
Abstract
Glioblastoma, the most common and aggressive form of primary adult brain tumor, is a devastating disease with a dismal two-year survival. Attempts to improve patient survival include a variety of treatment options, from monoclonal antibodies, vaccines, and microbubbles to exosomes and small-molecule inhibitors, all of which are in various stages of preclinical and clinical development. The most frequently tested type of novel therapeutics are the small-molecule inhibitors targeting key signaling pathways dysregulated in GBM, including TP53, retinoblastoma, and the receptor tyrosine kinase-driven EGF, PDGF, and c-MET pathways. This chapter will compare preclinical and clinical results for a subset of inhibitors targeting the receptor tyrosine kinase families EGF, VEGF, and PDGF along with the PI3K/Akt/mTor pathway and cell cycle inhibitors. In the discussion, potential resistance mechanisms which continue to pose significant barriers to effective small-molecule inhibition treatment of GBM will be discussed along with possible improvements.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
7.1 Introduction: Current Glioblastoma Therapy Options
The current standard of care for newly diagnosed patients with glioblastoma (GBM) includes surgery, radiation therapy (RT), and chemotherapy with the DNA methylating agent temozolomide (TMZ) [1]. Both TMZ and RT kill GBM cells by inducing DNA double-strand breaks (DSBs), which in turn activates apoptotic cell death [2]. Cells surviving the initial therapy rapidly proliferate leading to recurrent disease, for which known effective therapeutic options are limited [1]. FDA-approved treatments for recurrent GBM are limited to the antiangiogenic agent Avastin (bevacizumab) and the DNA alkylating drugs lomustine (CCNU) and carmustine (BCNU), while surgery done at recurrence has yielded equivocal results for patients [1]. The growth of both primary and recurrent tumors is primarily driven by vast signaling pathways activated or dysregulated in GBM. System-wide analysis conducted by The Cancer Genome Atlas in over 200 GBM patient samples reveals many commonly mutated and dysregulated pathways in GBM [3]. For instance, as shown in Fig. 7.1, 88 % of patients analyzed had altered signaling in the RTK/RAS/PI(3)K network with 45 % of patients showing either mutation or amplification of EGFR alone, while mutation and homozygous deletion of PTEN was found in 36 % of patients. Alterations in both the p53 and RB signaling pathways are also common (87 % and 78 %, respectively), with homozygous deletion of CDKN2A (ARF/P16/INK4A) and CDKN2B represented in approximately half of patient samples. The majority of the signaling pathways noted in this study provide a survival advantage either by decreasing apoptosis or increasing cell proliferation and angiogenesis, suggesting that key member proteins could provide novel targets for signaling modulation [4].

Commonly dysregulated pathways and pathway components in GBM patients. Sequencing of 206 glioblastoma patient tissue samples was performed to uncover the most commonly dysregulated pathways and pathway components present in GBM patient tissues. Reprinted by permission from Macmillan Publishers Ltd: Molecules [3], copyright (2009)
Similar system-wide genomic analysis performed in other cancer types has uncovered novel targets and pathways which could be used to provide direct clinical benefit [5, 6]. Although targeted chemotherapeutics created to modulate specific pathways identified in GBM patients could be similarly identified and targeted, no small-molecule inhibitor has been FDA approved for treatment of GBM patients. This is, at least in part, because of a lack of significant therapeutic benefit and/or toxicity that was observed in the initial preclinical and clinical studies [7, 8]. Significant complications continue to plague the development and testing of novel therapeutics in GBM, a few of which will be covered toward the end of this chapter in Sect. 7.3, Resistance Mechanisms to Small-Molecule Inhibitors .
7.2 Small-Molecule Novel Therapeutics
7.2.1 Receptor Tyrosine Kinase Inhibitors
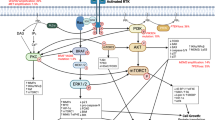
The receptor tyrosine kinases (RTKs) are frequently mutated and overexpressed in GBM, with a high frequency of alterations reported in the PDGF, EGF, and c-MET families, among others [9]. Mutational events in RTKs are often concurrent with other activating and silencing mutations, such as loss of the tumor suppressor genes phosphatase and tensin homolog (PTEN) and tumor protein (p53) [3]. Alterations in RTKs affect a wide range of downstream cellular pathways and processes including apoptosis evasion, growth, survival, and focal adhesion, as noted in Fig. 7.2. Figure 7.3 presents a simplified schematic of RTK/PI3K/Akt signaling and many of the chemotherapeutics directed against specific members of these pathways, a few of which will be discussed throughout this chapter. The following section will provide a detailed account of key inhibitors targeting the RTKs and pinpoint the observed clinical benefit, if any, in GBM.

Receptor tyrosine kinase pathway proteins and downstream signaling effects. The activation of receptor tyrosine kinases (RTKs), in addition to G-protein-coupled receptors (GPCRs), drives a multitude of downstream targets including Akt, mTor, and PI3K and their respective pathways, many of which are known to be dysregulated in various forms of cancer including GBM. Reprinted by permission from Macmillan Publishers Ltd: Nature Reviews Drug Discovery [56], copyright (2009)

Small-molecule inhibitors and their targets . Several small-molecule inhibitors targeting RTK, PI3K, and mTor have been tested preclinically and clinically for GBM and other cancer types. Only a select few examples from this extensive list are discussed in this chapter. Reprinted by permission from Macmillan Publishers Ltd: Nature Reviews Drug Discovery [56], copyright (2009)
7.2.1.1 Epidermal Growth Factor Inhibitors
One of the most extensively studied and frequently targeted of the RTKs across all cancer types is the erythroblastic leukemia viral oncogene homolog (ErbB) or epidermal growth factor (EGF) family of receptors [10, 11]. The EGF family consists of four members: EGFR/ErbB1/Her1, ErbB2/Her2, ErbB3/Her3, and ErbB4/Her4 [10]. Though only EGFR, Her3 and Her4 have known ligand-induced kinase activity, dimerization and oligimerization of all EGF family members allows for a wide variety of signaling pathway options [10, 11]. A key regulator of downstream RTK activity, PTEN was implicated as a possible negative regulator of the EGFR inhibitor response [12]. However, preclinical and clinical studies from other groups have repeatedly failed to confirm the importance of PTEN in receptor tyrosine kinase inhibitor (RTKi) efficacy [13, 14]. Thus, the relevance of PTEN status to EGFR inhibitor efficacy remains unknown.
EGFR targeting is a particularly attractive therapeutic strategy for GBM patients as approximately 34–63 % will have an amplification of EGFR and, of those, 25–64 % will also have an excision of exons 2–7, called EGFRvIII, which creates a constitutively activated protein kinase [15]. Deletion of exons 2–7 has been shown to drive EGFR addiction in cells; thus EGFR inhibition should be an effective therapeutic strategy for GBM patients [11, 16]. Activation of EGFR drives downstream signaling through a wide variety of pathways including JAK/STAT, RAF/MEK/ERK, and PI3K/AKT/mTor; thus uncontrolled or amplified EGFR activity is likely a critical tumorigenic event [11, 17]. Not surprisingly, there are a large number of EGF-directed chemotherapeutics which are in various stages of testing for GBM, as illustrated in Fig. 7.3. Three of the inhibitors included in Fig. 7.3, erlotinib, lapatinib, and gefitinib, have been extensively evaluated both preclinically and clinically for therapy in GBM, and the results of those studies are discussed below.
7.2.1.1.1 A. Erlotinib (Tarceva, Astellas Pharma)
Erlotinib is a selective and reversible, ATP-competitive intracellular kinase domain inhibitor of EGFR (ErbB1), FDA approved for treatment of non-small cell lung cancer patients (NSCLC) [16, 18]. Similar to GBM, a large number of NSCLC patients will have activating mutations in EGFR (approximately 40–80 %), with about 90 % of those mutations occurring in the kinase domain [19, 20].
7.2.1.1.1.1 Preclinical Evaluation
Xenograft studies in patient-derived orthotopic lines treated with erlotinib showed that EGFRvIII status is likely not a reliable predictor of single agent response [21]. One of only two responsive lines from the panel of 11 tested was an EGFR WT tumor. Although PTEN status did appear to predict for favorable response, since only two lines were responsive, the total number of lines tested were too few to make predictive statements on the role of PTEN status based on these data alone.
7.2.1.1.1.2 Clinical Evaluation
Erlotinib has been tested in five separate Phase II clinical trials (a sixth trial will be discussed in detail in the section for rapamycin). The first trial tested erlotinib in a nonrandomized and open-label study as a single agent in patients who had completed TMZ and RT treatments and were on their first relapse [22]. Forty-eight patients for the two-stage study were accrued, but low response rates (one complete response and three partial responses) in stage 1 led to the study being ended before the start of stage 2. Assessment of EGFR amplification did not indicate any significant survival difference between EGFR-amplified and non-amplified patients (progression-free survival (PFS) at 6 months 21.7 % vs 18.3 %, amplified and non-amplified, respectively). In the second trial, 96 patients were divided into two groups (group 1, 53 patients with recurrent GBM, oligodendroglioma, or anaplastic astrocytoma, and group 2, 43 GBM patients who were nonprogressive (NP) after RT) and treated with single agent erlotinib [8]. Group 1 patients were allowed no more than two prior relapses and two prior therapies. Group 2 patients were not allowed to have received any prior chemotherapy, including TMZ. Erlotinib treatment yielded a PFS at 6 months in recurrent GBM patients of only 3 % (27 % for recurrent AG patients), while estimated 1-year PFS was 9 % in the NP group. Pharmacokinetic analysis conducted in a subset of patients indicated that erlotinib penetration into tumors was insufficient to effectively inhibit EGFR phosphorylation [8, 23]. In the third trial, 97 newly diagnosed patients received erlotinib in combination with TMZ and RT and then continued erlotinib treatment with adjuvant TMZ [13]. No difference in median OS was seen in patients treated with erlotinib when compared to EORTC 26981/22981-NCIC historical controls (15.3 vs 15 months), and response had no correlation with PTEN or EGFR status. In the fourth study, 27 newly diagnosed patients were treated concurrently with TMZ, RT, and erlotinib, within 28 days of biopsy or resection [7]. PFS at 6 months was only 30 % for this study. The study authors also noted that the combination of RT, TMZ, and erlotinib had “unacceptable toxicity.” Finally in the fifth trial, 110 recurrent GBM patients were randomly assigned to receive either erlotinib (54 patients) or a control compound (either TMZ or BCNU (27 or 29 patients, respectively), depending upon whether the patient had been treated with TMZ previously) [24]. At 6 months, patients receiving erlotinib had a PFS of 11.4 %, while patients in the control arm had a PFS of 24.1 %. Collectively, the poor results from xenograft studies and clinical trials illustrate how little is known about the complexity of the EGFR signaling pathway and which factors are truly predictive of response to EGFR inhibition.
7.2.1.1.2 B. Lapatinib (Tykerb, Novartis)
A second EGFR inhibitor, lapatinib, also inhibits ErbB2 (Her2) [25]. Although Her2-driven pathways have not been implicated in GBM as they are in breast cancer, heterodimerization and oligimerization of all four EGF receptors are known signaling mechanisms; thus, dual inhibition of EGFR and Her2 as opposed to inhibition of only EGFR is a potential method to target and inhibit interactions that both receptors play roles in [10, 26]. Further, some studies have found that primary (de novo) GBM tumors have Her2 overexpression , though GBMs arising from lower-grade tumors (secondary GBM) do not appear to exhibit the same phenotype [27].
7.2.1.1.2.1 Preclinical Evaluation
Only one in vivo study of lapatinib treatment of GBM exists currently, and although five patient-derived xenografts were used in the study, only one xenograft line, GBM6, is noted by the authors as being responsive to in vivo treatment with lapatinib (placebo treatment vs lapatinib, p < 0.05) [25]. Interestingly, GBM6 was also included in the abovementioned in vivo study with erlotinib, and in that study, treatment of GBM6 with erlotinib did not produce a statistically significant survival benefit (p = 0.536) [21]. Thus, if the lapatinib results in GBM6 are reproducible, they may indicate that GBM6 is uniquely sensitive to Her2/neu inhibition or to the combination of EGFR and Her2/neu inhibition. Further, these results may hint at a certain subset of GBM tumors that are sensitive to dual inhibition of EGFR and Her2/neu.
7.2.1.1.2.2 Clinical Evaluation
Lapatinib has been tested in two Phase I/II trials, both of which were conducted in recurrent GBM patients. In the first Phase II trial, the best response was stable disease in four patients, while the remaining 13 patients had early progression; thus the study was ended prematurely [28]. Neither EGFRvIII nor PTEN status was predictive of outcome. The second trial tested the combination of pazopanib and lapatinib in 41 GBM patients at first or second recurrence . Patients were split into two groups: 1. EGFRvIII/PTEN positive and 2. EGFRvIII/PTEN negative [29]. This study also failed to meet primary endpoints with a PFS at 6 months of 0 % in the EGFRvIII/PTEN positive and 15 % in the EGFRvIII/PTEN negative groups. Although pharmacokinetic evaluation of pazopanib levels indicated effective concentrations reached, lapatinib doses achieved were subtherapeutic. Neither PTEN nor EGFRvIII status was predictive of outcome in this study. Although the preclinical results for lapatinib indicate a potential, select group of GBM patients that may respond to lapatinib, clinical studies have yet to effectively identify and benefit that specific patient population.
7.2.1.1.3 C. Gefitinib (Iressa, Astra-Zeneca, and Teva)
Gefitinib, similar to erlotinib, is an ATP-competitive selective inhibitor of the EGFR kinase domain [18]. Although there are slight differences between the two compounds (molecular weight and increased likelihood for adverse events (with erlotinib), etc), both retrospective and prospective studies conducted in NSCLC patients have failed to find a significant therapeutic difference [19, 30–32]. Similar comparative studies of erlotinib and gefitinib have not been conducted for GBM, either preclinically or clinically, so it is unclear if treatment with gefitinib and erlotinib in GBM patients will prove to yield essentially analogous results as well.
7.2.1.1.3.1 Preclinical Evaluation
Joshi et al. reported mixed results from in vivo studies performed in 9L rat gliosarcoma and the human GBM cell line 020913 with gefitinib [14]. Animals injected intracranially (IC) with 020913 and treated with gefitinib had a statistically significant survival benefit over placebo treated animals (p = 0.0001). However, rats implanted with 9L cells did not yield a similar benefit (p = 0.13). In their analysis of these results, the authors speculate that the 020913 growth conditions (stem cell media supplemented with EGF and FGF) may select for EGF dependence, whereas 9L cells, which are grown in complete media, are not similarly selected. Growth conditions have in fact been found to directly affect expression of EGFR [15]. Although the EGFR status of 9L cells is a point of contention, with some groups stating that 9L cells do not express EGFR, while others have noted that 9L cells do overexpress EGFR [33, 34], the relevance of EGFR status for inhibitor efficacy is unknown. Further, according to in vitro data noted by the authors, 9L cells treated with gefitinib were more sensitive than 020913 cells. Thus again, though differential effects with EGFR inhibition have been noted, no clear explanation has yet been found for these differences.
7.2.1.1.3.2 Clinical Evaluation
Gefitinib was tested in three Phase II trials, two conducted in recurrent and one in newly diagnosed GBM patients. The first study accrued 28 patients with mixed high-grade gliomas (grades III and IV) in which there was no appreciable efficacy [35]. PFS at 6 months for all patients was 14.3 % (12.5 % in the GBM patient subgroup). Five patients had stable disease and none had partial response. Neither EGFR expression nor gene status nor p-Akt level (a downstream target of EGFR signaling, discussed in greater detail in Sect. 7.2.2) predicted for outcome. In the second trial of gefitinib in recurrent patients, PFS at 6 months was 13 % with no objective tumor responses noted [36]. In the third trial, 96 newly diagnosed patients first underwent RT and then were treated with single agent gefitinib [37]. Patient results, when compared to historical controls, showed no significant difference in survival with gefitinib treatment (PFS at 12 months, post-RT vs historical controls 16.7 % and 30.3 %, respectively). Although there are slight differences between erlotinib and gefitinib, neither drug appears to provide significant benefit to GBM patients clinically.
7.2.1.2 Vascular Endothelial Growth Factor and Platelet-Derived Growth Factor Kinase Inhibitors
The vascular endothelial growth factor (VEGF) family consists of five members, VEGFA, B, C, D, and placental growth factor (though the best characterized is VEGFA) along with the three receptors, VEGFR1, 2, and 3 [38]. Initially identified as an endothelial cell mitogen, the VEGF family actually has functions in a wide variety of cell types and cellular functions, such as cancer stem cell function, tumorigenesis, and stimulation of epithelial to mesenchymal transition [38]. The platelet-derived growth factor (PDGF) family also has multiple components with five isoforms (PDGF-AA, -BB, -CC, -DD, and -AB). Binding of these factors leads to homo- or heterodimeric complexing of PDGFRα and PDGFRβ which ultimately drives downstream effects such as migration, cell survival, and growth [39]. Similar to EGF, both VEGF and PDGF expression have been found to be upregulated in GBM [3, 9]. PDGF and PDGFR are overexpressed in approximately 16 % of GBM, while VEGF-driven angiogenesis is considered a key factor in tumor growth and survival [40, 41]. Several inhibitors with activity against VEGF and PDGF are currently undergoing investigation for treatment of GBM , the results for two of which will be discussed in detail below.
7.2.1.2.1 A. Sorafenib (Nexavar, Bayer)
Sorafenib is a multi-targeted kinase inhibitor with activity against both the VEGF (VEGFR2 and 3) and PDGF (PDGFβ and KIT) families [42]. Sorafenib also inhibits both C-Raf and B-Raf along with MEK and ERK phosphorylation [42] and has been shown to induce apoptosis in a variety of cell lines through its downregulation of the pro-survival factor Mcl-1 [43].
7.2.1.2.1.1 Preclinical Evaluation
Available preclinical in vivo studies of sorafenib are limited, with only one study conducted in mice with orthotopically implanted U87 cells. Single agent sorafenib treatment in this study had a significant survival benefit over vehicle treatment (p < 0.05) [44].
7.2.1.2.1.2 Clinical Evaluation
Sorafenib has been tested in five clinical trials in GBM patients, the first of which, a Phase I/II, combined sorafenib with temsirolimus. However, the study was ended before the start of Phase II as no patients from Phase I made PFS at 6 months [45]. Median PFS in this study was 8 weeks. A separate Phase II trial with sorafenib in combination with bevacizumab conducted in recurrent GBM patients showed no benefit of the combination over bevacizumab historical controls (PFS at 6 months was 20.4 % for sorafenib and bevacizumab combination vs 16–24 % for bevacizumab historical controls) [46]. The third and fourth Phase II studies both tested the combination of sorafenib with low-dose TMZ in recurrent GBM patients, yet each had markedly different results. In both trials, patients received 800 mg/day of sorafenib and similar doses of TMZ (40 mg/m2/day vs 50 mg/m2/day). However the PFS at 6 months for each study is strikingly different. In the first study, the PFS at 6 months was only 9.4 %, while the second trial had a PFS at 6 months of 26 % [47, 48]. It is unclear why similar study designs yielded such dissimilar results. However, the results from both groups indicate that the combination of TMZ and sorafenib is of limited benefit to recurrent GBM patients. In the fifth study, 47 newly diagnosed patients who first underwent combined treatment with TMZ and RT were then treated with sorafenib and TMZ as maintenance therapy. Unfortunately, only about 60 % of patients successfully finished concurrent TMZ and RT and continued on to adjuvant TMZ and sorafenib. Though the PFS at 6 months for the entire treatment group was 50 %, the addition of sorafenib provided no survival benefit over historical controls [49]. Apparently, clinical sorafenib treatment does not have the same efficacy as what was found preclinically, especially when sorafenib efficacy is compared to historical controls.
7.2.1.2.2 B. Sunitinib (Sutent, Pfizer)
Sunitinib malate is a multi-targeted receptor tyrosine kinase inhibitor which inhibits PDGFRα and -β, VEGFR1 and 2, as well as RET, c-KIT, CSF-1R, and FLT3 [50]. Although multi-targeted kinases offer a wide range of inhibitor activity and thus provide more opportunities for efficacy, it becomes challenging, if not impossible, to thoroughly understand the mechanisms of action of a multi-targeted therapeutic. Differential efficacy can potentially be attributed to any or all of the known targets or the further downstream effects , making determining biomarkers and selecting patient populations extremely difficult.
7.2.1.2.2.1 Preclinical Evaluation
A 2007 study demonstrated a significant survival benefit in mice with U87 IC implanted tumors and treated with sunitinib at 80 mg/kg over placebo-treated mice (p < 0.0001) [50]. A second study by Joshi et al. reported that animals implanted with either 9L rat glioma or 020913 human GBM cells and treated with single agent sunitinib at 15 mg/kg did not have any survival benefit (p = 0.13 compared to placebo-treated mice) [14]. Although treatment with sunitinib in the first study showed survival benefit, the dose used is significantly higher than that used in patients in the clinical trials discussed below (37.5 mg), thus calling into question the reliability of those survival data.
7.2.1.2.2.2 Clinical Evaluation
Sunitinib has been tested in two Phase II trials, the first of which was conducted in newly diagnosed patients with unresectable GBM [51]. Patients were treated with sunitinib pre-RT and then concurrent with and post-RT treatment. Out of the 12 patients accrued for the study, only one (8.3 %) exhibited stable disease, while 11 (91.3 %) had disease progression on treatment. The lack of response in this patient population led to the study being terminated early. In a second Phase II study, patients with recurrent GBM or recurrent gliosarcoma were stratified by their previous exposure to bevacizumab into bevacizumab-resistant and bevacizumab-naïve groups [52]. Only 3/29 of the bevacizumab-naïve patients achieved radiographic response (10 %), while 0/29 of the bevacizumab-resistant patients did. Single agent sunitinib treatment provided no improvement in median time-to-progression or overall survival (1.6 and 3.8 months, respectively). Although the inhibition of multiple targets potentially provides more opportunities for target inhibition and treatment efficacy, neither sorafenib nor sunitinib appears to provide any significant benefit in GBM patients.
7.2.2 PI3K/mTor/Akt Pathway Inhibitors
Many of the downstream targets and pathways of the RTKs have been implicated as essential drivers in gliomagenesis, apoptosis evasion, and cell growth. One of the most targeted and dysregulated of those downstream pathways is the phosphoinositide 3-kinase/ mechanistic target of rapamycin (PI3K/mTor) pathway [53]. The PI3 kinases are subdivided into three different classes, though this chapter will only focus on the activity of the Class I PI3 kinases. Activation of a target receptor (either an RTK or G-protein-coupled receptor) induces interaction of PI3K, either directly or via a mediator, thus driving generation of phosphatidylinositol-3,4,5-trisphosphate (PIP3) [53, 54]. Figure 7.2 gives a simplified view of the signaling components involved in this pathway. Once generated, PIP3 interacts with Akt, causing conformational changes which expose the two activating phosphorylation sites, T308 and S473 [53]. Upon activation, Akt can then phosphorylate a wide range of proteins (as noted in Fig. 7.2), including tuberous sclerosis 2 (TSC2), a negative regulator of the mammalian target of rapamycin complex 1, or mTorc1 [53].
A component of both mTorc1 and mTorc2 , mTor is a serine/threonine kinase with broad activity in cell proliferation, growth, and survival [55]. Although mTor was initially named after its role in the cellular response to rapamycin, acute rapamycin treatment only affects mTor when complexed as mTorc1, a master regulator of protein synthesis (via 4EBP1 and S6K) and cellular nutrient response [55, 56]. mTorc2, on the other hand, is structurally affected only by chronic rapamycin treatment , although not all cell types respond uniformly [57]. mTorc2, unlike mTorc1, is insensitive to nutrient-driven signaling and instead plays a role in cell survival and cytoskeletal organization [55, 57].
7.2.2.1 A. Rapamycin (Sirolimus, Pfizer)
Rapamycin is a macrolide antibiotic with known activity as an antifungal, immunosuppressive, and antineoplastic [58]. All of these effects are due to rapamycin’s interactions with immunophilin FKBP12 , the complexing of which inhibits substrate recruitment and catalytic accessibility to mTor, though only when mTor is complexed as mTorc1 [58]. Rapamycin does have some marginal activity against mTorc2 as well [57].
7.2.2.1.1 Preclinical Evaluation
Fischer rats implanted IC with the RG2 cell line and treated with rapamycin had modest though statistically significant survival benefit (19.5 vs 24 days, p < 0.01) [59]. In a second in vivo study, CD1 nude mice implanted with U87MG cells also had a significant survival benefit with rapamycin treatment (p < 0.0001) [60]. Interestingly, mice implanted with patient-derived glioma lines and treated with single-agent rapamycin in studies from Zhuang et al. and Mendiburu-Eliçabe et al. failed to recapitulate the survival benefit noted in the U87 and RG2 studies, though Zhuang et al. did find that rapamycin may be a radiosensitizer as the combination of rapamycin and RT provided a significant survival benefit over RT alone (p < 0.005, RT vs rapamycin + RT) [61, 62].
7.2.2.1.2 Clinical Evaluation
An initial Phase I study in GBM patients selected for PTEN loss, which included specimen sampling to track drug concentration in the tumor, found that although tumor levels of rapamycin were sufficient to inhibit mTor, the magnitude of mTor inhibition achieved varied widely across all patients. Further, although Ki-67 expression (a marker of cell proliferation) was found to decrease in 7 of the 14 patients after treatment with rapamycin for 1 week, in direct correlation to the amount of mTor inhibition (p = 0.0047), the level of Ki-67 downregulation was not associated with intratumoral concentration of the drug [63]. A Phase II open-label study in 32 unselected, recurrent, heavily pretreated GBM patients treated with erlotinib and rapamycin showed no improvement in PFS (estimated PFS at 6 months was 3.1 %) or OS with the combination, and patient response was not correlated with PTEN expression [64]. Although response was correlated with p-Akt levels, the statistics were barely significant (p = 0.045). The comparison of preclinical and clinical rapamycin results indicates that the survival data from U87 and RG2 may not be as reliable as those data from the patient-derived lines.
7.2.2.2 B. Rad001 (Everolimus, Novartis)
Rad001 is a rapamycin ester analog (rapalog) which was designed to overcome rapamycin’s instability and insolubility. Similar to rapamycin, Rad001 is an immunosuppressant and an effective inhibitor of mTor activity , specifically when mTor is complexed as mTorc1 [59, 65]. In preclinical models Rad001 is well tolerated even at very high doses [66].
7.2.2.2.1 Preclinical Evaluation
In a panel of 17 orthotopically implanted, patient-derived lines, which included seven lines deficient in PTEN, only one line, GBM10, had a significant survival benefit with single-agent Rad001 treatment [67]. The only other line found to respond to Rad001 was a PTEN WT line (GBM22). Molecular analysis to determine specific mechanisms indicated that PTEN status was an insufficient biomarker for response of tumors to single agent Rad001. Although a second group found that single agent Rad001 treatment of animals with U87MG tumors provided significant survival benefit, the clinical relevance of immortalized lines to the patient experience is likely limited [68, 69].
7.2.2.2.2 Clinical Evaluation
Rad001 has been tested in one Phase II clinical trial, N057K, which added Rad001 to the combination of TMZ and RT along with adjuvant TMZ. The authors found that the inclusion of Rad001 in this newly diagnosed patient population provided no significant benefit when compared to historical controls (PFS at 6 months was 52 %) [70] (personal communication). Kreisl et al. in a Phase I study testing the combination of Rad001 and gefitinib in 22 recurrent patients found that, though the combination of the two drugs provided some stable disease (8) and partial responses (2), these responses were not durable (PFS at 6 months was 4.5 %). Although Rad001 is supposed to be an improved version of rapamycin, Rad001 fails to provide any more patient benefit than rapamycin does.
7.2.2.3 C. XL765 (Voxtalisib, Exelis)
XL765 is an ATP-competitive, selective, dual pan-Class I PI3K and mTor inhibitor undergoing testing in a wide variety of cancer types [71]. Although many mTor inhibitors , including the rapalogs, have been found to provide treatment benefit in some cancer types, selective inhibition of mTor often leads to compensatory upregulation of Akt, a consequence that is mitigated with usage of a dual PI3K/mTor inhibitor [54, 58, 72, 73].
7.2.2.3.1 Preclinical Evaluation
In vitro testing of XL765 conducted in five patient-derived xenograft lines treated with single agent XL765 showed excellent activity against all lines tested, while the combination of XL765 and TMZ showed evidence of synergistic activity in four out of the five lines tested [72]. Although in vivo testing of XL765 and TMZ in one line, GBM39, provided survival benefit over control (XL765 +TMZ vs control, 117 vs 55 days, p < 0.001), the combination of XL765 and TMZ failed to provide statistically significant benefit over the single agent TMZ arm (117 vs 83 days, p = 0.09).
7.2.2.3.2 Clinical Evaluation
XL765 has currently only been tested in one Phase I study in AGand GBM patients. Fifty-four patients who were either already receiving TMZ treatment (group 1) or were newly diagnosed (group 2) received XL765 either in combination with adjuvant TMZ (group 1) or along with RT and TMZ combined therapy dosing (group 2). Of the 47 evaluable patients, XL765 produced partial response in only two patients (overall response rate of 4 %) and stable disease in 32 patients (68 %) [71]. Inhibition of both PI3K and mTor does not appear to be a useful sensitization strategy, though further studies are necessary to completely rule out dual PI3K/mTor inhibitors as a possible treatment for GBM patients.
7.2.3 Inhibitors of Cell Cycle Progression
The cell cycle and DNA replication are tightly regulated by proteins that either inhibit or potentiate progression of cell division [74]. Retinoblastoma (pRb) and p53 are among the key proteins guiding cell cycle progression, with cell cycle dysregulation, either via the pRb or p53 pathway, a common occurrence in GBM and considered to be a critical step in gliomagenesis (Fig. 7.1) [3]. The high prevalence of cell cycle dysregulation in GBM has generated interest in creating pharmacological agents which can disturb various components of this complex system, though currently very few clinical studies have been conducted with cell cycle inhibitors in GBM.
7.2.3.1 G1/S Inhibitors
The retinoblastoma family consists of three members: Rb (pRb)/p105, p107, and Rb2/p130, all of which are cell cycle regulators via their inhibitory activity against the E2F family of transcription factors [75]. pRb, when hypophosphorylated, binds and inhibits the transcription factor E2F [75]. pRb inhibition is relieved by activated cyclin-dependent kinase 4/6 (CDK4/6) complexed with cyclin D1, which phosphorylates pRb, releasing E2F and ultimately allowing for progression through G1 into S-phase and finally cell division [75]. Inactivation or loss of pRb, which occurs in approximately 25 % of GBM, allows for persistent activation of the E2F family of transcripts and uninhibited cell division [75, 76]. In GBM with intact pRb, deletion of p16 and p14, negative regulators of the complexing of CDK4/6-cyclin D, along with amplification of CDK4/6 has been shown to mediate persistent hyperphosphorylation of pRb [3, 77]. Inhibition of G1/S components is an attractive sensitization strategy in GBM due to the high prevalence of cell cycle dysregulation found in these tumors. Several G1/S inhibitors, including roscovitine, palbociclib, abemaciclib, flavopiridol, and many others, have been tested in a variety of cancers, though only one, palbociclib, has been FDA approved for use in breast cancers.
7.2.3.1.1 A. Flavopiridol (Alvocidib, Tolero Pharmaceuticals)
Flavopiridol is a multi-targeted phosphokinase inhibitor with activity against several cyclin-dependent kinases (CDK1, 2, 4, 6, and 7) as well as some receptor tyrosine kinases and signal transducing kinases (i.e., Erk-1) [78]. Flavopiridol exhibits cytotoxicity against both actively dividing cells and resting cells [78]. Though the drug is currently not FDA approved for treatment of any neoplasm, it was designated as an orphan drug for acute myeloid leukemia by the FDA in 2014.
7.2.3.1.1.1 Preclinical Evaluation
Only one in vivo study in the murine GL261 line has been conducted with flavopiridol [79]. In this study, animals with IC GL261 tumors were treated days 7–11 after injection and then harvested on days 14, 21, and 28 for assessment of tumor volume, microvessel density, and apoptosis (by TUNEL). Though there was a significant decrease in tumor volume in mice treated with flavopiridol on day 14 post-injection (N = 6, p < 0.02, in comparison to control treated mice), by days 21 and 28, there was no longer any significant difference in tumor volume. Although these data indicate, as the authors note, that flavopiridol is capable of reaching the tumor and penetrating the brain, further long-term survival studies in primary human lines are necessary to accurately represent flavopiridol efficacy preclinically.
7.2.3.1.1.2 Clinical Evaluation
Currently, no clinical trials for GBM have been conducted with flavopiridol.
7.2.3.1.2 B. Palbociclib (PD0332991, Pfizer)
Palbociclib is an inhibitor of both CDK4 and CDK6-cyclin D1 kinase activity , with equal inhibition achieved for each kinase [80]. Tumors without pRb expression have been found to be resistant to palbociclib, while those that express pRb are potentially responsive to treatment [80].
7.2.3.1.2.1 Preclinical Evaluation
Single agent administration of palbociclib to mice with either orthotopically implanted U87 or GBM39 xenografts yielded significant benefit in comparison to placebo treatment (p < 0.001) [81]. In the same study, the combination of radiation and palbociclib in U87 implanted mice further improved survival, though only when RT was administered after palbociclib (p = 0.01 for single agent RT and palbociclib compared to the combination of palbociclib pretreatment and RT posttreatment). However, the combination of TMZ and palbociclib in a separate cohort of mice did not provide added benefit over the cyclical dosing of TMZ alone (78.1 vs 81.4 days, p = 0.970). A second study conducted in the patient-derived GBM6 line also showed some slight though statistically significant (p < 0.01) benefit with single agent treatment of palbociclib [82]. Data from these patient-derived lines indicate that palbociclib may have modest efficacy in GBM patients.
7.2.3.1.2.2 Clinical Evaluation
Only one clinical trial in recurrent GBM or gliosarcoma which is Rb positive is currently ongoing (ClinicalTrials.gov Identifier: NCT01227434). At this time no results are available.
7.2.3.2 G2/M Inhibitors
p53 is a key regulator of the cell cycle and a known tumor suppressor. Genotoxic and cytotoxic stress drive p53 to stall cell cycle progression, mainly at the G1/S checkpoint, or induce apoptosis [83]. Figure 7.4 is a simplified illustration of a few of the proteins responsible for DNA damage sensing and cell cycle progression, including p53. Mutation or deletion of p53 is found in approximately 35 % of GBM, though p53 pathway alterations are detected in 70 % of samples tested [3]. MDM2, a negative regulator of p53 function, keeps these cell cycle and proapoptotic pathways in check, and in the case of GBMs with intact p53, MDM2 amplification (found in approximately 11 % of GBMs) inhibits p53 activity [3]. p53-null tumors thus rely heavily on the G2/M checkpoint and the activity of the checkpoint proteins Wee1 and Myt1 to maintain genomic integrity [84–86].

The cell cycle and checkpoint proteins . Induction of DNA damage in the form of a DSB or ssDNA activates DNA damage repair proteins causing arrest at one of the cell cycle checkpoints via p53. Cells lacking p53 activity rely heavily on the cell cycle arrest at G2 driven by Wee1 to maintain genomic integrity [124]. Open access from InTechOpen
Wee1 and Myt1 are tyrosine kinases in the serine-threonine family of protein kinases that are key regulators of mitotic entry [85]. Wee1’s kinase function is specifically directed at phosphorylation of CDK1 at tyrosine 15, an inhibitory phosphorylation that halts cell cycle progression at G2/M, as illustrated in Fig. 7.4 [87]. Once the G2/M checkpoint is successfully completed, Cdc25 reactivates CDK1/Cyclin B by removal of the tyrosine 15 phosphorylation. CDK1/Cyclin B in turn targets Wee1 for hyperphosphorylation at threonine 293, marking it for translocation outside of the nucleus and degradation [87, 88].
In p53-null tumors , pharmacological inhibition of Wee1 may be particularly effective due to synthetic lethality [85]. The idea of synthetic lethality posits that loss or disruption of one gene, “A,” can be compensated for by a second gene “B.” However loss or disruption of both genes leads to cell death [89]. Thus p53-null tumors which are incapable of maintaining genomic integrity by arresting at the G1/S checkpoint along with pharmacological inhibition of Wee1 (leading to loss of a functional G2/M checkpoint) could potentially cause synthetic lethality. Additional cytotoxic damage introduced after loss of the G1 and G2 checkpoints could potentially allow for propagation of highly lethal adducts, such as those induced by TMZ and other cytotoxic agents. Thus, the combination of a cytotoxic agent like TMZ and a Wee1 inhibitor poses an attractive sensitization strategy to increase efficacy in GBM patients.
7.2.3.2.1 A. MK-1775 (AZD-1775, Astra-Zeneca)
MK-1775 is an ATP-competitive, selective inhibitor of Wee1 with single agent in vitro activity in a wide range of tumor cell lines [90], while the combination of MK-1775 and cytotoxic agents such as TMZ and gemcitabine further increases efficacy [91, 92]. Though results from Guertin et al. and Pokorny et al. indicate that p53 status did not predict for single agent or combination efficacy with TMZ, Rajeshkumar et al. did find that p53 status was relevant for the combination of gemcitabine and MK-1775 in pancreatic cancer lines.
7.2.3.2.1.1 Preclinical Evaluation
MK-1775 treatment of the clinically relevant xenografts (GBM22 and GBM12) implanted orthotopically failed to provide any survival benefit over placebo (36 vs 34 days p = 0.15) [91]. MALDI-MSI data from animals with either IC or flank tumors treated with a single dose of 200 mg/kg MK-1775 indicated that exposure levels in the brain were heterogeneous. Pharmacokinetic data further indicated that drug levels achieved in the brain (maximum 5 %) were likely insufficient to provide therapeutic benefit, even in the highly sensitive GBM22 line. Administration of MK-1775 to mice bearing GBM22 heterotopic tumors provided survival benefit over placebo (median survival 38 vs 30 days, p = 0.01), especially when combined with TMZ (median survival with TMZ of 91 days vs 240 days with the combination of TMZ and MK-1775 treated with a protracted dosing schedule, p = 0.02).
7.2.3.2.1.2 Clinical Evaluation
Although there are no Wee1 inhibitors FDA approved for treatment of any malignancy, over 18 clinical trials (one of which is being conducted in recurrent and newly diagnosed GBM patients) in a wide range of cancer types are listed on clinicaltrials.gov. Preclinical studies of Wee1 inhibitors as a single agent and in combination with FDA-approved cytotoxic agents have shown promising results; thus Wee1 inhibitors may very well be approved for clinical use in the near future [92, 93]. Understanding the Wee1 specific pathways and potential biomarkers as well as the reasons for differential response and lack of brain efficacy as noted from the study above will be essential for successful clinical application of Wee1 inhibitors in the future.
7.3 Resistance Mechanisms to Small-Molecule Inhibitors
Although several different compounds targeting a wide variety of pathways considered to be essential for GBM proliferation and survival have been tested both preclinically and clinically, none have provided benefit significant enough to warrant FDA approval. There are at least six main reasons for the lack of clinical efficacy seen among the drugs noted here. The first is the issue of pathway redundancy, while the second is the generation of secondary mutations with small-molecule treatment. The third highlights the difficulty of targeting extremely complex and incompletely understood pathways. The fourth point will focus on specific differences in EGFR mutations between NSCLC and GBM and why inhibitors, such as erlotinib and gefitinib, may be unrealistic options for GBM treatment. The fifth point covers the heterogeneous nature of GBM tumors, and the sixth will consider the unique environment presented by the brain and the obstacles that must be overcome when attempting to introduce novel therapeutics.
First, pathway redundancy is a key impediment to development of maximally effective drugs. Although novel therapeutics may target proteins and pathways considered key for cell survival, compensatory upregulation of untargeted pathways provides cells the ability to utilize alternatives which are still available. For instance, many NSCLC patients undergoing treatment with EGFR inhibitors regularly develop resistance when cells upregulate other pathways including c-MET, IGFR, VEGFR, and PDGFR [17]. Systems-level studies of the ErbB family and other key mitogenic factors commonly found to be upregulated in GBM have shown that these factors have “modularity” and “show redundancy of regulatory circuits” [94]. In fact, neither high brain accumulation of targeted drugs nor effective downregulation of drug target can guarantee patient efficacy [94]. Results from a Phase II trial definitively show that gefitinib crosses into the brain in very high concentrations (brain concentration 22 times higher than plasma) and is capable of efficiently reducing EGFR phosphorylation [94]. Yet EGFR perturbation did not lead to modulation of any downstream signal transducers, and in fact, EGFR phosphorylation was not even found to be a factor for “overall activation of the pathway” [94]. Similar issues have also been noted in hepatocellular carcinoma patient resistance to sorafenib, in prostate cancer patient resistance to Rad001, and in breast cancer patient resistance to lapatinib, in which cross talk and upregulation of untargeted or compensatory pathways are recognized as key mechanisms of acquired resistance [73, 95, 96]. If further analysis of the EGF pathway concludes that there is redundancy and modularity, those findings will significantly affect future attempts to target EGFR with single agents such as gefitinib and erlotinib.
A second mechanism of resistance is the generation of secondary mutations after initial tumor treatment. Along with upregulation of compensatory pathways, approximately 50 % of NSCLC patients treated with EGFR inhibitors will become resistant by emergence of a T790M mutation, which replaces the threonine for a bulkier methionine, preventing binding of the inhibitor, while maintaining catalytic activity [17, 97]. Similarly TMZ resistance in GBM patients has been linked to mutation of the mismatch repair gene, MSH6, while sunitinib resistance in gastrointestinal stromal tumors has been correlated with secondary mutation of KIT [98, 99]. Since regular biopsy of patient tumors is unrealistic in GBM, assessment of tumors at recurrence, when possible, along with generation of patient-derived xenografts with resistant phenotypes will allow for a better understanding of key mutations leading to therapeutic resistance.
A third mechanism of resistance is the complexity of the pathways targeted and the difficulty of correctly implicating biomarkers. In 2005, Mellinghoff et al. reported that PTEN status (WT or deleted) was a predictive marker for EGFR inhibitor efficacy. However in all of the EGFR and PI3K inhibitor trials described above in which PTEN status was noted, PTEN status failed to definitively correspond with inhibitor efficacy. Other groups have more recently implicated phosphorylation of PTEN at tyrosine 240 [100] and upregulation of EGFRvIII and PI3Kp110δ [101] as possible drivers of RTKi resistance as opposed to the more binary PTEN status. These findings indicate that our current level of pathway understanding, particularly for EGFR, is insufficient. In many of these failed clinical trials, a better understanding of molecular markers of response and the pathways targeted could lead to improved patient selection and study results.
A fourth resistance mechanism focuses specifically on the unique EGFR mutations found in GBM in comparison to those found in NSCLC tumors. Although erlotinib and gefitinib have both been FDA approved for treatment of NSCLC, a tumor with a high rate of EGFR activity, similar to GBM, efficacy achieved with these same inhibitors in GBM remains disappointing [19, 102, 103]. In-depth analysis of clinical lung cancer samples indicates that these tumors often have a high percentage of EGFR mutations, similar to GBM. However, the majority of EGFR mutations found in lung cancers are kinase domain (KD) mutations, as opposed to the extracellular domain (EC) mutations more commonly found in GBM [16]. Interestingly, a comparison of lung cancers that had either KD or EC mutations found that lines with EC mutations, when treated with gefitinib or erlotinib, were significantly more resistant to both erlotinib and gefitinib , in comparison to lines with KD mutations [16]. Although the idea of oncogene addiction posits that GBM cells that are EGFRvIII are addicted to the EGFR signaling pathway and EGFR inhibition should provide benefit, erlotinib and other similar inhibitors have not delivered [16]. The unique mutations found specifically in GBM could play an essential role in understanding this lack of efficacy.
Fifth, GBM is, by definition, a highly heterogeneous tumor [3, 4, 104]. Although whole genome sequencing and histopathology allow clinicians to categorize each tumor by its specific genetic abnormalities, sequencing and pathology results are limited by the samples taken [105, 106]. Unrepresented or underrepresented subpopulations of cells with different expression profiles which do not respond to the targeted treatment are a source for tumor resistance [107]. Although newer techniques such as single cell RNAseq allow a deeper understanding of tumor clonality and the possibility of improved chemotherapeutic targeting, significant cost and the issue of sampling bias mean that RNAseq is not yet ready for regular clinical use [105]. Further, all methods for categorizing tumors provide only a snapshot of the tumor as it is in the instant that the tissue samples are taken. Some of the studies noted above analyzed treatment efficacy based upon patient samples that were taken at initial surgery [29, 63, 64]. However a patient who presents at recurrence and is treated with a targeted therapy based upon limited and outdated pathology or sequencing data may not respond to treatment because the tumor has changed and the collected samples are not actually representative of the current tumor. Regular biopsy of GBMs is an unrealistic option; thus the development of assays that can be used to regularly monitor tumor expression patterns, via tumor-specific circulating DNA, for instance, is necessary to improve real-time molecular characteristics [108]. In essence, though targeted therapeutics are potentially promising options for GBM patients, limitations on histopathology and sequencing sample acquisition mean actual benefits are still quite limited.
The sixth and arguably one of the most important mechanisms of resistance is the unique environment of the brain and the blood-brain barrier (BBB) as significant impediments to effective drug delivery . Although the BBB provides essential protection to the normal brain against potential neurotoxins and harmful cells, it also acts as a safe haven for GBM tumors, keeping out commonly used chemotherapeutics [109]. Transcellular and paracellular passage into the brain parenchyma is regulated by two main mechanisms: the ATP binding cassette (ABC) transporters , located luminally on the endothelial cells, and tight junctions and adherens junctions present between the endothelial cells which constitute the brain vasculature, along with pericytes, astrocytes, and perivascular macrophages (illustrated in Fig. 7.5) [109]. Brain access is thus limited to diffusion of “very small or gaseous molecules (e.g., water, carbon dioxide)”; passive diffusion of larger solutes, which is limited by lipid solubility, electrical charge, and molecular weight; and active transport via specific solute carriers [109].

A diagram of various components of the blood-brain barrier. (a) Schematic of various cell types which line the blood vessels and constitute the blood-brain barrier, pericytes, endothelial cells, and astrocytes. (b) A detailed schematic of the luminal location of the drug pumps (p-gp and MRPs) along with the tight junctions (TJ) and adherens junctions (AJ) at the endothelial cell junctions which line the blood vessels. Reprinted with permission from Macmillan Publishers Ltd: Nature Reviews Drug Discovery [125], copyright (2007)
The ABC transporters p-glycoprotein (p-gp or multidrug resistance protein 1 (MDR1), also called permeability glycoprotein/ABCB1) and breast cancer resistance protein 1 (BCRP1/ABCG2) interact with and limit transcellular permeability of therapeutics present in the vasculature and prevent access to the brain by actively pumping out substrates. All but two (XL765 and MK-1775) of the small-molecule inhibitors discussed above have been shown to be a substrate for at least one of the drug pumps found in the brain (data summarized in Table 7.1) [66, 91, 102, 103, 110–116]. Inhibitors that are drug pump substrates may provide excellent therapeutic benefit in vitro (as noted for the Wee1 inhibitor MK-1775, [91]). However, substrates simply cannot accumulate in sufficient levels to have a meaningful therapeutic effect in the brain (hence the low brain accumulation of 5 % noted in PK results with MK-1775) [91]. Most GBM cells express P-gp at the level of the normal brain, with some evidence indicating that a subgroup of glioma cells express CD133, a proposed stem cell marker, along with increased expression of BCRP1 [107, 117]. Thus there may be a population of stem cells in the tumor which are able to evade drug effects by over-expression of drug pumps, ensuring propagation of the tumor even after chemotherapeutic treatment. Studies which take into consideration the basal expression of drug pumps in patients, as well as whether individual compounds bind to drug pumps, may be necessary for improved efficacy in the future.
The second component of the BBB, the tight junctions, also poses a significant barrier to effective chemotherapy delivery [109]. The tight junctions consist of claudin-5, occludin, and junctional adhesion molecules (JAMs) along with the intracellular adaptor proteins such as ZO-1, which link the transmembrane tight junction components with the actin cytoskeleton. The adherens junctions, of which VE-cadherin is one of the most important components, in combination with the tight junctions, connect the endothelial cells together and provide a relatively impermeable barrier in the normal brain [109, 118]. Although a compromised (“leaky”) BBB is considered to be a hallmark of GBM, careful studies of the brain environment actually indicate that patient tumors present with a heterogeneous distribution of BBB openness, with tumor cells found in areas of open and closed BBB [109]. Along with differential BBB integrity, Ortensi et al. also found that tumors often have increased expression of pro-invasive and stem cell markers in the tumor rim, the outer edge of tumor cells, in comparison to the more open and central tumor core [119]. It is possible that invading GBM cells are capable of adapting to the brain environment surrounding them and upregulating factors that improve their likelihood of survival [119]. It is likely these cells in the rim that prove to be difficult to reach and a significant barrier to effective small-molecule inhibitor treatment.
7.4 Future Perspectives
Considering the lack of promising results achieved with just the few chemotherapeutics described here, it is clear that new methods of drug delivery, discovery, and BBB penetration need to be created, along with a better understanding of the signaling pathways and factors found specifically in GBM tumors. To that end, a variety of modalities for improving brain access are being considered from microbubble injections targeted with focused ultrasound causing vibrations that can temporarily open the BBB [120] and bradykinin receptor agonists [121] to the drug pump inhibitors elacridar and tariquidar, which directly interact with and inhibit the substrate binding abilities of both p-gp and BCRP1 [122]. Although none of these modalities have yet been found to effectively overcome the BBB and allow for improved drug delivery, preclinical and clinical studies are still ongoing. Another promising option is to design inhibitors specifically for use in the brain with characteristics that allow for improved brain access and efficacy [123]. Regardless of the methodology, unless cancer researchers, clinicians, and drug developers can start to rethink the approach to GBM treatment, promising therapeutics will continue to fail at the clinical level.
Abbreviations
- AG:
-
Anaplastic glioma
- CDK:
-
Cyclin-dependent kinase
- DSBs:
-
Double-stranded breaks
- EGF:
-
Epidermal growth factor
- ErbB:
-
Erythroblastic leukemia viral oncogene homolog
- mTor:
-
Mammalian target of rapamycin
- mTorc1-2:
-
Mammalian target of rapamycin complex 1 and 2
- PDGF:
-
Platelet-derived growth factor
- PFS:
-
Progression-free survival
- PTEN:
-
Phosphatase and tensin homolog
- RT:
-
Radiation therapy
- RTK:
-
Receptor tyrosine kinase
- TSC2:
-
Tuberous sclerosis 2
References
Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma--are we there yet? Neuro Oncol. 2013;15:4–27.
Koukourakis GV, Kouloulias V, Zacharias G, Papadimitriou C, Pantelakos P, Maravelis G, Fotineas A, Beli I, Chaldeopoulos D, Kouvaris J. Temozolomide with radiation therapy in high grade brain gliomas: pharmaceuticals considerations and efficacy; a review article. Molecules. 2009;14:1561–77.
Network CGAR. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8.
Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, Agarwalla PK, Chheda MG, Campos B, Wang A, Brennan C, Ligon KL, Furnari F, Cavenee WK, Depinho RA, Chin L, Hahn WC. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012;26:756–84.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54.
Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, Lee C, Wagner SN, Li C, Golub TR, Rimm DL, Meyerson ML, Fisher DE, Sellers WR. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–22.
Peereboom DM, Shepard DR, Ahluwalia MS, Brewer CJ, Agarwal N, Stevens GH, Suh JH, Toms SA, Vogelbaum MA, Weil RJ, Elson P, Barnett GH. Phase II trial of erlotinib with temozolomide and radiation in patients with newly diagnosed glioblastoma multiforme. J Neurooncol. 2010;98:93–9.
Raizer JJ, Abrey LE, Lassman AB, Chang SM, Lamborn KR, Kuhn JG, Yung WK, Gilbert MR, Aldape KA, Wen PY, Fine HA, Mehta M, Deangelis LM, Lieberman F, Cloughesy TF, Robins HI, Dancey J, Prados MD; North American Brain Tumor Consortium. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro Oncol. 2010;12;95–103.
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O’Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L; TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77.
Appert-Collin A, Hubert P, Crémel G, Bennasroune A. Role of ErbB receptors in cancer cell migration and invasion. Front Pharmacol. 2015;6:283.
Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16.
Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH, Chute DJ, Riggs BL, Horvath S, Liau LM, Cavenee WK, Rao PN, Beroukhim R, Peck TC, Lee JC, Sellers WR, Stokoe D, Prados M, Cloughesy TF, Sawyers CL, Mischel PS. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–24.
Brown PD, Krishnan S, Sarkaria JN, Wu W, Jaeckle KA, Uhm JH, Geoffroy FJ, Arusell R, Kitange G, Jenkins RB, Kugler JW, Morton RF, Rowland Jr KM, Mischel P, Yong WH, Scheithauer BW, Schiff D, Giannini C, Buckner JC, North Central Cancer Treatment Group Study N0177. Phase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: North Central Cancer Treatment Group Study N0177. J Clin Oncol. 2008;26:5603–9.
Joshi AD, Loilome W, Siu IM, Tyler B, Gallia GL, Riggins GJ. Evaluation of tyrosine kinase inhibitor combinations for glioblastoma therapy. PLoS One. 2012;7, e44372.
Gan HK, Cvrljevic AN, Johns TG. The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBS J. 2013;280:5350–70.
Vivanco I, Robins HI, Rohle D, Campos C, Grommes C, Nghiemphu PL, Kubek S, Oldrini B, Chheda MG, Yannuzzi N, Tao H, Zhu S, Iwanami A, Kuga D, Dang J, Pedraza A, Brennan CW, Heguy A, Liau LM, Lieberman F, Yung WK, Gilbert MR, Reardon DA, Drappatz J, Wen PY, Lamborn KR, Chang SM, Prados MD, Fine HA, Horvath S, Wu N, Lassman AB, DeAngelis LM, Yong WH, Kuhn JG, Mischel PS, Mehta MP, Cloughesy TF, Mellinghoff IK. Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2012;2:458–71.
Luo M, Fu LW. Redundant kinase activation and resistance of EGFR-tyrosine kinase inhibitors. Am J Cancer Res. 2014;4:608–28.
Robinson KW, Sandler AB. EGFR tyrosine kinase inhibitors: difference in efficacy and resistance. Curr Oncol Rep. 2013;15:396–404.
Bronte G, Rolfo C, Giovannetti E, Cicero G, Pauwels P, Passiglia F, Castiglia M, Rizzo S, Vullo FL, Fiorentino E, Van Meerbeeck J, Russo A. Are erlotinib and gefitinib interchangeable, opposite or complementary for non-small cell lung cancer treatment? Biological, pharmacological and clinical aspects. Crit Rev Oncol Hematol. 2014;89:300–13.
Dowell JJ, Minna D, Kirkpatrick P. Erlotinib hydrochloride. Nat Rev Drug Discov. 2005;4:13–4.
Sarkaria JN, Yang L, Grogan PT, Kitange GJ, Carlson BL, Schroeder MA, Galanis E, Giannini C, Wu W, Dinca EB, James CD. Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel. Mol Cancer Ther. 2007;6:1167–74.
Yung WK, Vredenburgh JJ, Cloughesy TF, Nghiemphu P, Klencke B, Gilbert MR, Reardon DA, Prados MD. Safety and efficacy of erlotinib in first-relapse glioblastoma: a phase II open-label study. Neuro Oncol. 2010;12:1061–70.
Lassman AB, Rossi MR, Raizer JJ, Abrey LE, Lieberman FS, Grefe CN, Lamborn K, Pao W, Shih AH, Kuhn JG, Wilson R, Nowak NJ, Cowell JK, DeAngelis LM, Wen P, Gilbert MR, Chang S, Yung WA, Prados M, Holland EC. Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: tissue analysis from North American Brain Tumor Consortium Trials 01-03 and 00-01. Clin Cancer Res. 2005;11:7841–50.
van den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, Clement PM, Frenay M, Campone M, Baurain JF, Armand JP, Taphoorn MJ, Tosoni A, Kletzl H, Klughammer B, Lacombe D, Gorlia T. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol. 2009;27:1268–74.
Bareford MD, Hamed HA, Allegood J, Cruickshanks N, Poklepovic A, Park MA, Ogretmen B, Spiegel S, Grant S, Dent P. Lapatinib and obatoclax kill tumor cells through blockade of ERBB1/3/4 and through inhibition of BCL-XL and MCL-1. Mol Pharmacol. 2012;81:748–58.
Santa-Maria CA, Nye L, Mutonga MB, Jain S, Gradishar WJ. Management of Metastatic HER2-Positive Breast Cancer: Where Are We and Where Do We Go From Here? Oncology (Williston Park). 2016;30:148–55.
Mineo JF, Bordron A, Baroncini M, Maurage CA, Ramirez C, Siminski RM, Berthou C, Dam HP. Low HER2-expressing glioblastomas are more often secondary to anaplastic transformation of low-grade glioma. J Neurooncol. 2007;85:281–7.
Thiessen B, Stewart C, Tsao M, Kamel-Reid S, Schaiquevich P, Mason W, Easaw J, Belanger K, Forsyth P, McIntosh L, Eisenhauer E. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother Pharmacol. 2010;65:353–61.
Reardon DA, Groves MD, Wen PY, Nabors L, Mikkelsen T, Rosenfeld S, Raizer J, Barriuso J, McLendon RE, Suttle AB, Ma B, Curtis CM, Dar MM, de Bono J. A phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma. Clin Cancer Res. 2013;19:900–8.
Kim ST, Lee J, Kim JH, Won YW, Sun JM, Yun J, Park YH, Ahn JS, Park K, Ahn MJ. Comparison of gefitinib versus erlotinib in patients with nonsmall cell lung cancer who failed previous chemotherapy. Cancer. 2010;116:3025–33.
Lim SH, Lee JY, Sun JM, Ahn JS, Park K, Ahn MJ. Comparison of clinical outcomes following gefitinib and erlotinib treatment in non-small-cell lung cancer patients harboring an epidermal growth factor receptor mutation in either exon 19 or 21. J Thorac Oncol. 2014;9:506–11.
Yoshida T, Yamada K, Azuma K, Kawahara A, Abe H, Hattori S, Yamashita F, Zaizen Y, Kage M, Hoshino T. Comparison of adverse events and efficacy between gefitinib and erlotinib in patients with non-small-cell lung cancer: a retrospective analysis. Med Oncol. 2013;30:349.
Jacobs VL, Valdes PA, Hickey WF, De Leo JA. Current review of in vivo GBM rodent models: emphasis on the CNS-1 tumour model. ASN Neuro. 2011;3, e00063.
Qi L, Singh RP, Lu Y, Agarwal R, Harrison GS, Franzusoff A, Glode LM. Epidermal growth factor receptor mediates silibinin-induced cytotoxicity in a rat glioma cell line. Cancer Biol Ther. 2003;2:526–31.
Franceschi E, Cavallo G, Lonardi S, Magrini E, Tosoni A, Grosso D, Scopece L, Blatt V, Urbini B, Pession A, Tallini G, Crinò L, Brandes AA. Gefitinib in patients with progressive high-grade gliomas: a multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO). Br J Cancer. 2007;96:1047–51.
Rich JN, Reardon DA, Peery T, Dowell JM, Quinn JA, Penne KL, Wikstrand CJ, Van Duyn LB, Dancey JE, McLendon RE, Kao JC, Stenzel TT, Ahmed Rasheed BK, Tourt-Uhlig SE, Herndon 2nd JE, Vredenburgh JJ, Sampson JH, Friedman AH, Bigner DD, Friedman HS. Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol. 2004;22:133–42.
Uhm JH, Ballman KV, Wu W, Giannini C, Krauss JC, Buckner JC, James CD, Scheithauer BW, Behrens RJ, Flynn PJ, Schaefer PL, Dakhill SR, Jaeckle KA. Phase II evaluation of gefitinib in patients with newly diagnosed Grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int J Radiat Oncol Biol Phys. 2004;80:347–53.
Goel HL, Mercurio AM. VEGF targets the tumour cell. Nat Rev Cancer. 2013;13:871–82.
Heldin CH. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun Signal. 2013;11:97.
Nazarenko I, Hede SM, He X, Hedrén A, Thompson J, Lindström MS, Nistér M. PDGF and PDGF receptors in glioma. Ups J Med Sci. 2012;117:99–112.
Rubenstein JL, Kim J, Ozawa T, Zhang M, Westphal M, Deen DF, Shuman MA. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia. 2000;2:306–14.
Adnane L, Trail PA, Taylor I, Wilhelm SM. Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006;407:597–612.
Carra E, Barbieri F, Marubbi D, Pattarozzi A, Favoni RE, Florio T, Daga A. Sorafenib selectively depletes human glioblastoma tumor-initiating cells from primary cultures. Cell Cycle. 2013;12:491–500.
Siegelin MD, Raskett CM, Gilbert CA, Ross AH, Altieri DC. Sorafenib exerts anti-glioma activity in vitro and in vivo. Neurosci Lett. 2010;478:165–70.
Lee EQ, Kuhn J, Lamborn KR, Abrey L, DeAngelis LM, Lieberman F, Robins HI, Chang SM, Yung WK, Drappatz J, Mehta MP, Levin VA, Aldape K, Dancey JE, Wright JJ, Prados MD, Cloughesy TF, Gilbert MR, Wen PY. Phase I/II study of sorafenib in combination with temsirolimus for recurrent glioblastoma or gliosarcoma. North American Brain Tumor Consortium study 05-02. Neuro Oncol. 2012;14:1511–8.
Galanis E, Anderson SK, Lafky JM, Uhm JH, Giannini C, Kumar SK, Kimlinger TK, Northfelt DW, Flynn PJ, Jaeckle KA, Kaufmann TJ, Buckner JC. Phase II study of bevacizumab in combination with sorafenib in recurrent glioblastoma (N0776): a north central cancer treatment group trial. Clin Cancer Res. 2013;19:4816–23.
Reardon DA, Vredenburgh JJ, Desjardins A, Peters K, Gururangan S, Sampson JH, Marcello J, Herndon 2nd JE, McLendon RE, Janney D, Friedman AH, Bigner DD, Friedman HS. Effect of CYP3A-inducing anti-epileptics on sorafenib exposure: results of a phase II study of sorafenib plus daily temozolomide in adults with recurrent glioblastoma. J Neurooncol. 2011;101:57–66.
Zustovich F, Landi L, Lombardi G, Porta C, Galli L, Fontana A, Amoroso D, Galli C, Andreuccetti M, Falcone A, Zagonel V. Zagonel. Sorafenib plus daily low-dose temozolomide for relapsed glioblastoma: a phase II study. Anticancer Res. 2013;33:3487–94.
Hainsworth JD, Ervin T, Friedman E, Priego V, Murphy PB, Clark BL, Lamar RE. Concurrent radiotherapy and temozolomide followed by temozolomide and sorafenib in the first-line treatment of patients with glioblastoma multiforme. Cancer. 2010;116:3663–9.
de Boüard S, Herlin P, Christensen JG, Lemoisson E, Gauduchon P, Raymond E, Guillamo JS. Antiangiogenic and anti-invasive effects of sunitinib on experimental human glioblastoma. Neuro Oncol. 2007;9:412–23.
Balaña C, Gil MJ, Perez P, Reynes G, Gallego O, Ribalta T, Capellades J, Gonzalez S, Verger E. Sunitinib administered prior to radiotherapy in patients with non-resectable glioblastoma: results of a phase II study. Target Oncol. 2014;9:321–9.
Kreisl TN, Smith P, Sul J, Salgado C, Iwamoto FM, Shih JH, Fine HA. Continuous daily sunitinib for recurrent glioblastoma. J Neurooncol. 2013;111:41–8.
Akhavan D, Cloughesy TF, Mischel PS. mTOR signaling in glioblastoma: lessons learned from bench to bedside. Neuro Oncol. 2010;12:882–9.
Dienstmann R, Rodon J, Serra V, Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 2014;13:1021–31.
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93.
Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–44.
Schreiber KH, Ortiz D, Academia EC, Anies AC, Liao CY, Kennedy BK. Rapamycin-mediated mTORC2 inhibition is determined by the relative expression of FK506-binding proteins. Aging Cell. 2015;14:265–73.
Coppin C. Everolimus: the first approved product for patients with advanced renal cell cancer after sunitinib and/or sorafenib. Biologics. 2010;4:91–101.
Mineharu Y, Kamran N, Lowenstein PR, Castro MG. Blockade of mTOR signaling via rapamycin combined with immunotherapy augments antiglioma cytotoxic and memory T-cell functions. Mol Cancer Ther. 2014;13:3024–36.
Arcella A, Biagioni F, Antonietta Oliva M, Bucci D, Frati A, Esposito V, Cantore G, Giangaspero F, Fornai F. Rapamycin inhibits the growth of glioblastoma. Brain Res. 2013;1495:37–51.
Mendiburu-Eliçabe M, Gil-Ranedo J, Izquierdo M. Efficacy of rapamycin against glioblastoma cancer stem cells. Clin Transl Oncol. 2014;16:495–502.
Zhuang W, Li B, Long L, Chen L, Huang Q, Liang Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int J Cancer. 2011;129:2720–31.
Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, Hsueh T, Chen Y, Wang W, Youngkin D, Liau L, Martin N, Becker D, Bergsneider M, Lai A, Green R, Oglesby T, Koleto M, Trent J, Horvath S, Mischel PS, Mellinghoff IK, Sawyers CL. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5, e8.
Reardon DA, Desjardins A, Vredenburgh JJ, Gururangan S, Friedman AH, Herndon JE, Marcello J, Norfleet JA, McLendon RE, Sampson JH, Friedman HS. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J Neurooncol. 2010;96:219–30.
Witzke O, Sommerer C, Arns W. Everolimus immunosuppression in kidney transplantation: What is the optimal strategy? Transplant Rev (Orlando). 2016;30(p):3–12.
O’Reilly T, McSheehy PM. Biomarker development for the clinical activity of the mTOR inhibitor everolimus (RAD001): processes, limitations, and further proposals. Transl Oncol. 2010;3:65–79.
Yang L, Clarke MJ, Carlson BL, Mladek AC, Schroeder MA, Decker P, Wu W, Kitange GJ, Grogan PT, Goble JM, Uhm J, Galanis E, Giannini C, Lane HA, James CD, Sarkaria JN. PTEN loss does not predict for response to RAD001 (Everolimus) in a glioblastoma orthotopic xenograft test panel. Clin Cancer Res. 2008;14:3993–4001.
Alonso MM, Jiang H, Yokoyama T, Xu J, Bekele NB, Lang FF, Kondo S, Gomez-Manzano C, Fueyo J. Delta-24-RGD in combination with RAD001 induces enhanced anti-glioma effect via autophagic cell death. Mol Ther. 2008;16:487–93.
Huszthy PC, Daphu I, Niclou SP, Stieber D, Nigro JM, Sakariassen P, Miletic H, Thorsen F, Bjerkvig R. In vivo models of primary brain tumors: pitfalls and perspectives. Neuro Oncol. 2012;14:979–93.
Ma DJ, Galanis E, Anderson SK, Schiff D, Kaufmann TJ, Peller PJ, Giannini C, Brown PD, Uhm JH, McGraw S, Jaeckle KA, Flynn PJ, Ligon KL, Buckner JC, Sarkaria JN. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro Oncol. 2015;17:1261–9.
Wen PY, Omuro A, Ahluwalia MS, Fathallah-Shaykh HM, Mohile N, Lager JJ, Laird AD, Tang J, Jiang J, Egile C, Cloughesy TF. Phase I dose-escalation study of the PI3K/mTOR inhibitor voxtalisib (SAR245409, XL765) plus temozolomide with or without radiotherapy in patients with high-grade glioma. Neuro Oncol. 2015;17:1275–83.
Prasad G, Sottero T, Yang X, Mueller S, James CD, Weiss WA, Polley MY, Ozawa T, Berger MS, Aftab DT, Prados MD, Haas-Kogan DA. Inhibition of PI3K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro Oncol. 2011;13:384–92.
Tsaur I, Makarević J, Juengel E, Gasser M, Waaga-Gasser AM, Kurosch M, Reiter M, Wedel S, Bartsch G, Haferkamp A, Wiesner C, Blaheta RA. Resistance to the mTOR-inhibitor RAD001 elevates integrin α2- and β1-triggered motility, migration and invasion of prostate cancer cells. Br J Cancer. 2012;107:847–55.
Domínguez-Kelly R, Martín Y, Koundrioukoff S, Tanenbaum ME, Smits VA, Medema RH, Debatisse M, Freire R. 2011, Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J Cell Biol. 2011;194:567–79.
Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220–7.
Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–710.
Liu H, Dibling B, Spike B, Dirlam A, Macleod K. New roles for the RB tumor suppressor protein. Curr Opin Genet Dev. 2004;14:55–64.
Sedlacek HH. Mechanisms of action of flavopiridol. Crit Rev Oncol Hematol. 2001;38:139–70.
Newcomb EW, Tamasdan C, Entzminger Y, Arena E, Schnee T, Kim M, Crisan D, Lukyanov Y, Miller DC, Zagzag D. Flavopiridol inhibits the growth of GL261 gliomas in vivo: implications for malignant glioma therapy. Cell Cycle. 2004;3:230–4.
Cadoo KA, Gucalp A, Traina TA. Palbociclib: an evidence-based review of its potential in the treatment of breast cancer. Breast Cancer. 2014;6:123–33.
Michaud K, Solomon DA, Oermann E, Kim JS, Zhong WZ, Prados MD, Ozawa T, James CD, Waldman T. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010;70:3228–38.
Cen L, Carlson BL, Schroeder MA, Ostrem JL, Kitange GJ, Mladek AC, Fink SR, Decker PA, Wu W, Kim JS, Waldman T, Jenkins RB, Sarkaria JN. p16-Cdk4-Rb axis controls sensitivity to a cyclin-dependent kinase inhibitor PD0332991 in glioblastoma xenograft cells. Neuro Oncol. 2012;14:870–81.
Attardi LD, de Vries A, Jacks T. Activation of the p53-dependent G1 checkpoint response in mouse embryo fibroblasts depends on the specific DNA damage inducer. Oncogene. 2004;23:973–80.
Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–97.
Mueller S, Haas-Kogan DA. WEE1 kinase as a target for cancer therapy. J Clin Oncol. 2015;33:3485–7.
Russell MR, Levin K, Rader J, Belcastro L, Li Y, Martinez D, Pawel B, Shumway SD, Maris JM, Cole KA. Combination therapy targeting the Chk1 and Wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res. 2013;73:776–84.
Enders GH. Gauchos and ochos: a Wee1-Cdk tango regulating mitotic entry. Cell Div. 2010;5:12.
Watanabe N, Broome M, Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995;14:1878–91.
Kaelin WG. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–98.
Guertin AD, Li J, Liu Y, Hurd MS, Schuller AG, Long B, Hirsch HA, Feldman I, Benita Y, Toniatti C, Zawel L, Fawell SE, Gilliland DG, Shumway SD. Preclinical evaluation of the WEE1 inhibitor MK-1775 as single-agent anticancer therapy. Mol Cancer Ther. 2013;12:1442–52.
Pokorny JL, Calligaris D, Gupta SK, Iyekegbe DO, Mueller D, Bakken KK, Carlson BL, Schroeder MA, Evans DL, Lou Z, Decker PA, Eckel-Passow JE, Pucci V, Ma B, Shumway SD, Elmquist WF, Agar NY, Sarkaria JN. The efficacy of the Wee1 inhibitor MK-1775 combined with temozolomide is limited by heterogeneous distribution across the blood-brain barrier in glioblastoma. Clin Cancer Res. 2015;21:1916–24.
Rajeshkumar NV, De Oliveira E, Ottenhof N, Watters J, Brooks D, Demuth T, Shumway SD, Mizuarai S, Hirai H, Maitra A, Hidalgo M. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17:2799–806.
Kreahling JM, Foroutan P, Reed D, Martinez G, Razabdouski T, Bui MM, Raghavan M, Letson D, Gillies RJ, Altiok S. Wee1 inhibition by MK-1775 leads to tumor inhibition and enhances efficacy of gemcitabine in human sarcomas. PLoS One. 2013;8, e57523.
Hegi ME, Diserens AC, Bady P, Kamoshima Y, Kouwenhoven MC, Delorenzi M, Lambiv WL, Hamou MF, Matter MS, Koch A, Heppner FL, Yonekawa Y, Merlo A, Frei K, Mariani L, Hofer S. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib--a phase II trial. Mol Cancer Ther. 2011;10:1102–12.
D’Amato V, Raimondo L, Formisano L, Giuliano M, De Placido S, Rosa R, Bianco R. Mechanisms of lapatinib resistance in HER2-driven breast cancer. Cancer Treat Rev. 2015;41:877–83.
Zhai B, Sun XY. Mechanisms of resistance to sorafenib and the corresponding strategies in hepatocellular carcinoma. World J Hepatol. 2013;5:345–52.
Godin-Heymann N, Ulkus L, Brannigan BW, McDermott U, Lamb J, Maheswaran S, Settleman J, Haber DA. The T790M “gatekeeper” mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Mol Cancer Ther. 2008;7:874–9.
Guo T, Hajdu M, Agaram NP, Shinoda H, Veach D, Clarkson BD, Maki RG, Singer S, Dematteo RP, Besmer P, Antonescu CR. Mechanisms of sunitinib resistance in gastrointestinal stromal tumors harboring KITAY502-3ins mutation: an in vitro mutagenesis screen for drug resistance. Clin Cancer Res. 2009;15:6862–70.
Yip S, Miao J, Cahill DP, Iafrate AJ, Aldape K, Nutt CL, Louis DN. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res. 2009;15:4622–9.
Fenton TR, Nathanson D, Ponte de Albuquerque C, Kuga D, Iwanami A, Dang J, Yang H, Tanaka K, Oba-Shinjo SM, Uno M, Inda MM, Wykosky J, Bachoo RM, James CD, DePinho RA, Vandenberg SR, Zhou H, Marie SK, Mischel PS, Cavenee WK, Furnari FB. Resistance to EGF receptor inhibitors in glioblastoma mediated by phosphorylation of the PTEN tumor suppressor at tyrosine 240. Proc Natl Acad Sci U S A. 2012;109:14164–9.
Schulte A, Liffers K, Kathagen A, Riethdorf S, Zapf S, Merlo A, Kolbe K, Westphal M, Lamszus K. Erlotinib resistance in EGFR-amplified glioblastoma cells is associated with upregulation of EGFRvIII and PI3Kp110δ. Neuro Oncol. 2013;15:1289–301.
Agarwal S, Sane R, Gallardo JL, Ohlfest JR, Elmquist WF. Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J Pharmacol Exp Ther. 2010;334:147–55.
de Vries NA, Buckle T, Zhao J, Beijnen JH, Schellens JH, van Tellingen O. Restricted brain penetration of the tyrosine kinase inhibitor erlotinib due to the drug transporters P-gp and BCRP. Invest New Drugs. 2012;30:443–9.
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suvà ML, Regev A, Bernstein BE. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–401.
Navin NE. The first five years of single-cell cancer genomics and beyond. Genome Res. 2015;25:1499–507.
Schwartz SA, Weil RJ, Thompson RC, Shyr Y, Moore JH, Toms SA, Johnson MD, Caprioli RM. Proteomic-based prognosis of brain tumor patients using direct-tissue matrix-assisted laser desorption ionization mass spectrometry. Cancer Res. 2005;65:7674–81.
Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67.
Carlsson SK, Brothers SP, Wahlestedt C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol Med. 2014;6:1359–70.
van Tellingen O, Yetkin-Arik B, de Gooijer MC, Wesseling P, Wurdinger T, de Vries HE. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat. 2015;19:1–12.
Agarwal S, Sane R, Ohlfest JR, Elmquist WF. The role of the breast cancer resistance protein (ABCG2) in the distribution of sorafenib to the brain. J Pharmacol Exp Ther. 2011;336:223–33.
Anglicheau D, Pallet N, Rabant M, Marquet P, Cassinat B, Méria P, Beaune P, Legendre C, Thervet E. Role of P-glycoprotein in cyclosporine cytotoxicity in the cyclosporine-sirolimus interaction. Kidney Int. 2006;70:1019–25.
de Gooijer MC, Zhang P, Thota N, Mayayo-Peralta I, Buil LC, Beijnen JH, van Tellingen O. P-glycoprotein and breast cancer resistance protein restrict the brain penetration of the CDK4/6 inhibitor palbociclib. Invest New Drugs. 2015;33:1012–9.
Huang WC, Hsieh YL, Hung CM, Chien PH, Chien YF, Chen LC, Tu CY, Chen CH, Hsu SC, Lin YM, Chen YJ. BCRP/ABCG2 inhibition sensitizes hepatocellular carcinoma cells to sorafenib. PLoS One. 2013;8, e83627.
Polli JW, Humphreys JE, Harmon KA, Castellino S, O’Mara MJ, Olson KL, John-Williams LS, Koch KM, Serabjit-Singh CJ. The role of efflux and uptake transporters in [N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metab Dispos. 2008;36:695–701.
Yacyshyn BR, Bowen-Yacyshyn MB, Pilarski LM. Inhibition by rapamycin of P-glycoprotein 170-mediated export from normal lymphocytes. Scand J Immunol. 1996;43:449–55.
Zhou L, Schmidt K, Nelson FR, Zelesky V, Troutman MD, Feng B. The effect of breast cancer resistance protein and P-glycoprotein on the brain penetration of flavopiridol, imatinib mesylate (Gleevec), prazosin, and 2-methoxy-3-(4-(2-(5-methyl-2-phenyloxazol-4-yl)ethoxy)phenyl)propanoic acid (PF-407288) in mice. Drug Metab Dispos. 2009;37:946–55.
Demeule M, Shedid D, Beaulieu E, Del Maestro RF, Moghrabi A, Ghosn PB, Moumdjian R, Berthelet F, Béliveau R. Expression of multidrug-resistance P-glycoprotein (MDR1) in human brain tumors. Int J Cancer. 2001;93:62–6.
Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat. 2002;200:629–38.
Ortensi B, Setti M, Osti D, Pelicci G. Cancer stem cell contribution to glioblastoma invasiveness. Stem Cell Res Ther. 2013;4:18.
Liu HL, Fan CH, Ting CY, Yeh CK. Combining microbubbles and ultrasound for drug delivery to brain tumors: current progress and overview. Theranostics. 2014;4:432–44.
Côté J, Bovenzi V, Savard M, Dubuc C, Fortier A, Neugebauer W, Tremblay L, Müller-Esterl W, Tsanaclis AM, Lepage M, Fortin D, Gobeil F. Induction of selective blood-tumor barrier permeability and macromolecular transport by a biostable kinin B1 receptor agonist in a glioma rat model. PLoS One. 2012;7, e37485.
Pajeva IK, Sterz K, Christlieb M, Steggemann K, Marighetti F, Wiese M. Interactions of the multidrug resistance modulators tariquidar and elacridar and their analogues with P-glycoprotein. ChemMedChem. 2013;8:1701–13.
Salphati L, Heffron TP, Alicke B, Nishimura M, Barck K, Carano RA, Cheong J, Edgar KA, Greve J, Kharbanda S, Koeppen H, Lau S, Lee LB, Pang J, Plise EG, Pokorny JL, Reslan HB, Sarkaria JN, Wallin JJ, Zhang X, Gould SE, Olivero AG, Phillips HS. Targeting the PI3K pathway in the brain--efficacy of a PI3K inhibitor optimized to cross the blood-brain barrier. Clin Cancer Res. 2012;18:6239–48.
Lopez-Contreras AJ, Fernandez-Capetillo O. Signalling DNA damage, protein phosphorylation in human health, Dr. Cai Huang (Ed.) 2012, ISBN: 978-953-51-0737-8, InTech, DOI: 10.5772/50863. Available from: http://www.intechopen.com/books/protein-phosphorylation-in-human-health/signalling-dna-damage.
Cecchelli R, Berezowski V, Lundquist S, Culot M, Renftel M, Dehouck MP, Fenart L. Modelling of the blood-brain barrier in drug discovery and development. Nat Rev Drug Discov. 2007;6:650–61.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing AG
About this chapter
Cite this chapter
Pokorny, J.L., Kitange, G.J., Ma, D.J. (2016). Small-Molecule Inhibitors in Glioblastoma: Key Pathways and Resistance Mechanisms. In: Tivnan, A. (eds) Resistance to Targeted Therapies Against Adult Brain Cancers. Resistance to Targeted Anti-Cancer Therapeutics. Springer, Cham. https://doi.org/10.1007/978-3-319-46505-0_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-46505-0_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-46504-3
Online ISBN: 978-3-319-46505-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)