
Abstract
HIV-1 protease inhibitors (PIs) are competitive active-site inhibitors that mimic the transition state of the enzyme’s substrate, and are the most potent antiretroviral drugs against HIV infection. HIV-1 protease processes the viral polyproteins at specific cleavage sites and allows infectious mature virions and hence spread of the virus. Unfortunately rapid viral evolution combined with selective pressure of therapy causes selection of many drug-resistant variants that are no longer efficiently inhibited by the PIs. HIV-1 protease can tolerate extensive mutations, with close to half of the 99-residues making up each of the chains in the homodimeric protease and residues at substrate cleavage sites mutating to escape PI pressure. Structural and biophysical studies of many drug-resistant HIV-1 protease variants revealed insights into how mutations at and outside of the protease active site are able to confer PI resistance while still allowing recognition and processing of substrates, and why substrate mutations coevolve with primary protease mutations. We summarize the main molecular mechanisms underlying PI resistance due to primary, secondary, and substrate coevolved mutations and how this knowledge may guide the design of robust inhibitors to avoid resistance.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
- Direct-acting antiviral (DAA) drugs
- gag gene
- pol gene
- Active site mutation
- Protease substrate envelope
- Hydrophobic sliding
- Network hypothesis
- Coevolution
1 Introduction
HIV-1 protease inhibitors (PIs) are competitive active-site inhibitors that mimic the transition state of the enzyme’s substrate and are the most potent antiretroviral drugs against HIV infection. HIV-1 protease processes the viral polyproteins at specific cleavage sites and allows infectious mature virions and hence spread of the virus. Unfortunately rapid viral evolution combined with selective pressure of therapy causes selection of many drug-resistant variants that are no longer efficiently inhibited by the PIs. HIV-1 protease can tolerate extensive mutations, with close to half of the 99-residues making up each of the chains in the homodimeric protease and residues at substrate cleavage sites mutating to escape PI pressure. Structural and biophysical studies of many drug-resistant HIV-1 protease variants revealed insights into how mutations at and outside of the protease active site are able to confer PI resistance while still allowing recognition and processing of substrates, and why substrate mutations coevolve with primary protease mutations. We summarize the main molecular mechanisms underlying PI resistance due to primary, secondary, and substrate coevolved mutations and how this knowledge may guide the design of robust inhibitors to avoid resistance.
2 HIV-1 Protease as a Drug Target
In the fourth decade after the first reporting of what became the worldwide AIDS epidemic, a cure for HIV-1 still eludes the medical community. According to the recent reports published by UNAIDS, there are ~35 million people living with HIV/AIDS around the globe [1]. Although no permanent cure or vaccine for AIDS exists, there are over 30 direct-acting antiviral (DAA) drugs that belong to seven classes targeting various stages in the life cycle of HIV [2], including protease inhibitors (PIs). With the introduction of DAA combinations as highly active antiretroviral therapy (HAART), overall, the quality and life expectancy of HIV-infected patients have greatly improved [3–5]. However, low drug adherence, toxicity, and high pill burden with some second-line therapies, coupled with the error-prone mechanism of HIV reverse transcriptase, have led to the emergence of drug resistance in HIV-infected patients under therapy.
In the last 25 years, drug discovery efforts aided by structure-based design have led to the development of nine FDA-approved PIs (Fig. 35.1): saquinavir (SQV) [6], indinavir (IDV) [7], ritonavir (RTV) [8], nelfinavir (NFV) [9], amprenavir (APV) [10], lopinavir (LPV) [11], atazanavir (ATV) [12], tipranavir (TPV) [13], and darunavir (DRV) [14–16]. All PIs are competitive inhibitors that bind at the protease active site (Fig. 35.2). The active site of this homodimeric aspartyl protease is formed at the interface of two identical 99-residue monomers and contains the catalytic aspartic acid at residue 25 in both subunits [17, 18]. In unliganded state, the protease is symmetric with highly flexible flaps that open up to allow access to the active site, but close to cover and interact with the bound ligand (substrate or inhibitor). When bound, PIs interact mainly with the hydrophobic S2–S2′ pockets at the active site. The peptidomimetic (except tipranavir) inhibitors were designed to mimic the transition state intermediate of peptide substrate by forming critical interactions with the catalytic Asp25, and contain non-cleavable peptide isosteres as core scaffolds. PIs are the most potent anti-HIV drugs with IC50 values in the low picomolar range. The cooperative dose–response curves with high slopes allow for extraordinarily high level of inhibition at clinical concentrations, which are well above the IC50 [19, 20].

Chemical structures of FDA-approved HIV-1 protease inhibitors. The non-cleavable dipeptide isostere cores mimicking the transition state are hydroxyethylamine (blue), hydroxyaminopentane (red), and hydroxyethylene (magenta)

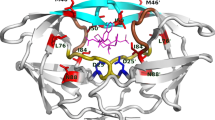
Structure of HIV-1 protease bound to inhibitor DRV (PDB 1T3R). (a) The enzyme is a homodimer of two non-covalently assembled 99-residue chains (in dark and light gray). Each monomer contributes a catalytic Asp (teal side chain) to the active site where the inhibitor (magenta) binds. The flaps close over the bound ligand. (b) Residues that mutate to confer resistance to protease inhibitors are depicted by colored side chains. Location of primary resistance mutations at the active site (D30, V32, I47, G48, I50, V82, I84; red), primary resistance mutations outside the active site (M46, F53, I54 in flaps and L24, L33, L76, N88, L90; orange), and secondary resistance mutations (L10, V11, K20, E35, K43, Q58, V71, G73, T74, N83, L89; blue)
3 HIV-1 Protease in the Viral Life Cycle
HIV infects and replicates in CD4+ immune cells by reverse-transcribing its single-stranded RNA genome. The viral genome includes gag and pol genes encoding polyprotein precursors Gag and Gag/Pol that need to be processed by HIV protease into individual viral proteins (Fig. 35.3a). Proteolytic cleavage of Gag yields the structural proteins matrix (MA), capsid (CA), nucleocapsid (NC), and p6. Gag/Pol is transcribed as a result of ribosomal frameshifting occurring ~10 % of the time near the end of the gag gene [21], and in addition to the Gag structural proteins includes viral enzymes protease (PR), reverse transcriptase (RT), RNase H (RH), and integrase (IN). The newly assembled budding HIV particles are released from the host cell as noninfectious immature virions that contain unprocessed Gag. Processing of Gag by the viral protease induces a major structural rearrangement and triggers the maturation of infectious virus. In total, HIV-1 protease recognizes and cleaves five sites in Gag including those between the viral proteins and spacer peptides p1 and p2 (Fig. 35.3b). The specific, sequential, and ordered processing of Gag by protease is essential for viral maturation and infectivity [22–25]. In addition to viral polyprotein precursors, HIV-1 protease cleaves host cell proteins, including translation initiation factors eIF4 and eIF3d, to inhibit host translation [26, 27].

HIV-1 protease substrates and the substrate envelope. (a) Processing of Gag to individual viral proteins at five specific sites allows viral maturation. (b) The amino acid sequences of cleavage sites within Gag and Pol polyproteins. Notice the lack of any conserved substrate recognition motif at the sequence level. (c) The overlay of cleavage site sequences in protease-bound crystal structures reveals the substrate envelope (blue volume). The inhibitors (below, red volume) protrude out the substrate envelope to contact protease residues (labeled) that mutate to confer resistance. Panel (c) reprinted from King et al. [35], Copyright (2004), with permission from Elsevier
4 PI Resistance Mutations In and Outside the Protease
The high replication rate of HIV coupled with the error-prone viral reverse transcriptase enables a highly heterogeneous viral population with different mutations. This preexisting diverse pool includes mutations that are expanded to confer resistance under the selective pressure of inhibitor therapy. Combinations of three or more DAAs have high enough selective pressure to minimize the emergence of resistance; however resistance has been observed for each of the HIV DAAs, including the PIs. Highly mutated viral variants can be selected under low plasma concentrations such as due to low patient adherence, or transmitted to newly infected individuals to cause therapy failure.
In viral sequences from patient isolates, up to 60–63 % of the HIV-1 protease sequences can vary, indicating very high tolerance to amino acid substitutions [28, 29] (Fig. 35.2b). Of the 99 positions in each monomer, only 37 are invariant (with mutation frequencies <0.5 %) and 17 are sites of nontreatment-related polymorphisms [28, 29]. The remaining 45 positions have been implicated in drug resistance. Mutations at 26 of these 45 positions can significantly decrease susceptibility to one or more PIs [29, 30], 16 of which are located outside the active site region and the flaps. In most cases, multiple mutations within and outside the protease active site coevolve to confer resistance to a particular inhibitor. Mutations that directly confer resistance—mostly located at the protease active site—are classified as primary mutations, while other mutations selected in the presence of primary mutations but that do not confer resistance by themselves are called secondary mutations. The most common primary resistance mutations include D30N, G48V, I50V/L, V82A/V/T, I84V (within the active site), and L90M (no direct contact with the inhibitor). The resistance pathway and accumulation of mutations depend on the HIV clade (and/or preexisting variants), and inhibitor(s) administered and therefore selected against. The first-generation PIs RTV, SQV, IDV, and NFV lose significant potency against drug-resistant variants and are susceptible to single “signature” active-site mutations. The latest and most potent PI, DRV, is active against most of the multidrug-resistant variants and typically up to 20 mutations need to coexist to confer high levels of DRV resistance.
HIV-1 PI resistance is also associated with coevolution of mutations in the viral genome outside the protease, particularly within the Gag cleavage sites NC/p1 and p1/p6 (reviewed in [31]). While avoiding inhibitor binding, the mutated protease needs to maintain its biological function of substrate recognition and cleavage. The coevolution of cleavage sites may compensate for lost efficiency due to primary protease resistance mutations. Several Gag substrate mutations have also been classified as primary resistance mutations as they confer PI resistance in the absence of any protease mutations [32–34].
5 Molecular Mechanisms of Resistance
Drug resistance in HIV-1 protease has been extensively studied at the molecular level, particularly by biophysical and structural analysis of various protease mutants, yielding a plethora of information on structural, enzymatic, and dynamic changes associated with inhibitor resistance [34, 36–49]. These data enabled formulating hypotheses on molecular mechanisms of resistance, which led to strategies for designing inhibitors that avoid resistance, and may be applicable to other disease targets where resistance quickly emerges.
5.1 Active-Site Mutations and the Substrate Envelope
The active site of HIV-1 protease is mainly formed by residues 25–32 (including the catalytic Asp25), 47–53, and 80–84 from both monomers. Active-site mutations at residues that directly contact the inhibitor are quickly selected under PI monotherapy (red in Fig. 35.2b). Although chemically different, the three-dimensional shape and electrostatic character of the HIV-1 PIs are fairly similar; therefore a small set of mutations can result in a protease variant with multidrug resistance. Nevertheless, in most cases, specific signature active-site mutations confer resistance to a given inhibitor. Why a specific mutation is selected against an inhibitor, and how the protease is able to maintain its biological function despite an active-site mutation, is effectively explained by the protease substrate envelope.
The cleavage site sequences are highly heterogeneous, and amino acid sequence alone cannot explain how protease is able to recognize its substrates with high specificity. High-resolution crystal structures of HIV-1 protease bound to peptides corresponding to these cleavage sites revealed that the substrates adopt a specific, conserved three-dimensional shape when bound at the active site (Fig. 35.3c) [50, 51]. This overlapping volume occupied by bound protease substrates and spanning P4′–P4 sites defines the substrate envelope. The P1–P3 region of the substrates forms a toroid, likely critical in specific recognition of asymmetrical ligands by the homodimeric protease. In addition to describing the structural substrate recognition motif of the protease, the substrate envelope serves as a template for contrasting the binding of inhibitors to that of the natural substrates in resistance development, and comparing substrates among themselves in relation to substrate coevolution.
Similar to the substrates, the chemically diverse HIV-1 PIs share a conserved inhibitor envelope in protease-bound structures [35, 51, 52] spanning P2′–P2 sites. Superposition of the two envelopes reveals locations where inhibitors protrude out the substrate envelope and contact protease active-site residues. Such protrusions render an inhibitor vulnerable to mutations, as protease contacts at these locations are more important for inhibitor binding compared to substrates. An amino acid substitution could differentially weaken inhibitor contacts without substantially affecting substrate binding. Accordingly, protease residues that contact inhibitors beyond the substrate envelope correspond to locations of major active-site resistance mutations.
Several primary mutations are signature for resistance to a particular inhibitor, such as D30N to NFV, I50V/L to APV/DRV/ATV, G48V to SQV/ATV, and V82A to SQV/RTV. These signature mutations also primarily correspond to locations where individual inhibitors protrude out the substrate envelope. As the protease active site is mostly hydrophobic, side-chain substitutions due to primary mutations mainly affect van der Waals contacts with the ligand. However, analysis of protease–inhibitor complex structures with both wild-type and resistant variants has revealed that structural changes are often more complex than a simple loss of van der Waals contact at the site of mutation [39, 41, 50]. Rather, drug resistance mutations often cause an overall rearrangement of contacts around the inhibitor at the active site.
The substrate envelope broadly defines the evolutionary constraints on the selection of active-site mutations to confer drug resistance from a structural viewpoint. Mutations that abrogate essential contacts with the substrates would be detrimental to biological function, and thus are selected against. Instead, mutations are selected to weaken inhibitor contacts while still maintaining functionally essential substrate interactions. Such mutations tip the competition between inhibitor binding versus substrate recognition/processing in favor of the substrates, thus conferring drug resistance.
In addition to physical contacts with the inhibitor, drug resistance mutations can also alter the conformational dynamics of HIV-1 protease. The protease is a highly flexible enzyme that undergoes major conformational changes involving the flaps and the hydrophobic core during ligand binding and release [53–56]. This concerted change requires extensive side-chain repacking at the hydrophobic core, or hydrophobic sliding, as revealed in molecular dynamics (MD) simulations [56]. Reversible cross-linking of core hydrophobic residues carefully chosen based on the MD results elegantly demonstrated that the core dynamics directly modulates the enzyme’s activity [57]. Considering drug resistance in the context of the balance between inhibitor binding and substrate processing, any dynamic change that disfavors the inhibitor over the substrates would contribute to conferring resistance. As the inhibitor needs to stay bound at the active site for efficient inhibition with the flaps closed, while the substrates need to get processed and released for efficient turnover, flap dynamics would differentially affect the two processes. Such changes in flap dynamics have been revealed in MD simulations as well as experimental NMR and EPR dynamics of HIV-1 protease drug-resistant variants [36, 58–60]. This resistance mechanism through changes in the protease conformational dynamics may be common to mutations both at and outside the active site.
5.2 Resistance Mutations Outside the Protease Active Site
In addition to the major mutations at the protease active site, many mutations elsewhere in the protease are selected in resistance to protease inhibitors. Some of these mutations are major resistance mutations, even though they are located outside the active site and do not physically contact the ligand (orange in Fig. 35.2b). Yet others have been classified as secondary or minor as they do not confer significant levels of resistance when present alone (blue in Fig. 35.2b), but may assist in recovering the enzyme fitness or stability lost due to primary mutations.
While the substrate envelope provides an efficient framework to rationalize the selection of active-site mutations, understanding the molecular mechanisms underlying resistance due to changes in a side chain not in physical contact with the inhibitor is more challenging. Recent studies suggest that protease conformational dynamics and changes therein may play a major role in propagating the effect of such mutations to the active site. HIV-1 protease variants with single- or double-secondary resistance mutations bound to DRV were characterized by crystal structures and MD simulations, and displayed changes both in dynamics and subtle but significant rearrangements in the structure around the active site [45]. Interestingly, secondary mutations located at different positions in the protease structure had a common mechanism of propagating their effects to the active site and altering mainly the interactions of residue 47 with DRV. The network hypothesis was proposed to explain how distal mutations are able to affect the interactions at the active site through common mechanisms (Fig. 35.4): Residues that undergo secondary resistance mutations and active-site residues affected by secondary mutations are all part of a hydrogen-bonded interaction network in the protease structure. Although much less is known on how the mutations outside the active site contribute to resistance, hydrophobic sliding in relation to conformational dynamics and the more recent network hypothesis have provided inroads that may lead to more detailed and perhaps unified hypothesis to explain the underlying molecular mechanisms by which mutations at these sites directly contribute to resistance—rather than being compensatory.

The network hypothesis postulates that the network of hydrogen bonds in the protease structure connects the distal drug resistance mutation sites to the active site. Mutation at residues outside the active site (colored magenta, green, red, and orange) are able to affect the interactions with the bound inhibitor and active-site dynamics through common mechanisms, as they are all part of this connected network. Adapted with permission from Ragland et al. [45]. Copyright (2014) American Chemical Society
5.3 Substrate Mutations and Coevolution
In addition to extensive mutations selected in HIV-1 protease under drug pressure to evade inhibition, the viral genome mutates elsewhere as well, especially at polyprotein Gag cleavage sites [31, 33, 61]. Evaluation of coevolution in terms of substrate envelope provided two mechanistic insights [62, 63]: (1) the two most divergent substrates with respect to fit within the envelope are the ones that are the most susceptible to mutations. Nc/p1 and p1/p2 protrude beyond the substrate envelope more than expected based on their size, and mutations therein are more frequent compared to the other substrates. (2) When protease resistance mutations abrogate the fit of a particular substrate within the consensus envelope, substrate mutations may help restore the fit within the substrate envelope. Thus, the substrate envelope is preserved by coevolution of protease and substrate.
Gag mutations have been thought to be compensatory mutations that rescue viral fitness lost due to protease mutations. However, some substrate mutations are able to directly confer PI resistance even in the absence of protease mutations and accumulating evidence suggests substrate mutations as an alternative pathway to resistance in patients failing therapy. Some of the most common substrate mutations at the NC/p1 and p1/p6 cleavage sites are classified as primary resistance mutations. A431V mutation at the NC/p1 cleavage site is the most frequent substrate mutation selected under PI pressure, and confers resistance to all PIs except DRV [33, 34]. Both A431V and I437V mutations at the NC/p1 cleavage site have been shown to have little effect on replicative capacity but instead directly confer antiviral resistance [32].
Statistical analysis of viral sequences specifically correlates primary drug resistance mutations in HIV-1 protease to substrate mutations, indicating coevolution [33]. A431V is often observed in combination with major protease mutations I50L, V82A, and I84V, while I437V correlates with I54V and I84V. Under drug pressure, the resistance mutations selected may differentially impair the protease activity on Gag cleavage sites, which would interfere with the ordered processing of Gag. Coevolution of substrates possibly restores proper Gag processing by more efficient cleavage by the protease.
Coevolved mutations of the substrate do not necessarily restore the specific protease–substrate interactions lost due to primary mutations. Structural analysis of coevolution for the Gag A431V and V82A protease mutations revealed the mechanism to be much more complex than a simple switch of A and V side-chain contacts (Fig. 35.5) [64]: V82A protease mutation causes loss of vdW contacts with F433 (not A431), while the A431V substrate mutation optimally fills the P2 pocket and reorients the substrate peptide to a more favorable conformation to stabilize overall interactions with the protease. Similarly, the coevolution mutations at the p1/p6 cleavage site (L449F or S451N) with NFV resistance mutations D30N/N88D do not restore the lost interactions of residue 30 but establish alternate contacts between the protease and substrate [65]. The individual coevolution mutations L449F and S451N enhance protease contacts and fit within the envelope. However, two large side chains together do not further improve contacts with the protease or fit within the substrate envelope, causing protrusions. This structural finding explains why, although frequently selected in correlation with protease NFV resistance mutations, L449F and S451N do not occur simultaneously at the p1/p6 cleavage site in viral sequences [33, 65].

Coevolution of NC-p1 cleavage site with V82A protease mutation. (a) Drug resistance mutation V82A causes loss of vdW contacts with Gag F433 (PheP1′), but not with A431 (AlaP2). Coevolution of the substrate to A431V does not enhance intermolecular vdW contacts at the mutation site. Rather, (b) the whole substrate peptide reorients (magenta versus cyan) and new water-mediated hydrogen bonds are formed between the peptide and protease (yellow dotted lines). Adapted from Prabu-Jeyabalan et al. [64] with permission. Copyright © 2004, American Society for Microbiology
Mutations at the p1/p6 cleavage site also coevolve with protease I50V major resistance mutation. I50V is commonly observed in patients failing therapy with APV and DRV, and also impairs protease catalytic efficiency. I50V often occurs together with the secondary mutation A71V, which compensates for protease efficiency [66, 67]. The substrate Gag L449F mutation rescues the protease activity by 10-fold, whereas P453L, although located distal from the catalytic site, causes a 23-fold enhancement [68]. The WT protease processes the mutated substrates more efficiently compared to the native substrate. This suboptimal cleavage efficiency at the p1/p6 site may be key for temporal regulation of Gag processing preventing premature viral maturation [23, 69]. A recent study with a series of crystal structures of I50V/A71V protease bound to p1/p6 substrate variants and MD simulations revealed molecular mechanisms underlying this coevolution [70]. The substrate residue Gag 453 is located away from the protease active site and does not make substantial contacts with the protease. Why P453L coevolution mutation is selected and how it may affect protease binding were not clear. P453L substrate mutation was demonstrated to induce a distal conformational change in one of the protease loops to enhance vdW contacts at residue 449 (Fig. 35.6). Reciprocally, L449F mutation propagates to a conformational change at residue 453, indicating interdependency between the two sites. In general, the coevolution mutations at the substrate do not directly restore interactions lost due to I50V, but instead establish other interactions that are not restricted to the site of mutation. The Gag mutations L449F and P453L enhance vdW interactions between the substrate and mutant protease by distal effects, whereas R452S results in an additional hydrogen bond.

Distal effects of p1–p6 substrate coevolution mutations in binding drug-resistant I50V/A71V protease. (a) The vdW contacts of residues in HIV-1 protease–substrate cocrystal structures colored blue to red for increasing contacts. The substrate mutation at P1′ position (L449F) enhances contacts at P5′ (Gag 453). (b) The distal substrate mutation PP5′L (P453L) causes a conformational change in the protease flap and alters substrate–protease interactions. The protease flaps are in cyan and yellow in complex structures with WT (navy blue) and P5′L (orange) substrates, respectively. Reprinted from Ozen et al. [70]
In addition to enhancing substrate–protease interactions, coevolution may restore conformational dynamics at the active site, which is crucial for substrate binding and processing. In the case of I50V protease with p1/p6 substrate coevolution, mutation of the protease or the native substrate alone disturbed the dynamics, which was restored to a wild-type-like state in all coevolved complexes bearing complementary mutations in both the protease and the substrate [70]. Hence, in addition to the specific shape adopted and shared by all substrates when bound to the HIV-1 protease, as defined by the substrate envelope, a conserved dynamic behavior around the active site may be an additional substrate recognition and selection constraint. This dynamic constraint may contribute to the selection of substrate coevolution mutations in response to the disturbed dynamics in mutated drug-resistant protease.
5.4 Thermodynamics of PI Binding to Resistant Variants
Design and development of potent HIV-1 protease inhibitors require maximizing the binding affinity to target, which is dictated by the free energy of binding composed of enthalpy and entropy change between the unbound and bound states. Binding enthalpy mainly depends on the favorable interactions between the ligand and the protease, while the change in degrees of freedom (of the ligand, target, and solvent) determines the binding entropy. The first-generation PIs were entropy-driven binders as the strategy was to design conformational constraints to preposition the compound in a binding-competent state, with additional favorable solvation entropy due to burial of hydrophobic groups and release of structured water molecules. Further optimization yielded more potent PIs with both favorable enthalpy and entropy of binding, such as DRV [71–73]. However, highly potent entropy-driven inhibitors are also possible, such as TPV [74]. The interplay between entropy and enthalpy of binding at the molecular level is not straightforward in drug design, and enhancing one may inadvertently affect the other, resulting in entropy–enthalpy compensation.
The enthalpic and entropic contributions to binding various drug-resistant variants of HIV-1 protease have been determined by isothermal titration calorimetry to understanding how mutations affect the energetics of inhibitor binding [39, 41–43, 71, 74]. In one variant with multiple mutations both within and outside the active site (L10I/G48V/I54V/V82A), the resistance mutations drastically altered the thermodynamics of binding, regardless of the PI tested [39]. Contrary to another variant (V82T/I84V) with similar levels of affinity loss, the first variant displayed extreme entropy–enthalpy compensation on the order of 10–15 kcal/mol. Thus drug resistance mutations in the protease can modulate the thermodynamics and hence affinity of binding. However, when the mutations in this variant are introduced individually or when the I54V mutation is replaced with I54A, this extreme entropy–enthalpy compensation no longer exists [75]. NMR and MD results suggested that alterations in protease conformational dynamics especially at the flap region may be underlying the observed thermodynamic behavior [58, 59], but a better understanding of the molecular mechanisms involved warrants further analysis, in particular of changes in water solvation. This complex and cooperative interdependency in altering thermodynamics of PI binding and conferring resistance presents an additional challenge in the rational design of robust drugs to avoid resistance.
6 Designing Robust Drugs to Avoid Resistance
HIV-1 protease is arguably the most extensively studied drug target to structurally and dynamically characterize how selected mutations confer resistance to inhibitors. We have learned critical insights, which should be transferable to other rapidly evolving disease targets where resistance emerges and impairs treatment options. Perhaps the main message from the HIV-1 protease drug resistance field to the drug design community is the need to shift the current paradigm of regarding resistance only as an afterthought, toward employing strategies to avoid resistance at the very first design and optimization steps of drug development. An effective approach to avoid susceptibility to major active-site mutations is to design inhibitors that stay within the substrate envelope. The highly potent and robust DRV provides a proof of concept for this strategy [14, 16, 71]. Additional libraries designed to stay within the envelope versus paired compound that protrude out provided additional support to validate this strategy [76]. In fact, exploiting the unused regions of the substrate envelope and exploring the chemical space while staying within the substrate envelope was successful in designing compounds even more potent and more robust than DRV [77]. More recently, the substrate envelope hypothesis and the related design strategy have been shown to hold true for HCV NS3/4A protease and its inhibitors as well [78, 79], and should be more generally applicable to other targets.
While we have some valuable insights into how mutations at the protease active site and elsewhere confer resistance, the molecular mechanisms of resistance due to the complex combination of mutations and interdependency in drug resistance are far more complex. To further our understanding of these molecular mechanisms underlying resistance to HIV-1 protease inhibitors, we need more comprehensive approaches unifying the structure, conformational dynamics, and energetics of inhibitor binding. Such an approach may lead to compounds that target and potently inhibit not only the wild-type enzyme but also a wide variety of variants that exist in patient populations. In the absence of a cure and considering the rapid evolution of the virus, the chances of replacing combination therapies with such a compound as single agent may be slim. Regardless, a detailed understanding of the wide variety of mutations and molecular mechanisms underlying resistance to HIV-1 protease inhibitors would provide the opportunity to develop design strategies to avoid drug resistance, by exploiting the biological and functional constraints on the evolution of the drug target.
References
UNAIDS. (2013). Global report: UNAIDS report on the global AIDS epidemic.
Mehellou Y, De Clercq E. Twenty-six years of anti-HIV drug discovery: where do we stand and where do we go? J Med Chem. 2010;53:521–38.
Hogg RS, Heath KV, Yip B, Craib KJP, O’Shaughnessy MV, Schechter MT, Montaner JSG. Improved survival among HIV-infected individuals following initiation of antiretroviral therapy. JAMA. 1998;279:450–4.
Hogg RS, OShaughnessy MV, Gataric N, Yip B, Craib K, Schechter MT, Montaner JSG. Decline in deaths from AIDS due to new antiretrovirals. Lancet. 1997;349:1294.
Palella FJ, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman DJ, Holmberg SD, Investigators HOS. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N Engl J Med. 1998;338:853–60.
Roberts NA, Martin JA, Kinchington D, Broadhurst AV, Craig JC, Duncan IB, Galpin SA, Handa BK, Kay J, Krohn A, et al. Rational design of peptide-based HIV proteinase inhibitors. Science. 1990;248:358–61.
Dorsey BD, Levin RB, McDaniel SL, Vacca JP, Guare JP, Darke PL, Zugay JA, Emini EA, Schleif WA, Quintero JC, et al. L-735,524: the design of a potent and orally bioavailable HIV protease inhibitor. J Med Chem. 1994;37:3443–51.
Kempf DJ, Marsh KC, Denissen JF, McDonald E, Vasavanonda S, Flentge CA, Green BE, Fino L, Park CH, Kong XP, et al. ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc Natl Acad Sci U S A. 1995;92:2484–8.
Kaldor SW, Kalish VJ, Davies 2nd JF, Shetty BV, Fritz JE, Appelt K, Burgess JA, Campanale KM, Chirgadze NY, Clawson DK, et al. Viracept (nelfinavir mesylate, AG1343): a potent, orally bioavailable inhibitor of HIV-1 protease. J Med Chem. 1997;40:3979–85.
Kim EE, Baker CT, Dwyer MD, Murcko MA, Rao BG, Tung RD, Navia MA. Crystal-structure of HIV-1 protease in complex with Vx-478, a potent and orally bioavailable inhibitor of the enzyme. J Am Chem Soc. 1995;117:1181–2.
Sham HL, Kempf DJ, Molla A, Marsh KC, Kumar GN, Chen CM, Kati W, Stewart K, Lal R, Hsu A, et al. ABT-378, a highly potent inhibitor of the human immunodeficiency virus protease. Antimicrob Agents Chemother. 1998;42:3218–24.
Robinson BS, Riccardi KA, Gong YF, Guo Q, Stock DA, Blair WS, Terry BJ, Deminie CA, Djang F, Colonno RJ, et al. BMS-232632, a highly potent human immunodeficiency virus protease inhibitor that can be used in combination with other available antiretroviral agents. Antimicrob Agents Chemother. 2000;44:2093–9.
Turner SR, Strohbach JW, Tommasi RA, Aristoff PA, Johnson PD, Skulnick HI, Dolak LA, Seest EP, Tomich PK, Bohanon MJ, et al. Tipranavir (PNU-140690): a potent, orally bioavailable nonpeptidic HIV protease inhibitor of the 5,6-dihydro-4-hydroxy-2-pyrone sulfonamide class. J Med Chem. 1998;41:3467–76.
De Meyer S, Azijn H, Surleraux D, Jochmans D, Tahri A, Pauwels R, Wigerinck P, de Bethune MP. TMC114, a novel human immunodeficiency virus type 1 protease inhibitor active against protease inhibitor-resistant viruses, including a broad range of clinical isolates. Antimicrob Agents Chemother. 2005;49:2314–21.
Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, et al. Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob Agents Chemother. 2003;47:3123–9.
Surleraux DL, Tahri A, Verschueren WG, Pille GM, de Kock HA, Jonckers TH, Peeters A, De Meyer S, Azijn H, Pauwels R, et al. Discovery and selection of TMC114, a next generation HIV-1 protease inhibitor. J Med Chem. 2005;48:1813–22.
Navia MA, Fitzgerald PM, McKeever BM, Leu CT, Heimbach JC, Herber WK, Sigal IS, Darke PL, Springer JP. Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature. 1989;337:615–20.
Wlodawer A, Miller M, Jaskolski M, Sathyanarayana BK, Baldwin E, Weber IT, Selk LM, Clawson L, Schneider J, Kent SB. Conserved folding in retroviral proteases: crystal structure of a synthetic HIV-1 protease. Science. 1989;245:616–21.
Jilek BL, Zarr M, Sampah ME, Rabi SA, Bullen CK, Lai J, Shen L, Siliciano RF. A quantitative basis for antiretroviral therapy for HIV-1 infection. Nat Med. 2012;18:446–51.
Sampah MES, Shen L, Jilek BL, Siliciano RF. Dose-response curve slope is a missing dimension in the analysis of HIV-1 drug resistance. Proc Natl Acad Sci U S A. 2011;108:7613–8.
Jacks T, Power MD, Masiarz FR, Luciw PA, Barr PJ, Varmus HE. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature. 1988;331:280–3.
Erickson-Viitanen S, Manfredi J, Viitanen P, Tribe DE, Tritch R, Hutchison 3rd CA, Loeb DD, Swanstrom R. Cleavage of HIV-1 gag polyprotein synthesized in vitro: sequential cleavage by the viral protease. AIDS Res Hum Retroviruses. 1989;5:577–91.
Pettit SC, Lindquist JN, Kaplan AH, Swanstrom R. Processing sites in the human immunodeficiency virus type 1 (HIV-1) Gag-Pro-Pol precursor are cleaved by the viral protease at different rates. Retrovirology. 2005;2:66.
Pettit SC, Sheng N, Tritch R, Erickson-Viitanen S, Swanstrom R. The regulation of sequential processing of HIV-1 Gag by the viral protease. Adv Exp Med Biol. 1998;436:15–25.
Wiegers K, Rutter G, Kottler H, Tessmer U, Hohenberg H, Krausslich HG. Sequential steps in human immunodeficiency virus particle maturation revealed by alterations of individual Gag polyprotein cleavage sites. J Virol. 1998;72:2846–54.
Jager S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, et al. Global landscape of HIV-human protein complexes. Nature. 2012;481:365–70.
Ventoso I, Blanco R, Perales C, Carrasco L. HIV-1 protease cleaves eukaryotic initiation factor 4G and inhibits cap-dependent translation. Proc Natl Acad Sci U S A. 2001;98:12966–71.
Rhee SY, Taylor J, Fessel WJ, Kaufman D, Towner W, Troia P, Ruane P, Hellinger J, Shirvani V, Zolopa A, et al. HIV-1 protease mutations and protease inhibitor cross-resistance. Antimicrob Agents Chemother. 2010;54:4253–61.
Wu TD, Schiffer CA, Gonzales MJ, Taylor J, Kantor R, Chou S, Israelski D, Zolopa AR, Fessel WJ, Shafer RW. Mutation patterns and structural correlates in human immunodeficiency virus type 1 protease following different protease inhibitor treatments. J Virol. 2003;77:4836–47.
Velazquez-Campoy A, Vega S, Freire E. Amplification of the effects of drug resistance mutations by background polymorphisms in HIV-1 protease from African subtypes. Biochemistry (Mosc). 2002;41:8613–9.
Fun A, Wensing AM, Verheyen J, Nijhuis M. Human Immunodeficiency Virus Gag and protease: partners in resistance. Retrovirology. 2012;9:63.
Dam E, Quercia R, Glass B, Descamps D, Launay O, Duval X, Krausslich HG, Hance AJ, Clavel F, Grp AS. Gag mutations strongly contribute to HIV-1 resistance to protease inhibitors in highly drug-experienced patients besides compensating for fitness loss. PLoS Pathog. 2009;5:e1000345.
Kolli M, Stawiski E, Chappey C, Schiffer CA. Human immunodeficiency virus type 1 protease-correlated cleavage site mutations enhance inhibitor resistance. J Virol. 2009;83:11027–42.
Nijhuis M, van Maarseveen NM, Lastere S, Schipper P, Coakley E, Glass B, Rovenska M, de Jong D, Chappey C, Goedegebuure IW, et al. A novel substrate-based HIV-1 protease inhibitor drug resistance mechanism. PLoS Med. 2007;4:e36.
King NM, Prabu-Jeyabalan M, Nalivaika EA, Schiffer CA. Combating susceptibility to drug resistance: lessons from HIV-1 protease. Chem Biol. 2004;11:1333–8.
Galiano L, Ding F, Veloro AM, Blackburn ME, Simmerling C, Fanucci GE. Drug pressure selected mutations in HIV-1 protease alter flap conformations. J Am Chem Soc. 2009;131:430–1.
Gulnik SV, Suvorov LI, Liu B, Yu B, Anderson B, Mitsuya H, Erickson JW. Kinetic characterization and cross-resistance patterns of HIV-1 protease mutants selected under drug pressure. Biochemistry (Mosc). 1995;34:9282–7.
Heaslet H, Kutilek V, Morris GM, Lin YC, Elder JH, Torbett BE, Stout CD. Structural insights into the mechanisms of drug resistance in HIV-1 protease NL4-3. J Mol Biol. 2006;356:967–81.
King NM, Prabu-Jeyabalan M, Bandaranayake RM, Nalam MN, Nalivaika EA, Ozen A, Haliloglu T, Yilmaz NK, Schiffer CA. Extreme entropy-enthalpy compensation in a drug-resistant variant of HIV-1 protease. ACS Chem Biol. 2012;7:1536–46.
Liu Z, Wang Y, Brunzelle J, Kovari IA, Kovari LC. Nine crystal structures determine the substrate envelope of the MDR HIV-1 protease. Protein J. 2011;30:173–83.
Mittal S, Bandaranayake RM, King NM, Prabu-Jeyabalan M, Nalam MN, Nalivaika EA, Yilmaz NK, Schiffer CA. Structural and thermodynamic basis of amprenavir/darunavir and atazanavir resistance in HIV-1 protease with mutations at residue 50. J Virol. 2013;87:4176–84.
Muzammil S, Ross P, Freire E. A major role for a set of non-active site mutations in the development of HIV-1 protease drug resistance. Biochemistry (Mosc). 2003;42:631–8.
Ohtaka H, Schon A, Freire E. Multidrug resistance to HIV-1 protease inhibition requires cooperative coupling between distal mutations. Biochemistry (Mosc). 2003;42:13659–66.
Perryman AL, Lin JH, McCammon JA. HIV-1 protease molecular dynamics of a wild-type and of the V82F/I84V mutant: possible contributions to drug resistance and a potential new target site for drugs. Protein Sci. 2004;13:1108–23.
Ragland DA, Nalivaika EA, Nalam MN, Prachanronarong KL, Cao H, Bandaranayake RM, Cai Y, Kurt-Yilmaz N, Schiffer CA. Drug resistance conferred by mutations outside the active site through alterations in the dynamic and structural ensemble of HIV-1 protease. J Am Chem Soc. 2014;136:11956–63.
Saskova KG, Kozisek M, Lepsik M, Brynda J, Rezacova P, Vaclavikova J, Kagan RM, Machala L, Konvalinka J. Enzymatic and structural analysis of the I47A mutation contributing to the reduced susceptibility to HIV protease inhibitor lopinavir. Protein Sci. 2008;17:1555–64.
Skalova T, Dohnalek J, Duskova J, Petrokova H, Hradilek M, Soucek M, Konvalinka J, Hasek J. HIV-1 protease mutations and inhibitor modifications monitored on a series of complexes. Structural basis for the effect of the A71V mutation on the active site. J Med Chem. 2006;49:5777–84.
Wilson SI, Phylip LH, Mills JS, Gulnik SV, Erickson JW, Dunn BM, Kay J. Escape mutants of HIV-1 proteinase: enzymic efficiency and susceptibility to inhibition. Biochim Biophys Acta. 1997;1339:113–25.
Yedidi RS, Proteasa G, Martinez JL, Vickrey JF, Martin PD, Wawrzak Z, Liu Z, Kovari IA, Kovari LC. Contribution of the 80s loop of HIV-1 protease to the multidrug-resistance mechanism: crystallographic study of MDR769 HIV-1 protease variants. Acta Crystallogr D Biol Crystallogr. 2011;67:524–32.
Prabu-Jeyabalan M, Nalivaika EA, Romano K, Schiffer CA. Mechanism of substrate recognition by drug-resistant human immunodeficiency virus type 1 protease variants revealed by a novel structural intermediate. J Virol. 2006;80:3607–16.
Prabu-Jeyabalan M, Nalivaika E, Schiffer CA. Substrate shape determines specificity of recognition for HIV-1 Protease: analysis of crystal structures of six substrate complexes. Structure. 2002;10:369–81.
Chellappan S, Kairys V, Fernandes MX, Schiffer C, Gilson MK. Evaluation of the substrate envelope hypothesis for inhibitors of HIV-1 protease. Proteins. 2007;68:561–7.
Freedberg DI, Ishima R, Jacob J, Wang YX, Kustanovich I, Louis JM, Torchia DA. Rapid structural fluctuations of the free HIV protease flaps in solution: relationship to crystal structures and comparison with predictions of dynamics calculations. Protein Sci. 2002;11:221–32.
Hornak V, Okur A, Rizzo RC, Simmerling C. HIV-1 protease flaps spontaneously open and reclose in molecular dynamics simulations. Proc Natl Acad Sci U S A. 2006;103:915–20.
Ishima R, Freedberg DI, Wang YX, Louis JM, Torchia DA. Flap opening and dimer-interface flexibility in the free and inhibitor-bound HIV protease, and their implications for function. Structure. 1999;7:1047–55.
Scott WR, Schiffer CA. Curling of flap tips in HIV-1 protease as a mechanism for substrate entry and tolerance of drug resistance. Structure. 2000;8:1259–65.
Mittal S, Cai Y, Nalam MN, Bolon DN, Schiffer CA. Hydrophobic core flexibility modulates enzyme activity in HIV-1 protease. J Am Chem Soc. 2012;134:4163–8.
Cai Y, Kurt Yilmaz N, Myint W, Ishima R, Schiffer CA. Differential flap dynamics in wild-type and a drug resistant variant of HIV-1 protease revealed by molecular dynamics and NMR relaxation. J Chem Theory Comput. 2012;8:3452–62.
Cai Y, Myint W, Paulsen JL, Schiffer CA, Ishima R, Kurt Yilmaz N. Drug resistance mutations alter dynamics of inhibitor-bound HIV-1 protease. J Chem Theory Comput. 2014;10:3438–48.
de Vera IMS, Smith AN, Dancel MCA, Huang X, Dunn BM, Fanucci GE. Elucidating a relationship between conformational sampling and drug resistance in HIV-1 protease. Biochemistry (Mosc). 2013;52:3278–88.
Kolli M, Lastere S, Schiffer CA. Co-evolution of nelfinavir-resistant HIV-1 protease and the p1-p6 substrate. Virology. 2006;347:405–9.
Ozen A, Haliloglu T, Schiffer CA. Dynamics of preferential substrate recognition in HIV-1 protease: redefining the substrate envelope. J Mol Biol. 2011;410:726–44.
Ozen A, Haliloglu T, Schiffer CA. HIV-1 protease and substrate coevolution validates the substrate envelope as the substrate recognition pattern. J Chem Theory Comput. 2012;8:703–14.
Prabu-Jeyabalan M, Nalivaika EA, King NM, Schiffer CA. Structural basis for coevolution of a human immunodeficiency virus type 1 nucleocapsid-p1 cleavage site with a V82A drug-resistant mutation in viral protease. J Virol. 2004;78:12446–54.
Kolli M, Ozen A, Kurt-Yilmaz N, Schiffer CA. HIV-1 protease-substrate coevolution in nelfinavir resistance. J Virol. 2014;88:7145–54.
Nijhuis M, Schuurman R, de Jong D, Erickson J, Gustchina E, Albert J, Schipper P, Gulnik S, Boucher CAB. Increased fitness of drug resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. AIDS. 1999;13:2349–59.
Rhee SY, Gonzales MJ, Kantor R, Betts BJ, Ravela J, Shafer RW. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003;31:298–303.
Maguire MF, Guinea R, Griffin P, Macmanus S, Elston RC, Wolfram J, Richards N, Hanlon MH, Porter DJT, Wrin T, et al. Changes in human immunodeficiency virus type 1 Gag at positions L449 and P453 are linked to 150V protease mutants in vivo and cause reduction of sensitivity to amprenavir and improved viral fitness in vitro. J Virol. 2002;76:7398–406.
Pettit SC, Henderson GJ, Schiffer CA, Swanstrom R. Replacement of the P1 amino acid of human immunodeficiency virus type 1 Gag processing sites can inhibit or enhance the rate of cleavage by the viral protease. J Virol. 2002;76:10226–33.
Ozen A, Lin KH, Kurt Yilmaz N, Schiffer CA. Structural basis and distal effects of Gag substrate coevolution in drug resistance to HIV-1 protease. Proc Natl Acad Sci U S A. 2014;111:15993–8.
King NM, Prabu-Jeyabalan M, Nalivaika EA, Wigerinck P, de Bethune MP, Schiffer CA. Structural and thermodynamic basis for the binding of TMC114, a next-generation human immunodeficiency virus type 1 protease inhibitor. J Virol. 2004;78:12012–21.
Velazquez-Campoy A, Luque I, Todd MJ, Milutinovich M, Kiso Y, Freire E. Thermodynamic dissection of the binding energetics of KNI-272, a potent HIV-1 protease inhibitor. Protein Sci. 2000;9:1801–9.
Velazquez-Campoy A, Todd MJ, Freire E. HIV-1 protease inhibitors: Enthalpic versus entropic optimization of the binding affinity. Biochemistry (Mosc). 2000;39:2201–7.
Muzammil S, Armstrong AA, Kang LW, Jakalian A, Bonneau PR, Schmelmer V, Amzel LM, Freire E. Unique thermodynamic response of tipranavir to human immunodeficiency virus type 1 protease drug resistance mutations. J Virol. 2007;81:5144–54.
Foulkes-Murzycki JE, Rosi C, Kurt Yilmaz N, Shafer RW, Schiffer CA. Cooperative effects of drug-resistance mutations in the flap region of HIV-1 protease. ACS Chem Biol. 2013;8:513–8.
Shen Y, Altman MD, Ali A, Nalam MN, Cao H, Rana TM, Schiffer CA, Tidor B. Testing the substrate-envelope hypothesis with designed pairs of compounds. ACS Chem Biol. 2013;8:2433–41.
Nalam MN, Ali A, Reddy GS, Cao H, Anjum SG, Altman MD, Yilmaz NK, Tidor B, Rana TM, Schiffer CA. Substrate envelope-designed potent HIV-1 protease inhibitors to avoid drug resistance. Chem Biol. 2013;20:1116–24.
Romano KP, Ali A, Royer WE, Schiffer CA. Drug resistance against HCV NS3/4A inhibitors is defined by the balance of substrate recognition versus inhibitor binding. Proc Natl Acad Sci U S A. 2010;107:20986–91.
Soumana DI, Ali A, Schiffer CA. Structural analysis of asunaprevir resistance in HCV NS3/4A protease. ACS Chem Biol. 2014;9:2485–90.
Acknowledgments
This work was supported by the National Institutes of Health Grants specifically from NIGMS P01 GM109767 and R01 GM65347. We acknowledge past and present members of the Schiffer research group, including Akbar Ali, for help with preparing Fig. 35.1 and Madhavi Kolli for useful suggestions.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Yilmaz, N.K., Schiffer, C.A. (2017). Drug Resistance to HIV-1 Protease Inhibitors: Molecular Mechanisms and Substrate Coevolution. In: Mayers, D., Sobel, J., Ouellette, M., Kaye, K., Marchaim, D. (eds) Antimicrobial Drug Resistance. Springer, Cham. https://doi.org/10.1007/978-3-319-46718-4_35
Download citation
DOI: https://doi.org/10.1007/978-3-319-46718-4_35
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-46716-0
Online ISBN: 978-3-319-46718-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)