
Abstract
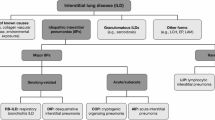
The classification of the idiopathic interstitial pneumonias (IIPs) has recently been updated by the ATS and ERS [1] (Table 6.1). Idiopathic pulmonary fibrosis (IPF) is the most common of the IIPs, and data from existing registries suggest that IPF accounts for 17–37% of all interstitial lung disease (ILD) diagnoses [2, 3]. Although estimates of the true incidence and prevalence of IPF are hampered by different methodologies used in epidemiological studies. IPF is a heterogeneous disease with some patients experiencing slow progressive disease, others a much more rapidly progressive disease and others still experiencing periods of stability punctuated by accelerated decline within acute exacerbations. Median survival is just 2–3 years [4]. It is appreciated that the development of pulmonary hypertension (PH) within IPF is common and its development has a dramatic effect both on morbidity and mortality. The desire to improve prognosis and quality of life in patients with IIP-associated PH (IIP–PH) who unfortunately at present have no clinically proven intervention to do so drives clinical research within this difficult area. The study of PH within IIP has predominantly focused upon IPF or mixed patient groups with IIP (which are predominantly made up of IPF patients). Therefore, this chapter focuses predominantly on PH within the IPF population.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
- Idiopathic Interstitial Pneumonia (IIP)
- Interstitial Lung Disease (ILD)
- Idiopathic Pulmonary Fibrosis (IPF)
- Heart Catheterisation
- Riociguat
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
The classification of the idiopathic interstitial pneumonias (IIPs) has recently been updated by the ATS and ERS [1] (Table 6.1). Idiopathic pulmonary fibrosis (IPF) is the most common of the IIPs, and data from existing registries suggest that IPF accounts for 17–37% of all interstitial lung disease (ILD ) diagnoses [2, 3]. Although estimates of the true incidence and prevalence of IPF are hampered by different methodologies used in epidemiological studies. IPF is a heterogeneous disease with some patients experiencing slow progressive disease, others a much more rapidly progressive disease and others still experiencing periods of stability punctuated by accelerated decline within acute exacerbations. Median survival is just 2–3 years [4]. It is appreciated that the development of pulmonary hypertension (PH) within IPF is common and its development has a dramatic effect both on morbidity and mortality. The desire to improve prognosis and quality of life in patients with IIP-associated PH (IIP–PH ) who unfortunately at present have no clinically proven intervention to do so drives clinical research within this difficult area. The study of PH within IIP has predominantly focused upon IPF or mixed patient groups with IIP (which are predominantly made up of IPF patients). Therefore, this chapter focuses predominantly on PH within the IPF population.
Prevalence
The prevalence of PH in interstitial lung disease ILD is difficult to quantify, both in IPF and in other ILDs . In IPF, it varies greatly according to the severity of pulmonary fibrosis in studied cohorts, with a range of 10–78% in published series. Series with a higher prevalence, ranging from 30 to 45%, consist of reports in patients listed for lung transplantation [5–8], with the presence of PH rising from 38% at initial investigation to 78% at the time of transplantation [8]. By contrast, in the patient cohort enrolled in the ARTEMIS placebo-controlled trial of ambrisentan in mild-to-moderate IPF, right heart catheterisation disclosed that 10% of patients had PH, mostly categorised as Group 3 PH [9]. Thus, PH in IPF includes a subgroup in which PH is a manifestation of advanced ILD , a second subgroup in which PH can be viewed as disproportionate to the severity of ILD, and a significant proportion in which this distinction cannot be made with confidence. It is telling that in IPF, and in ILD in general, the presence of PH has no overall association with the severity of pulmonary function impairment or the severity of pulmonary fibrosis on computed tomography (CT) [5, 10, 11].
The prevalence of PH has not been studied in the other idiopathic interstitial pneumonias (IIPs), but relevant observations have been made in interstitial lung disease associated with connective tissue disease (CTD-ILD ), a group of disorders in which all of the IIP histological patterns have been reported. In systemic sclerosis (SSc), the frequency of PH is strongly linked to autoantibody status. Disproportionate PH, akin to isolated PAH, is strongly associated with anti-centromere antibody positivity whereas PH secondary to extensive interstitial fibrosis occurs most often in SSc patients with anti-topoisomerase positivity [12]. This observation is highly relevant to IIPs other than IPF. A significant proportion of patients previously classified as having an IIP can be viewed as having an occult connective tissue disease (“interstitial pneumonitis with autoimmune features”), with the presence of disproportionate PH (“multi-compartment disease”) contributing to the definition of this syndrome [13].
The importance of selection bias in influencing PH prevalence in IPF, other IIPs and ILDs in general cannot be overstated. In 246 consecutive Japanese patients with sarcoidosis, the prevalence of pulmonary artery systolic pressure (PASP) >40 mm Hg on Doppler echocardiography was only 5.6% [14] but in a cohort of sarcoidosis patients limited by chronic exertional dyspnoea, the mean pulmonary artery pressure (mPAP) at right heart study exceeded 25 mmHg in 47% of 53 patients [15]. This dichotomy applies also to IPF and other IIPs in the distinction between unselected patients and those with major exercise limitation.
Another important limitation in current prognostic and diagnostic PH series is that in advanced interstitial lung disease, a definitive diagnosis of IPF using current diagnostic guideline criteria cannot be made in a significant proportion of IPF patients. Current series can be subdivided into those containing patients with definite IPF (i.e. with the exclusion of many IPF patients not meeting formal diagnostic criteria) and those containing patients with “fibrotic IIP” (i.e. IPF in most cases, but with minority subgroups of fibrotic NSIP and unclassifiable fibrotic disease). Both approaches have merits and studies of both types are cited in this review.
Prognostic Significance
PH is a malignant prognostic determinant in ILDs , with outcomes studied most frequently in IPF. In an echocardiographic series, median survival was 4.7 years and 4.1 years in IPF patients with echocardiographic PASP of 0–34 mmHg and 35–49 mmHg, respectively, but was only 0.7 years in patients with PASP>50 mmHg [16]. In a cohort containing IPF patients listed for lung transplantation, mortality was 28.8% in those with PH at right heart study compared to 5.5% in those without PH [5]. PH is particularly frequent and has a very poor outcome when IPF coexists with emphysema, probably due to the combined impact of two processes and consequent overall severity of lung disease. In reports of patients with combined pulmonary fibrosis and emphysema, survival of only 30% at 1 year and very poor survival associated with a PASP>75 mmHg have been reported [17, 18].
In patients with IPF undergoing right heart catheterisation, short- not intermediate-term mortality was most strongly linked to increases in pulmonary vascular resistance (PVR) and was also strongly associated with increased mean pulmonary artery pressure and the severity of right ventricular dysfunction on echocardiography (but not to other echocardiographic variables) [19]. These observations were mirrored in a cohort of ILD patients, including a minority with IPF: early mortality was much more strongly linked to a marked increase in PVR than to any other variable at right heart catheterisation or on echocardiography [20]. Recently, increased pulmonary vascular resistance estimated by Doppler echocardiography has been shown to predict mortality in ILD patients [21].
The importance of recognising left heart disease as a contributor to PH has yet to be studied definitively in IPF and other IIPs. However, accumulated clinical experience and reported experience in sarcoidosis PH, taken together, indicate the likely prognostic significance of concurrent cardiac disease. In one sarcoidosis series, outcomes were significantly better in patients with PH due to left heart disease than in the remaining patients with PH, reflecting the efficacy of therapies used to treat left ventricular dysfunction and underlining the importance of distinguishing between these two major PH subgroups [22]. In IPF patients with PH associated with mild-to-moderate disease, an important minority subgroup has WHO Group 2 PH (i.e. PH associated with left heart disease) [23].
Mortality is linked, both in IPF and in other ILDs , to a number of non-invasive markers associated with PH, other than echocardiography. The prognostic significance of elevated brain natriuretic peptide (BNP) levels was first explored in 176 patients with a mixture of chronic pulmonary diseases including a large subset with ILDs of various types [24]. Severe PH (mPAP >35 mm Hg) was diagnosed in over 25% of cases: increasing BNP levels were a risk factor for mortality, independent of pulmonary function impairment or hypoxaemia. In a cohort of 90 patients with a mixture of ILDs , higher BNP concentrations were associated with increased mortality independent of age, gender and pulmonary function impairment [25]. In this study, patients with BNP ≥ 20 pmol/L had a 14-fold increase in mortality over patients with BNP < 4 pmol/L. In a review of 131 IPF patients undergoing echocardiography and BNP measurement, increased BNP levels were predictive of mortality with no independent added prognostic value provided by echocardiographic data [26]. It should be stressed, however, that this marker of PH is also a marker of cardiac disease in general, with increased mortality associated with elevated BNP levels in all three series likely to reflect the combined impact of PH and other forms of cardiac disease.
Pulmonary artery size on high resolution computed tomography (HRCT ) appears to be a predictor of IPF mortality. In a population of 98 IPF patients, increases in the pulmonary artery diameter/ascending aorta diameter ratio (PA:A ratio) were associated with increased mortality [27]. Patients with a PA:A ratio >1 had a strikingly higher risk of death or transplant, with this threshold found to be an independent adverse prognostic indicator (hazard ratio approximately 4.0).
Pulmonary function variables influenced by the presence of PH also have major prognostic significance in IPF and in other ILDs . The malignant prognostic significance of resting hypoxia has long been recognised. More specific to PH is a reduction in DLco disproportionate to measurements of lung volume. In a fibrotic IIP series, baseline gas transfer coefficient levels (Kco, synonymous with DLco adjusted for VA) were associated with increased early and overall mortality [28]. In two fibrotic IIP series, a six-month decline in Kco was predictive of increased mortality, independent of FVC levels, in two fibrotic IIP series [28, 29] and was associated with an increased likelihood of the development of echocardiographic PH [28].
Severe desaturation during a six minute walk test has been associated with increased mortality in fibrotic IIP, with a desaturation threshold of 88% identified as a malignant prognostic determinant [30, 31]. Differences in mortality above and below this threshold appear to mirror differences in mortality in IPF patients with and without PH.
Diagnosis
The importance of right heart catheterisation (RHC ) as a reference standard for the diagnosis of PH applies to PH in patients with ILD . The considerable variability between centres in the threshold for performing RHC is likely to reflect the absence of evidence of treatment benefits from targeted PH treatments in the context of ILD . However, the prognostic significance of proven PH in IPF and other ILDs may be highly influential in determining the timing and priority of lung transplantation. Furthermore, a definite PH diagnosis allows patients to be considered for enrolment in treatment trials or for consideration of targeted therapy on compassionate grounds and in these contexts, the identification of concurrent left heart disease has major management implications. For all these reasons, accurate algorithms are needed in which non-invasive evaluation leads to the appropriate use of diagnostic RHC . The difficulty confronting the ILD clinician is that no single non-invasive test is sufficiently accurate, in isolation, to provide a confident prediction of findings at RHC . Therefore, it is necessary to integrate findings from a number of tests before proceeding to invasive evaluation.
Correlations between echocardiographic findings and RHC data have been examined in a number of series. In IPF, depending upon the PASP threshold examined against RHC data, positive predictive values for PH vary between 35% and 65% (46% if PASP>50 mmHg) with negative predictive values ranging from 65 to 80%. The high false-positive rate indicates that echocardiographic PH is more likely to represent a true positive when the pretest likelihood is high and does not support the routine use of echocardiography at baseline in screening for PH [32]. This conclusion is compatible with echocardiographic-RHC correlations in the largest series published in chronic lung disease, including a large patient subset with a mixture of ILDs [7]. Of 374 patients referred for consideration of lung transplantation, 25% with measurable PASP were considered to have echocardiographic PH (PASP>45 mmHg). At RHC , it transpired that the diagnosis of PH was falsely positive in 48% and that on other, echocardiography overstated PASP by approximately 10 mmHg, although underestimation also occurred in a minority. These findings are broadly similar to those reported in SSc, the other ILD in which echocardiographic-RHC correlations have been examined. Importantly, a consistent finding in a number of reports is that echocardiographic identification of right ventricular dilatation or dysfunction is an invaluable ancillary diagnostic sign, increasing the likelihood that PH is severe.
In an early PH diagnostic study of serum BNP in ILD , BNP levels were increased in 20/39 cases and correlated with increases in mPAP, and reductions in the six-minute walk distance and cardiac output [31]. A BNP threshold of 33 pg/mL had an ROC area under curve of 96% in identifying severe PH (mPAP>35 mm) but this pilot finding in an underpowered cohort requires further evaluation before BNP increases can be used with precision to nuance the perceived likelihood of PH. Currently, accumulated experience indicates that serum BNP estimation may be most helpful when levels are normal and, thus, well below thresholds generally associated with PH in published data. This view is strongly supported in a recent mixed ILD cohort, in which a NT-proBNP <95 ng/L at initial diagnostic evaluation precluded a positive echocardiographic screen for PH [33].
The ancillary CT signs of an increase in the absolute size of the main pulmonary artery [34, 35] and in the ratio of the pulmonary artery diameter to the diameter of the ascending aorta (the PA:A ratio) [36] have both been shown to correlate with increases in the mPAP in general PH cohorts. However, increases in pulmonary artery diameter are not reliably linked to the presence of PH in ILD . In a cross-sectional study of 65 patients with advanced IPF, the diameter of main pulmonary artery did not differ significantly according to the presence or absence of PH [11]. However, in a study of 77 patients with chronic lung disease, including 45 with various forms of ILD , mPAP levels correlated with the PA:A ratio and were most strongly linked to a composite index containing the PA:A ratio and echocardiographic PASP [37]. The discordance between these two series indicates that the reliability of ancillary HRCT signs of PH may depend upon the severity of underlying pulmonary fibrosis.
In IPF, most pulmonary function variables do not differ significantly between patients with and without PH. DLco levels are only a modestly reliable guide to the likelihood of PH as they are influenced by both pulmonary vasculopathy and pulmonary fibrosis. A reduction in DLco that is disproportionate to lung volumes, as captured by a reduction in Kco or an increase in the FVC/DLco ratio has yet to be explored definitively in IPF, although in one IPF cohort, PH was more frequently present when the DLco level was less than 30% and when the FVC level was >70% [6]. However, in SSc, increases in the FVC/DLco ratio have consistently been associated with an increased likelihood of PH, including in patients with overt pulmonary fibrosis [38–40]. This finding that was validated in the prospective DETECT study, in which an algorithm to screen for PH, with a view to selecting patient to undergo RHC , was developed (although it should be stressed that only a minority of patients in this study had clinically significant ILD ) [41]. More data are required to evaluate the role of the Kco and FVC/DLco ratio in providing ancillary evidence of PH in ILD , with concurrent smoking-related emphysema likely to be a major confounder [42], an important consideration in IPF.
Six-minute walk data, including the walk distance and severity of oxygen desaturation , are influenced both by pulmonary vasculopathy and by the severity of pulmonary fibrosis. In a cohort with advanced interstitial lung disease of various types, the six minute walk distance was observed to be significantly reduced, and the severity of oxygen desaturation significantly increased, in patients with PH at RHC [43]. Abnormal heart rate recovering following a six minute walk test was predictive of the presence of PH at right heart study in an IPF cohort [44]. However, no discrete diagnostic threshold in any single variable has been validated in a subsequent study. Six-minute walk data are probably most helpful diagnostically when there is an absence of major exercise intolerance or oxygen desaturation , reducing the likelihood of underlying PH.
Overall, the use of non-invasive tools to select ILD patients for RHC is an imprecise exercise. The most widely used approach currently is to base investigation on patients considered to be at higher risk of PH based on the severity of ILD and worsening exercise intolerance. It then appears logical to reconcile findings from a number of tests including echocardiography, HRCT , pulmonary function variables, six-minute walk data and serum BNP estimation.
Pathogenesis of PH in IIP
The current classification of PH associated with ILD (including IIP) is based on perceived common pathophysiology leading to the development of PH; it stresses the presence of lung fibrosis and hypoxia being the major contributory factors [45]. There are several criticisms of this simplified view: First, although ablation of pulmonary vessels (“vascular rarefaction”) due to parenchymal destruction is clearly important, there is no direct relationship between the level of fibrosis (and therefore rarefaction), as determined by pulmonary function tests [6, 46] or CT parameters [11], and haemodynamic measurements of pulmonary artery pressure or pulmonary vascular resistance (PVR). Second, most patients with IIP who develop PH are not sufficiently hypoxaemic to explain the level of rise of pulmonary pressure. The pathogenesis of PH in IIP (and other ILDs ) is therefore likely to be more complex and includes other causes such as the balance between rarefaction and angiogenesis, true pulmonary vascular remodelling, chronic pulmonary vasoconstriction (other than due to hypoxia), mechanical causes, such as shear stress, as well as important co-morbid conditions such as pulmonary embolism, sleep-disordered breathing and left-sided heart disease.
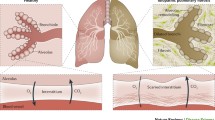
Rarefaction and Angiogenesis
The observation that vascular remodelling occurred in pulmonary fibrosis was first made in 1963 by Turner-Warwick who demonstrated anastomoses between the systemic and pulmonary micro-vasculature associated with neovascularisation within areas of fibrosis [47], while other studies have reported an overall reduced vascular density [48]. Subsequently, it has been accepted that both phenomena occur in different areas of the same lung [49, 50]. Increased vascularity is seen at the active interface between fibrosis and normal lung parenchyma, and decreased vascularity within areas of fibrosis with abnormally dilated vessels within areas of honeycombing [49]. Indeed, new vessel formation has been shown to be maladaptive with increased irregularity and dilatation [51], and lacking an elastin layer may therefore demonstrate decreased compliance [49], potentially contributing to an increased PVR.
It is not clear what role neovascularisation plays in progression of fibrosis, if any, or whether it remains a by-product of active inflammation. Neovascularisation itself appears to be controlled by a host of angiostatic and angiogenic factors perhaps under the influence of master regulators such as hypoxia initiation factor (HIF ) [52] and nuclear factor (NF)-κB [53]. The main factors include vascular endothelial growth factor (VEGF) [54], angiopoetin-1(Ang-1) [55], transforming growth factor (TGF-β1) [56], endostatin [57, 58], pigment epithelium-derived factor (PEDF ) [50] and angiopoetin-2 (Ang-2) [55], with their angiogenic or angiostatic roles in IIP summarised in Table 6.2. It is likely that angiogenesis in IIP occurs in response to the observed reduction in capillary density [47], which relates to rarefaction, hypoxic pulmonary vascular remodelling and other causes. This is supported by studies showing that increasing angiostatin levels resulted in reduction of VEGF and led to a worsening of hypoxic PH [59], whereas VEGF overexpression protected against hypoxic PH [60].
Pulmonary Vascular Remodelling
Pulmonary vascular remodelling refers to hyperplasia of the cellular components of the vascular wall, which results in structural wall changes and resulting decreased vascular distensibility and compliance. The layers involved include the intimal layer composed of endothelial cells, the smooth muscle layer or media and the interstitium composed of fibroblasts and extracellular matrix components. As mentioned earlier, hypoxic pulmonary vascular remodelling is likely in all patients with alveolar hypoxia, but further to this, there is heterogeneity in the pattern of remodelling seen in IPF. Farkas et al. reported vessels with isolated medial hyperplasia, vessels with intimal lesions, vessels obstructed with scar tissue and plexiform lesions [61]. Furthermore, it appears that the extent of these changes correlates with the disease activity in surrounding areas [61]. Although the exact pathogenesis of remodelling seen is not known it is likely to be related to a number of factors:
-
1.
Chemokines and cytokines released from damaged/apoptotic endothelial cells, such as endothelin (ET-1), platelet-derived growth factor (PDGF), angiotensin (AT) II and TGF-β1 [61–65];
-
2.
The same factors may be released from the surrounding fibrotic milieu;
-
3.
Increased levels of oxidative stress [66];
-
4.
Reduced production of vasodilators and anti-proliferative molecules such as nitric oxide (NO) and prostacyclin (PGI2) [61, 62, 64, 67];
-
5.
Increased production of vasoconstrictors and mitogens such as ET-1, thromboxane and AT-II [62, 64];
-
6.
Finally, there is evidence for endothelial cell to mesenchymal cell transition a source for increased numbers of myofibroblasts or vascular smooth muscle cells in remodelled vessels [68–70].
The relative contribution of remodelled vessels to the overall PH phenotype in patients with PH-IIP is crucial as these are potentially amenable to treatment in the same way that pulmonary vasodilators are used to treat pulmonary arterial hypertension (PAH). In fact, this hypothesis has led to a series of clinical trials with variable results to date (see Treatment Section).
Potential Vasoactive Mediators and Growth Factors
ET-1
ET-1 is responsible for both vasoconstriction and the growth of vascular smooth muscle cells [64]. Levels of ET-1 have found to be increased in the lungs of patients with IPF, with expression strongest in fibrotic areas [71, 72]. Furthermore, in the same patient group, there is a positive correlation of ET-1 concentration in peripheral blood samples of patients with pulmonary pressures [73].
VEGF
As its name suggests VEGF is an angiogenic growth factor responsible for endothelial cell migration during angiogenesis and is important in endothelial cell survival and proliferation; its transcription is mainly induced by hypoxia and TGF-β1 [74, 75]. In keeping with VEGF controlling neoangiogenesis in areas of vascular rarefaction, VEGF levels are increased on immunohistochemistry staining in these areas [49]. VEGF is however reduced in broncho-alveolar lavage fluid from fibrotic regions and especially in fibroblastic foci [49, 50, 76]. Indeed, endothelial cell apoptosis correlates with reduced vascular density and decreased capillary branching [48–50] in these areas.
TGF-β
TGF-β is a profibrotic growth factor, predominantly secreted by macrophages, epithelial cells and fibroblasts [77]. TGF-β1 is likely to contribute to local endothelial cell apoptosis and therefore vascular rarefaction. It also has the potential to cause muscularisation of local pulmonary arteries [50, 78–80].
PDGF
PDGF expression is present on fibroblasts, epithelial cells and platelets [81]. It causes fibroblast proliferation and migration, and also activation of vascular smooth muscle cells and fibroblasts which results in muscularisation and fibrosis of the intimal and adventitial layers [82].
Hypoxia-Related Mechanisms
Although we have argued that hypoxia is not the prime driver of PH in patients with IIP, it cannot be ignored and will certainly factor in patients who have severe parenchymal fibrotic disease, or in those having an acute exacerbation, and may occur intermittently during sleep-disordered breathing or physical exertion. Detailed review of the mechanisms of hypoxia-related vascular remodelling is out of the scope of this chapter and the reader is encouraged to read the recent review by Welsh and Peacock [83].
Sleep-Disordered Breathing
Nocturnal hypoxia is very common in IIP with the majority of prospective studies being performed in IPF; the incidence of obstructive sleep apnoea (OSA) varying from 59 to 90% [84–86]. In fact, nocturnal desaturation is common even in the absence of OSA [87]. Patients with IIP are vulnerable to nocturnal desaturation due to the fact that many patients are on the steep portion of the oxygen–haemoglobin dissociation curve and small changes in arterial oxygen tension result in a large decrease in oxygen saturation; more severe nocturnal hypoxaemia is seen in patients with lower daytime PaO2 and oxygen saturations [88–90]. Episodes of alveolar hypoventilation induced, in particular, by REM sleep may also play a role in increasing nocturnal hypoxia [91]. Disproportionate nocturnal desaturation has been found in patients with mild ILD and correlated with signs of PH on echocardiogram [92], as well as being an independent predictor of prognosis [84, 92].
Elevated levels of ET-1 have been demonstrated in the blood of patients with ILD and significantly higher levels were found in patients with elevated pulmonary pressures. Levels of ET-1 were measured during sleep and rose in all patients during episodes of desaturation below 90% and correlated with PaO2 and pulmonary arterial pressure which was measured simultaneously [93]. Repetitive short episodes of hypoxaemia have been demonstrated to increase PVR [94], which may lead to vascular remodelling and propagation of PH. Intermittent nocturnal hypoxia may also reset peripheral chemoreceptors lowering the hypoxic drive and worsening daytime hypoxia, further exacerbating the development of PH.
Left-Sided Heart Disease
The prevalence of left ventricular systolic and diastolic dysfunction increases with advancing age and advancing co-morbidities. Diastolic dysfunction is often overlooked although it has been shown to have a median prevalence of 36% (range 15.8–52.8%) in individuals over the age of 60 [95]. It is an important consideration in patients with IIP given the demographic of patients with the condition. Limited studies have looked specifically in IIP. However, a small, echo-based study (n = 44) identified significant LV diastolic dysfunction in 91% of patients with IPF and found no evidence of LV diastolic dysfunction in controls, who were age and sex matched [96]. Furthermore, in a study evaluating the prevalence of pulmonary hypertension in a lung transplant population with IPF (mean FVC 54.6% ± 17.3%) 16.1% demonstrated an elevated pulmonary capillary wedge pressure on RHC [6]. In terms of an underlying aetiology, in patients under evaluation for lung transplantation, 28.8% of 73 patients with IPF had evidence of coronary artery disease, which was associated with worse outcome [97].
Thrombosis
Large epidemiological studies have suggested an association between IPF and vascular thrombotic diseases such as deep vein thrombosis (DVT ) and pulmonary embolism (PE) [98–100]. For example, a recent large-scale epidemiological study analysing mortality data demonstrated a 34% higher risk of a venous thromboembolism (VTE ) in IPF patients above the background population, and IPF patients with a VTE died earlier than those with IPF alone [74.3 versus 77.4 years in females (p < 0.0001); 72.0 versus 74.4 years in males (p < 0.0001)] [100]. Supporting these studies, there appears to be a hypercoagulable state in animal models of IPF [101] and patients with IPF [102]. Tissue factor, a trigger of the extrinsic clotting pathway, is raised in BAL of patients with biopsy-proven IPF [102, 103]; plasminogen activator inhibitors are also raised, indicating reduced thrombolysis [103]. In addition, platelets are activated in patients with IPF [104] and d-dimer levels raised [105, 106], suggesting ongoing activation of coagulation and fibrinolysis. A small prospective trial performed baseline and follow-up CT pulmonary angiograms (CTPA ) at 3 months in IIP patients without symptoms of PE, where one-third of the patients had evidence of pulmonary emboli on either their baseline or follow-up CTPA [107]. Although no studies as yet document the prevalence of PE in patients presenting with PH-IIP, the observed increase in PE in IIP suggests that this co-morbidity should certainly be excluded in any IIP patient being worked up for PH.
Treatment of IIP–PH
Background
Recent progress has been made in the management of IPF with the use of pirfenidone and nintedanib which reduces the rate of decline in FVC by approximately half in patients with mild-to-moderate disease [108, 109] and a lower risk of subsequent decline in FVC or death in individuals who have progressed on treatment [110]. Unfortunately, at present there is no specific therapy approved for PH associated with IIP, and the evaluation of pulmonary vasodilators in IIP–PH has been punctuated by clinical trials showing a lack of effect on primary outcomes.
The Fifth World Symposium of Pulmonary Hypertension and the European Society of Cardiology/European Respiratory Society PH guidelines [45], and the ATS/ERS guideline for the management of IPF do not advocate the routine use of pulmonary vasodilators [111] in IIP–PH, but suggest further clinical trials before recommendations can be made; however, both advocate optimisation of the underlying disease process and oxygen therapy as the main clinical interventions. In addition, measures to identify and treat co-morbidities that may contribute to a pulmonary hypertensive phenotype should be made.
Basic Principles of Treatment of PH Associated with IIP
Oxygen Therapy
Results from the landmark oxygen trials conducted in patients with COPD [112, 113] have been extrapolated to formulate recommendations for oxygen therapy in many chronic respiratory conditions such as ILD and PH [114]. Without any more specific, up to date studies these recommendations apply therefore to patients with IIP and PH. Patients with IIP–PH should be evaluated for the need for oxygen therapy at rest, on exercise and overnight. As a general rule oxygen saturations should be kept above 90% at all times. In addition, patients should be warned about the risk of travelling to high altitudes (>1500 m) and should be considered for fitness to fly tests for air travel.
Prevent and Treat Exacerbations
Acute exacerbation in IIP represents a period of rapid worsening of symptoms and decline in pulmonary function and is the most common cause of deterioration and death in IPF [115, 116]. Many of the consequences of an acute exacerbation, such as worsening gas exchange, and cytokine release from infection, will potentially lead to increases in pulmonary pressures as well as right ventricular dysfunction. In addition, pulmonary hypertension has been demonstrated to be associated with a higher risk of developing an acute exacerbation and poorer survival [117]. The cause of an acute exacerbation is often not clear and is usually attributed to infection [118], be it viral or bacterial, although an association with air pollution [119] and micro-aspiration [120] has also been demonstrated.
The treatment of an acute exacerbation in IPF is focused upon supportive measures (accepting that no proven intervention exists), such as oxygen therapy, non-invasive ventilation, broad-spectrum antibiotic therapy and careful control of fluid balance. The international evidence-based guidelines on the management of IPF make a weak recommendation for using corticosteroids in an acute exacerbation of IPF [4], stating that they should be used in the majority of patients based upon anecdotal reported benefits and the extremely high mortality associated with an acute exacerbation of IPF. Antifibrotic therapy should be continued if already in use although not commenced during an acute exacerbation [121]. It is desirable although challenging to try and prevent future exacerbations through a combination of annual influenza vaccination in addition to strep pneumonia vaccination and consideration of prophylactic antibiotics when infections are recurrent.
Recognition and Treatment of Coexistent Co-morbidities
Sleep-Discorded Breathing/Nocturnal Desaturation
As mentioned in the Pathogenesis Section, nocturnal desaturation and OSA are common in IIP and likely contribute to the development of PH. Overnight oximetry should be performed in all patients with a clinical suspicion of IIP–PH and sleep studies performed where the history/examination is suggestive of coexistent OSA.
Left Heart Disease
Similarly, ischaemic heart disease and systemic hypertension are common in the patients with IIP–PH. This may lead to left ventricular diastolic dysfunction, which may be a significant driver of PH in these patients. Therefore, all attempts should be made through history, examination and investigations to recognise and treat causes of left heart dysfunction in patients with IIP–PH. ECG and echocardiography are particularly important in terms of investigation as well as careful review of systemic blood pressure. Patients with suspected ischaemic heart disease should be referred to a cardiologist. Systemic blood pressure should be kept under meticulous control and addition of a diuretic may be necessary.
Pulmonary Embolism
Due to the increased prevalence of VTE in IIP as described earlier, it is advisable that IIP patients presenting with pulmonary hypertension, or those with clinical deterioration but stability in fibrosis, be investigated for occult pulmonary emboli. Clinical risk scores to predict PE in large populations include the Wells score, with an area under curve (AUC) of 0.778 (95% CI 0.740–0.818, p = <0.001), and the revised Geneva score with an AUC of 0.693 (95% CI 0.653–0.736, p = <0.001) [122]. The same scores evaluated in a small study in patients with ILD (n = 57, 27 of which had IIP), demonstrate an AUC 0.720 ± 0.083(CI 0.586–0.831), and 0.704 ± 0.081(CI 0.568–0.817) for the Wells score, and revised Geneva score, respectively [107]. The findings suggest clinical risk scores may play a role in predicting pretest probability of coexistent PE, although larger studies in the IIP population are required. The appropriate modality of imaging unfortunately remains undefined in this population, and most patients undergo a CTPA . In a retrospective review of 130 patients with diffuse interstitial lung disease, CTPA was demonstrated to provide adequate opacification of the pulmonary arteries down to the segmental level, whereas in controls without significant parenchymal lung disease was adequate to the sub-segmental level [123]. A recent, small retrospective study evaluated the concordance between CTPA and ventilation perfusion Single Photon Emission Computed Tomography imaging (V/Q-SPECT) in 22 patients with ILD who had clinically deteriorated. In this small study, CTPA detected proximal PE reliably, and the negative VQ scans were also negative on CTPA . V/Q picked up more sub-segmental defects than on CTPA , some of which may have related to areas of fibrosis, although the addition of low dose CT did not, at least in this study, improve the diagnostic accuracy [124]. In practice and previous studies do suggest, however, that the comparison of VQ with areas of fibrosis on CT is useful to determine the aetiology of smaller perfusion defects [125].
In terms of management, at present there is debate as to whether patients (who have a “normal cardiorespiratory reserve”) with single sub-segmental defects might benefit from anticoagulation, and a wide variation in clinical practice occurs. A recent general review suggests that withholding anticoagulation may be appropriate in patients who are informed, have negative lower limb Doppler’s, are low risk and can have close follow up [126]. Patients with IIP–PH are a high-risk group and anticoagulation is advised. Unfortunately, the choice of anticoagulant is also contentious. Of note, a double-blind, randomised, placebo-controlled trial evaluated a potential survival benefit with warfarin (INR target 2.0 to 3.0) in IPF patients (all-comers). An early safety review revealed not only a lack of benefit, but also that warfarin carried a significantly higher risk of death or decline in FVC by 10% or greater [106]. This increased mortality related to warfarin is postulated to relate to an increased risk of acute exacerbations [127].
We recommend that IIP patients who develop PH due to IIP be evaluated with CTPA (where no contraindications exist) in their workup. In some cases, especially in those with less extensive fibrosis, consider additional V/Q SPECT where clinical suspicion remains high, accepting that there may be false positives especially in sub-segmental vessels. We advocate anticoagulation with novel oral anticoagulants (where appropriate) for all individuals with PE including isolated sub-segmental PE, especially in view of minimising drug interactions. Anticoagulation should be long term unless a clear and reversible provoking factor is present.
Evidence for the Use of Pulmonary Vasodilators
Although the current guidelines do not recommend the routine use of pulmonary vasodilators in patients with IIP–PH, it is worth reviewing the current data. This is summarised in Table 6.3.
Phosphodiesterase Type-5 Inhibitors (PDE-5)
PDE-5 inhibitors (sildenafil and tadalafil) inhibit the degradation of cyclic guanosine monophosphate, which is synthesised by soluble guanylate cyclase in response to nitric oxide (NO), leading to pulmonary vasodilation [22].
There is relevant recent basic science evidence supporting the use of sildenafil in IIP. An ex vivo study of the pulmonary arteries of 18 healthy donors, 9 IPF patients, 8 IPF-PH patients and 4 PH patients was performed to evaluate the vascular effects of sildenafil [128]. Sildenafil relaxed pre-contracted pulmonary arteries in healthy donors and non-PH IPF samples more than in IPF-PH and PH samples. This effect was increased in the presence of an intact endothelium. In addition, sildenafil prevented a TGF-β-induced mesenchymal/myofibroblast phenotype in human pulmonary artery endothelial cells and human pulmonary artery smooth muscle cells, with associated down-regulation of endothelial markers (including eNOS, VEGF) and upregulation of pulmonary PDE5 expression . The same authors also demonstrated in a rat model of bleomycin-induced pulmonary fibrosis and pulmonary hypertension that administration of sildenafil did not worsen ventilation perfusion matching [128].
Sildenafil has been evaluated in several small, non-randomised, open label populations with proven IIP–PH. First, a small open-label trial (16 patients, 7 with IPF) compared the vasodilatory effects of oral sildenafil and IV prostacyclin after nebulised nitric oxide (10–20 ppm) and demonstrated a 32.5% reduction in PVR (CI: −10.2 to −54.1), with sildenafil. The use of IV prostacyclin was associated with an increased V/Q mismatch and decreased arterial oxygenation, whereas oral sildenafil (and inhaled nitric oxide) maintained V/Q matching and increased arterial oxygenation [129]. Second, another small open-label trial evaluated the effect of sildenafil on 6-minute walk distance (6MWD), and included 14 patients with IPF and PH confirmed by RHC . Eleven patients were able to complete the screening and post-treatment 6MWT. More than half (57%) of the patients improved their 6MWD by > 20%, although there was no control group for comparison. The treatment (over a median follow up of 91 days) was generally well tolerated [130]. Third, a small retrospective review of was also performed in 15 patients with ILD and PH confirmed by either RHC or echocardiography. Following 6 months of sildenafil serum BNP levels were significantly lower and 6MWD improved although there was no change in echocardiographic systolic pulmonary pressure, arterial oxygen saturation or pulmonary function tests [131]. Finally, a small observational pilot study of PDE-5 inhibitors sildenafil or tadalafil in ILD (10 patients, 6 with IPF), importantly in patients with severe degrees of PH but milder degrees of ILD , with evaluation of invasive haemodynamics both at baseline and follow up. Cardiac index was shown to increase significantly and PVR fall with treatment, although no difference was seen in 6MWD, BNP or PFT with treatment [132]. Although a small study, this is an important one to demonstrate haemodynamic improvement in the ‘severe PH-milder lung disease’ phenotype.
The largest experience of sildenafil in IIP to date has been the double-blind, randomised, placebo-controlled STEP-IPF study . PH was not formally tested for, but the advanced nature of the IPF (DLCO <35%) made its coexistence likely. The primary outcome in this study was a 20% improvement in 6MWT, which was not met. However, several secondary outcome measures were met including an improvement in DLCO, partial pressure of oxygen and oxygen saturations, as well as quality of life measures. An improvement in the former three suggests that sildenafil was having a direct effect on the pulmonary vasculature [133].
All patients underwent a pre-enrolment echocardiogram to exclude significant aortic stenosis as an exclusion factor. Evaluation of the pre-enrolment echo was possible in 119 of 180 patients, and the interaction between right ventricular systolic dysfunction (RVSD ) and the effect of sildenafil was evaluated. Patients with RVSD who were on sildenafil experienced a lesser drop (99.3 m p = 0.01) in their 6MWD than patients with RVSD who were on placebo [134]. (The minimal clinical important difference for 6MWD in IPF has been demonstrated to be 24–45 m [135]). As well as preserving 6MWD, quality of life scores (St Georges Respiratory Questionnaire) also improved in the sildenafil-treated group [134].
Recently, a retrospective study (using an international registry COMPERA) evaluated patients with IIP and compared them with IPAH patients. There were 151 incident IIP diagnosed patients, who were significantly older than the IPAH patients and had more severely affected lung function (mean DLCO 28.5% predicted for IIP patients versus 50.1% in IPAH). Patients with IIP had lower mean pulmonary artery pressure 37 mmHg versus 45 mmHg, than patients with IPAH, although 79% of the IIP–PH patients met the “severe” PH criteria. Ninety-five per cent of the IIP–PH patients were treated with a single pulmonary vasodilator, 88% of which were a PDE5i. Treatment was associated with a 24.5 m improvement in 6MWD in IIP–PH versus 30 m in IPAH patients , and functional class improved in 22.4% in IIP–PH and 29.5% of IPAH. Patients who improved their 6MWD by at least 20 m or improved in functional class had a better prognosis than patients who did not despite almost identical haemodynamics at baseline . Patients with severe PH were no more likely to show improvements with treatment than patients with lower invasive pulmonary pressures. Interestingly, the authors of this study point out that the primary endpoint from STEP-IPF (increase in 6MWD by 20%) would have been met by 31% within the IIP–PH cohort [136].
Endothelin Receptor Antagonists
Endothelin-1 (ET-1) is a potent vasoconstrictor and promoter of vascular smooth muscle cell proliferation; its role in the pathogenesis of pulmonary arterial hypertension is firmly established [137]. ET-1 is also profibrotic [138], and elevated levels have been demonstrated in patients with IIP [73, 139] and levels have been shown to correlate with pulmonary arterial pressure and in a negative fashion with arterial oxygen content in a small group of patients [73]. ET-1 therefore seems like a very attractive target to prevent progression of the underlying fibrotic process within the lungs and attenuate the development of pulmonary vascular disease.
Endothelin receptor antagonists (ERAs ) have been evaluated in a similar fashion to sildenafil in an attempt to prevent time to deterioration in IPF in patients without PH. BUIILD-3 was a large randomised placebo-controlled trial which showed bosentan to be well tolerated in HRCT and biopsy confirmed IPF although no difference was demonstrated with placebo in time to IPF worsening or death (hazard ratio, 0.85 95% CI, 0.66–1.10) [140]. Another study (MUSIC) evaluated 178 patients with biopsy diagnosed IPF (FVC > 50% predicted and DLCO > 30%) in a prospective randomised double-blind placebo-controlled study using macitentan. There was no difference in the primary outcome (change in FVC from baseline up to month 12) or any of the secondary outcomes [141]. Artemis-IPF was a randomised double-blind placebo-controlled trial evaluating the role of ambrisentan in IPF and IPF-PH. The study was stopped following interim analysis as ambrisentan-treated patients were more likely to meet the pre-specified criteria for disease progression. Ten per cent of the group had pulmonary hypertension and sub-analysis of this group demonstrated similar findings although the study was not fully powered for all endpoints in this subgroup [9].
The bosentan in pulmonary hypertension-associated fibrotic idiopathic interstitial pneumonia (B-PHIT) was the first randomised, double-blind placebo-controlled study evaluating PH-specific treatment in IIP–PH. The study failed to show any difference in invasive pulmonary haemodynamics, functional capacity, 6MWD or QOL scores between placebo and bosentan, and subgroup analysis could not demonstrate a group that benefited [142]. However, there was no deterioration in oxygen saturation or oxygen requirement in the study period.
These trials demonstrate a lack of benefit with bosentan and macitentan and the potential for harm with ambrisentan and they are therefore not recommended for use in IIP or IIP–PH.
Prostanoids
Prostacyclin (PGI2) and its analogues are members of the prostanoid family. PGI2 inhibits platelet activation and acts as a potent vasodilator. PGI2 also displays anti-inflammatory and anti-proliferative properties.
Studies involving prostanoids have been small, non-randomised and limited to a short follow-up period with focus on invasive haemodynamics. In one study with 8 ILD patients (only one of which had IPF) with severe underlying pulmonary fibrosis found that inhaled iloprost caused pulmonary vasodilatation with maintenance of gas exchange and systemic arterial pressure whereas intravenous prostacyclin resulted in a significant drop in systemic arterial pressure and a marked increase in ventilation–perfusion mismatching [143].
Guanylate Cyclase Stimulators
Riociguat is a soluble guanylate cyclase stimulator that can synergise with endogenous NO or act independently of NO. It has been shown to improve exercise capacity and haemodynamics in patients with PAH [144]. In a pilot study (open-label, non-blinded, non-randomised) to assess safety and tolerability in patients with mild-to-moderate ILD but moderate-to-severe PH (n = 23, 82% of patients had underlying IIP), riociguat was well tolerated, with 2 of the 23 patients discontinuing the treatment prematurely. In terms of efficacy, PVR decreased, and cardiac output increased, with mean pulmonary pressure remaining unchanged, likely due to a higher cardiac output offsetting the effect of pulmonary vasodilation [145]. As a result of these promising findings a randomised, double-blind placebo-controlled trial on efficacy and safety of riociguat in IIP–PH (RISE-IIP ) was commenced in 2014 [146]. Unfortunately, this study has recently been halted prematurely due to increased mortality in the treatment arm. Bayer has recommended that riociguat is not used in this patient group.
In summary, the treatment of IIP–PH is challenging, and evidence of benefit with specific interventions is eagerly awaited to improve patient outcome. PDE-5 inhibitors appear to be the most likely class of drug to improve outcome in IIP–PH, although which patients stand to benefit and at which the stage of the disease (i.e. prior to development of PH or with onset of right ventricular dysfunction) remain unclear.
Conclusion
PH is commonly encountered in IIP and the likelihood of coexistent PH increases as the underlying disease progresses, although its presence is not reliably linked to disease characteristics, which confounds non-invasive detection and underscores the importance of invasive evaluation for confirmation where appropriate. The development of PH is associated with decline in functional status and dramatically worsens prognosis. The underlying aetiology of PH in IIP remains poorly defined and is multifactorial; further study to help develop novel treatment options is highly desirable. The management of PH within IIP at present is predominantly supportive in terms of evaluating for and treating hypoxaemia and other contributory causes of PH. At present there is no evidence to support the use of pulmonary vasodilators, although this remains a very active research area.
References
Travis WD et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–48.
Thomeer M, Demedts M, Vandeurzen K. Registration of interstitial lung diseases by 20 centres of respiratory medicine in Flanders. Acta Clin Belg. 2001;56(3):163–72.
Tinelli C et al. The Italian register for diffuse infiltrative lung disorders (RIPID): a four-year report. Sarcoidosis Vasc Diffuse Lung Dis. 2005;22(Suppl 1):S4–8.
Raghu G et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824.
Lettieri CJ et al. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2006;129(3):746–52.
Nathan SD et al. Pulmonary hypertension and pulmonary function testing in idiopathic pulmonary fibrosis. Chest. 2007;131(3):657–63.
Arcasoy SM et al. Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. Am J Respir Crit Care Med. 2003;167(5):735–40.
Nathan SD et al. Serial development of pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respiration. 2008;76(3):288–94.
Raghu G et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med. 2013;158(9):641–9.
The effect of diffuse fibrosis on reliability of ct signs of ph.pdf.
Zisman DA et al. High-resolution chest computed tomography findings do not predict the presence of pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2007;132(3):773–9.
Nihtyanova SI et al. Prediction of pulmonary complications and long-term survival in systemic sclerosis. Arthritis Rheumatol. 2014;66(6):1625–35.
Fischer A et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46(4):976–87.
Handa T et al. Incidence of pulmonary hypertension and its clinical relevance in patients with sarcoidosis. Chest. 2006;129(5):1246–52.
Baughman RP, Engel PJ, Nathan S. Pulmonary hypertension in sarcoidosis. Clin Chest Med. 2015;36(4):703–14.
Nadrous HF et al. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Chest. 2005;128(4):2393–9.
Cottin V et al. Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur Respir J. 2010;35(1):105–11.
Mejia M et al. Idiopathic pulmonary fibrosis and emphysema: decreased survival associated with severe pulmonary arterial hypertension. Chest. 2009;136(1):10–5.
Rivera-Lebron BN et al. Echocardiographic and hemodynamic predictors of mortality in idiopathic pulmonary fibrosis. Chest. 2013;144(2):564–70.
Corte TJ et al. Pulmonary vascular resistance predicts early mortality in patients with diffuse fibrotic lung disease and suspected pulmonary hypertension. Thorax. 2009;64(10):883–8.
Yasui K et al. Pulmonary vascular resistance estimated by Doppler echocardiography predicts mortality in patients with interstitial lung disease. J Cardiol. 2016;68(4):300–7.
Baughman RP et al. Survival in sarcoidosis-associated pulmonary hypertension: the importance of hemodynamic evaluation. Chest. 2010;138(5):1078–85.
Raghu G et al. Pulmonary hypertension in idiopathic pulmonary fibrosis with mild-to-moderate restriction. Eur Respir J. 2015;46(5):1370–7.
Leuchte HH et al. Brain natriuretic peptide is a prognostic parameter in chronic lung disease. Am J Respir Crit Care Med. 2006;173(7):744–50.
Corte TJ et al. Elevated brain natriuretic peptide predicts mortality in interstitial lung disease. Eur Respir J. 2010;36(4):819–25.
Song JW, Song JK, Kim DS. Echocardiography and brain natriuretic peptide as prognostic indicators in idiopathic pulmonary fibrosis. Respir Med. 2009;103(2):180–6.
Shin S et al. Pulmonary artery size as a predictor of outcomes in idiopathic pulmonary fibrosis. Eur Respir J. 2016;47(5):1445–51.
Corte TJ et al. Pulmonary function vascular index predicts prognosis in idiopathic interstitial pneumonia. Respirology. 2012;17(4):674–80.
Peelen L et al. Fibrotic idiopathic interstitial pneumonias: mortality is linked to a decline in gas transfer. Respirology. 2010;15(8):1233–43.
Lama VN et al. Prognostic value of desaturation during a 6-minute walk test in idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 2003;168(9):1084–90.
Eaton T et al. Six-minute walk, maximal exercise tests: reproducibility in fibrotic interstitial pneumonia. Am J Respir Crit Care Med. 2005;171(10):1150–7.
Nathan SD et al. Right ventricular systolic pressure by echocardiography as a predictor of pulmonary hypertension in idiopathic pulmonary fibrosis. Respir Med. 2008;102(9):1305–10.
Andersen C et al. NT-proBNP <95 ng/l can exclude pulmonary hypertension on echocardiography at diagnostic workup in patients with interstitial lung disease. Eur Clin Respir J. 2016;3:32027.
Kuriyama K et al. CT-determined pulmonary artery diameters in predicting pulmonary hypertension. Investig Radiol. 1984;19(1):16–22.
Edwards PD, Bull RK, Coulden R. CT measurement of main pulmonary artery diameter. Br J Radiol. 1998;71(850):1018–20.
Ng CS, Wells AU, Padley SP. A CT sign of chronic pulmonary arterial hypertension: the ratio of main pulmonary artery to aortic diameter. J Thorac Imaging. 1999;14(4):270–8.
Devaraj A et al. Detection of pulmonary hypertension with multidetector CT and echocardiography alone and in combination. Radiology. 2010;254(2):609–16.
Steen V et al. Exercise-induced pulmonary arterial hypertension in patients with systemic sclerosis. Chest. 2008;134(1):146–51.
Hsu VM et al. Assessment of pulmonary arterial hypertension in patients with systemic sclerosis: comparison of noninvasive tests with results of right-heart catheterization. J Rheumatol. 2008;35(3):458–65.
Launay D et al. Clinical characteristics and survival in systemic sclerosis-related pulmonary hypertension associated with interstitial lung disease. Chest. 2011;140(4):1016–24.
Coghlan JG et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis. 2014;73(7):1340–9.
Antoniou KM et al. Combined pulmonary fibrosis and emphysema in scleroderma-related lung disease has a major confounding effect on lung physiology and screening for pulmonary hypertension. Arthritis Rheumatol. 2016;68(4):1004–12.
Kawut SM et al. Exercise testing determines survival in patients with diffuse parenchymal lung disease evaluated for lung transplantation. Respir Med. 2005;99(11):1431–9.
Swigris JJ et al. Heart rate recovery after six-minute walk test predicts pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respirology. 2011;16(3):439–45.
Galiè N et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2015;46(4):903–75.
Hamada K et al. Significance of pulmonary arterial pressure and diffusion capacity of the lung as prognosticator in patients with idiopathic pulmonary fibrosis. Chest. 2007;131(3):650–6.
Turner-Warwick M. Precapillary systemic-pulmonary anastomoses. Thorax. 1963;18:225–37.
Renzoni EA et al. Interstitial vascularity in fibrosing alveolitis. Am J Respir Crit Care Med. 2003;167(3):438–43.
Ebina M et al. Heterogeneous increase in CD34-positive alveolar capillaries in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2004;169(11):1203–8.
Cosgrove GP et al. Pigment epithelium–derived factor in idiopathic pulmonary fibrosis a role in aberrant angiogenesis. Am J Respir Crit Care Med. 2004;170:242–51.
Kwon KY, Park KK, Chang ES. Scanning electron microscopic study of capillary change in bleomycin-induced pulmonary fibrosis. J Korean Med Sci. 1991;6(3):234–45.
Tzouvelekis A et al. Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1alpha in disease pathogenesis. Am J Respir Crit Care Med. 2007;176(11):1108–19.
Strieter RM. Masters of angiogenesis. Nat Med. 2005;11(9):925–7.
Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9(6):669–76.
Margaritopoulos GA et al. Investigation of angiogenetic axis Angiopoietin-1 and -2/Tie-2 in fibrotic lung diseases: a bronchoalveolar lavage study. Int J Mol Med. 2010;26(6):919–23.
Pertovaara L et al. Vascular endothelial growth factor is induced in response to transforming growth factor-beta in fibroblastic and epithelial cells. J Biol Chem. 1994;269(9):6271–4.
Hanumegowda C, Farkas L, Kolb M. Angiogenesis in pulmonary fibrosis: too much or not enough? Chest. 2012;142(1):200–7.
Sumi M et al. Increased serum levels of endostatin in patients with idiopathic pulmonary fibrosis. J Clin Lab Anal. 2005;19(4):146–9.
Pascaud MA et al. Lung overexpression of angiostatin aggravates pulmonary hypertension in chronically hypoxic mice. Am J Respir Cell Mol Biol. 2003;29(4):449–57.
Partovian C et al. Adenovirus-mediated lung vascular endothelial growth factor overexpression protects against hypoxic pulmonary hypertension in rats. Am J Respir Cell Mol Biol. 2000;23(6):762–71.
Farkas L et al. VEGF ameliorates pulmonary hypertension through inhibition of endothelial apoptosis in experimental lung fibrosis in rats. J Clin Invest. 2009;119(5):1298–311.
Sakao S et al. Apoptosis of pulmonary microvascular endothelial cells stimulates vascular smooth muscle cell growth. Am J Physiol Lung Cell Mol Physiol. 2006;291(3):L362–8.
Nathan SD, Noble PW, Tuder RM. Idiopathic pulmonary fibrosis and pulmonary hypertension: connecting the dots. Am J Respir Crit Care Med. 2007;175(9):875–80.
Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109(2):159–65.
Ask K et al. Targeting genes for treatment in idiopathic pulmonary fibrosis: challenges and opportunities, promises and pitfalls. Proc Am Thorac Soc. 2006;3(4):389–93.
Teng RJ et al. Increased superoxide production contributes to the impaired angiogenesis of fetal pulmonary arteries with in utero pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2009;297(1):L184–95.
Smith AP, Demoncheaux EA, Higenbottam TW. Nitric oxide gas decreases endothelin-1 mRNA in cultured pulmonary artery endothelial cells. Nitric Oxide. 2002;6(2):153–9.
Hashimoto N et al. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2010;43(2):161–72.
Arciniegas E et al. Perspectives on endothelial-to-mesenchymal transition: potential contribution to vascular remodeling in chronic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293(1):L1–8.
Sakao S et al. VEGF-R blockade causes endothelial cell apoptosis, expansion of surviving CD34+ precursor cells and transdifferentiation to smooth muscle-like and neuronal-like cells. FASEB J. 2007;21(13):3640–52.
Giaid A et al. Expression of endothelin-1 in lungs of patients with cryptogenic fibrosing alveolitis. Lancet. 1993;341(8860):1550–4.
Saleh D et al. Elevated expression of endothelin-1 and endothelin-converting enzyme-1 in idiopathic pulmonary fibrosis: possible involvement of proinflammatory cytokines. Am J Respir Cell Mol Biol. 1997;16(2):187–93.
Trakada G, Spiropoulos K. Arterial endothelin-1 in interstitial lung disease patients with pulmonary hypertension. Monaldi Arch Chest Dis. 2001;56(5):379–83.
Voelkel NF, Vandivier RW, Tuder RM. Vascular endothelial growth factor in the lung. Am J Physiol Lung Cell Mol Physiol. 2006;290(2):L209–21.
Ramirez-Bergeron DL et al. HIF -dependent hematopoietic factors regulate the development of the embryonic vasculature. Dev Cell. 2006;11(1):81–92.
Meyer KC, Cardoni A, Xiang ZZ. Vascular endothelial growth factor in bronchoalveolar lavage from normal subjects and patients with diffuse parenchymal lung disease. J Lab Clin Med. 2000;135(4):332–8.
Farkas L et al. Pulmonary hypertension and idiopathic pulmonary fibrosis: a tale of angiogenesis, apoptosis, and growth factors. Am J Respir Cell Mol Biol. 2011;45(1):1–15.
Khalil N et al. Enhanced expression and immunohistochemical distribution of transforming growth factor-beta in idiopathic pulmonary fibrosis. Chest. 1991;99(3 Suppl):65s–6s.
Bergeron A et al. Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-beta and IL-10. Eur Respir J. 2003;22(1):69–76.
Zaiman AL et al. Role of the TGF-beta/Alk5 signaling pathway in monocrotaline-induced pulmonary hypertension. Am J Respir Crit Care Med. 2008;177(8):896–905.
Fredriksson L, Li H, Eriksson U. The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004;15(4):197–204.
Antoniades HN et al. Platelet-derived growth factor in idiopathic pulmonary fibrosis. J Clin Invest. 1990;86(4):1055–64.
Welsh DJ, Peacock AJ. Cellular responses to hypoxia in the pulmonary circulation. High Alt Med Biol. 2013;14(2):111–6.
Kolilekas L et al. Sleep oxygen desaturation predicts survival in idiopathic pulmonary fibrosis. J Clin Sleep Med. 2013;9(6):593–601.
Pihtili A et al. Obstructive sleep apnea is common in patients with interstitial lung disease. Sleep Breath. 2013;17(4):1281–8.
Mermigkis C et al. How common is sleep-disordered breathing in patients with idiopathic pulmonary fibrosis? Sleep Breath. 2010;14(4):387–90.
Perez-Padilla R et al. Breathing during sleep in patients with interstitial lung disease. Am Rev Respir Dis. 1985;132(2):224–9.
Midgren B et al. Oxygen desaturation during sleep and exercise in patients with interstitial lung disease. Thorax. 1987;42(5):353–6.
Clark M et al. A survey of nocturnal hypoxaemia and health related quality of life in patients with cryptogenic fibrosing alveolitis. Thorax. 2001;56(6):482–6.
Midgren B. Oxygen desaturation during sleep as a function of the underlying respiratory disease. Am Rev Respir Dis. 1990;141(1):43–6.
Fletcher EC et al. Pulmonary vascular hemodynamics in chronic lung disease patients with and without oxyhemoglobin desaturation during sleep. Chest. 1989;95(4):757–64.
Corte TJ et al. Elevated nocturnal desaturation index predicts mortality in interstitial lung disease. Sarcoidosis Vasc Diffuse Lung Dis. 2012;29(1):41–50.
Trakada G et al. Endothelin-1 levels in interstitial lung disease patients during sleep. Sleep Breath. 2003;7(3):111–8.
Talbot NP et al. Two temporal components within the human pulmonary vascular response to approximately 2 h of isocapnic hypoxia. J Appl Physiol (1985). 2005;98(3):1125–39.
van Riet EE et al. Epidemiology of heart failure: the prevalence of heart failure and ventricular dysfunction in older adults over time. A systematic review. Eur J Heart Fail. 2016;18(3):242–52.
Papadopoulos CE et al. Left ventricular diastolic dysfunction in idiopathic pulmonary fibrosis: a tissue Doppler echocardiographic [corrected] study. Eur Respir J. 2008;31(4):701–6.
Nathan SD et al. Prevalence and impact of coronary artery disease in idiopathic pulmonary fibrosis. Respir Med. 2010;104(7):1035–41.
Hubbard RB et al. The association between idiopathic pulmonary fibrosis and vascular disease: a population-based study. Am J Respir Crit Care Med. 2008;178(12):1257–61.
Sode BF et al. Venous thromboembolism and risk of idiopathic interstitial pneumonia: a nationwide study. Am J Respir Crit Care Med. 2010;181(10):1085–92.
Sprunger DB et al. Pulmonary fibrosis is associated with an elevated risk of thromboembolic disease. Eur Respir J. 2012;39(1):125–32.
Wygrecka M et al. Cellular origin of pro-coagulant and (anti)-fibrinolytic factors in bleomycin-injured lungs. Eur Respir J. 2007;29(6):1105–14.
Fujii M et al. Relevance of tissue factor and tissue factor pathway inhibitor for hypercoagulable state in the lungs of patients with idiopathic pulmonary fibrosis. Thromb Res. 2000;99(2):111–7.
Kotani I et al. Increased procoagulant and antifibrinolytic activities in the lungs with idiopathic pulmonary fibrosis. Thromb Res. 1995;77(6):493–504.
Fahim A et al. Increased platelet binding to circulating monocytes in idiopathic pulmonary fibrosis. Lung. 2014;192(2):277–84.
Kinder BW, Collard HR, King Jr TE. Anticoagulant therapy and idiopathic pulmonary fibrosis. Chest. 2006;130(1):302–3.
Noth I et al. A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186(1):88–95.
Luo Q et al. Prevalence of venous thromboembolic events and diagnostic performance of the wells score and revised geneva scores for pulmonary embolism in patients with interstitial lung disease: a prospective study. Heart Lung Circ. 2014;23(8):778–85.
Noble PW et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779):1760–9.
Richeldi L et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosi. N Engl J Med. 365(12):1079–87.
Nathan SD et al. Effect of continued treatment with pirfenidone following clinically meaningful declines in forced vital capacity: analysis of data from three phase 3 trials in patients with idiopathic pulmonary fibrosis. Thorax. 2016;71(5):429–35.
Raghu G et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192(2):e3–19.
Long term domiciliary oxygen therapy in chronic hypoxic cor pulmonale complicating chronic bronchitis and emphysema. Report of the Medical Research Council Working Party. Lancet. 1981;1(8222):681–6.
Continuous or nocturnal oxygen therapy in hypoxemic chronic obstructive lung disease: a clinical trial. Nocturnal Oxygen Therapy Trial Group. Ann Intern Med. 1980;93(3):391–8.
Hardinge M et al. British Thoracic Society guidelines for home oxygen use in adults. Thorax. 2015;70(Suppl 1):i1–43.
Kim DS et al. Acute exacerbation of idiopathic pulmonary fibrosis: frequency and clinical features. Eur Respir J. 2006;27(1):143–50.
Song JW et al. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J. 2011;37(2):356–63.
Judge EP et al. Acute exacerbations and pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Eur Respir J. 2012;40(1):93–100.
Huie TJ et al. A detailed evaluation of acute respiratory decline in patients with fibrotic lung disease: aetiology and outcomes. Respirology. 2010;15(6):909–17.
Johannson KA et al. Acute exacerbation of idiopathic pulmonary fibrosis associated with air pollution exposure. Eur Respir J. 2014;43(4):1124–31.
Lee JS et al. Bronchoalveolar lavage pepsin in acute exacerbation of idiopathic pulmonary fibrosis. Eur Respir J. 2012;39(2):352–8.
Maher TM et al. Development of a consensus statement for the definition, diagnosis, and treatment of acute exacerbations of idiopathic pulmonary fibrosis using the delphi technique. Adv Ther. 2015;32(10):929–43.
Shen JH et al. Comparison of the Wells score with the revised Geneva score for assessing suspected pulmonary embolism: a systematic review and meta-analysis. J Thromb Thrombolysis. 2016;41(3):482–92.
Wijesekera NT et al. Image quality of computed tomographic pulmonary angiography for suspected pulmonary embolus in patients with diffuse interstitial lung disease. J Thorac Imaging. 2012;27(3):156–63.
Leuschner G et al. Suspected pulmonary embolism in patients with pulmonary fibrosis: discordance between ventilation/perfusion SPECT and CT pulmonary angiography. Respirology. 2016;21(6):1081–7.
Strickland NH et al. Cause of regional ventilation-perfusion mismatching in patients with idiopathic pulmonary fibrosis: a combined CT and scintigraphic study. AJR Am J Roentgenol. 1993;161(4):719–25.
Peiman S et al. Subsegmental pulmonary embolism: a narrative review. Thromb Res. 2016;138:55–60.
Alagha K et al. Warfarin should be banned in ipf.pdf. Am J Respir Crit Care Med. 2015;191:958–60.
Milara J et al. Vascular effects of sildenafil in patients with pulmonary fibrosis and pulmonary hypertension: an ex vivo/in vitro study. Eur Respir J. 2016;47(6):1737–49.
Ghofrani HA et al. Sildenafil for treatment of lung fibrosis and pulmonary hypertension: a randomised controlled trial. Lancet. 2002;360(9337):895–900.
Collard HR, Anstrom KJ, Schwarz MI, Zisman DA. Sildenafil improves walk distance in idiopathic pulmonary fibrosis. Chest. 2007;131(3):897–9.
Corte TJ et al. The use of sildenafil to treat pulmonary hypertension associated with interstitial lung disease. Respirology. 2010;15(8):1226–32.
Zimmermann GS et al. Haemodynamic changes in pulmonary hypertension in patients with interstitial lung disease treated with PDE-5 inhibitors. Respirology. 2014;19(5):700–6.
Zisman DA et al. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med. 2010;363(7):620–8.
Han MK et al. Sildenafil preserves exercise capacity in patients with idiopathic pulmonary fibrosis and right-sided ventricular dysfunction. Chest. 2013;143(6):1699–708.
du Bois RM et al. Six-minute-walk test in idiopathic pulmonary fibrosis: test validation and minimal clinically important difference. Am J Respir Crit Care Med. 2011;183(9):1231–7.
Hoeper MM et al. Pulmonary hypertension in patients with chronic fibrosing idiopathic interstitial pneumonias. PLoS One. 2015;10(12):e0141911.
Giaid A et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328(24):1732–9.
Ross B, D’Orleans-Juste P, Giaid A. Potential role of endothelin-1 in pulmonary fibrosis: from the bench to the clinic. Am J Respir Cell Mol Biol. 2010;42(1):16–20.
Uguccioni M et al. Endothelin-1 in idiopathic pulmonary fibrosis. J Clin Pathol. 1995;48(4):330–4.
King Jr TE et al. BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(1):92–9.
Raghu G et al. Macitentan for the treatment of idiopathic pulmonary fibrosis: the randomised controlled MUSIC trial. Eur Respir J. 2013;42(6):1622–32.
Corte TJ et al. Bosentan in pulmonary hypertension associated with fibrotic idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 2014;190(2):208–17.
Olschewski H et al. Inhaled prostacyclin and iloprost in severe pulmonary hypertension secondary to lung fibrosis. Am J Respir Crit Care Med. 1999;160(2):600–7.
Ghofrani HA et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369(4):330–40.
Hoeper MM et al. Riociguat for interstitial lung disease and pulmonary hypertension: a pilot trial. Eur Respir J. 2013;41(4):853–60.
Bayer. Efficacy and safety of riociguat in patients with symptomatic pulmonary hypertension (PH) associated with idiopathic interstitial pneumonia’s (IIP) (RISE-IIP). https://clinicaltrials.gov/ct2/show/, NCT02138825.
King Jr TE et al. BUILD-1: a randomized placebo-controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177(1):75–81.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Bax, S., Wells, A., Price, L., Wort, J. (2017). Pulmonary Hypertension in Idiopathic Interstitial Pneumonias. In: Baughman, R., Carbone, R., Nathan, S. (eds) Pulmonary Hypertension and Interstitial Lung Disease. Springer, Cham. https://doi.org/10.1007/978-3-319-49918-5_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-49918-5_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-49916-1
Online ISBN: 978-3-319-49918-5
eBook Packages: MedicineMedicine (R0)