckenstock 2006). Other metabolites identified in culture N47 were e.g. 2-naphthoic acid (Meckenstock et al. 2000). Moreover, enzymatic activities of several reactions of the 2-methylnaphthalene degradation pathway were measured in naphthalene-grown cells i.e. naphthyl-2-methyl-succinate synthase, naphthyl-2-methyl-succinyl-CoA transferase, and naphthyl-2-methenyl-succinyl-CoA dehydrogenase (Safinowski and Meckenstock 2004, 2006). Naphthyl-2-methyl-succinate synthase (Nms) catalyzes fumarate addition to the methyl group leading to the formation of naphthyl-2-methyl-succinate (Annweiler et al. 2000). Nms is a glycyl radical enzyme analogous to the benzylsuccinate synthase (Bss) involved in anaerobic toluene degradation (Leuthner et al. 1998). The enzyme and metabolite studies supported a methylation of naphthalene to 2-methylnaphthalene and the subsequent degradation via the 2-methylnaphthalene pathway to the central metabolite 2-naphthoyl-CoA for the sulfate-reducing culture N47. The utilization of 2-methylnaphthalene of naphthalene-adapted N47 cells without induction phase further supported methylation as possible activation reaction (Safinowski and Meckenstock 2006). Recently, the complete gene cluster of 2-methylnaphthalene degradation and its expression in 2-methylnaphthalene-grown N47 cells were described (Selesi et al. 2010).
The second putative anaerobic degradation mechanism of naphthalene activation was for the first time proposed by Zhang and Young who observed the incorporation of 13C-labeled bicarbonate from the buffer into the carboxyl group of 2-naphthoic acid (Zhang and Young 1997). This finding was interpreted as a carboxylation as the initial activation reaction of naphthalene. A similar incorporation of 13C from the bicarbonate buffer into the carboxyl group was shown for culture N47 (Meckenstock et al. 2000). Comparative proteome analysis after electrophoretic separation of the cell extracts from sulfate-reducing cultures NaphS2, NaphS3, and NaphS6 growing on naphthalene and methylnaphthalene revealed Nms as a specific 2-methylnaphthalene-induced protein band that was apparently absent in naphthalene-grown cells (Musat et al. 2009). During simultaneous growth of culture NaphS2 on D8-labeled naphthalene and unlabeled 2-methylnaphthalene, a high abundance of deuterated 2-naphthoyl-CoA and a comparatively very low portion of unlabeled naphthyl-2-methyl-succinate were formed during growth (Musat et al. 2009). These results rather supported a direct carboxylation of naphthalene instead of methylation. The additional formation of trace amounts of labeled naphthyl-2-methyl-succinate during this experiment was explained by reverse reactions of the 2-methylnaphthalene degradation pathway starting from 2-naphthoyl-CoA (Musat et al. 2009). This could be as well the reason for the detection of 2-methlynaphthalene degradation–specific metabolites in naphthalene-grown N47 cells. In culture N47, the regulation of the naphthalene and 2-methylnaphthalene pathways is obviously different from culture NaphS2 as both pathways seem to be similarly expressed during growth on naphthalene. In contrast to the results of Safinowski and Meckenstock, the exposure of naphthalene-grown cells to 2-methylnaphthalene resulted in an extended lag phase (Musat et al. 2009). So far, the activity of a putative carboxylating enzyme could not be measured in crude cell extracts.
Both putative pathways have in common that 2-naphthoyl-CoA is the central intermediate compound. 2-Naphthoyl-CoA is subjected to ring reduction by a putative 2-naphthoyl-CoA reductase whose gene sequence has been recently deduced from the genome sequence of culture N47 (Selesi et al. 2010).
Here, we performed a proteomic study to identify enzymes involved in the initial activation reaction of anaerobic naphthalene degradation. In a differential expression study with naphthalene, 2-methylnaphthalene, and 2-naphthoic acid as growth substrates, the whole proteomes of naphthalene-grown N47 cells were screened for the presence of proteins of anaerobic aromatic hydrocarbon degradation. In combination with the genome sequence information, we were able to identify several genes coding for a putative carboxylating enzyme in naphthalene-grown N47 cultures.
Materials and methods
Growth conditions for the enrichment culture N47
The sulfate-reducing culture N47 was enriched from a former coal gasification site and cultivated as reported previously (Safinowski and Meckenstock 2004). The substrates naphthalene and 2-methylnaphthalene were added as 1.5% solutions in 2,2,4,4,6,8,8-heptamethylnonane (Sigma–Aldrich, Steinheim, Germany) to the cultivation bottles after autoclaving (1 ml/50 ml medium). 2-naphthoic acid was provided as solid crystals to the bottles before autoclaving to reach a final concentration of 600 μM. Inoculum of the enrichment culture N47 was added to the growth media in 1:10 dilutions. Substrate utilization was monitored by colorimetric measurement of sulfide production (Cline 1969).
Terminal restriction fragment length polymorphism
To assure that on both substrates the composition of the enrichment culture was identical, a sample was taken from every bottle for analysis of terminal restriction fragment length polymorphism (T-RFLP). Samples were washed 3 times with 0.5× PBS (phosphate-buffered saline). DNA extraction was done with FastDNA® SPIN Kit for Soil purchased from MP Biomedicals (Illkirch, France) following the delivered manual. In the first step instead of 978 μl of sodium phosphate buffer, only 878 μl was used. Additionally, 100 μl 10 mM potassium citrate was added, to get rid of the iron(II)sulfide, produced during growth. T-RFLP analysis of bacterial 16S rRNA gene amplicons was done as described elsewhere (Lueders et al. 2006) using primers Ba27f-FAM and 907r (Weisburg et al. 1991; Muyzer et al. 1995) and digestion by restriction enzyme MspI (Fermentas, St. Leon-Rot, Germany). Primary electropherogram evaluation was performed using GeneMapper 5.1 software (Applied Biosystems).
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis
For whole proteome analysis, triplicate cultures of N47 grown on naphthalene or 2-methylnaphthalene were harvested when 3.5 mM sulfide accumulated, 2-naphthoic acid grown cells at 2.5 mM sulfide concentration. Cell harvesting, protein purification, and SDS–PAGE of the 800 ml sample were performed as described elsewhere (Selesi et al. 2010). The visualization of protein bands in SDS–PAGE was done by Coomassie brilliant blue staining (Zehr et al. 1989).
Linear quadrupole ion trap-Orbitrap mass spectrometry with nano-electrospray ionization
Tryptic cleavage
Every lane of the gels was cut into 10 slices using a scalpel. Subsequently, each was washed for 10 min with 200 μl H2O. The destaining was done by washing the slices thrice with 200 μl 60% acetonitrile (ACN) for 15 and 10 min with 200 μl H2O. For protein reduction and alkylation 100 μl 5 mM dithiothreitol (DTT) was added and incubated for 15 min at 60°C. Then, the DTT solution was removed, 100 μl 25 mM iodacetamide was added and incubated for 15 min in the dark. Subsequently, the slice was washed with 200 μl H2O for 5 min. The next three washing steps were all done for 15 min and with a volume of 200 μl in the order: 100% ACN, 50 mM ammonium bicarbonate, and 40% ACN. As next washing step, 200 μl 100% ACN were used for 5 min. The supernatant was discarded and the resting gel pieces were air-dried. The gel slices were overlaid with 0.01 μg/μl trypsin in 50 mM ammonium bicarbonate. After 10 min, 25 mM ammonium bicarbonate solution was added to completely cover the slices with liquid. The digestion proceeded over night at 37°C. Then, 1–2 μl 0.5% trifluoroacetic acid (TFA) was added and shaken for 15 min. 50–100 μl 50% ACN and 0.1% TFA were added and shaken for 15 min. Afterward 50–100 μl 99.9% ACN together with 0.1% TFA was added and shaken for 15 min. The samples were completely dried in a speed vac. The samples were redissolved in 2% ACN/0.5% TFA immediately before the analysis.
MS analysis and data processing
The digested peptides were separated by reversed phase chromatography (PepMap, 15 cm × 75 μm ID, 3 μm/100Å pore size, LC Packings) operated on a nano-HPLC (high performance liquid chromatography) (Ultimate 3000, Dionex) with a nonlinear 120 min gradient using 2% acetonitrile in 0.1% formic acid in water (A) and 0.1% formic acid in 98% acetonitrile (B) as eluents with a flow rate of 250 nl/min. The gradient settings were subsequently: 0–90 min: 2–30% B, 90–105 min: 31–99% B, 106–115 min: Stay at 99% B. The nano-LC was connected to a linear quadrupole ion trap-Orbitrap (LTQ Orbitrap XL) mass spectrometer (ThermoFisher, Bremen, Germany) equipped with a nano-ESI (electrospray ionization) source. The mass spectrometer (MS) was operated in the data-dependent mode to automatically switch between Orbitrap-MS and LTQ-MS/MS acquisition. Survey full scan MS spectra (from m/z 300 to 1,500) were acquired in the Orbitrap with resolution R = 60,000 at m/z 400 (after accumulation to a target of 1,000,000 charges in the LTQ). The method used allowed sequential isolation of the ten most intense ions, depending on signal intensity, for fragmentation on the linear ion trap using collision-induced dissociation at a target value of 100,000 ions. High-resolution MS scans in the orbitrap and MS/MS scans in the linear ion trap were performed in parallel. Target peptides already selected for MS/MS were dynamically excluded for 30 s. General mass spectrometry conditions were electrospray voltage, 1.25–1.4 kV; no sheath and auxiliary gas flow. Ion selection threshold was 500 counts for MS/MS, and an activation Q-value of 0.25 and activation time of 30 ms were also applied for MS/MS.
All MS/MS samples were analyzed using Mascot (Matrix Science (Version: 2.2.06). Mascot was set up to search the N47 Database (Number of residues: 1,353,040, Number of sequences: 5,244) assuming the digestion enzyme trypsin and with a fragment ion mass tolerance of 1 Da and a parent ion tolerance of 10 PPM. Iodoacetamide derivative of cysteine as stable modification and oxidation of methionine, deamidation of arginine and glutamine as well as hydroxyl modification of lysine, arginine and histidines as variable modifications were specified in Sequest and Mascot as variable modifications.
Criteria for identification
Scaffold (version Scaffold_2_02_03, Proteome Software Inc., Portland, Oregon) was used to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 80.0% probability as specified by the Peptide Prophet algorithm (Keller et al. 2002). Protein identifications were accepted if they could be established at greater than 95% probability and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm (Nesvizhskii et al. 2003). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony.
Nevertheless, it is a great challenge to identify all expressed proteins due to multiple reasons. For example the large number of genes encoded in organisms, the dynamic range of proteins is very large and often beyond the intrinsic limitation of instrument sensitivity and furthermore posttranslational modifications further increasing protein complexity (Wu and Han 2006).
Two-dimensional gel electrophoresis
For comparative proteome analysis of naphthalene- and 2-methylnaphthalene-grown N47 cultures, cells from 1 l cultures were harvested, and proteins were extracted and quantified as mentioned earlier. Aliquots of triplicate samples containing 50 μg proteins were separated in the first dimension by isoelectric focusing (IEF) and in the second dimension by SDS–polyacrylamide gel electrophoresis (SDS–PAGE). IEF was performed using 18 cm immobilized nonlinear pH gradient strips (pH 3–10; GE Healthcare Europe GmbH, Freiburg, Germany). The strips were rehydrated for 16 h at room temperature in the presence of lysis buffer in an Immobiline DryStrip reswelling tray (GE Healthcare). The strips were placed in a Manifold tray on the Ettan IPGphor II unit (GE Healthcare) and covered with DryStrip cover fluid (GE Healthcare). Voltage was increased stepwise from 150 to 8,000 V over 5.5 h and held at this value for 2 h resulting in 32 kVh. Prior to the second dimension, strips were equilibrated in buffer containing 6 M urea, 2% SDS, 30% glycerol, 100 mM Tris, pH 8.8, 0.01% bromophenol blue, reduced with 60 mM 1,4-dithioerythritol and subsequently alkylated with 220 mM iodoacetamide. The stripes were immediately applied on top of a 12% SDS–PAGE, and electrophoresis was carried out at 50 V for 30 min and then increased to 90 V until the bromophenol blue dye migrated to the bottom of the gel. The proteins were visualized by silver staining (Rappsilber et al. 2000), scanned and analyzed using Proteomweaver 2-D Analysis Software (Bio-Rad Laboratories, Hercules, USA).
Matrix-assisted laser desorption ionization-time of flight mass spectrometry
Tryptic cleavage
Protein spots from 2D-gels were manually cut out, destained and washed with buffer containing 50 mM NH4HCO3 in 30% ACN and equilibrated in 10 mM NH4HCO3 prior to proteolytic digestion. Gel pieces were shrunk with 100% v/v ACN and rehydrated in 10 mM NH4HCO3. This treatment was repeated, followed by the addition of 0.1–0.2 μg of modified trypsin (SIGMA) per piece. Digestion was carried out overnight at 37°C. The supernatant was collected and combined with the eluates of subsequent elution steps with 80% v/v ACN, 1% v/v TFA. The combined eluates were dried in a SpeedVac centrifuge. The dry samples were dissolved in 20 μl 50% v/v ACN, 0.1% v/v TFA for the subsequent MALDI preparation. Therefore, 0.5 μl of a 1:1 mixture of sample and a matrix solution consisting of 5 mg/ml CHCA (Bruker, Bremen, Germany) were spotted on a MALDI target.
MS analysis and data processing
Mass spectra were acquired using a Proteomics Analyzer 4700 (MALDI-TOF/TOF) mass spectrometer (Applied Biosystems, Framingham, USA). Measurements were performed with a 355 nm Nb:YAG laser in positive reflector mode with a 20-kV acceleration voltage. For each MS and MS/MS spectrum, 3,000 shots were accumulated. For each spot on a MALDI plate, the eight most intense peptides were selected for additional MS/MS analysis. The acquired MS/MS spectra were searched against the UniRef100 databases (updated April 2009) using an in-house version of Mascot with the following parameters: As taxon we chose human and as enzyme trypsin. We set carbamidomethylation as fixed modification and oxidized methionine as variable modifications.
Criteria for identification
The GPS Explorer 2 software reports two different scores: The Mascot best ion score, the highest score of a single peptide, and a total ion score, the sum of all peptide scores of one protein. The significance level for a peptide score is usually higher than 20 and for a protein score higher than 50–60. Because different database searches have different Mascot significance levels due to different databases sizes and different numbers of masses submitted for a search, scores cannot be compared directly. For this reason, the software calculates a confidence interval from Mascot protein scores or ion scores, and the Mascot significance level for each search is defined as the 95% confidence level. Therefore, the total ion score confidence level is a reliable and comparable parameter for the significance of a database search.
Results and discussion
Culture composition
For differential expression experiments with the enrichment culture N47, it was essential to assure that the microbial composition was not changing when growing on different substrates. Therefore, T-RFLP analysis was done for every grown culture. All T-RFLP fingerprints (Fig. 1) showed a dominant 513-bp T-RF assigned to Deltaproteobacteria and a minor 207-bp T-RF belonging to members of Spirochaetes which agrees totally with the figure given earlier by Selesi et al. (2010). As the culture composition did not change, we could directly assign differences in protein expression to the utilization of naphthalene, 2-methylnaphthalene, or 2-naphthoic acid as growth substrate. Furthermore, all proteins measured by MS/MS peptide sequencing were identified by mapping them to the database of the genome of Deltaproteobacterium N47, which has been recently annotated (Bergmann et al. 2010). This assures that all identified enzymes involved in the anaerobic degradation of naphthalene belong to the Deltaproteobacterium N47. Therefore, the further conclusions were drawn as dealing with a pure culture.

T-RFLP analysis of bacterial 16S rRNA gene sequence of culture N47 grown on naphthalene (a), 2-methylnaphthalene (b) and 2-naphthoic acid (c). The length of major T-RFs is indicated
Shotgun proteome analysis of naphthalene- and 2-methylnaphthalene- grown N47 cells
Comparing the bands for naphthalene and 2-methylnaphthalene of the Coomassie-stained one-dimensional SDS–PAGE, no significant difference was visible. However, the MS/MS peptide sequencing identified 854 proteins when growing on naphthalene as sole carbon source and 782 proteins with 2-methylnaphthalene. In both lanes, 689 proteins were identified, whereas 165 unique proteins were exclusively detected with naphthalene and 93 with 2-methylnaphthalene.
Nevertheless, the detection of proteins meant to be exclusive for a certain substrate has to be treated with caution. As already mentioned in the material and methods section, if a protein could not be detected, this does not necessarily imply that it is not expressed upon growth (Wu and Han 2006). Moreover, there is a certain loss during protein purification. However, none of the proteins exclusively detected in cells grown with either naphthalene or 2-methylnaphthalene as substrate could be related to aromatic hydrocarbon degradation. For some proteins, a potential function in aromatics degradation could not be excluded completely because they did not show the slightest homology to known proteins and their function could not be assigned, therefore.
For both substrates, naphthalene and 2-methylnaphthalene, all enzymes for the complete anaerobic 2-methylnaphthalene degradation pathway (Selesi et al. 2010) were expressed, which agrees with earlier results (Safinowski and Meckenstock 2006) and might also support the methylation hypothesis for the initial reaction step of anaerobic naphthalene degradation.
In contrast to this study, SDS–PAGE studies of strain NaphS2 showed strain specific induction of a nms-synthase with 2-methylnaphthalene as growth substrate but not with naphthalene (Musat et al. 2009). Therefore, methylation as initial reaction mechanism for anaerobic degradation of naphthalene by NaphS2 was excluded by the authors.
Proteome analysis using 2-DGE
To quantitatively compare proteomic differences in expression levels during growth on naphthalene and 2-methylnaphthalene, we employed two-dimensional gel electrophoresis (2-DGE). For both substrates, the protein patterns showed more than 900 spots, sharing similarities in distribution and intensity (Fig. 2). A closer inspection revealed 19 spots exclusively expressed with naphthalene and 65 proteins which were up-regulated with naphthalene compared to 2-methylnaphthalene as growth substrate. Even though there are a number of limitations, it is commonly accepted that a two-fold change or larger is sufficient to state significant differences in expression (Ting et al. 2009). Among the exclusively expressed or up-regulated proteins with naphthalene as growth substrate, potential candidate enzymes for the initial step in anaerobic naphthalene degradation should be present.

Silver-stained 2-DGE gel of proteins from naphthalene- (left) and 2-methylnaphthalene-grown cells (right). The marked spots were sequenced by MS/MS
However, it was only possible to identify 2 of the exclusively expressed spots by MALDI-TOF–MS/MS and assign them to the genome sequence: ORF N47_E42520 (spot nr. 1) and ORF N47_E41950 (spot nr. 11). These sequences indicated homology to a hypothetical cytosolic protein, putative uncharacterized protein and an ATPase (AAA + superfamily)-like protein. Nonetheless, from neither of the identified protein sequences, a conclusion could be drawn on a potential role in naphthalene degradation. Adjacent genes of both ORFs showed weather no similarity (downstream) or similarity with uncharacterized proteins (upstream) and did also not allow concluding for possible functions of the proteins represented by spots 1 and 11.
Therefore, 18 randomly chosen spots of the up-regulated proteins of naphthalene-grown cultures were sequenced which resulted in the identification of 8 distinct proteins (Table 1). The determined sequence of spot nr 28, which matched ORF N47_A08840 and spot nr 37, which matched ORF N47_F16090, had both homologies to hypothetical proteins only.
ORF N47_E41500 was retrieved when sequencing spot nr 20. This was identified as enoyl-CoA hydratase, as the alignment with the best BLAST hit resulted in 47% identity with 95% sequence coverage with the sequence of Thermosinus carboxydivorans Nor1. Enoyl-CoA hydratase is known to hydrate the double bond between the alpha and beta carbon atom of acyl-CoA, essential in fatty acid metabolism. Furthermore, it has been shown that an enoyl-CoA hydratase is leading to the hydrolytic aromatic ring cleavage in anaerobic benzoate degradation (Boll et al. 2002; Rabus et al. 2005). Sequenced spot nr 36 matched ORF N47_E41480, which is coding for the beta-subunit of 2-naphthoyl-CoA reductase, identified by Selesi et al. (2010). Additionally, the delta-subunit of 2-naphthoyl-CoA reductase (ORF N47_E41460) was hit by the sequence of spot nr 22.
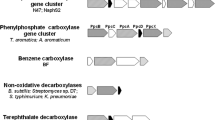
The sequences of spots nr 30, 35, and 41 matched an identical ORF (N47_K27480), which had 29% identity with 90% sequence coverage to 3-octaprenyl-4-hydroxybenzoate decarboxylase of Halorhodospira halophila SL1 based on amino acid sequence alignment. The same enzyme was also hit with 30% identity by ORF N47_K27500, which was matched by the sequenced spots nr 21 and 43. Spots nr 23 and 26 were assigned to ORF N47_K27520, which showed homology to an ATPase-like protein (involved in chromosome partitioning) with 38% identity. Another six sequenced spots (25, 29, 32, 34, 40, and 44) hit the same ORF N47_K27540, with amino acid sequence homology to alpha-subunit of phenylphosphate carboxylase with 45% identity, which is sufficient to assign functional and structural properties to the enzyme.
Verification of putative candidates by homology studies
A further BLAST (Altschul et al. 1997) comparison on amino acid level of ORF N47_K27540 of N47 (putative carboxylase alpha-subunit) to recently discovered putative anaerobic benzene carboxylase of the iron-reducing benzene-degrading culture BF (Abu Laban et al. 2010) resulted in 48% identity, and covered nearly 99% of the N47 ORF length. However, the function of anaerobic benzene carboxylase was deduced from sequence homology to the enzyme phenylphosphate carboxylase (Schuhle and Fuchs 2004; Abu Laban et al. 2010). Phenylphosphate carboxylase catalyzes the carboxylation of phenylphosphate to 4-hydroxybenzoate during anaerobic phenol degradation (Schuhle and Fuchs 2004). The putative carboxylase ORF N47_K27540 of N47 showed 45% protein sequence identity when blasting it against the alpha-subunit of phenylphosphate carboxylase of A. aromaticum EbN1 which was also the best hit when blasting the N47 sequence against all non-redundant peptide sequence databases. The calculated molecular masses as well as the poly peptide chain length of the putative N47 carboxylase and the phenylphosphate carboxylase of strain EbN1 were in the same range (53,992 Da (EbN1) vs 53,226 Da (N47) and 485 aa`s (EbN1) vs 481 aa`s (N47)). As the alignment values were sufficient for functional and structural consistency, we conclude that ORF N47_K27540 codes for the putative alpha-subunit of an anaerobic naphthalene carboxylase.
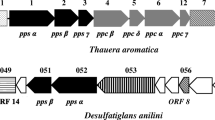
In addition to a putative carboxylase alpha-subunit, we identified four ORFs for a putative beta-subunit on the same contig FR695876: N47_K27500, N47_K27390, N47_K27480 and N47_K27460. These ORFs are located in relative neighborhood to the putative alpha-subunit on the genome and exhibit significant homology to the gene of the phenylphosphate carboxylase beta-subunit (ppcB) of A. aromaticum EbN1 (Fig. 3). Putative ppcA and ppcB (except N47_K27390) genes for naphthalene carboxylase are expressed on both substrates naphthalene and 2-methylnaphthalene.

Genes identified on contig FR695876 in the genome of sulfate-reducer N47 encoding putative proteins of anaerobic polycyclic aromatic hydrocarbon degradation
Notably, similar to anaerobic benzene carboxylase, we could not identify an ORF on the N47 genome which might be similar to the ppcC or ppcD genes coding for the two other subunits (gamma and delta) of phenylphosphate carboxylase (Table 2). In anaerobic phenol degradation, the delta-subunit catalyzes the phosphatase reaction of phenolphosphate (Schuhle and Fuchs 2004). Similar to anaerobic benzene carboxylation such a phosphatase reaction would not make sense for carboxylation of naphthalene because there is simply no activated phosphate bound to the molecule. Furthermore, this finding excludes that the putative carboxylase protein which is expressed during growth on naphthalene (N47_K27540) might be involved in degradation of phenol or other phenolic compounds because here the gamma- and delta-subunits would be needed. We also exclude a carboxylation mechanism similar to the ATP-independent phenol carboxylation used by other anaerobic phenol degraders, because those enzymes contain 6 subunits (He and Wiegel 1995) which were not found in N47. Only the genes identified for putative naphthalene carboxylase subunits show similarity to this 4-hydroxybenzoate decarboxylase, but it is significantly smaller than the similarity to the phenylphosphate carboxylase alpha- and beta-subunit.
This is further supported when growing culture N47 on phenol. The T-RFLP analysis of the microbial community showed that the dominant 513-bp T-RF (assigned to Deltaproteobacteria) for the naphthalene-grown culture became much smaller and the formerly marginal 207-bp T-RF (belonging to members of Spirochaetes) became dominant for the phenol-grown culture (data not shown). This indicates that the phenol degradation was performed by the spirochaetal members of the culture and not by the sulfate-reducing deltaproteobacteria.
2-Naphthoate-CoA-ligase
In contrast to the anaerobic benzene carboxylation gene cluster of culture BF where a putative benzoate-CoA-ligase is located directly downstream of the carboxylase genes on the genome, no putative 2-naphthoate-CoA-ligase gene could be identified in the genomic neighborhood of the proposed N47 naphthalene carboxylase genes. However, we have been able to measure a specific ATP-dependent 2-naphthoate-CoA-ligase activity in crude cell extracts of culture N47 grown on naphthalene (data not shown). The amino acid sequence of benzoate-CoA ligase of Rhodopseudomonas palustris (Gibson et al. 1994) was blasted against the N47 genome to identify putative candidates (Table 3).
However, one of the ORFs given in Table 3 is probably the 2-naphthoate-CoA-ligase needed for anaerobic naphthalene degradation, even if not located on the same contig as the carboxylase. The most promising ORF for a putative 2-naphthoate-CoA-ligase is N47_B20660, because it is expressed exclusively on naphthalene and 2-naphthoic acid according to the one-dimensional SDS–PAGE proteomic approach. Similar to growth on naphthalene, during growth on 2-naphthoic acid an activation of the carboxyl group to the coenzyme A thioester 2-naphthoyl-CoA would be essential. Nevertheless, also ORFs N47_J03730 and N47_J06130 are possible candidates, as both were expressed in cells grown on all three tested substrates. However, there was no information about possible up-regulation available because the respective spots could not been identified in the 2D gels. Therefore, a specific 2-naphthoate-CoA-ligase could not be identified yet.
Conclusion
Until now two hypotheses for the initial step in anaerobic naphthalene degradation are discussed. On the one hand, a methylation of naphthalene to 2-methylnaphthalene was proposed (Meckenstock et al. 2000; Safinowski and Meckenstock 2006). The 2-methylnaphthalene would then be further degraded by the recently described 2-methylnaphthalene degradation pathway (Annweiler et al. 2000; Safinowski and Meckenstock 2004; Selesi et al. 2010). On the other hand, a direct carboxylation of naphthalene to 2-naphthoic acid was suggested (Zhang and Young 1997; Musat et al. 2009).
Our results lead to another line of evidence that in anaerobic degradation, naphthalene is activated by a carboxylase reaction producing 2-naphthoic acid. This conclusion is based on (1) the specific up-regulation of a carboxylase-related polypeptide during growth on naphthalene as compared to 2-methylnaphthalene, (2) a striking similarity of the sequence and the gene cluster structure to the genes encoding anaerobic benzene carboxylase which catalyze a chemically similar reaction, and (3) the absence of gamma- and delta-subunits of the putative naphthalene carboxylase which distinguishes this enzyme from phenylphosphate carboxylase, the closest-related enzyme where the two subunits gamma and delta are needed for the phosphatase reaction.
The similarity of the carboxylase enzymes for the non-substituted aromatic hydrocarbons naphthalene and benzene suggests a new class of carboxylases catalyzing this novel reaction. The anaerobic activation of non-substituted aromatic hydrocarbons presents an unprecedented reaction in biochemistry which remains open to be elucidated in detail.
References
Abu Laban N, Selesi D, Rattei T, Tischler P, Meckenstock RU (2010) Identification of enzymes involved in anaerobic benzene degradation by a strictly anaerobic iron-reducing enrichment culture. Environ Microbiol 12:2783–2796
Altschul SF et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Annweiler E et al (2000) Anaerobic degradation of 2-methylnaphthalene by a sulfate-reducing enrichment culture. Appl Environ Microb 66:5329–5333
Bergmann FD, Selesi D, Weinmaier T, Tischler P, Rattei T, Meckenstock RU (2010) Genomic insights into the metabolic potential of the polycyclic aromatic hydrocarbon degrading sulphate-reducing Deltaproteobacterium N47. Environ Microbiol
Boll M, Fuchs G, Heider J (2002) Anaerobic oxidation of aromatic compounds and hydrocarbons. Curr Opin Chem Biol 6:604–611
Cline JD (1969) Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol Oceanogr 14:454–458
Galushko A, Minz D, Schink B, Widdel F (1999) Anaerobic degradation of naphthalene by a pure culture of a novel type of marine sulphate-reducing bacterium. Environ Microbiol 1:415–420
Gibson J, Dispensa M, Fogg GC, Evans DT, Harwood CS (1994) 4-Hydroxybenzoate-coenzyme A ligase from Rhodopseudomonas palustris: purification, gene sequence, and role in anaerobic degradation. J Bacteriol 176:634–641
Habe H, Omori T (2003) Genetics of polycyclic aromatic hydrocarbon metabolism in diverse aerobic bacteria. Biosci Biotech Bioch 67:225–243
He Z, Wiegel J (1995) Purification and characterization of an oxygen-sensitive reversible 4-hydroxybenzoate decarboxylase from Clostridium hydroxybenzoicum. Eur J Biochem 229:77–82
Keller A, Nesvizhskii AI, Kolker E, Aebersold R (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74:5383–5392
Leuthner B et al (1998) Biochemical and genetic characterization of benzylsuccinate synthase from Thauera aromatica: a new glycyl radical enzyme catalysing the first step in anaerobic toluene metabolism. Mol Microbiol 28:615–628
Lueders T, Kindler R, Miltner A, Friedrich MW, Kaestner M (2006) Identification of bacterial micropredators distinctively active in a soil microbial food web. Appl Environ Microb 72:5342–5348
Meckenstock RU, Annweiler E, Michaelis W, Richnow HH, Schink B (2000) Anaerobic naphthalene degradation by a sulfate-reducing enrichment culture. Appl Environ Microb 66:2743–2747
Musat F et al (2009) Anaerobic degradation of naphthalene and 2-methylnaphthalene by strains of marine sulfate-reducing bacteria. Environ Microbiol 11:209–219
Muyzer G, Teske A, Wirsen C, Jannasch H (1995) Phylogenetic relationships of Thiomicrospira species and their identification in deep-sea hydrothermal vent samples by denaturing gradient gel electrophoresis of 16S rDNA fragments. Arch Microbiol 164:165–172
Nesvizhskii AI, Keller A, Kolker E, Aebersold R (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75:4646–4658
Rabus R et al (2005) The genome sequence of an anaerobic aromatic-degrading denitrifying bacterium, strain EbN1. Arch Microbiol 183:27–36
Rappsilber J, Siniossoglou S, Hurt EC, Mann M (2000) A generic strategy to analyze the spatial organization of multi-protein complexes by cross-linking and mass spectrometry. Anal Chem 72:267–275
Rockne KJ, Chee-Sanford JC, Sanford RA, Hedlund BP, Staley JT, Strand SE (2000) Anaerobic naphthalene degradation by microbial pure cultures under nitrate-reducing conditions. Appl Environ Microb 66:1595–1601
Safinowski M, Meckenstock RU (2004) Enzymatic reactions in anaerobic 2-methylnaphthalene degradation by the sulphate-reducing enrichment culture N47. FEMS Microbiol Lett 240:99–104
Safinowski M, Meckenstock RU (2006) Methylation is the initial reaction in anaerobic naphthalene degradation by a sulfate-reducing enrichment culture. Environ Microbiol 8:347–352
Schuhle K, Fuchs G (2004) Phenylphosphate carboxylase: a new C-C lyase involved in anaerobic in phenol metabolism in Thauera aromatica. J Bacteriol 186:4556–4567
Selesi D et al (2010) Combined genomic and proteomic approaches identify gene clusters involved in anaerobic 2-methylnaphthalene degradation in the sulfate-reducing enrichment culture N47. J Bacteriol 192:295–306
Ting L, Cowley MJ, Hoon SL, Guilhaus M, Raftery MJ, Cavicchioli R (2009) Normalization and statistical analysis of quantitative proteomics data generated by metabolic labeling. Mol Cell Proteomics 8:2227–2242
Walter MC et al (2009) PEDANT covers all complete RefSeq genomes. Nucleic Acids Res 37:D408–D411
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697–703
Wu LF, Han DK (2006) Overcoming the dynamic range problem in mass spectrometry-based shotgun proteomics. Expert Rev Proteomic 3:611–619
Zehr BD, Savin TJ, Hall RE (1989) A one-step, low background Coomassie staining procedure for polyacrylamide gels. Anal Biochem 182:157–159
Zhang XM, Young LY (1997) Carboxylation as an initial reaction in the anaerobic metabolism of naphthalene and phenanthrene by sulfidogenic consortia. Appl Environ Microb 63:4759–4764
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Joerg Overmann.
Rights and permissions
About this article
Cite this article
Bergmann, F.D., Selesi, D. & Meckenstock, R.U. Identification of new enzymes potentially involved in anaerobic naphthalene degradation by the sulfate-reducing enrichment culture N47. Arch Microbiol 193, 241–250 (2011). https://doi.org/10.1007/s00203-010-0667-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-010-0667-4