
Abstract
The last 35 years of clinical research on aspirin have proven its value as a lifesaving drug for the treatment and prevention of atherothrombosis. The place of low-dose aspirin in the therapeutic armamentarium has not been substantially altered by the successful development of new antiplatelet agents, including P2Y12 and PAR-1 receptor blockers. The demonstration of additive beneficial effects resulting from effective blockade of the platelet ADP and/or thrombin receptor combined with thromboxane suppression in high-risk patients is consistent with the multifactorial nature of atherothrombosis. Besides becoming an essential component of the antithrombotic strategy in high-risk settings, low-dose aspirin has also provided a mechanistic insight into the participation of platelets in other pathophysiologic processes, including colorectal cancer. The aim of this chapter is to review the mechanism of action and clinical pharmacology of aspirin as an antiplatelet agent, as well as the randomized trial evidence supporting its efficacy and safety. Moreover, more recent findings on additional long-term benefits of aspirin therapy will be discussed and new developments put into perspective.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
- Antiplatelet Therapy
- Antiplatelet Effect
- Major Bleeding Complication
- Major Vascular Event
- Antiplatelet Regimen
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Historical Perspective
Although synthesized in an industrial environment in 1897 and marketed in 1899, acetylsalicylic acid (aspirin) remains a cornerstone of antiplatelet therapy for the treatment of acute ischemic syndromes, both coronary and cerebrovascular, as well as for their secondary prevention (Amsterdam et al. 2014; Lansberg et al. 2012; Parekh et al. 2013; Roffi et al. 2016; Vandvik et al. 2012). In contrast, its role in the primary prevention of atherothrombosis has been the subject of debate during the past 10 years and remains highly controversial (Patrono 2013). Historically, aspirin has evolved from a prototypic nonsteroidal anti-inflammatory drug (NSAID) to a highly targeted antiplatelet drug (Born and Patrono 2006), largely as a consequence of fundamental discoveries on its mechanism of action in the early seventies (Vane 1971; Hamberg et al. 1975; Roth et al. 1975) (Fig. 1). These include the discovery by J. Vane (1971) that aspirin inhibits prostaglandin (PG) synthesis, the discovery by M. Hamberg and B. Samuelsson (1975) of thromboxane (TX) A2 as the major arachidonic acid (AA) metabolite in human platelets responsible for the pro-aggregating effect of AA, and the characterization by P. Majerus and his colleagues (1975) of acetylation of platelet PG-synthase as the molecular mechanism responsible for permanent inactivation of its cyclooxygenase (COX) activity. These pivotal studies provided the basis for the development of mechanism-based biomarkers of TXA2 inhibition in man, i.e., serum TXB2 (Patrono et al. 1980) and urinary TX metabolite (TXM) excretion (Roberts et al. 1981). The availability of these biomarkers allowed characterizing the dose and time dependence of the inhibitory effects of aspirin on platelet TXA2 production in healthy subjects (Patrignani et al. 1982; FitzGerald et al. 1983). The consistent demonstration of saturability of the platelet-inhibiting effect of aspirin at low doses given once daily and the characterization of its biochemical selectivity in sparing prostacyclin (PGI2) biosynthesis (Patrignani et al. 1982; FitzGerald et al. 1983) provided the rationale for testing the clinical efficacy and safety of a low-dose aspirin regimen in a variety of high cardiovascular (CV) risk settings (Patrono 1994) including acute myocardial infarction (MI) (ISIS-2 Collaborative Group, 1988). The publication of the International Study of Infarct Survival (ISIS)-2 (1988), demonstrating a remarkable protective effect of low-dose aspirin against vascular mortality at 5 weeks after a suspected acute MI (Fig. 2), represented the turning point for the use of aspirin as an antithrombotic agent and provided perhaps the most convincing evidence for an important role of platelet TXA2 in the pathophysiology of coronary atherothrombosis (Davì and Patrono 2007).

Fundamental discoveries on the mechanism of action of aspirin. The figure illustrates the discovery by J. Vane (1971) that aspirin inhibits prostaglandin (PG) synthesis; the discovery by B. Samuelsson’s Group (1975) of thromboxane (TX) A2 as the major arachidonic acid (AA) metabolite in human platelets responsible for the pro-aggregating effect of AA; and the characterization by P. Majerus’ Group (1975) of acetylation of platelet PG-synthase as the molecular mechanism responsible for permanent inactivation of its cyclooxygenase (COX) activity

Effects of streptokinase, low-dose aspirin, both or neither on 5-week vascular mortality in 17,187 patients with suspected acute myocardial infarction. Reproduced from ISIS-2 Collaborative Group (1988) with permission from The Lancet
Pharmacokinetics
Aspirin is rapidly absorbed in the stomach and upper small intestine, primarily by passive diffusion of non-dissociated acetylsalicylic acid across gastrointestinal (GI) membranes. Drug plasma levels peak at 30–40 min after the ingestion of uncoated aspirin (Pedersen and FitzGerald 1984). In contrast, it can take up to 4 or 5 h for plasma aspirin levels to peak after the administration of enteric-coated formulations (Patrignani et al. 2014). Orally administered aspirin undergoes substantial presystemic hydrolysis to salicylic acid in the gut content, the gut wall, and the liver (Pedersen and FitzGerald 1984). The systemic bioavailability of regular aspirin tablets is approximately 45–50 % over a wide range of doses (20–1300 mg) (Pedersen and FitzGerald 1984). However, lower bioavailability of enteric-coated preparations due to poor absorption from the higher pH environment of the small intestine may result in inadequate platelet inhibition (Cox et al. 2006; Grosser et al. 2013), particularly in obese subjects (Cox et al. 2006). Aspirin first comes into contact with platelets in the gut capillaries during absorption, and, as a consequence, platelets are exposed to higher drug levels than are present in the systemic circulation (Pedersen and FitzGerald 1984). This may explain, at least in part, the relative biochemical selectivity of low-dose aspirin (75–100 mg once daily) in sparing vascular PGI2 production (Pedersen and FitzGerald 1984).
Aspirin has a half-life of 15–20 min in the bloodstream (Pedersen and FitzGerald 1984; Patrignani et al. 2014). Despite the rapid clearance of aspirin from the circulation, its antiplatelet effect lasts for the platelet life-span (8–10 days) owing to the permanent inactivation of platelet PGH-synthase (PGHS)-1, an effect that can be reversed only through the generation of new platelets. Given the short half-life of aspirin in the systemic circulation, the long-lasting duration of its antiplatelet effect is ensured by acetylation of PGHS-1 in the bone marrow megakaryocytes and limited new protein synthesis in anucleate platelets. Thus, there is a complete dissociation between the pharmacokinetics and pharmacodynamics of aspirin, allowing the use of a once-a-day regimen for antiplatelet therapy. However, reduced systemic bioavailability of the drug or faster renewal of the drug target may reduce the duration of its full antiplatelet effect and require a different (e.g., twice daily) dosing regimen (Rocca et al. 2012; Pascale et al. 2012) (see below).
Mechanism of Action
Aspirin inhibits platelet function through a molecularly sophisticated mechanism of action that appears ideally suited to target anucleate cell fragments, i.e., by rapidly inducing an irreversible modification in a critical platelet protein that cannot be repaired during the 24-h dosing interval. The limited systemic bioavailability of the active moiety, acetylsalicylic acid, and its short half-life contribute to restraining the extent and duration of any extra-platelet effects of the drug.
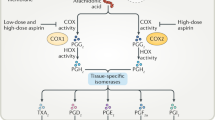
Aspirin exerts its unique antiplatelet effects by selectively acetylating a single serine residue (Ser-529) of PGHS-1 (DeWitt et al. 1990). This causes permanent inactivation of the COX activity of the enzyme, which catalyzes the conversion of AA to PGG2, but does not appreciably affect its peroxidase activity responsible for reducing PGG2 to PGH2. Aspirin inhibits the COX activity of PGHS-1 by placing a larger than normal side chain at position 529 thereby interfering with arachidonate binding to the COX active site (DeWitt et al. 1990). Acetylation of the PGHS-1 and PGHS-2 by aspirin is regulated by the catalytic activity of the peroxidase which yields a higher oxidative state of the enzyme (Bala et al. 2008). In cells with high levels of hydroperoxy-fatty acids, the efficacy of aspirin in acetylating PGHS-2 is markedly decreased as compared to cells with low levels of hydroperoxides (Bala et al. 2008). This finding may explain, at least in part, the differential dose requirements for the anti-inflammatory effects (largely dependent on PGHS-2 inhibition, as with other NSAIDs) as compared to the antiplatelet effects of aspirin (entirely dependent on PGHS-1 inhibition), despite comparable potency in inhibiting the two purified enzymes in vitro (Bala et al. 2008). Presystemic acetylation of platelet PGHS-1 (Pedersen and FitzGerald 1984), as noted above, as well as the cumulative nature of inactivation of platelet COX-1 activity upon repeated daily dosing (Patrignani et al. 1982) may also contribute to the relative biochemical selectivity of low-dose aspirin in vivo (Patrignani et al. 1982; FitzGerald et al. 1983).
PGHS-1 and PGHS-2 are homodimers composed of identical subunits, but it has been shown that only one subunit is active at a time during catalysis; moreover, many NSAIDs bind to a single subunit of a PGHS dimer to inhibit the COX activity of the entire dimer (Shimokawa and Smith 1992). However, 50 % acetylation of platelet PGHS-1 is not sufficient to fully suppress the maximal capacity to synthesize TXA2, and partial acetylation of the second subunit is required in order to suppress platelet COX-1 activity completely (Patrignani et al. 2014; Li et al. 2014).
Permanent inactivation of platelet COX-1 by low-dose aspirin represents the best characterized mechanism of action to explain its clinical efficacy as an antithrombotic agent (Patrono 1994). The finding that the acetylation of platelet PGHS-1 (Patrignani et al. 2014), the inhibition of TXA2 production (Patrignani et al. 1982), and the clinical efficacy of aspirin in reducing the risk of major vascular events in high-risk patients (ATT Collaboration 2002) are saturable at low doses (i.e., even much higher doses are not more effective) allows establishing a cause-effect relationship between this selective acetylation process and its clinical readout (s) (Patrono 2015). Although aspirin can acetylate a number of plasma proteins (e.g., fibrinogen or prothrombin), enzymes, and DNA in vitro (Patrono 2015), this usually requires millimolar concentrations that are approximately 100–1000 times higher than those achievable after oral dosing of low-dose aspirin (Patrignani et al. 2014). Thus, the apparently heterogeneous therapeutic effects of low-dose aspirin may well reflect the multifaceted consequences of platelet COX-1 inhibition on pathophysiological processes (e.g., response to injury and tissue repair) that participate in such diverse diseases as coronary atherothrombosis and colorectal cancer, rather than “pleiotropic” effects on different drug targets (Patrono 2015).
Pharmacodynamics
The pharmacodynamics of aspirin as an antiplatelet agent can be adequately described by serum TXB2 measurements (Patrono et al. 1980, 1985; Patrignani et al. 1982). Platelet TXA2 production during whole blood clotting, as reflected by serum TXB2, represents a highly specific and sensitive index of the maximal biosynthetic capacity of circulating platelets in response to endogenously formed thrombin (Patrono et al. 1980). This biomarker is ideally suited to characterize the pharmacological inhibition of platelet COX-1 activity, when the inhibitor is added in vitro or administered in vivo (Patrono et al. 1980; Patrignani et al. 1982). After oral dosing of aspirin in healthy subjects, serum TXB2 concentrations are reduced in a dose-dependent fashion with a maximal inhibitory effect achieved after a single dose of 100 mg (Patrono et al. 1980; Patrignani et al. 1982). However, because of the cumulative nature of platelet COX-1 inactivation upon repeated daily dosing (Patrignani et al. 1982), the same ceiling effect of virtually 100 % suppression of serum TXB2 can be obtained with daily doses as low as 30 mg (Patrignani et al. 1982). Earlier studies using the ability of ingested aspirin to inhibit subsequent in vitro acetylation of PGHS-1 in washed platelets by [3H-acetyl]-aspirin (Burch et al. 1978) underestimated the potency of aspirin in inhibiting COX-1 activity because full acetylation of the enzyme is not required in order to inhibit completely TXA2 production (Patrignani et al. 2014; Li et al. 2014). Burch et al (1978) reported that a daily dose of 325 mg was required to inhibit by >95 % subsequent acetylation of the platelet enzyme by [3H-acetyl]-aspirin but acknowledged that “the degree to which cyclooxygenase must be inhibited to alter thrombosis is unknown.” Similarly, in the study of FitzGerald et al (1983), urinary TXM excretion was maximally inhibited at doses of 325 mg daily and above, most likely because extra-platelet sources contribute appreciably to TXM excretion and are less sensitive to the inhibitory effect of aspirin.
Upon discontinuing aspirin after a brief course of 325 mg per day, no new enzyme appeared in circulating platelets for approximately 2 days (Burch et al. 1978). The authors interpreted the “lag” in the return of unacetylated enzyme to the circulation as evidence that aspirin acetylates PGHS-1 in the bone marrow megakaryocyte (Burch et al. 1978). In fact, human megakaryocytes express both PGHS-1 and PGHS-2 (Rocca et al. 2002). Similarly, Patrignani et al (1982) observed a 2-day lag in the recovery of serum TXB2 following discontinuation of a 30-day regimen of 30 mg daily. Thereafter, both the unacetylated enzyme (Burch et al. 1978) and serum TXB2 (Patrignani et al. 1982) returned toward pre-aspirin levels with a linear time course consistent with platelet turnover (life-span 8.2±2 days). Therefore, bone marrow megakaryocytes represent an important drug target contributing to the long-lasting antiplatelet effect of aspirin, inasmuch as the new platelets released during 24–48 h after dosing most likely carry PGHS-1 that has been acetylated in their progenitors. However, substantial interindividual variability in the rate of recovery of platelet COX-1 activity has been described in aspirin-treated patients with vascular disease (Rocca et al. 2012). Accelerated platelet turnover, as seen in association with type 2 diabetes mellitus (T2DM) and following coronary artery bypass graft (CABG) surgery, as well as reduced systemic bioavailability of enteric-coated aspirin in obese subjects, may shorten the duration of the full inhibitory effect on platelet TXA2 production to less than 24 h and require more frequent dosing (Rocca et al. 2012; Pascale et al. 2012; Patrono et al. 2013; Paikin et al. 2015) (Fig. 3). Variable duration of the antiplatelet effect of aspirin, noncompliance, or a pharmacodynamic interaction with other NSAIDs (see below) may all be responsible for less than complete inhibition of serum TXB2 and TXA2-dependent platelet function measured 24 h after dosing. However, these phenomena should not be erroneously labeled as aspirin “resistance,” because they all have a plausible explanation and can be easily diagnosed and corrected (Patrono and Rocca 2007; Grosser et al. 2013). If “resistance” is defined in classical pharmacological terms, i.e., as the failure of actual aspirin intake to fully inactivate platelet COX-1 (Patrono and Rocca 2007), then aspirin resistance is either a very rare phenomenon (Santilli et al. 2009; Grosser et al. 2013) or does not exist. A relatively large study of 400 healthy volunteers failed to identify a single case of true drug resistance (Grosser et al. 2013).

Model of altered aspirin pharmacodynamics in type 2 diabetes mellitus. Under conditions of normal megakaryopoiesis, low-dose aspirin acetylates COX isozymes in both circulating platelets and bone marrow megakaryocytes, and only negligible amounts of unacetylated enzymes are resynthesized within the 24-h dosing interval. This pharmacodynamic pattern is associated with virtually complete suppression of platelet TXA2 production in peripheral blood, as reflected by serum TXB2, throughout the dosing interval. Under conditions of abnormal megakaryopoiesis, an accelerated rate of COX-isozyme resynthesis is biologically plausible in bone marrow megakaryocytes, accompanied by faster release of immature platelets with unacetylated enzyme(s) during the aspirin dosing interval, and in particular between 12 and 24 h after dosing. This pharmacodynamic pattern is associated with incomplete suppression of platelet TXA2 production in peripheral blood and time-dependent recovery of TXA2-dependent platelet function during the 24-h dosing interval. Twice daily administration of low-dose aspirin can reverse this abnormal biochemical phenotype. Immunohistochemistry panels depict megakaryocytes stained for COX-1 and COX-2, and peripheral washed platelets stained for COX-1
Because of its unique pharmacokinetic and pharmacodynamic features, aspirin has a lesser inhibitory effect on PGI2 than on TXA2 biosynthesis at all doses, reaching a ceiling effect on the former at 650–1300 mg daily (FitzGerald et al. 1983). Substantial sparing of in vivo PGI2 production, as reflected by PGI2 metabolite (PGIM) excretion, has been reported during administration of aspirin 100 mg daily both in health (Capone et al. 2004) and disease (Cavalca et al. 2014). It is likely that substantial inhibition of PGI2 biosynthesis at higher aspirin doses reflects dose-dependent acetylation of vascular (both endothelial and smooth muscle cell) PGHS-2 (McAdam et al. 1999; Yu et al. 2012). The clinical significance of a PGI2-sparing regimen of aspirin administration remained largely hypothetical for about 25 years after the original proposal of endothelium-derived PGI2 representing an important mediator of vessel-wall thromboresistance (Moncada and Vane 1979). The clinical development of the coxibs as selective inhibitors of COX-2 (FitzGerald and Patrono 2001) and their placebo-controlled, long-term trials have provided convincing evidence that inhibition of vascular PGI2 biosynthesis unaccompanied by adequate inhibition of platelet TXA2 production is associated with a doubling of the risk of major coronary events (Kearney et al. 2006; CNT Collaboration 2013). Meta-analyses of the coxib trials have also provided evidence that some traditional COX-2 inhibitors (e.g., diclofenac and ibuprofen), which only incompletely and transiently inhibit platelet COX-1 activity because of their reversible mechanism of action and short half-life, may increase the risk of major vascular events to a similar extent as the coxibs (Kearney et al. 2006; CNT Collaboration 2013), thereby reinforcing the importance of virtually complete and persistent blockade of platelet COX-1 activity throughout the dosing interval in order to exert cardioprotective effects. In fact, the relationship between inhibition of serum TXB2 and suppression of TXA2-dependent platelet activation in vivo is strikingly nonlinear (Reilly and FitzGerald 1987; Santilli et al. 2009), with >97 % inhibition of the former being required for full suppression of the latter (Santilli et al. 2009) (Fig. 4).

Nonlinear relationship between inhibition of platelet COX-1 activity, as reflected by serum TXB2, and inhibition of TXA2 biosynthesis in vivo, as reflected by urinary 11-dehydro-TXB2 excretion. Reproduced from Santilli et al. (2009) with permission from the American College of Cardiology
Drug–Drug Interactions
Aspirin has been reported to interfere with the antihypertensive effect of blood pressure-lowering drugs, particularly angiotensin-converting enzyme (ACE) inhibitors (Patrono et al. 2008).
This represents a mechanism-based pharmacodynamic interaction that is shared by all NSAIDs and is largely attributed to COX-2 inhibition in the renal cortex and medulla, resulting in increased vascular resistance and sodium retention, respectively (Patrono et al. 2008). This pro-hypertensive effect was not apparent with once daily administration of low-dose (75 mg) aspirin in intensively treated hypertensive patients, in whom this antiplatelet regimen did not impair blood pressure control or the need for antihypertensive therapy as compared to placebo (Hansson et al. 1998).
A subgroup analysis of the PLATO trial, a randomized comparison of ticagrelor vs. clopidogrel in aspirin-treated patients with acute coronary syndromes (ACS) (Wallentin et al. 2009), revealed a potential interaction between aspirin and ticagrelor (Mahaffey et al. 2011). Thus, the higher the daily dose of aspirin (allowed range, 75–325 mg), the lower the benefit of ticagrelor vs. clopidogrel. Although the mechanism of this apparent interaction remains elusive, the FDA has approved ticagrelor for aspirin-treated ACS patients with a warning to maintain the daily aspirin dose at/or lower than 100 mg.
Some NSAIDs, i.e., ibuprofen (Catella-Lawson et al. 2001; Renda et al. 2006) and naproxen (Capone et al. 2005; Li et al. 2014), interfere with the antiplatelet effect of low-dose aspirin by competing for a common docking site (Arginine-120) within the COX-channel of PGHS-1, to which aspirin binds reversibly with low affinity prior to acetylating Ser-529. Analgesic drugs which have been reported not to share this pharmacodynamic interaction with low-dose aspirin include paracetamol (Catella-Lawson et al. 2001), diclofenac (Catella-Lawson et al. 2001), and celecoxib (Renda et al. 2006), i.e., COX-2 inhibitors with some degree of COX-2 selectivity (FitzGerald and Patrono 2001). The use of these NSAIDs in high-risk patients treated with low-dose aspirin should be restricted to the lowest effective dose for the shortest duration necessary for arthritic symptom control, or else aspirin should be replaced with clopidogrel if the patient is not on dual antiplatelet therapy (Patrono and Baigent 2014).
Clinical Efficacy and Safety
The efficacy and safety of aspirin have been evaluated in several populations, ranging from patients presenting with an acute MI or an acute ischemic stroke to apparently healthy persons at low risk of CV events (Patrono et al. 2005). The highly variable benefit/risk profile of the same antiplatelet strategy in different clinical settings reflects the variable incidence of ischemic vs. hemorrhagic complications in the treated population, as well as the relative importance of TXA2-dependent platelet activation in coronary vs. cerebrovascular atherothrombosis and in primary hemostasis of the GI tract vs. the brain (Davì and Patrono 2007).
In patients with acute MI, low-dose aspirin (162.5 mg daily) started within 24 h after the onset of symptoms reduced vascular mortality (primary endpoint) by 23 %, as well as recurrent MI or stroke by 50 % (ISIS-2 Collaborative Group 1988). Treatment of 1000 such patients for about 5 weeks resulted in 40 fewer major vascular events with no statistically significant increase in major bleeding complications. In patients with acute ischemic stroke, aspirin (160–300 mg daily) started within 48 h after the onset of symptoms reduced overall mortality by approximately 10 % and improved functional outcome at 30 days (Lansberg et al. 2012). Treatment of 1000 such patients for about 3 weeks resulted in 9 fewer deaths and 7 more patients with a good functional outcome at the expense of 4 more major (nonfatal) extracranial bleeds (Lansberg et al. 2012). Despite the development of newer antiplatelet agents, low-dose aspirin is still recommended with the highest grade and level of evidence (1A) as initial treatment and long-term therapy of patients with ACS (Roffi et al. 2016) or acute ischemic stroke (Lansberg et al. 2012).
In stable patients with atherothrombotic vascular disease, both individual studies (Patrono et al. 2008) and meta-analyses of randomized trials of antiplatelet therapy (ATT Collaboration 2002) indicate that low-dose aspirin reduces the risk of recurrence of a serious vascular event by approximately 25 %. Indirect comparisons of the results of these high-risk trials provide no evidence of a dose-dependent effect within a wide range of aspirin daily doses (30–1500 mg) (ATT Collaboration 2002). Moreover, a limited number of direct, randomized comparisons of higher vs. lower doses of aspirin in patients with cerebrovascular disease or ACS showed no evidence of superiority of higher vs. lower doses, consistent with saturability of the antiplatelet effect at low doses (Patrono 2015) (Fig. 5).

Acetylation of platelet PGHS-1, inhibition of TXA2 production, and reduction of vascular events by aspirin. The left panel illustrates the saturability of the acetylation process measured in vitro and ex vivo following oral dosing with 100 mg daily in healthy subjects (Patrignani et al. 2014); the middle panel depicts saturability of the inhibitory effect on platelet COX-1 activity, as reflected by serum TXB2 measurements after single oral doses of aspirin in healthy subjects (Patrignani et al. 1982); the right panel illustrates saturability of the clinical effect of aspirin in reducing risk of serious vascular events in high-risk patients (ATT Collaboration 2002)
Among a wide range of high-risk patients, in whom the annual rate of a serious vascular event ranges between 4 and 6 %, low-dose aspirin may prevent approximately 10–15 fatal and nonfatal ischemic events for every 1000 patients treated for 1 year (number needed to treat [NNT]: 67–100) (Patrono et al. 2005). This substantial benefit is obtained at the expense of causing 1–2 major extracranial (mostly GI) bleeding complications per 1000 patients (number needed to harm [NNH]: 500–1000) and 1–2 hemorrhagic strokes per 10,000 patients (NNH: 5000–10,000) (Patrono et al. 2005). Therefore, for most patients with a prior vascular event (at average bleeding risk) taking low-dose aspirin, the number in whom the recurrence of a serious vascular event would be avoided clearly outweighs the number in whom aspirin would cause a major bleeding complication (Patrono et al. 2005; Patrono 2015).
In contrast, among asymptomatic subjects without a prior vascular event, the balance of benefits and risks of long-term therapy with low-dose aspirin is substantially uncertain because the risks without aspirin, and hence the absolute benefits of antiplatelet prophylaxis, are about an order of magnitude lower than in secondary prevention (Patrono 2013). Moreover, in contrast to secondary prevention trials, in which a clear benefit of antiplatelet prophylaxis was demonstrated for both fatal and nonfatal coronary and cerebrovascular events, the benefit of aspirin in primary prevention trials was limited to reducing the risk of nonfatal MI (ATT Collaboration 2009) (Fig. 6). On the basis of a meta-analysis of individual participant data from the six largest primary prevention trials of aspirin in approximately 90,000 subjects at relatively low vascular risk (0.6 % per year), the ATT Collaboration (2009) has shown that, irrespective of age or sex, the absolute reduction in serious vascular events would be only about twice as large as the absolute increase in nonfatal GI bleeding. Moreover, the predicted 5-year absolute effects of allocation to aspirin in subjects at low to high coronary risk (5-year risk from <5 % to >10 %) would yield a relatively constant ratio between the calculated NNH (from 1000 to 100, respectively) and NNT values (from 500 to 50, respectively) (ATT Collaboration 2009). This is not surprising, as the main risk factors for coronary events (with the exception of hypercholesterolemia) were also associated with bleeding complications (ATT Collaboration 2009). The current uncertainty on the benefit/risk profile is reflected by conflicting guidelines on the use of aspirin in primary prevention, as well as by its heterogeneous regulatory status in different countries (Patrono 2015).

Serious vascular events in primary prevention trials and proportional effects of aspirin allocation. Actual numbers for aspirin-allocated trial participants, and adjusted numbers for control-allocated trial participants, are presented, together with the corresponding mean yearly event rate (in parentheses). Rate ratios (RRs) for all trials are indicated by squares and their 99 % CIs by horizontal lines. Subtotals and their 95 % CIs are represented by diamonds. Squares or diamonds to the left of the solid line indicate benefit. MI myocardial infarction, CHD coronary heart disease. (asterisk) Myocardial infarction, stroke, or vascular death. Vascular death is coronary heart disease death, stroke death, or other vascular death (which includes sudden death, death from pulmonary embolism, and death from any hemorrhage but in the primary prevention trials excludes death from an unknown cause). Reproduced from ATT Collaboration (2009) with permission from The Lancet
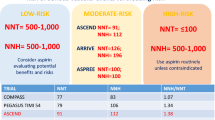
Based on the available results of primary and secondary prevention trials, one can identify three areas of CV risk (Fig. 7): (1) one with an annual risk up to 1.5 %, where evidence from five randomized trials allows a reliable calculation of both NNT and NNH values to guide a personalized approach to antiplatelet prophylaxis; (2) an intermediate zone between 1.5 and 3 % annual CV risk, where evidence from a single trial is insufficient to reliably assess benefit and harm and new trials are needed (see below) to address this knowledge gap; and (3) an area of annual risk >3 %, starting with patients with chronic stable angina, where the evidence from many trials shows that the benefit of aspirin treatment clearly outweighs any potential harm resulting from bleeding complications (Patrono 2015).

The numbers of vascular events avoided and episodes of major bleeding caused per 1000 patients treated with aspirin per year are plotted from the results of individual placebo-controlled trials of aspirin in different patient populations characterized by various degrees of cardiovascular risk, as noted on the abscissa. Number needed to treat (NNT) and number needed to harm (NNH) values are given for subjects in three categories of risk on the basis of the presence or absence of randomized controlled trials. ACCEPT-D Aspirin and Simvastatin Combination for Cardiovascular Events Prevention Trials in Diabetes, ASCEND A Study of Cardiovascular Events in Diabetes, ARRIVE Aspirin to Reduce Risk of Initial Vascular Events, ASPREE ASPirin in Reducing Events in the Elderly, BDT British Doctors Trial, HOT Hypertension Optimal Treatment Study, PHS Physicians’ Health Study, PPP Primary Prevention Project, SAPAT Swedish Angina Pectoris Aspirin Trial, TPT Thrombosis Prevention Trial, WHS Women’s Health Study. Reproduced from Patrono (2015) with permission from the American College of Cardiology
Several lines of evidence support a chemopreventive effect of aspirin against cancer of the GI tract, particularly colorectal cancer (Thun et al. 2012), through a mechanism of action possibly related to platelet inhibition (Patrignani and Patrono 2016) (Fig. 8). It has been argued that even a 10 % reduction by low-dose aspirin in overall cancer incidence over 5 years would yield an absolute benefit comparable to the absolute reduction in serious vascular events; these combined benefits would outweigh the absolute excess of major bleeding complications by three- to fivefold, depending on age and gender (Thun et al. 2012). The US Preventive Services Task Force (Bibbins-Domingo et al 2016) has recently published a recommendation statement addressing the possibility that the same antiplatelet regimen of low-dose aspirin may be recommended for the primary prevention of both CV disease and colorectal cancer in adults ages 50–59 years who have a 10 % or greater 10-year CVD risk, are not at increased risk for bleeding, and have a life expectancy of at least 10 years.

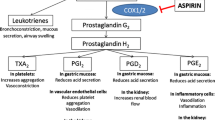
A wide repertoire of lipid and protein mediators, which may contribute to several clinical syndromes possibly responsive to low-dose aspirin therapy, are released upon platelet activation and aggregation. The figure illustrates inhibition of platelet prostanoid production by aspirin and its functional and clinical consequences. The lines of evidence supporting the protective effects of aspirin are summarized below each panel. AA arachidonic acid, ASPREE ASPirin in Reducing Events in the Elderly, RCT randomized controlled trial(s), PGE 2 prostaglandin E2, PGH 2 prostaglandin H2, TXA 2 thromboxane A2. Reproduced from Patrono (2015) with permission from the American College of Cardiology
Low-dose aspirin has been reported to produce additional benefits in the prevention of venous thromboembolism (Patrono 2015) and preeclampsia (Mol et al. 2015) that may well reflect the participation of TXA2-dependent platelet activation in their pathophysiology (Fig. 8).
Ongoing Trials
Several ongoing, placebo-controlled trials may help assess the benefit/risk profile of low-dose aspirin in preventing CV complications and other outcomes (including dementia and cancer) in approximately 50,000 participants at higher CV risk than in the earlier trials because of diabetes mellitus (ASCEND [A Study of Cardiovascular Events in Diabetes], British Heart Foundation 2015; and ACCEPT-D [Aspirin and Simvastatin Combination for Cardiovascular Events Prevention Trials in Diabetes], De Berardis et al. 2007), advanced age (ASPREE [ASPirin in Reducing Events in the Elderly], ASPREE Investigator Group 2013), or a cluster of CV risk factors (not including diabetes) expected to confer a 10-year risk >15 % (ARRIVE [Aspirin to Reduce Risk of Initial Vascular Events], Bayer HealthCare 2015). The inclusion of colorectal cancer as a prespecified secondary endpoint in these trials will allow collecting a large amount of prospective data on the chemopreventive effect of low-dose aspirin during a 5- to 7.5-year follow-up (Patrono 2015). In addition, a number of adjuvant trials in cancer patients (e.g., ADD-Aspirin trial) are currently ongoing or planned in which participants undergone primary treatment with curative intent for an early-stage common solid tumor (e.g., colorectal, gastroesophageal, breast, or prostate) are randomized to receive aspirin (one or two daily doses in the range 100–300 mg) or placebo for 1–8 years and will be assessed for disease-free survival or overall survival (Patrono 2015) (Fig. 9).

Design of the ADD-Aspirin trial, four parallel phase-3 trials in patients with colorectal, breast, gastroesophageal, or prostate cancer. Courtesy of Prof. Ruth Langley, University College London, UK
Gaps in Knowledge and Future Directions for Research
The last 35 years of clinical research on aspirin have proven its value as a lifesaving drug for the treatment and prevention of atherothrombosis (Patrono et al. 2005). The place of low-dose aspirin in the therapeutic armamentarium has not been substantially altered by the successful development of at least ten new antiplatelet agents, including P2Y12 and PAR-1 receptor blockers (Patrono and Rocca 2010). The demonstration of additive beneficial effects resulting from effective blockade of the platelet ADP and/or thrombin receptor on top of TXA2 suppression in high-risk patients is consistent with the multifactorial nature of atherothrombosis and the nonredundant nature of these different pathways of platelet activation (Davì and Patrono 2007). However, the size of the residual CV risk in patients with ACS (about 10 % experiencing a major vascular event at 1 year) despite optimal pharmacological treatment, including low-dose aspirin, effective P2Y12 inhibitors and statins, calls for reappraisal of the pathophysiology of these adverse outcomes and innovative preventive strategies (Roffi et al. 2016). Furthermore, several randomized trials are underway to determine whether aspirin can be dropped from combined antiplatelet regimens for the long-term management of patients who are treated with one of the new antiplatelet drugs (Vranckx et al. 2015).
Besides becoming an essential component of the antithrombotic strategy in high-risk settings, low-dose aspirin has also provided a mechanistic insight into the participation of platelets in other pathophysiologic processes. The hypothesis that platelet activation during the repair process of intestinal mucosal lesions may trigger an early event in colorectal carcinogenesis, i.e., COX-2 induction in adjacent nucleated cells and increased PGE2 production, was formulated on the basis of the lack of a dose effect in the apparent protection against cancer development and death associated with aspirin use in observational studies (Patrono et al. 2001), a finding supported by post hoc analyses of CV prevention trials of aspirin showing saturability of the apparent chemopreventive effect at low doses (Thun et al. 2012). This hypothesis is being tested prospectively in the ongoing primary prevention and adjuvant trials mentioned above (Patrono 2015).
Similarly, the potential role of platelet-derived inflammatory mediators in neurodegeneration and cognitive decline is currently being explored by the ASPREE Investigator Group (2013) in a placebo-controlled aspirin trial of 19,000 elderly subjects with no diabetes or CVD who are being followed for 5 years with death, dementia, or significant disability as the primary endpoint. Measurement of noninvasive biomarkers of platelet activation in substudies of the ongoing trials may allow further characterization of the role of platelet activation and inhibition in atherothrombotic and other important disorders sharing common mechanisms of disease.
Take-Home Messages
-
Low-dose aspirin remains the cornerstone of antiplatelet therapy in the treatment of acute ischemic syndromes and in the secondary prevention of atherothrombosis.
-
The place of aspirin in primary prevention remains controversial because of the uncertain balance of benefits vs risks, requiring a personalized approach to therapy that takes into consideration current knowledge as well as patient’s values and preferences.
-
Additional research is needed to characterize the mechanism of action and optimal dose of aspirin in the chemoprevention of colorectal cancer, a potential additional benefit of long-term antiplatelet therapy.
References
Amsterdam EA, Wenger NK, Brindis RG, ACC/AHA Task Force Members et al (2014) 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 130:e344–e426
Aspirin to Reduce Risk of Initial Vascular Events (ARRIVE) Bayer HealthCare. http://www.arrive-study.com/EN/. Accessed 16 Nov 2015
ASPREE Investigator Group (2013) Study design of ASPirin in Reducing Events in the Elderly (ASPREE): a randomized, controlled trial. Contemp Clin Trials 36:555–564
ATT Collaboration (2002) Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 324:71–86
ATT Collaboration (2009) Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet 373:1849–1860
Bala M, Chin CN, Logan AT et al (2008) Acetylation of prostaglandin H2 synthases by aspirin is inhibited by redox cycling of the peroxidase. Biochem Pharmacol 75:1472–1481
Bibbins-Domingo K on behalf of the U.S. Preventive Services Task Force (2016) Aspirin use for the primary prevention of cardiovascular disease and colorectal cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 164:836–845
Born G, Patrono C (2006) Antiplatelet drugs. Br J Pharmacol 147(Suppl 1):S241–S251
British Heart Foundation. A Study of Cardiovascular Events in Diabetes (ASCEND) Trial Web-site. http://www.ctsu.ox.ac.uk/ascend. Accessed 16 Nov 2015
Burch JW, Stanford N, Majerus PW (1978) Inhibition of platelet prostaglandin synthetase by oral aspirin. J Clin Invest 61:314–319
Capone ML, Tacconelli S, Sciulli MG et al (2004) Clinical pharmacology of platelet, monocyte, and vascular cyclooxygenase inhibition by naproxen and low-dose aspirin in healthy subjects. Circulation 109:1468–1471
Capone ML, Sciulli MG, Tacconelli S et al (2005) Pharmacodynamic interaction of naproxen with low-dose aspirin in healthy subjects. J Am Coll Cardiol 45:1295–1301
Catella-Lawson F, Reilly MP, Kapoor SC et al (2001) Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med 345:1809–1817
Cavalca V, Rocca B, Squellerio I et al (2014) In vivo prostacyclin biosynthesis and effects of different aspirin regimens in patients with essential thrombocythaemia. Thromb Haemost 112:118–127
Cox D, Maree AO, Dooley M et al (2006) Effect of enteric coating on antiplatelet activity of low-dose aspirin in healthy volunteers. Stroke 37:2153–2158
Coxib and traditional NSAID Trialists’ (CNT) Collaboration (2013) Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet 382:769–779
Davì G, Patrono C (2007) Platelet activation and atherothrombosis. N Engl J Med 357:2482–2494
De Berardis G, Sacco M, Evangelista V, ACCEPT-D Study Group et al (2007) Aspirin and Simvastatin Combination for Cardiovascular Events Prevention Trials in Diabetes (ACCEPT-D): design of a randomized study of the efficacy of low-dose aspirin in the prevention of cardiovascular events in subjects with diabetes mellitus treated with statins. Trials 8:21
DeWitt DL, El-Harith EA, Kraemer SA et al (1990) The aspirin and heme-binding sites of ovine and murine prostaglandin endoperoxide synthases. J Biol Chem 265:5192–5198
FitzGerald GA, Patrono C (2001) The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med 345:433–442
FitzGerald GA, Oates JA, Hawiger J et al (1983) Endogenous biosynthesis of prostacyclin and thromboxane and platelet function during chronic administration of aspirin in man. J Clin Invest 71:676–788
Grosser T, Fries S, Lawson JA et al (2013) Drug resistance and pseudoresistance: an unintended consequence of enteric coating aspirin. Circulation 127:377–385
Hamberg M, Svensson J, Samuelsson B (1975) Thromboxanes: a new group of biologically active compounds derived from prostaglandin endoperoxides. Proc Natl Acad Sci U S A 72:2994–2998
Hansson L, Zanchetti A, Carruthers SG et al (1998) Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. HOT Study Group. Lancet 351:1755–1762
ISIS-2 (Second International Study of Infarct Survival) Collaborative Group (1988) Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 2:349–360
Kearney PM, Baigent C, Godwin J et al (2006) Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ 332:1302–1308
Lansberg MG, O’Donnell MJ, Khatri P et al (2012) Antithrombotic and thrombolytic therapy for ischemic stroke: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 141(Suppl):e601S–e636S
Li X, Fries S, Li R et al (2014) Differential impairment of aspirin-dependent platelet cyclooxygenase acetylation by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci U S A 111:16830–16835
Mahaffey KW, Wojdyla DM, Carroll K et al (2011) PLATO Investigators. Ticagrelor compared with clopidogrel by geographic region in the Platelet Inhibition and Patient Outcomes (PLATO) trial. Circulation 124:544–554
McAdam BF, Catella-Lawson F, Mardini IA et al (1999) Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A 96:272–277
Mol BW, Roberts CT, Thangaratinam S et al (2015) Pre-eclampsia. Lancet. doi:10.1016/S0140-6736(15)00070-7
Moncada S, Vane JR (1979) Arachidonic acid metabolites and the interactions between platelets and blood-vessel walls. N Engl J Med 300:1142–1147
Paikin JS, Hirsh J, Ginsberg JS et al (2015) Multiple daily doses of acetyl-salicylic acid (ASA) overcome reduced platelet response to once-daily ASA after coronary artery bypass graft surgery: a pilot randomized controlled trial. J Thromb Haemost 13:448–456
Parekh AK, Galloway JM, Hong Y et al (2013) Aspirin in the secondary prevention of cardiovascular disease. N Engl J Med 368:204–205
Pascale S, Petrucci G, Dragani A et al (2012) Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewal of the drug target. Blood 119:3595–3603
Patrignani P, Patrono C (2016) Aspirin and cancer. J Am Coll Cardiol 68:967-976
Patrignani P, Filabozzi P, Patrono C (1982) Selective cumulative inhibition of platelet thromboxane production by low-dose aspirin in healthy subjects. J Clin Invest 69:1366–1372
Patrignani P, Tacconelli S, Piazuelo E et al (2014) Reappraisal of the clinical pharmacology of low-dose aspirin by comparing novel direct and traditional indirect biomarkers of drug action. J Thromb Haemost 12:1320–1330
Patrono C (1994) Aspirin as an antiplatelet drug. N Engl J Med 330:1287–1294
Patrono C (2013) Low-dose aspirin in primary prevention: cardioprotection, chemoprevention, both, or neither? Eur Heart J 34:3403–3411
Patrono C (2015) The multifaceted clinical readouts of platelet inhibition by low-dose aspirin. J Am Coll Cardiol 66:74–85
Patrono C, Baigent C (2014) Nonsteroidal anti-inflammatory drugs and the heart. Circulation 129:907–916
Patrono C, García Rodríguez LA, Landolfi R, Baigent C (2005) Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med 353:2373–2383
Patrono C, Rocca B (2007) Drug insight: aspirin resistance—fact or fashion? Nat Clin Pract Cardiovasc Med 4:42–50
Patrono C, Rocca B (2010) The future of antiplatelet therapy in cardiovascular disease. Annu Rev Med 61:49–61
Patrono C, Ciabattoni G, Pinca E et al (1980) Low dose aspirin and inhibition of thromboxane B2 production in healthy subjects. Thromb Res 17:317–327
Patrono C, Ciabattoni G, Patrignani P et al (1985) Clinical pharmacology of platelet cyclooxygenase inhibition. Circulation 72:1177–1184
Patrono C, Patrignani P, García Rodríguez LA (2001) Cyclooxygenase-selective inhibition of prostanoid formation: transducing biochemical selectivity into clinical read-outs. J Clin Invest 108:7–13
Patrono C, Baigent C, Hirsh J et al (2008) Antiplatelet drugs: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th edition). Chest 133:199S–233S
Patrono C, Rocca B, De Stefano V (2013) Platelet activation and inhibition in polycythemia vera and essential thrombocythemia. Blood 121:1701–1711
Pedersen AK, FitzGerald GA (1984) Dose-related kinetics of aspirin. Presystemic acetylation of platelet cyclooxygenase. N Engl J Med 311:1206–1211
Reilly IA, FitzGerald GA (1987) Inhibition of thromboxane formation in vivo and ex vivo: implications for therapy with platelet inhibitory drugs. Blood 69:180–186
Renda G, Tacconelli S, Capone ML et al (2006) Celecoxib, ibuprofen, and the antiplatelet effect of aspirin in patients with osteoarthritis and ischemic heart disease. Clin Pharmacol Ther 80:264–274
Roberts LJ 2nd, Sweetman BJ, Oates JA (1981) Metabolism of thromboxane B2 in man. Identification of twenty urinary metabolites. J Biol Chem 256:8384–8393
Rocca B, Secchiero P, Ciabattoni G et al (2002) Cyclooxygenase-2 expression is induced during human megakaryopoiesis and characterizes newly formed platelets. Proc Natl Acad Sci U S A 99:7634–7639
Rocca B, Santilli F, Pitocco D et al (2012) The recovery of platelet cyclooxygenase activity explains interindividual variability in responsiveness to low-dose aspirin in patients with and without diabetes. J Thromb Haemost 10:1220–1230
Roffi M, Patrono C, Collet JP et al (2016) 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: Task Force for the Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST-Segment Elevation of the European Society of Cardiology (ESC). Eur Heart J 37:267–315
Roth GJ, Stanford N, Majerus PW (1975) Acetylation of prostaglandin synthase by aspirin. Proc Natl Acad Sci U S A 72:3073–3076
Santilli F, Rocca B, De Cristofaro R et al (2009) Platelet cyclooxygenase inhibition by low-dose aspirin is not reflected consistently by platelet function assays: implications for aspirin “resistance”. J Am Coll Cardiol 53:667–677
Shimokawa T, Smith WL (1992) Prostaglandin endoperoxide synthase. The aspirin acetylation region. J Biol Chem 267:12387–12392
Thun MJ, Jacobs EJ, Patrono C (2012) The role of aspirin in cancer prevention. Nat Rev Clin Oncol 9:259–267
Vandvik PO, Lincoff AM, Gore JM et al (2012) Primary and secondary prevention of cardiovascular disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 141(Suppl):e637S–e668S
Vane JR (1971) Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol 231:232–235
Vranckx P, Valgimigli M, Windecker S et al (2015) Long-term ticagrelor monotherapy versus standard dual antiplatelet therapy followed by aspirin monotherapy in patients undergoing biolimus-eluting stent implantation: rationale and design of the GLOBAL LEADERS trial. EuroIntervention 11(7). pii: 20150318-06. doi:10.4244/EIJY15M11_07. [Epub ahead of print]
Wallentin L, Becker RC, Budaj A, Investigators PLATO et al (2009) Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 361:1045–1057
Yu Y, Ricciotti E, Scalia R et al (2012) Vascular COX-2 modulates blood pressure and thrombosis in mice. Sci Transl Med 4:132ra54
Acknowledgments
Dr. Patrono’s studies were supported by grants from the European Commission (FP6 EICOSANOX Consortium and FP7 SUMMIT Consortium) and from the Italian Ministry of University and Research (PRIN Project 2013). The expert editorial assistance of Ms. Daniela Basilico and Ms. Patrizia Barbi is gratefully acknowledged. Dr. Patrono has received consulting fees and an institutional grant from Bayer AG for investigator-initiated research on platelet activation in diabetes mellitus and is a member of the scientific advisory board of the Aspirin Foundation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Patrono, C. (2017). Aspirin. In: Gresele, P., Kleiman, N., Lopez, J., Page, C. (eds) Platelets in Thrombotic and Non-Thrombotic Disorders. Springer, Cham. https://doi.org/10.1007/978-3-319-47462-5_83
Download citation
DOI: https://doi.org/10.1007/978-3-319-47462-5_83
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-47460-1
Online ISBN: 978-3-319-47462-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)