
Abstract
Sigma1 (also known as this sigma-1 receptor) is an unusual and enigmatic transmembrane protein implicated in a diverse array of biological processes ranging from neurodegenerative disease to cancer. Despite decades of research, the molecular architecture of Sigma1 is only beginning to become clear. Recent work has established that Sigma1 is an oligomer, and crystallographic studies have now offered the first high-resolution views of its molecular structure. For the first time, these results provide a detailed framework to understand mutagenesis data and the molecular pharmacology of Sigma1 ligands. Structural data also raise new questions surrounding the mechanisms of ligand activity and the molecular basis for interactions between Sigma1 and other proteins. As Sigma1 research enters the structural era, the field is poised for new discoveries and reevaluation of old data and old models.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Sigma1 (also known as the sigma-1 receptor) is an unusual transmembrane receptor first discovered 40 years ago and subsequently studied extensively for its pharmacological properties, which in some respects resemble those of opioid receptors (Walker et al. 1990). However, the development of photoaffinity probes and enantiomerically pure ligands later showed that Sigma1 resemblance to opioid receptors is largely superficial. Unlike opioid receptors, Sigma1 has a molecular weight of ~25 kDa, and it shows a strong preference for the (+) enantiomers of benzomorphan ligands. In contrast, true opioid receptors selectively bind the (−) enantiomer (Martin et al. 1984). Moreover, the effects of Sigma1 ligands are not antagonized by the opioid antagonists naltrexone and naloxone (Largent et al. 1987), which block agonist activity at the μ, δ, and κ opioid receptors. Finally, unlike the opioid receptors, Sigma1 does not appear to signal through heterotrimeric G proteins (Lupardus et al. 2000), further confirming its lack of relation to opioid receptors.
While Sigma1 was extensively characterized with respect to its pharmacology in the 1980s and early 1990s, the first structural insights regarding Sigma1 came only in 1996 when the protein was cloned from guinea pig liver (Hanner et al. 1996), and shortly thereafter from a human cell line (Kekuda et al. 1996) as well as mouse (Seth et al. 1997) and rat tissues (Seth et al. 1998). Remarkably, the sequence of Sigma1 shows no similarity to any other mammalian protein, but it possesses clear homology (30% sequence identity) to the yeast C8–C7 sterol isomerase Erg2p. Hydrophobicity analysis of the guinea pig Sigma1 protein sequence showed a single highly hydrophobic segment with a potential ER-retention signal at its amino terminus, suggesting a single-pass transmembrane topology with a short luminal tail at the amino-terminus and the majority of the protein located on the cytosolic side of the ER membrane. As discussed below, this single-pass transmembrane model was less widely accepted than later proposed architectures placing both the amino- and carboxy-termini in the ER lumen, although the prediction of a single-pass fold was ultimately shown to be correct.
The determination of the receptor’s primary sequence led to extensive prediction and speculation regarding the three-dimensional structure of Sigma1. One report (Aydar et al. 2002) proposed a two-pass transmembrane architecture on the basis of antibody staining with Sigma1 fused to amino- and carboxy-terminal green fluorescent protein (GFP). An important caveat to this work, however, is that GFP is known to be poorly secreted (Li et al. 2002), and the amino-terminal fusion may have altered the insertion topology of the protein. The two-pass model was widely accepted as established fact, serving as the basis for efforts to map the position of the putative second transmembrane domain (Ortega-Roldan et al. 2015) and other studies using molecular modeling to predict the receptor’s structure computationally (Laurini et al. 2011). In contrast, efforts to determine the Sigma1 structure experimentally lagged behind, hindered by the myriad challenges associated with biochemical manipulation of integral membrane proteins.
2 Crystal Structure of Human Sigma1
Beginning in 2007, a series of technological innovations revolutionized membrane protein crystallography, particularly for G protein-coupled receptors (GPCRs). Advances include the use of fusion proteins like T4 lysozyme (Rosenbaum et al. 2007), lipid mesophase crystallization techniques (Caffrey et al. 2012), microdiffraction (Smith et al. 2012), and novel detergents (Chae et al. 2010). Together, these methods have led to dozens of GPCR structures, and these approaches are increasingly finding application in membrane protein crystallography more generally.
To determine the structure of Sigma1, a GPCR-inspired approach was employed (Schmidt et al. 2016). The receptor was expressed using Sf9 insect cells to produce high levels of functional protein, which was then purified using lauryl maltose neopentyl glycol (LMNG), a protein-stabilizing detergent that has been widely used in the GPCR field. This detergent has an exceptionally low critical micelle concentration (Chung et al. 2012), and it has been shown to enhance stability of monomeric membrane proteins as well as preserve the stability of multi-protein complexes (Chae et al. 2010).
Purification of the full-length Sigma1 protein was achieved by antibody affinity chromatography, allowing rapid isolation of Sigma1 in high biochemical purity (Schmidt et al. 2016). Purified protein was then crystallized using the lipidic cubic phase (LCP) method (Caffrey et al. 2012). This approach entails reconstituting the protein in a liquid crystalline lipid bilayer and performing crystallization experiments with Sigma1 embedded in this membrane throughout the crystallogenesis process. Consequently, LCP crystals typically show arrays of protein molecules in flat membrane bilayers stacked together to form the crystal lattice. In the case of Sigma1, this approach led to structures (Schmidt et al. 2016) of the receptor bound to two chemically distinct ligands: PD144418, a high-affinity, selective antagonist (Lever et al. 2014; Akunne et al. 1997), and 4-IBP, a high-affinity ligand with an incompletely understood efficacy profile (John et al. 1994). These ligands were chosen by screening a small collection of compounds with 1 nM or higher affinity in crystallization trials, with PD144418 and 4-IBP showing the largest improvements in crystal size and appearance. While Sigma1 structure agrees closely with prior pharmacological data, it shows little similarity to previous computational models (Laurini et al. 2011), highlighting the challenges of predicting membrane protein structure ab initio.
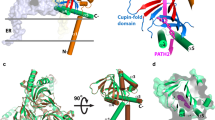
The overall structure of the protein is unusual, with no significant resemblance to any other protein of known structure. Contrary to the prevailing view that the receptor possessed a two-pass transmembrane architecture, the structure reveals definitively that Sigma1 possesses only a single transmembrane domain, located at its amino terminus and encompassing residues 8 to 32. The remainder of the structure forms a single domain, with the ligand-binding site at its center. In the crystal, the receptor exists in a trimeric arrangement in which three protomers are intimately associated to form a flat triangle, with a transmembrane domain at each corner (Fig. 1 a, b). The entire membrane-proximal surface is flat and hydrophobic, and the presence of ordered lipid molecules indicates that this surface is likely buried within the membrane. The receptor thus contains structural elements typical of both transmembrane receptors and peripheral membrane proteins.

Overall structure of Sigma1: The overall fold of Sigma1 is unusual, with three protomers forming a triangular structure with a single transmembrane helix at each corner. The majority of the protein is located carboxy-terminal to the transmembrane domain, on the cytoplasmic surface of the membrane. (a) The structure is shown viewed parallel to the membrane plane. (b) The triangular architecture of the receptor is apparent when viewed through the membrane plane facing the cytosol
The carboxy-terminal cytosolic domain of Sigma1 comprises the bulk of the protein, encompassing residues from 33 to 223. The overall fold of this region is unlike any other protein crystallized to date, but at its core contains a cupin-like β-barrel which encloses the bound ligand (Fig. 2). The cupin fold is a conserved structural motif found in a wide variety of proteins, many of which are bacterial metalloenzymes. In most of these proteins, the barrel-like cupin fold is essential for binding to small-molecule substrates and catalytic metal ions (Dunwell et al. 2001). In most cases, these enzymes perform redox chemistry on small-molecule metabolites. In the case of Sigma1, this fold serves to envelope the ligand, occluding it entirely from solvent. The carboxy-terminal domain also contains the entire oligomerization interface, mediating the threefold non-crystallographic symmetry. Although each of the three protomers in the structure is crystallographically independent, they show no significant differences other than in orientation of the transmembrane domain. Lattice contacts are mostly mediated by these transmembrane domains, which pack in both parallel and antiparallel configurations to form the crystal.

Structure of the carboxy-terminal domain: The carboxy-terminal domain of Sigma1 makes up the bulk of the protein. (a) Viewed from the side (i.e., parallel to the membrane) the cupin domain at the core of the protein is shaded in blue. The bound antagonist PD144418 is shown in spheres at the center of the cupin domain. (b) Viewed from the top (membrane-facing side) the major structural elements are visible. (c) A bacterial cupin protein (light blue, PDB ID 3BCW) is superimposed on Sigma1, showing the high degree of structural conservation. (d) A cluster of ordered lipids are observed in the crystal structure at the junction of the carboxy-terminal domain and the transmembrane helix (shown in yellow sticks, oxygen atoms in red). The flexible tails of the lipids are not crystallographically resolved
3 Structural Basis for Ligand Recognition
The structures of Sigma1 bound to two drug-like ligands offer the first detailed views of ligand recognition by the receptor. The two structures are remarkably similar to one another, with few differences in ligand/receptor interactions (Schmidt et al. 2016). Previous work using site-directed mutagenesis and radioligand-binding assays identified many of the key residues that are essential for ligand-binding activity. Among these, Asp126 and Glu172 are particularly notable, as mutation of either results in a profound loss of ligand-binding activity (Seth et al. 2001). The crystal structures show that Glu172 serves as a counterion to the protonated ligand amine, directly interacting via a hydrogen bond. Like Glu172, Asp126 is also essential for high-affinity ligand binding. Unlike Glu172 however, it does not interact directly with the ligand. Instead, Asp126 engages in a 2.6 Å hydrogen bond with Glu172, indicating that it must be protonated and resulting in overall charge neutrality in the binding pocket when the ligand is bound. With the exception of Asp126 and Glu172, the ligand-binding site is hydrophobic overall, and is largely composed of aromatic residues. Figure 3 shows the structure of the ligand-binding site, highlighting residues in contact with the bound ligand.

Structure of the ligand-binding site: The ligand-binding site of Sigma1 is shown in two different views. (a) An overall view of the binding site reveals the largely hydrophobic contacts between the ligand and Sigma1, with the exception of a salt bridge to Glu172. (b) The chemical structure of the bound ligand PD144418. (c) A close-up of the binding site shows the extended polar network including Asp126 and Tyr103 as hydrogen-bonding partners to Glu172
The solvent-occluded charge–charge interaction in the binding site closely resembles similar ligand-binding modes observed in biogenic amine GPCRs, including receptors for acetylcholine (Haga et al. 2012; Kruse et al. 2012), dopamine (Chien et al. 2010), histamine (Shimamura et al. 2011), and serotonin (Wang et al. 2013; Wacker et al. 2013). In these receptors, the conserved residue Asp3.32 (Ballesteros–Weinstein numbering (Ballesteros and Weinstein 1995)) engages in a salt bridge with the protonated ligand amine, paralleling the role of Glu172 in Sigma1. Although Sigma1 is unrelated to the aminergic GPCRs in sequence, the close parallels in binding site structure offer an explanation for the cross-reactivity of ligands like haloperidol, which binds both D2 dopamine receptor and Sigma1 with nanomolar potency. It is possible that the similar binding modes arise from convergent evolution of biogenic amine recognition sites, which could imply that Sigma1 acts as a receptor for a neurotransmitter or similar small molecule. However, as discussed below it remains unknown if Sigma1 indeed responds to any endogenous agonist.
Surprisingly, the ligand-binding site in both of the crystal structures is entirely occluded from solvent, offering no possible path for ligand entry or egress. This shows that the protein must be able to undergo dynamic changes to allow ligands to bind and dissociation from the receptor, but the path of ligand entry and exit remains unknown. The enclosed ligand-binding site offers a clear explanation for the very slow binding kinetics of most sigma ligands (Itzhak 1989), contrasting with opioid receptors which show rapid binding kinetics at their highly exposed ligand-binding sites (Cassel et al. 2005).
In addition to drug-like small molecules, Sigma1 has also been shown to bind to a range of lipids, including sterols like progesterone as well as sphingolipids (Ramachandran et al. 2009). Because crystallization experiments were conducted in the absence of these molecules the structural basis for their interaction with Sigma1 remains unknown. Nonetheless, a cluster of four bound monoolein lipid molecules are observed in the structure in a cleft between the transmembrane domain and the carboxy-terminal domain. This region is flanked by Gln33, Leu100, Trp121, Val177, and Leu214, and may serve as a site for lipid regulation of Sigma1 activity.
4 Oligomerization
Recent work from multiple labs has shown evidence that the Sigma1 receptor is likely to function as an oligomer, with possible regulation of oligomerization state by small-molecule ligands (Mishra et al. 2015; Gromek et al. 2014). The crystal structures further support this idea, showing an intimately associated trimer formed by the carboxy-terminal domain of each protomer. The interaction surface is extensive, involving more than 30 residues in each protomer, primarily along loops of the adjacent β-strands in the cupin domain, particularly along the cytosolic face (Fig. 4a). The residues in the oligomerization interface are largely hydrophobic, although some hydrophilic amino acids are present at the periphery of the interface. Within the oligomerization interface Trp136 is among the most extensively engaged residues, embedded deeply within a hydrophobic pocket on the adjacent protomer formed by Phe83, Ala110, Leu111, and Trp169. A hydrogen-bonding network centered on Arg119 also links adjacent protomers (Fig. 4b), as does a threefold symmetric aromatic stacking interaction among Phe191 residues from each protomer (Fig. 4c). Importantly, the oligomerization interface is highly conserved in sequence, attesting to its functional importance.

Oligomerization: The interactions among Sigma1 protomers are extensive, involving dozens of amino acids primarily on the cytosolic face of the trimer. Viewed from the cytosolic face most of the oligomerization contact residues are resolved, shown as sticks
Size-exclusion experiments with multi-angle light scattering (SEC-MALS) showed that purified Sigma1 in detergent exists in a range of oligomeric states, with molecular weights ranging from at least 140 up to 400 kDa (Schmidt et al. 2016). Similarly, other biochemical and pharmacological work has shown that Sigma1 exists in a range of oligomeric states in detergents, with high-molecular-weight species being stabilized by ligands (Gromek et al. 2014). Cell-based fluorescence resonance energy transfer (FRET) studies have shown similar effects, as well as revealed agonist stabilization of low-molecular-weight species (Mishra et al. 2015). Taken together, these data indicate that oligomerization is an important feature of Sigma1 function, although the mechanistic details of oligomerization changes and any regulation thereof remain to be fully elucidated.
5 Implications for Sigma1 Function
While the Sigma1 receptor has been extensively studied by ligand-binding assays, the identity of the endogenous ligand, if any, remains unclear. Proposed ligands include dimethyltryptamine (Fontanilla et al. 2009) (DMT), but this has been called into question (Keiser et al. 2009) because of the much higher affinity of DMT at serotonergic receptors including the 5-HT2A receptor, which binds to DMT with 100-fold higher affinity than does Sigma1. Moreover, the behavioral effects of DMT administration are abrogated in 5-HT2A receptor knockout mice (Keiser et al. 2009). Given these results, the identity of any endogenous Sigma1 ligand remains unclear.
Indeed, it is possible that the term “receptor” is something of a misnomer, and Sigma1 may function in an altogether different manner than conventional receptor families. One intriguing possibility is suggested by the close sequence homology between Sigma1 and the fungal sterol isomerase ERG2. While it has been demonstrated that Sigma1 cannot complement ERG2 gene deletion in yeast (Hanner et al. 1996), it remains possible that Sigma1 possesses enzymatic activity that has yet to be discovered. Alternatively, Sigma1 may be the result of evolutionary repurposing of an enzyme to a receptor, converting an active site into a ligand-binding site for regulation of receptor function.
In addition to pharmacological research, Sigma1 has been extensively probed in cell biological studies. This work has offered insight into a wide variety of aspects of Sigma1 activity, suggesting a possible role as a multifunctional regulator of transmembrane signaling. Interactions have been reported with GPCRs (Kim et al. 2010), ion channels (Aydar et al. 2002; Balasuriya et al. 2014), and chaperone proteins (Hayashi and Su 2007), among many others (Su et al. 2016). Current structural data are insufficient to comment substantively on the nature of such interactions, but this will doubtless be an important area for future research. Importantly, the advent of structural data now allows more rational construct design and analysis for cellular work. In particular, many previous experiments used constructs designed based on incorrect topological models that identified residues 100–223 as the carboxy-terminal domain. We now know that residues 33–99 are also integral parts of this domain, contributing two out of ten beta strands and two out of four alpha helices. Accordingly, the interpretation of research that used only partial fragments of this domain may need to be reconsidered.
6 Outlook
With the advent of high-resolution structural data for Sigma1, the field is poised for new insights and reconsideration of previous models. The discovery that Sigma1 possesses only a single transmembrane domain in particular highlights the risks associated with overreliance on a single model, and should now guide informed design of modified receptor constructs in future work. Despite the important insights offered by recent structures, other key questions remain unanswered and several areas for future research are highlighted below. In the long term, a complete understanding of Sigma1 biology will require a molecular understanding of ligand binding, efficacy, and regulation of interactions with other proteins. In each respect, structural biology is likely to play a pivotal role.
6.1 Relationship to Erg2p and Enzymatic Activity
The sequencing of Sigma1 20 years ago offered the first clear connection to a protein of well-described function, revealing sequence similarity to the yeast sterol isomerase Erg2p. In fungi, this enzyme plays an essential role in ergosterol biosynthesis, catalyzing the transfer of a double bond between the C8 and C9 positions to a new site between the C8 and C7 carbons. In humans, the analogous reaction is catalyzed by the emopamil-binding protein (EBP), which is unrelated to Sigma1 and Erg2p in primary sequence (Moebius et al. 1997). The sequence similarity between Sigma1 and Erg2p implies that the latter is likely to possess a similar membrane-embedded fold. This would allow the hydrophobic lipid substrate (fecosterol) to access the catalytic site, while the equally hydrophobic product (episterol) can then escape directly into the bilayer.
Despite their sequence similarity and conservation of the catalytic/ligand-binding glutamate (Glu174 in yeast Erg2p, Glu172 in human Sigma1), the Sigma1 receptor fails to complement ERG2 gene deletion in yeast (Hanner et al. 1996). This fact has been taken as evidence for a lack of Sigma1 catalytic activity. Nonetheless, Sigma1 has not been directly assessed for its ability to catalyze the analogous transformation in cholesterol, and it remains possible that an enzymatic activity like that of EBP may in fact be present. The development of robust procedures for purification of homogenous, functional Sigma1 should now allow straightforward assessment of this possibility in the near future.
6.2 Molecular Efficacy and Oligomerization
Many Sigma1 ligands have been classified as agonists and antagonists on the basis of their effects on animals (Nguyen et al. 2015). However, little is known regarding the molecular basis for ligand efficacy in terms of the specific receptor conformation(s) stabilized by agonists vs. antagonists. Cellular FRET data suggest a possible role for at least some Sigma1 antagonists in stabilizing high-molecular-weight oligomers, while certain agonists suppress oligomerization (Mishra et al. 2015). Nonetheless, the molecular mechanistic basis for these effects remains unknown. While it is increasingly apparent that oligomerization is a key aspect of Sigma1 function, its exact role and connection to ligand efficacy are likely to be important areas of research in years to come. In particular, elucidation of distinct conformations/oligomerization states is poised to be an important area for Sigma1 structural biology.
6.3 Interactions with Other Proteins
Sigma1 interactions with other proteins have been the subject of intense investigation for decades, with a wide range of proteins proposed as interaction partners (Su et al. 2016). Key areas for future work include validating these interactions with purified proteins, mapping sites of interaction, and determining the molecular basis for regulation of Sigma1/effector protein interactions. Recent advances in high-resolution electron microscopy (Liao et al. 2013) are particularly exciting, as these techniques may allow investigation of Sigma1 complexes with high-molecular-weight putative binding partners like inositol phosphate receptors, for which electron microscopy structural data has recently become available (Fan et al. 2015).
References
Akunne HC, Whetzel SZ, Wiley JN, Corbin AE, Ninteman FW, Tecle H, Pei Y, Pugsley TA, Heffner TG (1997) The pharmacology of the novel and selective sigma ligand, PD 144418. Neuropharmacology 36(1):51–62
Aydar E, Palmer CP, Klyachko VA, Jackson MB (2002) The sigma receptor as a ligand-regulated auxiliary potassium channel subunit. Neuron 34(3):399–410
Balasuriya D, D’Sa L, Talker R, Dupuis E, Maurin F, Martin P, Borgese F, Soriani O, Edwardson JM (2014) A direct interaction between the sigma-1 receptor and the hERG voltage-gated K+ channel revealed by atomic force microscopy and homogeneous time-resolved fluorescence (HTRF(R)). J Biol Chem 289(46):32353–32363. doi:10.1074/jbc.M114.603506
Ballesteros J, Weinstein H (1995) Integrated methods for modeling G-protein coupled receptors. Methods Neurosci 25:366–428
Caffrey M, Li D, Dukkipati A (2012) Membrane protein structure determination using crystallography and lipidic mesophases: recent advances and successes. Biochemistry 51(32):6266–6288. doi:10.1021/bi300010w
Cassel JA, Daubert JD, DeHaven RN (2005) [(3)H]Alvimopan binding to the mu opioid receptor: comparative binding kinetics of opioid antagonists. Eur J Pharmacol 520(1–3):29–36. doi:10.1016/j.ejphar.2005.08.008
Chae PS, Rasmussen SG, Rana RR, Gotfryd K, Chandra R, Goren MA, Kruse AC, Nurva S, Loland CJ, Pierre Y, Drew D, Popot JL, Picot D, Fox BG, Guan L, Gether U, Byrne B, Kobilka B, Gellman SH (2010) Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nat Methods 7(12):1003–1008. doi:10.1038/nmeth.1526
Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC (2010) Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330(6007):1091–1095. doi:10.1126/science.1197410
Chung KY, Kim TH, Manglik A, Alvares R, Kobilka BK, Prosser RS (2012) Role of detergents in conformational exchange of a G protein-coupled receptor. J Biol Chem 287(43):36305–36311. doi:10.1074/jbc.M112.406371
Dunwell JM, Culham A, Carter CE, Sosa-Aguirre CR, Goodenough PW (2001) Evolution of functional diversity in the cupin superfamily. Trends Biochem Sci 26(12):740–746
Fan G, Baker ML, Wang Z, Baker MR, Sinyagovskiy PA, Chiu W, Ludtke SJ, Serysheva II (2015) Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature 527(7578):336–341. doi:10.1038/nature15249
Fontanilla D, Johannessen M, Hajipour AR, Cozzi NV, Jackson MB, Ruoho AE (2009) The hallucinogen N,N-dimethyltryptamine (DMT) is an endogenous sigma-1 receptor regulator. Science 323(5916):934–937
Gromek KA, Suchy FP, Meddaugh HR, Wrobel RL, LaPointe LM, Chu UB, Primm JG, Ruoho AE, Senes A, Fox BG (2014) The oligomeric states of the purified sigma-1 receptor are stabilized by ligands. J Biol Chem 289(29):20333–20344. doi:10.1074/jbc.M113.537993
Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T, Kobayashi T (2012) Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 482(7386):547–551. http://www.nature.com/nature/journal/v482/n7386/abs/nature10753.html#supplementary-information
Hanner M, Moebius FF, Flandorfer A, Knaus HG, Striessnig J, Kempner E, Glossmann H (1996) Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc Natl Acad Sci U S A 93(15):8072–8077
Hayashi T, Su TP (2007) Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131(3):596–610
Itzhak Y (1989) Multiple affinity binding states of the sigma receptor: effect of GTP-binding protein-modifying agents. Mol Pharmacol 36(4):512–517
John CS, Vilner BJ, Bowen WD (1994) Synthesis and characterization of [125I]-N-(N-benzylpiperidin-4-yl)-4-iodobenzamide, a new sigma receptor radiopharmaceutical: high-affinity binding to MCF-7 breast tumor cells. J Med Chem 37(12):1737–1739
Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, Whaley R, Glennon RA, Hert J, Thomas KL, Edwards DD, Shoichet BK, Roth BL (2009) Predicting new molecular targets for known drugs. Nature 462(7270):175–181. doi:10.1038/nature08506
Kekuda R, Prasad PD, Fei YJ, Leibach FH, Ganapathy V (1996) Cloning and functional expression of the human type 1 sigma receptor (hSigmaR1). Biochem Biophys Res Commun 229(2):553–558
Kim FJ, Kovalyshyn I, Burgman M, Neilan C, Chien CC, Pasternak GW (2010) Sigma 1 receptor modulation of G-protein-coupled receptor signaling: potentiation of opioid transduction independent from receptor binding. Mol Pharmacol 77(4):695–703. doi:10.1124/mol.109.057083
Kruse AC, Hu J, Pan AC, Arlow DH, Rosenbaum DM, Rosemond E, Green HF, Liu T, Chae PS, Dror RO, Shaw DE, Weis WI, Wess J, Kobilka BK (2012) Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 482(7386):552–556. http://www.nature.com/nature/journal/v482/n7386/abs/nature10867.html#supplementary-information
Largent BL, Wikstrom H, Gundlach AL, Snyder SH (1987) Structural determinants of sigma receptor affinity. Mol Pharmacol 32(6):772–784
Laurini E, Col VD, Mamolo MG, Zampieri D, Posocco P, Fermeglia M, Vio L, Pricl S (2011) Homology model and docking-based virtual screening for ligands of the sigma1 receptor. ACS Med Chem Lett 2(11):834–839. doi:10.1021/ml2001505
Lever JR, Miller DK, Fergason-Cantrell EA, Green CL, Watkinson LD, Carmack TL, Lever SZ (2014) Relationship between cerebral sigma-1 receptor occupancy and attenuation of cocaine’s motor stimulatory effects in mice by PD144418. J Pharmacol Exp Ther 351(1):153–163. doi:10.1124/jpet.114.216671
Li J, Xu H, Bentley WE, Rao G (2002) Impediments to secretion of green fluorescent protein and its fusion from Saccharomyces cerevisiae. Biotechnol Prog 18(4):831–838. doi:10.1021/bp020066t
Liao M, Cao E, Julius D, Cheng Y (2013) Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504(7478):107–112. doi:10.1038/nature12822
Lupardus PJ, Wilke RA, Aydar E, Palmer CP, Chen Y, Ruoho AE, Jackson MB (2000) Membrane-delimited coupling between sigma receptors and K+ channels in rat neurohypophysial terminals requires neither G-protein nor ATP. J Physiol 526(Pt 3):527–539
Martin BR, Katzen JS, Woods JA, Tripathi HL, Harris LS, May EL (1984) Stereoisomers of [3H]-N-allylnormetazocine bind to different sites in mouse brain. J Pharmacol Exp Ther 231(3):539–544
Mishra AK, Mavlyutov T, Singh DR, Biener G, Yang J, Oliver JA, Ruoho A, Raicu V (2015) The sigma-1 receptors are present in monomeric and oligomeric forms in living cells in the presence and absence of ligands. Biochem J 466(2):263–271. doi:10.1042/BJ20141321
Moebius FF, Striessnig J, Glossmann H (1997) The mysteries of sigma receptors: new family members reveal a role in cholesterol synthesis. Trends Pharmacol Sci 18(3):67–70
Nguyen L, Lucke-Wold BP, Mookerjee SA, Cavendish JZ, Robson MJ, Scandinaro AL, Matsumoto RR (2015) Role of sigma-1 receptors in neurodegenerative diseases. J Pharmacol Sci 127(1):17–29. doi:10.1016/j.jphs.2014.12.005
Ortega-Roldan JL, Ossa F, Amin NT, Schnell JR (2015) Solution NMR studies reveal the location of the second transmembrane domain of the human sigma-1 receptor. FEBS Lett 589(5):659–665. doi:10.1016/j.febslet.2015.01.033
Ramachandran S, Chu UB, Mavlyutov TA, Pal A, Pyne S, Ruoho AE (2009) The sigma1 receptor interacts with N-alkyl amines and endogenous sphingolipids. Eur J Pharmacol 609(1–3):19–26
Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK (2007) GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 318(5854):1266–1273. doi:10.1126/science.1150609
Schmidt HR, Zheng S, Gurpinar E, Koehl A, Manglik A, Kruse AC (2016) Crystal structure of the human sigma1 receptor. Nature 532(7600):527–530. doi:10.1038/nature17391
Seth P, Leibach FH, Ganapathy V (1997) Cloning and structural analysis of the cDNA and the gene encoding the murine type 1 sigma receptor. Biochem Biophys Res Commun 241(2):535–540
Seth P, Fei YJ, Li HW, Huang W, Leibach FH, Ganapathy V (1998) Cloning and functional characterization of a sigma receptor from rat brain. J Neurochem 70(3):922–931
Seth P, Ganapathy ME, Conway SJ, Bridges CD, Smith SB, Casellas P, Ganapathy V (2001) Expression pattern of the type 1 sigma receptor in the brain and identity of critical anionic amino acid residues in the ligand-binding domain of the receptor. Biochim Biophys Acta 1540(1):59–67
Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, Kobayashi T, Stevens RC, Iwata S (2011) Structure of the human histamine H1 receptor complex with doxepin. Nature 475(7354):65–70. doi:10.1038/nature10236
Smith JL, Fischetti RF, Yamamoto M (2012) Micro-crystallography comes of age. Curr Opin Struct Biol 22(5):602–612. doi:10.1016/j.sbi.2012.09.001
Su TP, Su TC, Nakamura Y, Tsai SY (2016) The sigma-1 receptor as a pluripotent modulator in living systems. Trends Pharmacol Sci. doi:10.1016/j.tips.2016.01.003
Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, Liu W, Xu HE, Cherezov V, Roth BL, Stevens RC (2013) Structural features for functional selectivity at serotonin receptors. Science 340(6132):615–619. doi:10.1126/science.1232808
Walker JM, Bowen WD, Walker FO, Matsumoto RR, De Costa B, Rice KC (1990) Sigma receptors: biology and function. Pharmacol Rev 42(4):355–402
Wang C, Jiang Y, Ma J, Wu H, Wacker D, Katritch V, Han GW, Liu W, Huang XP, Vardy E, McCorvy JD, Gao X, Zhou XE, Melcher K, Zhang C, Bai F, Yang H, Yang L, Jiang H, Roth BL, Cherezov V, Stevens RC, Xu HE (2013) Structural basis for molecular recognition at serotonin receptors. Science 340(6132):610–614. doi:10.1126/science.1232807
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing AG
About this chapter
Cite this chapter
Kruse, A. (2016). Structural Insights into Sigma1 Function. In: Kim, F., Pasternak, G. (eds) Sigma Proteins: Evolution of the Concept of Sigma Receptors. Handbook of Experimental Pharmacology, vol 244. Springer, Cham. https://doi.org/10.1007/164_2016_95
Download citation
DOI: https://doi.org/10.1007/164_2016_95
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-65851-3
Online ISBN: 978-3-319-65853-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)