
Abstract
Jatropha curcas L. is a potential plant species for biodiesel production. However, its seed yield is too low for profitable production of biodiesel. To improve the productivity, genetic improvement through breeding is essential. Marker-assisted selection (MAS) has the huge potential to accelerate genetic improvement. We mapped 506 markers (216 microsatellites and 290 SNPs from ESTs) onto 11 linkage groups. However, genetic analysis of the yield traits has not been done in jatropha. Quantitative trait loci (QTL) mapping was conducted to identify genetic factors controlling growth and seed yield in jatropha. We identified a total of 28 QTLs for 11 growth and seed traits using a population of 296 backcrossing jatropha trees. QTL and expression QTL analyses were applied to identify genetic factors that are relevant to seed oil traits in jatropha. We screened key genes in auxin pathway including ARF and IAA families and downstream effectors to identify candidate genes controlling seed size in jatropha. JcARF19 was mapped in the major QTL region and significantly associated with seed length.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
The increasing demand for diesel coupled with the continuous rise in price of crude oil has forced us to search for an ecologically sustainable alternative energy source (Fairless 2007). Biodiesel from vegetable oil emerged as a viable alternative, particularly nonedible vegetable oil. Jatropha curcas L., also called Physic nut, is a perennial poisonous shrub belonging to the Euphorbiaceous family (Heller 1996). This plant originating from Mexico and central America has been spread to other tropical and subtropical countries and is mainly grown in Asia and Africa. People claimed that J. curcas is resistant to a high degree of drought and does not directly compete with food crops (Openshaw 2000; Fairless 2007). The generation interval of J. curcas was only 6 months in tropical regions and its genome consists of 11 chromosome pairs (Carvalho et al. 2008). Its seeds contain ca. 30% oil that is usable in a standard diesel engine (Shah et al. 2005). Therefore, J. curcas is regarded as a promising candidate for producing biodiesel (Fairless 2007; Jain and Sharma 2010).
However, J. curcas has been an uncultivated wild-species, and until recently, little (Wang et al. 2011b) is known about its genetics and genome. Although some genes involved in pathways for biosynthesis of fatty acids and lipids have been cloned recently (Zhang et al. 2007; Carvalho et al. 2008; Gu et al. 2011), methods for gene silencing have been established (Ye et al. 2009), and the genome was sequenced (Sato et al. 2011). J. curcas has never been domesticated and bred for producing oil in large scale. Detailed selective breeding has not been extensively carried out yet. To make the production of J. curcas profitable and sustainable, genetic improvement of oil yield and quality, as well as diseases and pests resistance is demanded.
Molecular breeding, also called marker-assisted selection (MAS), refers to the procedure of the use of DNA markers which are tightly linked to traits to assist phenotypic selection. In comparison with the traditional breeding, molecular breeding possesses several advantages such as selection at seedling stage, no influence of environment, and selection of preferred homozygotes, thus accelerating the genetic improvement. With the rapid development of next-generation sequencing (NGS) technologies, it is now easy to detect and characterize a large number of DNA markers using NGS and polymerase chain reaction (PCR). Genomic resources such as molecular markers, linkage maps, ESTs and genome sequences, as well as mapped quantitative trait loci (QTL) for important traits, are powerful tools to speed up genetic improvement for oil yield and quality through MAS.
2 Microsatellite- and SNP-Based Linkage Map of Jatropha
A genetic linkage map is an essential tool in molecular breeding for genetic improvement (Guimarães 2007). Such a map will facilitate genome mapping, genetic dissection of QTL and positional cloning of important genes and provide a scaffold for assembling physical maps and an indispensable tool for functional genomics (Harushima et al. 1998; Meksem and Kahl 2006). Linkage maps have been constructed in a number of economically important species (Harushima et al. 1998; Hayashi et al. 2001; Hwang et al. 2009; Li et al. 2009; Ren et al. 2009; Wang et al. 2011a; Xia et al. 2010) using different markers, such as protein polymorphisms, RAPD, RFLP, AFLP, microsatellites (also termed as single sequence repeats: SSRs), and SNPs in reference families. Although a rather large variety of markers exists, each possessing its own advantages and drawbacks, microsatellites are the most preferred markers for linkage mapping (Goldstein and Schlotterer 1999). Recently, single nucleotide polymorphism (SNP) markers have attracted significant attention in creating dense genetic linkage maps and genome-wide association studies (Wang et al. 2005) because SNPs are the most abundant class of polymorphisms in genomes and can be genotyped cost-effectively (Rafalski 2002).
With the elucidation of genes involved in many biochemical pathways, information generated in the model species, Arabidopsis thaliana, holds enormous potential for application in breeding of other crops (Panjabi et al. 2008). Establishment of syntonic relationships between A. thaliana and other crops through comparative mapping would be beneficial for the identification of candidate genes contributing to agronomic traits from corresponding regions in A. thaliana and also serve as a resource to generate more markers for fine mapping in syntonic regions of other crops (Brown 2007).
2.1 Identifying and Genotyping of Microsatellite and SNP Markers
In order to facilitate genetic improvement of jatropha, we constructed a first-generation linkage map comprising 216 microsatellites and 290 SNPs and spanning 1440.9 cM. We generated a comparative map between J. curcas and A. thaliana containing 192 marker loci derived from expressed sequence tags (ESTs). This linkage map represents the first linkage map of Jatropha and could provide an indispensable and powerful tool for QTL analysis, gene mapping, and marker-assisted selection (MAS) in breeding.
A total of 245 microsatellite loci were informative in the mapping panel 296 SNPs were identified which showed polymorphism among the parental lines of the mapping population. To our surprise, in the two J. curcas individuals (i.e., PZM16 and ZS-2), all 245 microsatellites were homozygous, while in the J. integerrima individual (S001), 65% of these microsatellites were heterozygous, and the remaining microsatellites were homozygous, but the genotypes of the J. integerrima individual were different from these of the two J. curcas individuals. Therefore, two F1 hybrid individuals (i.e., CI7041 and CI7018) were all heterozygous at all 245 microsatellite loci. The B1CF1 families were highly informative for constructing a linkage map.
2.2 Linkage Map
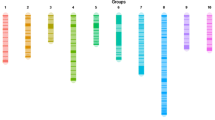
For each of the 245 microsatellite and 296 SNP markers, genotypes were obtained for all 93 offspring. Genotype data of the markers were passed forward into linkage analysis. As a result, 216 microsatellites and 290 SNPs were mapped into 11 linkage groups. The remaining 29 informative microsatellites and 6 informative SNPs were not mapped to the linkage map. The length of the 11 linkage groups ranged from 84.9 to 187.5 cM (Table 2.1; Figs. 2.1 and 2.2). The linkage map covered 1440.9 cM with average marker spacing of 2.8 cM ranging from 1.2 to 4.3 cM. The number of markers on each linkage group ranged from 22 for linkage group 5 (LG 5) to 36 for LG 6 (see Table 2.1; Figs. 2.1, 2.2). LG 11 possessed the highest density of markers with marker spacing shorter than 2 cM, while LGs 1, 4, and 8 had the relatively lower density of markers with marker spacing shorter than 4 cM. The 506 DNA markers were located in 324 discrete positions on 11 linkage groups. Therefore, the average space of discrete positions was 4.4 cM ranging from 2.7 for LG 11 to 5.7 cM for LG 1. The most spaces between two discrete positions of markers were smaller than 20 cM on the linkage map. However, there were still a few spaces where the distances between two discrete positions of markers were larger than 20 cM, such as eSNP0815-Jatr650-Jcuint220 on LG 1, eSNP0081-SNP005-1 on LG 2, eSNP0271-SNP032-1 on LG 3, Jcuint302-eSNP0193 and Jatr600-eSNP0086 on LG 4, and eSNP0114-eSNP0045 and eSNP0195-eSNP0979 on LG 8. Most of the large spaces were located in the end of the linkage groups. Most of the segregation distortion markers were clustered on LGs 1, 6, 9, and 10. It can be deduced that the suggested segregation distortion loci at these marker regions might link to deleterious alleles. If these loci in the regions are removed from the analysis, <5% of the remaining markers show significant distortion, as expected by chance. Similar phenomena were reported in the linkage maps we constructed for Asian sea bass (Wang et al. 2011a).

A genetic linkage map of Jatropha. Estimates of map distances between markers are indicated in Kosambi centimorgans (Wang et al. 2011b)

A genetic linkage map of Jatropha. Estimates of map distances between markers are indicated in Kosambi centimorgans (continued) (Wang et al. 2011b)
To conduct the comparative genome analysis to identify conserved syntenies, BlastX searches were performed for 222 ESTs containing mapped SSR or SNP markers. Highest percentage of the marker sequences of jatropha could be assigned to 215 ESTs (96.8%) in castor bean, followed by poplar 202 (91.0%) whose genomic sequence has been determined. As searches retrieved 192 (86.5%) sequences from Arabidopsis, we compared the Jatropha linkage maps to sequence maps of the Arabidopsis chromosomes which are available. An uneven distribution of Ath loci originating from each Arabidopsis chromosome was observed in the genome of jatropha. Among the 11 LGs of jatropha, all the LGs except 3, 4, 9, 10, and 11 contained Ath loci from each of the five Arabidopsis chromosomes (Ath Chr1–Ath Chr5). LGs 3 and 10 were devoid of loci from AthChr2 and AthChr4, respectively. LGs 4, 9, and 11 did not contain any locus from AthChr3. The conserved blocks, which were defined as regions that contained at least two Ath loci from the same block region, were drawn on Figs. 2.3 and 2.4. As a result, 176 (79.2%) of the markers were corresponded to sequences mapping to loci in the Arabidopsis genome. A total of 38 genomic blocks from Arabidopsis genome were identified in the genome of jatropha with an average of 2.8 paralogous blocks for each jatropha linkage group. Figures 2.3 and 2.4 show the comparative map of the 11 jatropha linkage groups and the 5 Arabidopsis chromosomes. Conserved synteny blocks were identified in all 11 jatropha linkage groups, each of which contained from 2 to 5 Arabidopsis chromosomal blocks. The largest synteny block conserved between jatropha and Arabidopsis was found in LG 6 with 24 markers spanning 128.7 cM in the Jatropha linkage group and their best matches spanning 3 fragments in chromosomes 1, 3, and 4 of the Arabidopsis genome.

A comparative map between Jatropha (Jcu) and Arabidopsis (Ath). Orthologous Jcu and Ath chromosomes are shown with lines connecting orthologous markers (Wang et al. 2011b)

A comparative map between Jatropha (Jcu) and Arabidopsis (Ath). Orthologous Jcu and Ath chromosomes are shown with lines connecting orthologous markers (continued) (Wang et al. 2011b)
3 Mapping QTLs for Oil Traits and EQTLs for Oleosin Genes
Oil containing a high amount of unsaturated fatty acid can find an application as biodiesel feed stock. To make the production of jatropha profitable and sustainable, genetic improvement of oil yield and quality is demanded.
Among the fatty acid present in the jatropha seed oil, linoleic acid (18:2), oleic acid (18:1), palmitic acid (16:0), and stearic acid (18:0) are dominant compositions. Oleic and linoleic acids are the major constituents of jatropha oil (Costa et al. 2010). The breeding goal for jatropha seed oil trait improvement is to increase total oil content and oleic acid and decrease palmitic content (Chhetri et al. 2008).
QTL analysis has been performed to detect the genetic bases of important agronomic or physiological traits, providing valuable information for trait improvement. Genetic markers have made it possible to detect QTLs that are significantly associated with traits and made selection more effective (Wang et al. 2011a). Genetic response can be improved by including the QTLs in marker-assisted selection, which is a method of selection that makes use of phenotypic, genotypic, and pedigree data (Varshney and Tuberosa 2007). This approach treats mRNA expression levels as quantitative traits in a segregating population and maps expression QTL (eQTL) that control expression levels in vivo. For almost any gene analyzed in a segregating population, eQTL analysis can identify the genomic regions influencing its expression level. eQTL that maps to the same genetic location as the gene whose transcript is being measured generally indicates the presence of a cis-acting regulatory polymorphism in the gene (cis-eQTL). eQTL that maps distant to the location of the gene being assayed most likely identifies the location of trans-acting regulators (trans-eQTL) that may control the expression of a number of genes elsewhere in the genome. The genetical genomics approach has been employed for identifying eQTL regulating gene expression (Sladek and Hudson 2006; Yin et al. 2010).
As detailed above, a first-generation genetic linkage map of jatropha has been established, thus providing a necessary tool for a whole-genome scan for QTLs and eQTLs affecting economically important traits including seed oil traits.
The linkage map, covering 663.0 cM of the genome, converged into 11 linkage groups consisting of 95 DNA markers. The average distance between markers was 7.0 cM. Most of the linkage groups were consistent with those described above. QTL analyses were performed on the means of fatty acid composition, total oil content, and expression levels of OleI, OleII, and OleIII (Table 2.2; Fig. 2.5). We detected 18 QTLs and 3 eQTLs for all traits examined. Individual eQTL or QTL were detected with percentage of variation explained (PVE or r 2) 0.1–36.0%, and 5 of them had PVE exceeding 10%. QTLs or eQTLs with positive and negative allelic effects were identified, with a positive effect implying a higher value for the trait conferred by the allele from PZM16 and vice versa (Fig. 2.6).

Whole-genome scan for QTL for oil traits and Oleosin gene expression in Jatropha. a QTL scans of oil traits on linkage maps. Horizontal line indicates 5% LOD significance thresholds (2.5) based on permutation. b QTL scans of OleI, OleII and OleIII expressions on linkage maps. Horizontal line indicates LOD significance threshold (2.0) (Liu et al. 2011)

Summary of QTL (eQTL) locations detected on the genome of jatropha. QTLs (eQTLs) represented by bars are shown on the left of the linkage groups, close to their corresponding markers. The lengths of the bars are proportional to the confidence intervals of the corresponding QTLs (eQTLs) in which the inner line indicates position of maximum LOD score (Liu et al. 2011)
Eighteen QTLs were identified dispersed among all the linkage groups except LGs 3 and 11. A QTL of highly significant effect was determined to be located on LG 1 explaining 36% of variation of C18:1 composition and was found to be associated with C18:2 compositions (Table 2.2; Fig. 2.6). Interestingly, another QTL on LG10 explained 5.9% of variation of C18:1 composition was also associated with C18:2 compositions. Higher values for C18:1 were conferred by the allele from PZM16, while higher values for C18:2 from Hybrid CI7041.
Four QTLs were detected underlying total oil content. At the three QTLs on LGs 1, 2, and 4, respectively, the alleles from hybrid CI7041 contributed high total oil content. The most effective QTL was spotted on LG 4 explaining 11.1% of the variation, whose higher value for total oil content was conferred by the allele from hybrid CI7041.
There were strong QTLs for C18:1 and total oil content detected on LGs 1 and 4, respectively. Mean phenotypic values of each trait were calculated for those progeny with the alternate alleles of the microsatellite markers, inherited from the J. integerrima (aa) or J. curcas (AA). A two-way ANOVA was performed on the progeny using two allelic combinations (AA, Aa) from markers linked to QTLs in order to investigate associations between phenotypic traits and genotypes of the QTLs. The phenotype values of each allelic combination of the QTLs are listed in Fig. 2.7. Significant differences of phenotype means among different allelic combinations were identified, revealing the effects of alternative alleles inherited from the parents.

C18:1 composition (left) and total oil content (right) of plants with different genotypes. Favorite alleles for C18:1 composition are AA from J. curcas, and those for total oil content are Aa from hybrid of J. integerrima and J. curcas (right) (Liu et al. 2011)
Progenies with AA genotype at the marker Jcuint057 located in qC18:1-1 showed the higher C18:1 content (43.0%) than Aa (30.9%). By contrast, progeny with Aa genotype at the marker Jatr872 located in qOilC-4 showed the higher total oil content (38.0%) than AA (33.7%) (Fig. 2.7). These results suggested the effect of the two QTLs are opposite on these two key oil traits and favorite alleles were differentially from J. curcas and J. integerrima.
SNP markers were developed in OleI, OleII and OleIII genes (Table 2.3), which were mapped on LGs 5, 3, and 5 respectively (Fig. 2.6). OleI and OleIII were mapped on LG5 where the QTLs qC18:0-5, qC18:1-5, and qOleIII underlying C18:0, C18:1, and OleIII expression clustered. Negative additive effect value of qOleIII-5 indicated that J. curcas alleles were positive for OleIII expressions, of which LOD score was 3.1. This eQTL of OleIII was localized near OleIII gene and overlapped with the QTLs controlling C18:0 and C18:1, revealing a cis- or trans-element for OleIII which subsequently controlling the C18:0 and C18:1. One eQTL on LG 8 qOleI-8 was detected underlying OleI expression with LOD 1.9 (Table 2.2; Figs. 2.6, 2.7). Additive effect value of qOleI-8 was positive, indicating that J. integerrima alleles were positive for OleI expressions. To find as many putative QTLs (eQTLs) as possible, and to obtain a clearer understanding of the relationships among examined traits, a threshold eQTL of 1.9 for declaring a suggestive eQTL was employed. Low thresholds may not be useful in plant breeding programs, but they have been shown to help in understanding relationships among traits (Thumma et al. 2001). OleII was located on LG 3. One eQTL for OleII was detected on LG 6 with LOD 2.6, which closed to qC18:0-6. It is suggested that a trans-element for OleII could harbor in this region which controlling the C18:0. Additive effect values indicated that J. curcas alleles were negative, indicating that the effect of J. curcas alleles was positive for OleII expressions.
4 An Approach for Jatropha Improvement Using Pleiotropic QTLs
Higher seed yield is one of the objectives of jatropha breeding. However, genetic analysis of the yield traits has not been done in jatropha. QTL mapping was conducted to identify genetic factors controlling growth and seed yield in jatropha, a promising biofuel crop.
As in other crops, almost all the economically important traits in jatropha, such as seed yield, biotic or abiotic stress resistance, are quantitative and determined by multiple genes with minor effects, which are described as QTL. In contrast, jatropha had not yet undergone a careful breeding program with systematic selection and improvement of suitable germplasm.
Growth and seed traits were measured in a QTL mapping population, and the frequency distributions of all traits in the progeny showed a continuous distribution. The distribution of phenotypic values showed bidirectional transgressive segregation (Table 2.4), revealing complex genetic bases of these traits. While seed yield in J. curcas was higher than that in J. integerrima, branch number in J. integerrimais significantly higher than that in J. curcas. The data implied that J. integerrima germplasm could be applied for hybrid breeding to improve agronomic traits, such as branch number in the 4th and 10th months, and the female flower number. Correlation analysis among these traits was performed (Table 2.5), and total seed weight showed a significant correlation with total branch number, female flower number and fruit number, with coefficients 0.364, 0.294, and 0.308, respectively. Therefore, these agronomic traits were suggested to be key factors for seed yields.
QTL analyses were performed on the means of growth traits, branch number, female flower and fruit number, and seed yield (Table 2.6; Fig. 2.8). We have detected 28 QTLs for all traits examined with LOD threshold 2.0–2.5 determined by permutations. Individual QTLs were detected with percentage of variation explained (PVE or R 2) 3–21.16%, and four of them had PVE exceeding 10%. QTLs with positive and negative additive effects were identified, with a positive effect implying a higher value for the trait conferred by the allele from J. curcas, and negative from J. integerrima (Table 2.6).

Summary of QTL locations detected. QTL represented by bars is shown on the left of the linkage groups, close to their corresponding markers. The lengths of the bars are proportional to the confidence intervals of the corresponding QTL in which the inner line indicates position of maximum LOD score. The confidence intervals of QTL are shown in green color, and two QTL clusters are highlighted in red (Sun et al. 2012)
4.1 QTLs for Growth Traits
Sixteen QTLs were identified and dispersed among all the linkage groups except LGs 2 and 8. Four QTLs over-lapping on the lower part of LG 5, namely qH10m-5, qD4m-5, qD10m-5, and qTBN-5, were detected underlying plant height in the 10th month, stem diameter in the 4th and 10th months, and total branch number, respectively (Fig. 2.8). Additive effects of these QTLs were positive, indicating that the alleles from J. curcas increased these trait values.
Conversely, two QTLs, namely qBN4m-6 and qBN10m-6, were detected on the lower part of LG 6 controlling branch number with negative additive values, indicating J. integerrima allele increased branch number.
4.2 QTLs for Seed Traits
On LGs 5 and 7, two QTLs of qTSW-5 and qTSW-7 were detected controlling total seed weight, which is one of the most economically important traits. Interestingly, QTLs underlying yield related traits were clustered at these two QTLs. At qWT-5, four QTLs underlying plant height, stem diameter, branch number, and female flower number were detected. Near qTSW-7, three QTLs of qH4m-7, qTBN-7, and qFruitNo-7 were detected, controlling plant height, total branch number, and fruit number, respectively. It was noteworthy that two QTL clusters were detected on LGs 5 and 7, respectively. Five QTLs were detected on the lower part of LG 5 (Fig. 2.9a), and four QTL clusters were detected on lower part of LG 7 (Fig. 2.9b).

QTL clusters on LGs 5 and 7. QTL scans of growth on linkage maps. Horizontal line indicates 5% LOD significance thresholds (2.0) based on permutation. a LG 5; b LG 7 (Sun et al. 2012)
4.3 Favored Alleles Originated from Two Parents
Two QTL clusters were detected consisting of five and four QTLs, controlling total seed weight, plant height, stem diameter, female flower number, and fruit number. The positive additive effects indicated higher values for the traits conferred by the allele from J. curcas. Meanwhile, five QTLs on LG 6, namely qH4m-6, qBN4m-6, qBN10m-6 qFFN-6, and qFruitNo-6, controlling plant height, branch number (in 4th and 10th months post-seed germination), female flower number, and fruit number, respectively, were detected with negative additive effects indicating higher values conferred by J. integerrima (Table 2.6).
4.4 Major Effects of QTSW-5 and QTSW-7
A two-way analysis of variance (ANOVA) was carried out to assess genetic effects and interactions of the two QTLs of qTSW-5 and qTSW-7 controlling total seed weight. The values of different genotypes are shown in Fig. 2.10. Total seed weight was significantly increased in the presence of these two QTLs. When qTSW-5 presented, total seed weight was improved from 16.66 ± 7.26 to 42.00 ± 5.06 g, and qTSW-7, from 15.97 ± 6.36 to 42.69 ± 6.16 g (Fig. 2.10a).

Total seed weight (g) and related traits of plants with different genotypes of qTSW-5 (AA, Aa) and qTSW-7 (BB, Bb); N denotes sample number of each genotypic classes; Error bars denote standard errors (SEs). a Significant major effects of the two QTLs on seed yield; b the two QTLs with pleiotropic roles in regulating plant growth and seed yield. Significant at P < 0.01 of Bonferroni test (Sun et al. 2012)
Interestingly, we found that the two QTLs for seed yield overlapped with other QTLs for other agronomic traits than seed yield itself. ANOVA showed that the QTL qTSW-5 for seed yield affected significantly plant height, stem diameter, new branch number per branch, and female flower number, while qTSW-7 affected plant height, total branch number, and fruit number (Fig. 2.10b).
4.5 Effect of Pyramiding QTSW-5 and QTSW-7
The interaction between marker effects for qTSW-5 and qTSW-7 was nonsignificant with a relatively low P value (0.14) (Table 2.7), while the marker effects for qTSW-5 and qTSW-7 were nonadditive (Fig. 2.11). This could be caused by the lack of power in the ANOVA due to an unequal distribution of genotypic classes (Fig. 2.10). Despite the nonsignificance of the interaction of the two QTLs, total seed weight was significantly increased in the presence of the two QTLs. Lines carrying both QTLs produced an average 61.93 ± 7.31 g of seeds, nearly three times as much as any other genotype combinations (Fig. 2.11). Therefore, although total seed weight could be improved by introducing the two QTLs, there would be advantages to be gained by pyramiding the two QTLs.

Effects of pyramiding the two QTLs of qTSW-5 (genotypes of AA, Aa) and qTSW-7 (BB, Bb) on seed yield. Error bars denote SEs (Sun et al. 2012)
5 Identification of Candidate Genes JcARF19 and JcIAA9 Associated with Seed Size Traits
Larger seeds provide the germinating seedling a larger supply of nutrients; thus, it increases its competitiveness during seedling establishment and tolerance toward adverse environmental condition (Harper et al. 1970).
The big size of jatropha seeds can be utilized to produce high-quality biodiesel fuel. Interest in using jatropha to help alleviate the energy crisis is increasing and, in fact, jatropha is becoming one of the world’s key crops for biodiesel production (Ye et al. 2012) because of its high content of oil in its seeds, tolerance to drought, and ability to thrive in arid soil. Woody bioenergy plants such as jatropha have competitive economic advantages as many years’ economic returns after one-time planting investment. Since its short life cycle time, jatropha has been regarded as a potential model woody plant. Therefore, it has been widely used to rehabilitate wastelands, improve the environment, and enhance the quality of rural life by providing economic value for marginal farmlands (Wu et al. 2011). Furthermore, the reduction of greenhouse gas emission for generating 1 GJ of energy by using biodiesel can be at least 40 and up to 107% with respect to fossil diesel according to three independent investigations in South America, Europe, and Asia (Almeida et al. 2011; Bailis and Baka 2010; Kumar et al. 2012). It is, however, a nondomesticated plant with poor and unpredictable seed productivity in large-scale plantation, and little is known about seed size- and yield-determining genes (Sanderson 2009).
Phytohormones play pivotal roles in the developmental processes of diverse yield traits. Plant growth and development are sustained by continuous cell division, which is mainly regulated by auxin (Durbak et al. 2012). Interaction between auxin and other hormones, such as cytokinin, and how they determine a specific developmental output are still poorly understood, especially in the reproductive organs that are important in seed yield traits (Ioio et al. 2008; Durbak et al. 2012; Ruan et al. 2012). Auxin response factors (ARFs) are transcription factors that play a crucial role in auxin signaling pathway. AUX/IAA (indole-3-acetic acid) proteins control the transcription of auxin-inducible genes by binding to the paired ARFs (Berleth et al. 2004). ARF2 is a key regulator of seed size in Arabidopsis (Okushima et al. 2005a). In arf2 mutants, the ovule and seed cavity are enlarged compared with the wild type due to extra cell divisions in the integument. The reduction of seed numbers prevents its application in agriculture (Okushima et al. 2005a). In rice, another ARF family gene ARF8 appears to be associated with seed development, indicating that different ARF family members may control seed development in various plants (Yang et al. 2006; Xue et al. 2009). Arabidopsis mutations of ARF2, ARF9, ARF13, and ARF14 had weak effects on auxin-inducible gene expression, whereas mutations in ARF3, ARF4, ARF6, ARF7, ARF10, and ARF19 had strong effects. This suggested that some ARFs play a major role in auxin-inducible gene expression, while others may play an auxiliary or redundant role at the young seedling stage (Lee et al. 2009). On the other hand, ten gain-of-function AUX/IAA mutants were identified, namely IAA1/AXR5, IAA3/SHY2, IAA6/SHY1, IAA7/AXR2, IAA12/BDL, IAA14/SLR, IAA17/AXR3, IAA18, IAA19/MSG2, and IAA28, with each exhibiting reduced auxin response in various aspects of development and growth (Berleth et al. 2004). ARF7 and ARF19 activate the transcription of several lateral organ boundaries-domain (LBD) genes, including LBD16 and LBD29. As a transcriptional regulator, LBD16 may also activate the downstream transcriptional network for lateral root (LR) initiation (Okushima et al. 2007). The lists of auxin-regulated genes of which expression is inhibited in the mutants contain putative downstream targets of ARF7 and ARF19 (Okushima et al. 2005b).
5.1 QTL and Candidate Gene Mapping
We identified seven ARFs, four Aux/IAA, and four downstream genes from a cDNA library of jatropha seed (Gu et al. 2012). We identified the genomic sequence of these genes from databases of public (Sato et al. 2011) and Temasek Life Sciences Laboratory (unpublished data, Dr. Yan Hong, Temasek Life Sciences Laboratory, Singapore). Alignment and phylogenetic analyses were conducted with references to ARF and Aux/IAA proteins from Arabidopsis and related known proteins from other plants. We named the seven ARF genes as ARF1, ARF2, ARF4, ARF5, ARF7, ARF9, and ARF19, the four IAA genes as IAA3, IAA9, IAA14, and IAA19, and four downstream genes as LBD16, LBD18, LBD19, and ARGOS, individually, with the support of alignment and phylogenetic analysis data. SNP markers were further developed for these genes. All these genes were mapped onto the linkage map (Fig. 2.12a). Seed traits were measured in the QTL mapping population. Seed size in J. curcas was much bigger than in J. integerrima. Correlation analysis among these traits was performed; we observed significant correlations among the seed traits, including single seed weight, seed length, seed width, and seed height (Ye et al. 2014). We performed QTL analyses on seed traits (Fig. 2.12) and detected 21 QTLs for all the traits. Most importantly, we detected a major QTL qSL11-a controlling seed length with a high LOD score of 16.69 and PVE 29.6% on LG 11, where harboring ARF19 gene. The peak of the major QTL qSL11-a was located at the position of ARF19, controlling seed length with the highest contribution rate in this study. The result implied that ARF19 is a strong candidate for the major factors affecting seed length (Fig. 2.12a, b).

Candidate gene mapping. a Jatropha ARF and Aux/IAA genes on the linkage map. b qSL11-a with LOD 17.1 on LG11, the QTL with strong effect on seed length, was detected in the region harboring ARF19. Horizontal line indicates 5% LOD significance thresholds (2.5) based on permutation. c LD mapping of nucleotide polymorphisms in qSL11-a and seed length across a diverse set of Jatropha accessions. An SNP within the ARF19 gene shows significant signal (Ye et al. 2014)
5.2 Candidate Gene LD Mapping
We carried out a LD mapping in this QTL region. Fifteen amplicons were genotyped in the vicinity of the ARF19 gene. The ARF19 gene was mapped between two SNP markers, eSNP0536 and KASII [Ketoacylacyl carrier protein (ACP) synthase II gene] (Fig. 2.12b). KASII was found next to the marker Jcuint020. A striking level of linkage disequilibrium was observed among amplicons across the QTL region (Fig. 2.12c), with most of r 2 and D′ values higher than 0.8. Association tests were performed in the 15 informative markers (Fig. 2.12c). The pattern of statistical association in the simple regression model indicated generally high P values in the 15 amplicons, with the highest [−log10(P) = 13.67 and contribution rate R 2 = 56.32] in the ARF19 gene (Ye et al. 2014). After controlling for population structure, a single nucleotide polymorphism (SNP) within the ARF19 gene still showed significant signals (Fig. 2.12c).
5.3 IAA9, Involved in the Auxin Signal Transduction, Defines Seed Length
Expressions of these genes were observed in the mapping population. We found that IAA9 was mapped near a QTL qSL-4 of seed length affecting seed length. To further investigate the effects of ARF19 and IAA9 on seed size, we searched eQTLs for the two genes by investigating their expressions in the QTL mapping population. Expressions of ARF19 and IAA9 genes in leaf were higher in J. integerrima than in J. curcas but lower in J. integerrima seed. The expression levels of genes were correlated with each other in both of leaf and seed. The gene expressions were highly correlated with seed length and plant height. A total of four eQTLs were found to control ARF19 and IAA9 expression (Table 2.8 and Fig. 2.13). We detected an eQTL cluster controlling ARF19 and IAA9 gene expressions on LG 9, which harbors a QTL controlling plant growth and seed size (Sun et al. 2012). The results indicated that ARF19 and IAA9 together with their QTLs/eQTLs significantly affected plant growth and seed size.

Summary of QTL (eQTL) locations detected. QTL (eQTL) represented by bars are shown on the left of the linkage groups, close to their corresponding markers. The lengths of the bars are proportional to the confidence intervals of the corresponding QTLs (eQTLs) in which the inner line indicates position of maximum LOD score. ARF and Aux/IAA genes were highlighted in bold, italic, underline, and green. eQTL regions are dark on LGs (Ye et al. 2014)
5.4 ARF19 and IAA9 Interaction
The SNP was found in the domain IV of ARF19 where a highly conserved amino acid serine (Ser1079) in J. curcas was mutated to glycine in J. integerrima due to a G/A mutation. Average seed length of plants with serine in the ARF19 gene (1.68 ± 0.01 cm) was significantly longer than that with glycine (1.49 ± 0.02 cm) (Fig. 2.14e). Because of the positive seed length/SNP correlation and the critical position of SNP in domain IV, we deduced that the SNP mutation was crucial to seed size of jatropha. We further investigated the interaction of ARF19 and IAA9 on seed length through ANOVA (Fig. 2.14f). Both genes affected seed size significantly (Ye et al. 2014). Seed length in plants with JcIAA9 (1.60 ± 0.01 cm) was significantly longer than that with JiIAA9 (1.56 ± 0.02 cm), suggesting a mild effect of IAA9 on seed length. Further analysis on the difference of seed length among plants with a combination of IAA9 and ARF19 showed that a plant with JcIAA9 and JcARF19 (from J. curcas) was even longer (1.70 ± 0.02 cm), whereas plant with JiIAA9 and JiARF19 (from J. integerrima) was even shorter (1.47 ± 0.03 cm). This evidence suggested that ARF19 and IAA9 interacted with each other, affecting seed length phenotype, consistent with the results in eQTL analysis.

Variants of ARF19 and IAA9 in Jatropha species differing in their seed length. a ARF19 gene structure and one key natural variation between alleles from J. curcas and J. Integerrima. Unfilled box: UTR; filled box: coding exon; lines connecting the boxes: intron. b Occurrence of SNP was identified in ARF19 in Jatropha with longer and shorter seed length. SNP was identified in ARF19 of J. curcas and J. integerrima. Amino acid alignment of ARF19 (c) and IAA9 (d). The red star in c and d showed the mutation of JiARF19 and JcIAA9 on conserved domains. JcJatrophacurcas, JiJatrophaintegerrima, RcRicinuscommunis, VvVitisvinifera, PtPopulustrichocarpa, SlSolanumlycopersicum, NtNicotianatabacum. e Seed lengths of Jatropha plants with the serine or glycine in ARF19. f Seed lengths in plants with JcIAA9 and JiIAA9, and interaction between ARF19 and IAA9 (Ye et al. 2014)
5.5 Gene Structure of ARF19 and IAA9
The intron–exon structure of this genomic region of ARF19 in jatropha was conserved in fabids plants (Fig. 2.14a). Sequencing of the two jatropha full-length ARF19cDNAs (JcARF19 and JiARF19) revealed the occurrence of SNPs in ARF19 domain IV, which was the protein–protein interaction site to mediate auxin signal transduction (Fig. 2.14b). A phylogenic tree was generated and showed that ARF19 genes identified from Jatropha are homologs of Arabidopsis thaliana ARF19 (Ye et al. 2014). The SNP present in the J. curcas and J. integerrima (Fig. 2.14b) and amino acid affected in key domains were indicated (Fig. 2.14c). Meanwhile, one SNP in IAA9 was found in the linker region between domains II and III of IAA9 (Fig. 2.14d). The amino acid involved in the SNP of IAA9 conserved in many plants including J. integerrima as Alane, while encoded Glyine in J. curcas (Fig. 2.14d).
In conclusion, we provided evidences that ARF19 and IAA9 contributed to seed length phenotype. ARF19 and IAA9, involved in auxin pathway signal transduction, are conserved in higher plants. It will be feasible to increase crop yield by integrating favored alleles of ARF19 and IAA9 into elite varieties of seed crops including jatropha.
References
Almeida J, Achten WMJ, Duarte MP, Mendes B, Muys B (2011) Benchmarking the environmental performance of the Jatropha biodieselsystem through a generic life cycle assessment. Environ Sci Technol 45:5447–5453
Bailis RE, Baka JE (2010) Greenhouse gas emissions and land use change from Jatropha curcas-based jet fuel in Brazil. Environ Sci Technol 44:8684–8691
Berleth T, Krogan NT, Scarpella E (2004) Auxin signals—turning genes on and turning cells around. Curr Opin Plant Biol 7(5):553–563
Brown JR (2007) Comparative genomics: basic and applied research. CRC Press, Boca Raton
Carvalho CR, Clarindo WR, Praça MM, Araújo FS, Carels N (2008) Genome size, base composition and karyotype of Jatropha curcas L., an important biofuel plant. Plant Sci 174(6):613–617. doi:10.1016/j.plantsci.2008.03.010
Chhetri AB, Tango MS, Budge SM, Watts KC, Islam MR (2008) Non-edible plant oils as new sources for biodiesel production. Int J Mol Sci 9(2):169–180
Costa GG, Cardoso KC, Del Bem LE, Lima AC, Cunha MA, de Campos-Leite L, Vicentini R, Papes F, Moreira RC, Yunes JA (2010) Transcriptome analysis of the oil-rich seed of the bioenergy crop Jatropha curcas L. BMC Genom 11(1):462
Durbak A, Yao H, McSteen P (2012) Hormone signaling in plant development. Curr Opin Plant Biol 15(1):92–96
Fairless D (2007) Biofuel: the little shrub that could–maybe. Nature 449(7163):652–655
Goldstein DB, Schlotterer C (eds) (1999) Microsatellites: evolution and applications
Gu K, Chiam H, Tian D, Yin Z (2011) Molecular cloning and expression of heteromeric ACCase subunit genes from Jatropha curcas. Plant Sci 180(4):642–649
Gu K, Yi C, Tian D, Sangha JS, Hong Y, Yin Z (2012) Expression of fatty acid and lipid biosynthetic genes in developing endosperm of Jatropha curcas. Biotechnol Biofuels 5(1):47
Guimarães EP (2007) Marker-assisted selection: current status and future perspectives in crops, livestock, forestry and fish. Food and Agriculture Organization, Rome (incomplete)
Harper JL, Lovell P, Moore K (1970) The shapes and sizes of seeds. Annu Rev Ecol Syst 1:327–356
Harushima Y, Yano M, Shomura A, Sato M, Shimano T, Kuboki Y, Yamamoto T, Lin SY, Antonio BA, Parco A (1998) A high-density rice genetic linkage map with 2275 markers using a single F2 population. Genetics 148(1):479–494
Hayashi M, Miyahara A, Sato S, Kato T, Yoshikawa M, Taketa M, Hayashi M, Pedrosa A, Onda R, Imaizumi-Anraku H (2001) Construction of a genetic linkage map of the model legume Lotus japonicus using an intraspecific F2 population. DNA Res 8(6):301–310
Heller J (1996) Physic nut. Jatropha curcas L. promoting the conservation and use of underutilized and neglected crops, 1. IBPGR, Roma (in full)
Hwang T-Y, Sayama T, Takahashi M, Takada Y, Nakamoto Y, Funatsuki H, Hisano H, Sasamoto S, Sato S, Tabata S (2009) High-density integrated linkage map based on SSR markers in soybean. DNA Res:dsp010
Ioio RD, Nakamura K, Moubayidin L, Perilli S, Taniguchi M, Morita MT, Aoyama T, Costantino P, Sabatini S (2008) A genetic framework for the control of cell division and differentiation in the root meristem. Science 322(5906):1380–1384
Jain S, Sharma MP (2010) Biodiesel production from Jatropha curcas oil. Renew Sustain Energy Rev 14(9):3140–3147. doi:10.1016/j.rser.2010.07.047
Kumar S, Singh J, Nanoti SM, Garg MO (2012) A comprehensive life cycle assessment (LCA) of Jatropha biodiesel production in India. Bioresource Technol 110:723–729
Lee DJ, Park JW, Lee HW, Kim J (2009) Genome-wide analysis of the auxin-responsive transcriptome downstream of iaa1 and its expression analysis reveal the diversity and complexity of auxin-regulated gene expression. J Exp Bot 60(13):3935–3957
Li F, Kitashiba H, Inaba K, Nishio T (2009) A Brassica rapa linkage map of EST-based SNP markers for identification of candidate genes controlling flowering time and leaf morphological traits. DNA Res 16(6):311–323
Liu P, Wang CM, Li L, Sun F, Liu P, Yue GH (2011) Mapping QTLs for oil traits and eQTLs for oleosin genes in jatropha. BMC Plant Biol 11:132
Meksem K, Kahl G (eds) (2006) The handbook of plant genome mapping: genetic and physical mapping. Wiley, Oxford
Okushima Y, Fukaki H, Onoda M, Theologis A, Tasaka M (2007) ARF7 and ARF19 regulate lateral root formation via direct activation of LBD/ASL genes in Arabidopsis. Plant Cell Online 19(1):118–130
Okushima Y, Mitina I, Quach HL, Theologis A (2005a) AUXIN RESPONSE FACTOR 2 (ARF2): a pleiotropic developmental regulator. Plant J 43(1):29–46
Okushima Y, Overvoorde PJ, Arima K, Alonso JM, Chan A, Chang C, Ecker JR, Hughes B, Lui A, Nguyen D (2005b) Functional genomic analysis of the AUXIN RESPONSE FACTOR gene family members in Arabidopsis thaliana: unique and overlapping functions of ARF7 and ARF19. Plant Cell Online 17(2):444–463
Openshaw K (2000) A review of Jatropha curcas: an oil plant of unfulfilled promise. Biomass Bioenergy 19(1):1–15
Panjabi P, Jagannath A, Bisht NC, Padmaja KL, Sharma S, Gupta V, Pradhan AK, Pental D (2008) Comparative mapping of Brassica juncea and Arabidopsis thaliana using intron polymorphism (IP) markers: homoeologous relationships, diversification and evolution of the A, B and C Brassica genomes. BMC Genom 9(1):113
Rafalski A (2002) Applications of single nucleotide polymorphisms in crop genetics. Curr Opin Plant Biol 5(2):94–100
Ren Y, Zhang Z, Liu J, Staub JE, Han Y, Cheng Z, Li X, Lu J, Miao H, Kang H (2009) An integrated genetic and cytogenetic map of the cucumber genome. PLoS One 4(6):e5795
Ruan Y-L, Patrick JW, Bouzayen M, Osorio S, Fernie AR (2012) Molecular regulation of seed and fruit set. Trends Plant Sci 17(11):656–665
Sanderson K (2009) Wonder weed plans fail to flourish. Nature 461:328–329
Sato S, Hirakawa H, Isobe S, Fukai E, Watanabe A, Kato M, Kawashima K, Minami C, Muraki A, Nakazaki N, Takahashi C, Nakayama S, Kishida Y, Kohara M, Yamada M, Tsuruoka H, Sasamoto S, Tabata S, Aizu T, Toyoda A, Shin-i T, Minakuchi Y, Kohara Y, Fujiyama A, Tsuchimoto S, Kajiyama S, Makigano E, Ohmido N, Shibagaki N, Cartagena JA, Wada N, Kohinata T, Atefeh A, Yuasa S, Matsunaga S, Fukui K (2011) Sequence analysis of the genome of an oil-bearing tree, Jatropha curcas L. DNA Res 18(1):65–76
Shah S, Sharma A, Gupta MN (2005) Extraction of oil from Jatropha curcas L. seed kernels by combination of ultrasonication and aqueous enzymatic oil extraction. Bioresour Technol 96(1):121–123. doi:10.1016/j.biortech.2004.02.026
Sladek R, Hudson TJ (2006) Elucidating cis-and trans-regulatory variation using genetical genomics. Trends Genet 22(5):245–250
Sun F, Liu P, Ye J, Lo LC, Cao S, Li L, Yue GH, Wang CM (2012) An approach for jatropha improvement using pleiotropic QTLs regulating plant growth and seed yield. Biotechnol Biofuels 5:1–10
Thumma BR, Naidu BP, Chandra A, Cameron DF, Bahnisch LM, Liu C (2001) Identification of causal relationships among traits related to drought resistance in Stylosanthes scabra using QTL analysis. J Exp Bot 52(355):203–214
Varshney RK, Tuberosa R (2007) Genomics-assisted crop improvement, vol 1. Springer, Berlin
Wang CM, Bai ZY, He XP, Lin G, Xia JH, Sun F, Lo LC, Feng F, Zhu ZY, Yue GH (2011a) A high-resolution linkage map for comparative genome analysis and QTL fine mapping in Asian seabass, Lates calcarifer. BMC Genom 12(1):174
Wang CM, Liu P, Yi C, Gu K, Sun F, Li L, Lo LC, Liu X, Feng F, Lin G (2011b) A first generation microsatellite-and SNP-based linkage map of Jatropha. PLoS One 6(8):e23632
Wang WY, Barratt BJ, Clayton DG, Todd JA (2005) Genome-wide association studies: theoretical and practical concerns. Nat Rev Genet 6(2):109–118
Wu J, Liu Y, Tang L, Zhang F, Chen F (2011) A study on structural features in early flower development of Jatropha curcas L. and the classification of its inflorescences. Afr J Agr Res 6:275–284
Xia JH, Liu F, Zhu ZY, Fu J, Feng J, Li J, Yue GH (2010) A consensus linkage map of the grass carp (Ctenopharyngodon idella) based on microsatellites and SNPs. BMC Genom 11(1):135
Xue L-J, Zhang J-J, Xue H-W (2009) Characterization and expression profiles of miRNAs in rice seeds. Nucleic Acids Res 37(3):916–930
Yang JH, Han SJ, Yoon EK, Lee WS (2006) Evidence of an auxin signal pathway, microRNA167-ARF8-GH3, and its response to exogenous auxin in cultured rice cells. Nucleic Acids Res 34(6):1892–1899
Ye J, Qu J, Bui HTN, Chua NH (2009) Rapid analysis of Jatropha curcas gene functions by virus-induced gene silencing. Plant Biotechnol J 7:964–976
Ye J, Hong Y, Qu J, Wang C (2012) Improvement of Jatropha oil bygenetic transformation. Springer Science Publishers, New York
Ye J, Liu P, Zhu CS, Qu J, Wang XH, Sun YW, Sun F, Jiang YL, Yue GH, Wang CM (2014) Identification of candidate genes JcARF19 and JcIAA9 associated with seed size traits in Jatropha. Funct Integr Genom 14(4):66–757
Yin Z, Meng F, Song H, Wang X, Xu X, Yu D (2010) Expression quantitative trait loci analysis of two genes encoding rubisco activase in soybean. Plant Physiol 152(3):1625–1637
Zhang Y, Wang Y, Jiang L, Xu Y, Wang Y, Lu D, Chen F (2007) Aquaporin JcPIP2 is involved in drought responses in Jatropha curcas. Acta Biochim Biophys Sin 39(10):787–794
Acknowledgements
The work is part of the project “Genetic Improvement of Jatropha” initiated and coordinated by Professor Nam-Hai Chua, Rockefeller University, USA. We thank Weijie Tang, State Key Laboratory of Crop Genetics and Germplasm Enhancement, Nanjing Agricultural University, 210095, Nanjing, China, for his editing work on this chapter. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of this chapter.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter

Cite this chapter
Ye, J., Wang, C., Yue, G. (2017). Linkage Mapping and QTL Analysis. In: Tsuchimoto, S. (eds) The Jatropha Genome. Compendium of Plant Genomes. Springer, Cham. https://doi.org/10.1007/978-3-319-49653-5_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-49653-5_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-49651-1
Online ISBN: 978-3-319-49653-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)