
Abstract
The sigma-1 receptor is an enigmatic ER-resident transmembrane protein linked to a variety of human diseases. Although the receptor was first cloned 20 years ago, the molecular structure of the protein and the mechanistic basis for its interaction with drug-like small molecules have remained unclear until recently. The determination of the first crystal structure of human sigma-1 offered the first detailed views of the sigma-1 architecture, and revealed an unusual overall fold with a single transmembrane helix in each protomer. The structure shows an overall trimeric receptor arrangement, and each protomer binds a single ligand molecule at the center of its carboxy-terminal domain. These results offer detailed molecular views of receptor structure, oligomerization, and ligand recognition, providing a framework for the next era of sigma-1 research.
Authors (Assaf Alon, Hayden Schmidt, Sanduo Zheng) contributed equally and are listed in alphabetical order.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
2.1 Introduction
The sigma-1 receptor is an unusual transmembrane protein implicated in a broad range of cellular functions and with possible roles in both normal and disease states in humans [1]. Since its discovery decades ago, the sigma-1 receptor has been implicated in a diverse array of pathophysiological conditions ranging from neurodegenerative disease [2] to cancer [3], and it has been reported to interact with numerous proteins including chaperones [4], ion channels [5, 6], and GPCRs [7]. Like the true opioid receptors, sigma-1 shows high affinity for benzomorphan compounds and on this basis it was originally classified as a member of this family [8]. However, subsequent studies with enantiomerically pure probe compounds showed that sigma-1 exhibits a preference for (+) benzomorphans, while true opioid receptors bind with high affinity only to the (−) enantiomer [9]. The endogenous ligand of sigma-1, if any, remains unclear. Although the hallucinogen N,N-dimethyl tryptamine (DMT) has been reported as a possible ligand [10], a subsequent study has cast doubt on this idea [11].
Although the pharmacology and cell biology of sigma-1 has been extensively studied for decades, it was not until 1996 that the first information regarding the molecular architecture of the protein became available when the receptor was cloned. Sigma-1 was first cloned from guinea pig [12], using classical biochemical techniques to isolate the receptor by tracking binding activity and then using degenerate oligonucleotide probes to clone the receptor for a cDNA library. The receptor was subsequently cloned from a human placental choriocarcinoma cDNA library [13], as well as from mouse [14] and rat [15] tissues.
The amino acid sequence of the receptor showed no similarity to any other mammalian protein, although it resembled that of the fungal sterol isomerase Erg2p. Hydrophobicity analysis of the sequence showed a highly hydrophobic segment at the receptor amino terminus, predicted to be a transmembrane domain. Initially it was proposed that this was the sole transmembrane helix in the receptor [12], although later a two-pass transmembrane model came to be more widely embraced. The latter model was supported primarily by a report of immunostaining experiments with antibodies to green fluorescent protein (GFP) fused to either the amino- or carboxy-terminus of the receptor [16]. An important caveat, however, is the fact that GFP is often poorly secreted, and the fusion protein may have exhibited aberrant membrane insertion properties. Nonetheless, the two-pass transmembrane model was widely embraced, and served as the basis for molecular modeling studies [17] and efforts to map the putative second transmembrane helix [18]. As discussed below, however, the crystal structure of the receptor shows only a single transmembrane domain, consistent with the earliest structural models rather than those that followed.
2.2 Approach to Structure Determination
Recent advances in membrane protein structural biology have revolutionized structure determination for human membrane proteins [19], particularly G protein-coupled receptors (GPCRs). Key advances include the widespread use of lipid-based crystallization methods [20] and concomitant improvements in X-ray diffraction methods for microcrystals [21]. Taken together, these techniques allow crystallization of membrane proteins in a lipid bilayer system similar to their biological milieu, improving the stability of the proteins and allowing examination of their structural interactions with lipids. Other important advances including the use of new detergents [22] have also had a major impact on membrane protein biochemistry, allowing straightforward manipulation of otherwise intractable receptors.
In approaching structural analysis of the sigma-1 receptor, a GPCR-inspired approach was used. While previous methods for sigma-1 biochemistry involved bacterial expression [23] and the use of harsh detergents like Triton X-100, our crystallization effort focused instead on expression in eukaryotic cells and purification in milder maltoside detergents. In brief, this entailed use of Sf9 insect cells and baculovirus transduction to produce receptor at high levels, followed by extraction in detergent and purification by antibody affinity chromatography [24]. This approach yielded pure and almost monodisperse receptor with minimal modifications to the receptor sequence. Following proteolitic removal of the amino-terminal FLAG epitope tag, the resulting crystallization sample contained only a four amino amino acid modification, “GPGS”, at the receptor’s amino terminus, with all other parts matching the wildtype human sigma-1 sequence.
Following purification, crystallization of the receptor was straightforward using the lipidic cubic phase technique, and with optimization crystallographic datasets were obtained for sigma-1 bound to PD144418, a high affinity antagonist [25], and the compound 4-IBP, which has an incompletely understood efficacy profile [26]. Structure determination was hindered by the lack of related structures for phase calculation, but after an extensive screening campaign a suitable dataset was obtained by soaking with tantalum bromide clusters [27], allowing SIRAS phase calculation [24].
2.3 Overall Structure of Sigma-1
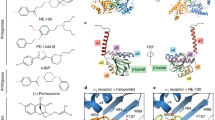
The crystal structures of the human sigma-1 receptor revealed an unusual fold, unique among known protein structures , and confirmed a single-pass transmembrane topology, in contrast to most previous models [24]. The protein crystallizes as an intimately associated triangular trimer with a transmembrane domain at each corner (Fig. 2.1). Residues 6–31 comprise the single transmembrane helix, with residues 32–223 forming a carboxy-terminal/cytosolic domain consisting of a β-barrel (residues 81–176) and flanking α-helices. This β-barrel constitutes both the ligand-binding site and the oligomerization interface. An unusual and striking feature of the structure is the presence of two α-helices (residues 177–223), which cover the membrane-proximal opening of the β-barrel. These helices have hydrophobic amino acids pointing toward the membrane surface, suggesting that the membrane-adjacent face of the trimer may be embedded within the lipid bilayer (Fig. 2.2). In addition, three arginine residues on the outer helix of each protomer are positioned in a way that would allow their positively charged side chains to interact with the negatively charged phospholipid head groups present in the cell membrane. In the crystal, these residues interact with sulfate ions in the crystallization buffer. Ordered monoolein lipids are also resolved, defining the boundary of the membrane plane. Thus, the crystal structure suggests that while the sigma-1 receptor only has one transmembrane domain, the membrane-proximal region of the protein formed by these two helices is partially embedded in the cytosolic side of the ER membrane, allowing this surface to dock against the lipid bilayer.

The overall structure of the human sigma-1 receptor. From the side, the receptor is observed to sit with the membrane-proximal surface partially embedded in the membrane, which is depicted in grey. The membrane boundary was determined using the PPM prediction server [42]. Viewing the receptor normal to the membrane from the ER surface shows the trimeric arrangement and overall architecture of the sigma-1 receptor

The membrane-proximal region of the sigma-1 receptor. A close-up view of a single protomer shows lipids observed in the crystal structure (depicted as yellow sticks; most of the lipid tails are disordered and not resolved). Shading indicates the location of the membrane. Arginine residues (orange) are well positioned to interact with phospholipid headgroups in the membrane, and many of the hydrophobic residues in the membrane-proximal helices (grey) are positioned in such a way as to be embedded in the hydrophobic membrane interior
Despite the unusual structure of the sigma-1 receptor as a whole, the β-barrel region bears significant structural similarity to cupin family proteins, with a root mean square deviation of 2.4–3.0 Å for most cupin domains [24], most of which are bacterial enzymes that also exhibit oligomerization . While there is no evidence of direct functional similarities between these proteins and sigma-1 receptor, the ligand-binding site of sigma-1 appears to be analogous to the active site of these proteins, which may suggest that the sigma-1 receptor descended from an enzyme that was evolutionarily repurposed. Indeed, the sigma-1 receptor’s closest homolog of well described function is the yeast sterol isomerase Erg2p [12].
Conservation analysis provides clues to the functional importance of the different regions of the sigma-1 receptor. The transmembrane helix is rather poorly conserved, with a relatively high degree of variation in the sequence among sigma-1 homologs (Fig. 2.3). The only sequence constraint on this helix appears to be the need for hydrophobicity, which suggests that the transmembrane helix is primarily an anchor to the membrane with little other function. In contrast, the β-barrel region, which includes the ligand-binding site and oligomerization interface, is almost perfectly conserved (Fig. 2.3). This suggests that both the ligand-binding site and the oligomerization interface are integral to sigma-1 receptor function.

Conservation of the sigma-1 receptor protomer. The receptor is shown with highly conserved regions colored in green and poorly conserved regions colored in red. Conservation analysis was performed using the ConSurf web server [43], using the most similar 300 sequences to the human sigma-1 receptor. The conservation map shows that the β-barrel region including the ligand-binding site and oligomerization interface is highly conserved, while the transmembrane domain is relatively poorly conserved
2.4 Oligomerization
In the crystal structure, sigma-1 is arranged as a triangular trimer with a ligand binding site in each protomer. Each interface between protomers buries roughly 9300 Å [2] of surface area, and the homotrimer interface is highly conserved across different species suggesting trimerization is physiologically relevant and not merely due to crystal packing (Fig. 2.4). The interface comprises a mix of polar and hydrophobic contacts. In particular, GxxxG motif (G87–G91), which was proposed to be part of a putative second transmembrane domain [16, 18], actually forms a beta-hairpin structure in the cytosolic domain buried deeply inside the center of interface and required for oligomerization and ligand binding [28]. From structural view, the distance between Cα atoms of G88 in each protomer is about 6 Å, consequently mutation of glycine to a large side chain residue would introduce a clash inside the interface, accounting for the observation that the G88L mutation favors the monomeric state over higher oligomeric states. Interestingly, the G88L mutant also exhibited a significant decrease in ligand binding , suggesting that the GxxxG-mediated oligomerization is likely important in either binding or protein folding, since G88 is distant from ligand binding site [28]. The correlation between oligomerization and ligand binding was also supported by the observation that oligomeric sigma-1 retained ligand binding while monomeric forms lost binding ability [28].

Oligomerization interface. (a) One protomer is shown as a surface and colored by sequence conservation, with residues more than 98 % and 80 % conservation highlighted in red and magenta, respectively. (b) A detailed view of interface with residues shown as sticks. The dashed lines indicate hydrogen bond and salt bridge interactions
However, despite the availability of the crystal structure of sigma-1 in a trimeric form, its oligomerization state in vivo remains uncertain. Detergent solubilized human sigma-1 in the presence of antagonist showed a broad range of oligomerization states as revealed by size-exclusion chromatography with multi-angle light scattering (SEC-MALS) as well as Native PAGE analysis [24]. In addition, several high molecular weight bands corresponding to tetramer and pentamer were identified using sigma-1 in rat liver membrane photoaffinity labeled by a radioiodinated ligand [29]. Taken together, these data suggest the sigma-1 trimer observed crystallographically may represent only one of many diverse oligomerization states existing in vivo which is prone to crystallization .
A related and important question regards the relationship between oligomerization and receptor activation. How do agonists and antagonists induce distinct cellular effects through the sigma-1 receptor? A cell-based study using FRET approaches revealed that in the absence of ligand, sigma-1 existed as a combination of different oligomeric states, while antagonist stabilized higher order oligomer, agonist instead favored small oligomers [30]. However, little difference in oligomerization was observed among ligand-free, agonist and antagonist bound sigma-1 when solubilized in detergent [24]. However, detergents do not perfectly mimic native membrane environments, thus these conditions may not reflect the actual state in vivo. A full understanding of sigma-1 oligomerization will require further biophysical and structural studies, including techniques like cryo-electron microscopy and NMR using sigma-1 reconstituted in lipid bilayers .
2.5 Ligand Recognition
The sigma-1 receptor has been shown to bind with high affinity and specificity to a variety of structurally diverse compounds [8]. Numerous structure-activity relationship (SAR) studies have been performed in an attempt to develop a common pharmacophore model, but the only common features shared by virtually all high-affinity sigma-1 ligands are a cationic amine and at least one aromatic ring, typically with three intervening methylenes [31]. Both sigma-1 crystal structures include bound ligands, offering a structural view of the sigma-1 receptor’s unique pharmacology.
The binding pocket of sigma-1 receptor is located in the center of a cone-shaped β-barrel that is gated at its wider side by two hydrophobic, membrane-parallel helices as discussed above (Fig. 2.1). In the crystal structure, the binding pocket is completely occluded from the solvent, and it remains unclear how ligands access the active site. The two possible pathways are either from the membrane through the two gating membrane-adjacent helices, or from the cytoplasm, through the narrow polar opening obstructed primarily by Gln135.
The sigma-1 receptor binding site is a wide and oblong cavity in the heart of the cytoplasmic domain. The binding pocket is lined with aromatic and hydrophobic residues, mirroring the hydrophobic nature of typical sigma-1 ligands. The only exceptions to the general hydrophobic character are the acidic residues Glu172 and Asp126. The former is highly conserved, and mutations in this position completely abrogate ligand binding as it serves to coordinate the positive charge of the ligand’s cationic amine [32]. Asp126, probably in a protonated form, and Tyr103 are positioned to stabilize and fix the orientation of Glu172. The relative scarcity of polar residues in the binding site and the many flexible hydrophobic residues such as leucine and methionine likely contribute to the pharmacological promiscuity of the receptor.
Two structures of sigma-1 receptor were solved, one of the receptor bound to PD144418, a sigma-1 antagonist [25], and another with the receptor bound to 4-IBP, a high-affinity ligand with a poorly characterized efficacy profile [26, 33]. These two compounds are chemically divergent, sharing only an elongated shape and a central cationic amine. Despite this, they bind to sigma-1 in a very similar manner (Fig. 2.5). The binding mode of these molecules is in agreement with phamacophore models of sigma-1 that predicted two hydrophobic sites on both sides of the cationic amine [31, 34], a primary site 6–10 Å from the cationic amine and a secondary site at a distance of 2.5–4 Å. The binding pocket also contains two tryptophan residues (Trp89 and Trp164), offering an explanation for the observed attenuation in binding upon exposure of sigma-1 to UV radiation [35].

Ligand recognition by sigma-1. The structures of sigma-1 bound to PD144418 (left) and 4-IBP (right) are shown. The interactions with the ligand in each case are predominantly hydrophobic in character, with the exception of a salt bridge interaction between the ligand amine and receptor Glu172. The two compounds bind with very similar overall poses, despite only modest chemical similarity
2.6 Disease-Associated Mutations
A number of mutations in sigma-1 have been linked to neurodegenerative disease in humans. Some of these occur in untranslated regions and may affect protein abundance [36, 37], while other mutations occur in the protein coding sequence [38–40]. For the latter class, the availability of structural information now offers new insight into the molecular mechanisms of sigma-1 dysfunction (Fig. 2.6).

Disease-associated mutations in sigma-1. (a) Overall view of the receptor with sites for human disease-associated point mutations in labeled boxes. (b) E102Q mutation associated with ALS likely disrupts hydrogen bond network. (c) E138Q mutation similarly prevents formation of salt bridge and hydrogen bond network with R117, which links receptor protomers together. (d) The surface-exposed E150K mutation is more enigmatic, with no clear reason for structural disruption upon mutation
One mutation, E102Q, was identified in a consanguineous family in Saudi Arabia and causes a juvenile-onset ALS -like neurodegenerative disease [39]. This mutation was subsequently shown to alter sigma-1 localization and mobility in cells [41]. Glu102 is highly conserved residue, and the crystal structure reveals an unusual role as a double hydrogen bond acceptor to Val36 and Phe37 backbone amines (Fig. 2.6a, b). Mutation of this to glutamine would disrupt one of these interactions, resulting in an unfavorable apposition of two hydrogen bond donors. A second mutation, E138Q, shows a similar hydrogen bonding network (Fig. 2.6c), and likewise is associated with autosomal-recessive distal hereditary motor neuropathy [40].
A third mutation in the coding sequence, E150K, is associated with a similar hereditary motor dysfunction [40]. Unlike E102Q and E138Q however, the molecular basis for the effect of this mutation remains unclear. Glu150 is a surface-exposed residue interacting largely with solvent, and mutation to lysine is unlikely to significantly alter receptor folding (Fig. 2.6d). Instead, this residue may play a role in sigma-1 interaction with effector proteins, or in some other as yet uncharacterized process.
2.7 Outlook
With the availability of high quality structural information, our understanding of sigma-1 function is poised for transformation. The detailed views of the ligand binding site will allow rational design of new sigma-1 ligands, with potentially unexpected properties, and the overall structure will enable rational design of engineered receptor constructs. However, many other important questions remain. The molecular distinction between agonists and antagonists , as well as the mechanisms of receptor activation are likely to be particularly important areas for understanding the molecular basis of sigma-1 function in years to come. In addition, a full understanding of sigma-1 activity will require studies of the receptor in complex with effector proteins, as well as further investigation of the role of oligomerization and its potential regulation by small molecule compounds.
References
Su TP, Su TC, Nakamura Y, Tsai SY (2016) The sigma-1 receptor as a pluripotent modulator in living systems. Trends Pharmacol Sci 37(4):262–278. doi:10.1016/j.tips.2016.01.003
Mavlyutov TA, Guo LW, Epstein ML, Ruoho AE (2015) Role of the sigma-1 receptor in Amyotrophic Lateral Sclerosis (ALS). J Pharmacol Sci 127:10–16. doi:10.1016/j.jphs.2014.12.013
Palmer CP, Mahen R, Schnell E, Djamgoz MB, Aydar E (2007) Sigma-1 receptors bind cholesterol and remodel lipid rafts in breast cancer cell lines. Cancer Res 67:11166–11175. doi:10.1158/0008-5472.can-07-1771
Hayashi T, Su TP (2007) Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131:596–610. doi:10.1016/j.cell.2007.08.036
Balasuriya D, Stewart AP, Edwardson JM (2013) The sigma-1 receptor interacts directly with GluN1 but not GluN2A in the GluN1/GluN2A NMDA receptor. J Neurosci 33:18219–18224. doi:10.1523/jneurosci.3360-13.2013
Gao XF, Yao JJ, He YL, Hu C, Mei YA (2012) Sigma-1 receptor agonists directly inhibit Nav1.2/1.4 channels. PLoS One 7:e49384. doi:10.1371/journal.pone.0049384
Kim FJ et al (2010) Sigma 1 receptor modulation of G-protein-coupled receptor signaling: potentiation of opioid transduction independent from receptor binding. Mol Pharmacol 77:695–703. doi:10.1124/mol.109.057083
Walker JM et al (1990) Sigma receptors: biology and function. Pharmacol Rev 42:355–402
Martin BR et al (1984) Stereoisomers of [3H]-N-allylnormetazocine bind to different sites in mouse brain. J Pharmacol Exp Ther 231:539–544
Fontanilla D et al (2009) The hallucinogen N,N-dimethyltryptamine (DMT) is an endogenous sigma-1 receptor regulator. Science 323:934–937. doi:10.1126/science.1166127
Keiser MJ et al (2009) Predicting new molecular targets for known drugs. Nature 462:175–181. doi:10.1038/nature08506
Hanner M et al (1996) Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc Natl Acad Sci U S A 93:8072–8077
Kekuda R, Prasad PD, Fei YJ, Leibach FH, Ganapathy V (1996) Cloning and functional expression of the human type 1 sigma receptor (hSigmaR1). Biochem Biophys Res Commun 229:553–558. doi:10.1006/bbrc.1996.1842
Seth P, Leibach FH, Ganapathy V (1997) Cloning and structural analysis of the cDNA and the gene encoding the murine type 1 sigma receptor. Biochem Biophys Res Commun 241:535–540. doi:10.1006/bbrc.1997.7840
Seth P et al (1998) Cloning and functional characterization of a sigma receptor from rat brain. J Neurochem 70:922–931
Aydar E, Palmer CP, Klyachko VA, Jackson MB (2002) The sigma receptor as a ligand-regulated auxiliary potassium channel subunit. Neuron 34:399–410
Laurini E et al (2011) Homology model and docking-based virtual screening for ligands of the sigma1 receptor. ACS Med Chem Lett 2:834–839. doi:10.1021/ml2001505
Ortega-Roldan JL, Ossa F, Amin NT, Schnell JR (2015) Solution NMR studies reveal the location of the second transmembrane domain of the human sigma-1 receptor. FEBS Lett 589:659–665. doi:10.1016/j.febslet.2015.01.033
Caffrey M, Li D, Dukkipati A (2012) Membrane protein structure determination using crystallography and lipidic mesophases: recent advances and successes. Biochemistry 51:6266–6288. doi:10.1021/bi300010w
Caffrey M, Cherezov V (2009) Crystallizing membrane proteins using lipidic mesophases. Nat Protoc 4:706–731
Smith JL, Fischetti RF, Yamamoto M (2012) Micro-crystallography comes of age. Curr Opin Struct Biol 22:602–612. doi:10.1016/j.sbi.2012.09.001
Chae PS et al (2010) Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nat Methods 7:1003–1008. doi:10.1038/nmeth.1526
Gromek KA et al (2013) Improved expression and purification of sigma 1 receptor fused to maltose binding protein by alteration of linker sequence. Protein Expr Purif 89:203–209. doi:10.1016/j.pep.2013.03.013
Schmidt HR et al (2016) Crystal structure of the human sigma1 receptor. Nature 532:527–530. doi:10.1038/nature17391
Lever JR et al (2014) Relationship between cerebral sigma-1 receptor occupancy and attenuation of cocaine's motor stimulatory effects in mice by PD144418. J Pharm Exp Ther 351:153–163. doi:10.1124/jpet.114.216671
John CS, Vilner BJ, Bowen WD (1994) Synthesis and characterization of [125I]-N-(N-benzylpiperidin-4-yl)-4- iodobenzamide, a new sigma receptor radiopharmaceutical: high-affinity binding to MCF-7 breast tumor cells. J Med Chem 37:1737–1739
Knablein J et al (1997) Ta6Br(2+)12, a tool for phase determination of large biological assemblies by X-ray crystallography. J Mol Biol 270:1–7
Gromek KA et al (2014) The oligomeric states of the purified sigma-1 receptor are stabilized by ligands. J Biol Chem 289:20333–20344. doi:10.1074/jbc.M113.537993
Pal A et al (2007) Identification of regions of the sigma-1 receptor ligand binding site using a novel photoprobe. Mol Pharmacol 72:921–933. doi:10.1124/mol.107.038307
Mishra AK et al (2015) The sigma-1 receptors are present in monomeric and oligomeric forms in living cells in the presence and absence of ligands. Biochem J 466:263–271. doi:10.1042/bj20141321
Glennon RA et al (1994) Structural features important for sigma 1 receptor binding. J Med Chem 37:1214–1219
Seth P et al (2001) Expression pattern of the type 1 sigma receptor in the brain and identity of critical anionic amino acid residues in the ligand-binding domain of the receptor. Biochim Biophys Acta 1540:59–67
Bermack JE, Debonnel G (2005) Distinct modulatory roles of sigma receptor subtypes on glutamatergic responses in the dorsal hippocampus. Synapse 55:37–44. doi:10.1002/syn.20085
Ablordeppey SY, Fischer JB, Law H, Glennon RA (2002) Probing the proposed phenyl-A region of the sigma-1 receptor. Bioorg Med Chem 10:2759–2765
Bowen WD, Hellewell SB, McGarry KA (1989) Evidence for a multi-site model of the rat brain sigma receptor. Eur J Pharmacol 163:309–318
Luty AA et al (2010) Sigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration-motor neuron disease. Ann Neurol 68:639–649. doi:10.1002/ana.22274
Ullah MI et al (2015) In silico analysis of SIGMAR1 variant (rs4879809) segregating in a consanguineous Pakistani family showing amyotrophic lateral sclerosis without frontotemporal lobar dementia. Neurogenetics 16:299–306. doi:10.1007/s10048-015-0453-1
Li X et al (2015) A SIGMAR1 splice-site mutation causes distal hereditary motor neuropathy. Neurology 84:2430–2437. doi:10.1212/wnl.0000000000001680
Al-Saif A, Al-Mohanna F, Bohlega S (2011) A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann Neurol 70:913–919. doi:10.1002/ana.22534
Gregianin E et al. (2016) Loss-of-function mutations in the SIGMAR1 gene cause distal hereditary motor neuropathy by impairing ER-mitochondria tethering and Ca2+ signalling. Hum Mol Genet. doi:10.1093/hmg/ddw220
Wong AY et al (2016) Aberrant subcellular dynamics of sigma-1 receptor mutants underlying neuromuscular diseases. Mol Pharmacol 90:238–253. doi:10.1124/mol.116.104018
Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL (2012) OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res 40:D370–D376. doi:10.1093/nar/gkr703
Ashkenazy H et al (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44:W344–W350. doi:10.1093/nar/gkw408
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG (outside the USA)
About this chapter
Cite this chapter
Alon, A., Schmidt, H., Zheng, S., Kruse, A.C. (2017). Structural Perspectives on Sigma-1 Receptor Function. In: Smith, S., Su, TP. (eds) Sigma Receptors: Their Role in Disease and as Therapeutic Targets. Advances in Experimental Medicine and Biology, vol 964. Springer, Cham. https://doi.org/10.1007/978-3-319-50174-1_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-50174-1_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-50172-7
Online ISBN: 978-3-319-50174-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)