
Abstract
Lung cancer will be diagnosed in 230,000 patients in the U.S. in 2013. Adenocarcinoma will be the most common histology, and 10 % of lung cancers will be diagnosed in never or former light smokers. These patients will be those most likely to harbor targetable mutations, in particular, mutations in epidermal growth factor (EGFR). Preclinical work beginning in the 1980s led to the development of EGFR-targeted therapy in lung cancer patients. Analysis of the responders to gefitinib and erlotinib led to the discovery of activating mutations underlying sensitivity to EGFR-directed treatment. Although EGFR-mutant patients have higher response rates, better quality of life, and longer progression free survival, all patients eventually develop resistance. Mutations in the tyrosine kinase domain that render tumors resistant to erlotinib and gefitinib are the most common mechanism of resistance. A second generation of EGFR inhibitors are now making their way to the clinic, with hopes of thwarting these resistance mechanisms or providing more durable responses via irreversible inhibition, as well as targeting of additional HER receptors. Here we review the evolution of EGFR as a target in lung cancer, and the second generation of EGFR inhibitors in development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In 2013, approximately 230,000 new cases of lung cancer will be diagnosed, and lung cancer will cause close to 160,000 deaths [1]. 80 % of these cases will be non-small cell lung cancer (NSCLC). Histology now plays a significant role in the diagnosis of lung cancer, as the optimal treatment will differ for patients with adenocarcinoma versus squamous cell carcinoma. Patients diagnosed with adenocarcinoma are also more likely to harbor mutations that could predict benefit from targeted therapy. Identification of the activating mutations of the epidermal growth factor (EGFR) tyrosine kinase led to the usage of EGFR tyrosine kinase inhibitors (TKI) in the first-line treatment setting. Integration of EGFR inhibition in this population has led to doubling of survival rates in these patients when compared to patients without activating mutations. However, all patients eventually develop resistance, and require therapy beyond EGFR inhibition. Building on the success of the initial reversible EGFR TKI’s, erlotinib and gefitinib, multiple new compounds that irreversibly bind EGFR are currently in development. Here, we will review the development of the EGFR pathway as a target in NSCLC, establishment as a first-line therapy with EGFR TKI, development of resistance, and the new inhibitors of EGFR that are on the way.
EGFR Review
EGFR is a member of the ErbB, or HER, family of receptors (EGFR, HER-2, HER-3, HER-4) [2, 3]. It is a transmembrane protein that has an internal tyrosine kinase domain. Activation by one of its many ligands, such as EGF or transforming growth factor alpha, leads to dimerization of EGFR, either with another EGFR (homodimerization) or one of the other receptors from the HER family (heterodimerization), which activates the catalytic system of the tyrosine kinase. This results in the activation of multiple pathways that promote survival, proliferation, angiogenesis and metastasis. These activities are mediated primarily through the mitogen activated protein kinase (MAPK) and PI3K pathways. In the mid 1980s, EGFR was first suggested as a potential target for cancer-directed therapy because of its overexpression in epithelial malignancies [3]. Two decades of preclinical work led to multiple agents targeting the EGFR pathway. Two monoclonal antibodies, one chimeric (cetuximab) and one fully humanized (panitumumab), were developed to block activation of EGFR in cancer cells. Two small molecule reversible inhibitors of the EGFR tyrosine kinase were also developed, one for intravenous use (gefitinib) and one orally available (erlotinib) [2]. Further study of the EGFR pathway showed tumor dependence on EGFR was increased in tumors with high gene copy number and in those with activating mutations [4–7].
Development of EGFR Tyrosine Kinase Inhibitors
The first generation of EGFR TKI’s moved from the bench to the clinic around the year 2000. Gefitinib and erlotinib both work in competitively inhibiting the ATP site of the EGFR tyrosine kinase domain [2]. Although highly selective, this inhibition is reversible.
They were both initially studied in the second-line and third-line setting in unselected populations of patients who had progressed on chemotherapy.
Phase II results of gefitinib used in two studies in this population led to Food and Drug Administration (FDA) approval in the third-line treatment for NSCLC. Fukuoka et al. reported their study involving 210 patients treated with either 250 mg or 500 mg [8]. Response rates (18.4 % vs. 19 %) and symptom improvement (40 % vs. 37 %) were similar for the two dosages, with the 250 mg group having fewer drug-related toxicities. Kris et al, also examined these two doses for gefinitib in patients with NSCLC previously treated with chemotherapy [9]. Again, the lower dose was better tolerated, with no difference in symptom reduction or radiographic response from the higher dose. Among the combined population, treated at both dose levels, the radiographic response rate was 10 %. Response rates were noted to be higher in patients who were women, had light or never smoking histories, or had adenocarcinoma histology. Based on objective response rates, the FDA granted gefitinib accelerated approval for the treatment of locally advanced or metastatic lung cancer patients who progressed following treatment with a platin-based doublet and docetaxel [10].
Phase III investigations of gefitinib tempered the enthusiasm for its use in the U.S. Based on the phase II studies, the ISEL investigators studied the effect of the 250 mg dose of gefitinib on overall survival in a placebo-controlled trial in patients refractory to chemotherapy [11]. Treatment with gefitinib in the heterogeneous population was unable to show a benefit compared to placebo, with overall survival of 5.6 months versus 5.1, (CI 0.77–1.02) p = 0.087. However, in a preplanned subgroup analysis, patients who were never smokers or were of Asian origin had significantly longer survival (8.9 vs. 6.1 months, and 9.5 vs. 5.5 months, respectively). Maruyama et al. also studied gefitinib compared to docetaxel in the second-line setting [12]. The primary outcome of the study, non-inferiority of overall survival, was not reached. Following these two negative studies, the FDA withdrew the drug from further use, except for patients who were deemed by their doctors to be benefitting from treatment with gefitinib [13].
Following promising phase II data, erlotinib moved into phase III trials in refractory NSCLC patients. In the BR.21 study, Shepard et al. studied erlotinib versus placebo in the second-line and third-line setting [14]. This study also enrolled all NSCLC patients, with 50 % having adenocarcinoma histology, and 29.5 % of patients having squamous cell carcinoma. Twenty-one percent of the patients were never smokers. The results were modest, with a response rate of only 8.9 %. The duration of response was significant, 7.9 months with erlotinib, versus 3.7 with placebo. BR.21 also achieved its primary endpoint, demonstrating a survival benefit of 6.7 months versus 4.7 months with placebo (p < 0.001). Subgroup analyses of the responses to EGFR TKI showed they were more likely to be in patients who were never smokers or were light smokers, of Asian origin, and with adenocarcinoma histology. BR.21 led to FDA approval in the United States for erlotinib in the second-line or third-line setting.
Identification of Activating Mutations in EGFR
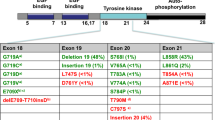
Further analysis into the patients who experienced greater responses to gefitinib led to the identification of activating mutations that engendered sensitivity to EGFR TKI therapy. Two groups reported on somatic mutations identified in gefitinib responders and not present in non-responders [5, 6]. Lynch et al. performed mutational analysis of the entire EGFR coding sequence on tumor specimens, along with matched normal tissue from nine patients who achieved a response with gefitinib and specimens from seven patients treated with gefitinib who did not have a response. The tumors from 25 patients who were not exposed to gefitinib were also examined. The nine specimens were from patients treated at Massachusetts General Hospital with single agent gefitinib, and represented 36 % of their group with response out of 275 total patients treated with gefinitib. EGFR mutations were found in eight of the nine responding patients and in zero of the seven patients who did not respond (P < 0.001). The majority of the responders were women with little or no smoking history, in line with the clinical characteristics of the responding patients in earlier gefitinib studies. The median survival of the responding group was 18 months. The mutations discovered were all involved in the coding of the tyrosine kinase domain of EGFR. Four of the eight mutations involved frame deletions in exon 19. Amino acid substitutions in exon 21 were the driving mutation in three patients, two of whom had L858R, and one with L861Q. The eighth patient was found to have a missense mutation in exon 18. Mutations in EGFR were also identified in two of the 25 patients not treated with gefitinib.
Paez et al. made similar discoveries at the same time as the Lynch group [6]. They first examined the tumor specimens of 119 patients, 58 from Japan and 61 from Boston. Somatic mutations were found in 15 of the 58 tumors of Japanese patients, and in one of 61 in the Boston sample. The mutations were similar to those identified by Lynch’s group, involving L858R, as well as missense and deletion mutations all within exons 18-21. Paez et al. then looked for EGFR mutations in patients treated with gefitinib. The patients were all Caucasians from the U.S., five of whom responded to gefinitib and four who experienced progression. All five pretreatment tumor specimens from responders showed mutations in the EGFR kinase domain, while none of the four non-responders had mutations.
A study by Pao et al. followed shortly after demonstrating that patients experiencing greater benefit to treatment with erlotinib also harbored these same activating mutations [7]. Five of the seven tumors from patients who responded to erlotinib had somatic mutations, whereas zero of ten tumors from non-responders had them (p = 0.003).
Subsequent studies have shown that deletions in exon 19 and the L858R substitution make up the majority, 85–90 %, of activating EGFR mutations in patients responding to EGFR TKI. These landmark discoveries paved the way for the establishment of personalized medicine for lung cancer patients.
The studies of Lynch and Paez showed an incidence of EGFR mutations in their untreated patients that was similar to the response rates seen in the initial studies of gefitinib.
Treatment Based on Phenotype and Genotype
The identification of activating mutations being the key determinant to response to EGFR TKI launched several phase II studies to see if prospective testing to personalize therapy was feasible and could lead to better outcomes. These studies were limited to patients with EGFR mutations. All patients were treated with gefitinib (two studies), or erlotinib (one study). Inoue et al. showed high response rate, 75 %, and a progression free survival (PFS) of 9 months, in 16 patients with EGFR mutations [15]. This compared favorably with the responses to chemotherapy in previous studies. Sequist et al. treated 31 patients with gefitinib with the primary outcome of response rate [16]. Thirty-four out of 98 patients were found to have EGFR mutations. Of these, 79 % had exon 19 deletions or the L858R mutation, the most common activating mutations. These patients had a 55 % response rate, and also showed a similar 9-month PFS.
Rosell et al. performed a similar prospective selection, treating patients with erlotinib [17]. In a nationwide study conducted by the Spanish Lung Cancer Study Group, they screened 2,105 patients with NSCLC and identified 350 patients with activating mutations, the majority having exon 19 (62.2 %), or L858R (37.8 %) mutations. A total of 217 patients were treated with erlotinib, achieving a PFS of 14 months, and overall survival of 27 months.
Mok et al. followed up these encouraging results with a phase III randomized trial comparing front-line treatment with gefitinib to carboplatin and paclitaxel (First Line Iressa versus Carboplatin/Paclitaxel in Asia [Iressa Pan-Asia Study, IPASS]) [18•]. Although they did not prospectively identify mutation status, their population was enriched by enrolling patients with the clinical characteristics that more often respond to EGFR TKI—Asians, never or former light smokers, and the group was predominantly female. The primary endpoint of IPASS was PFS with overall survival (OS) as a secondary endpoint, with a pre-planned subgroup analysis of the role of EGFR mutation on treatment efficacy. IPASS achieved its primary endpoint, showing an improved 12-month PFS with gefitinib treatment in this enriched population, 24.9 % vs. 6.7 % for chemotherapy, (hazard ratio [HR] for progression or death, 0.74; 95 % CI, 0.65–0.85, P < 0.001). Most importantly, the IPASS study showed that treatment with EGFR TKI should be based on mutation status rather than clinical features. Out of the 1,217 patients studied, EGFR mutation analysis was available from 437 patients. Sixty percent of the tumor specimens were positive for EGFR mutation, with the majority being exon 19 deletion or the L858R mutation. Patients with EGFR mutations had a significantly better PFS when treated with gefitinib compared with carboplatin and paclitaxel (HR for progression, 0.48; 95 % CI, 0.36–0.64; P < 0.001). Conversely, those without mutations had inferior outcomes with gefitinib, (HR 2.85; 95 % CI, 2.05–3.98; P < 0.001). The benefit of prospectively selecting patients for treatment based on EGFR mutations with gefitinib was further demonstrated by two additional studies performed in Asian populations [19, 20]. In the phase III OPTIMAL study, Zhou et al. treated Chinese patients harboring EGFR mutations with erlotinib in the front line setting, showing similar benefits to gefitinib in patients with activating mutations [21]. Rosell et al. prospectively treated European patients based on activating EGFR mutations with erlotinib in a phase III study compared to chemotherapy (EURTAC) [22]. The EURTAC authors showed the benefit of testing for EGFR in a non-Asian population. Multiple studies have further determined significant percentages of patients harboring activating mutations in populations that were non-Asian, male, and had smoking histories [23–25]. This has led to the recommendation that all patients with adenocarcinoma of the lung be tested for EGFR mutations [26].
Although these studies led to improved PFS, as well as quality of life, none of the studies have shown an improvement in overall survival. This is most likely the result of crossover of patients from the chemotherapy group to receive EGFR-directed therapy at the time of progression. Because of the lack of an overall survival benefit, it is unclear whether EGFR TKI should primarily be used in the first line setting or after progression. The concern involved with this approach is a potentially decreased response to EGFR TKI, and the decline of patients prior to receiving EGFR TKI. In review of the studies of gefitinib versus chemotherapy, upwards of 30 % of patients who received chemotherapy upfront were unable to receive this treatment with EGFR-directed therapy [20, 22].
Resistance to EGFR TKI
Beneficial as treatment with EGFR TKI is, all patients’ tumors will eventually develop resistance. Multiple methods of resistance have been demonstrated: development of T790M “gatekeeper” mutation, HER2 amplification, involvement of the MET pathway, and transformation into small cell or neuroendocrine tumor [27–32].
As the response rates in patients with activating mutations is not 100 %, 20–50 % of patients will have primary resistance to EGFR TKI’s. Multiple groups have reported various rates of the presence of T790M mutation in pre-treatment specimens, although this could be dependent on the method of testing [33, 34]. Alternate pathways to EGFR may also promote tumor growth. Some patients, because of either drug–drug interactions, or pharmacokinetics, may not achieve the necessary drug level for inhibition of EGFR.
For those that do respond but eventually progress, multiple mechanisms of resistance have been described. In up to 60 %, a secondary EGFR mutation may develop, the majority of which are the T790M mutation [31]. Studying the tumor from a patient who initially achieved a complete response and then experienced disease progression after 2 years of treatment with gefitinib, Kobayashi et al. identified the substitution of methionine for threonine at position 790 in exon 20 of EGFR [27]. Like the activating mutations, this mutation is in the tyrosine kinase domain, although in this case, it leads to steric hindrance of EGFR TKI’s, and leads to increased affinity for ATP. Pao et al. also showed the presence of T790M in two out of five patients with progression after initial response to TKI [35•]. Pao and his colleagues found no evidence of T790M in 157 tumors tested, and in only one of 1300 tumors that were analyzed for mutations in EGFR exons 18–21. Further studies have reported various percentages of T790M incidence, which may depend on the testing method. The presence of pre-treatment T790M has been shown to lead to inferior outcomes in patients treated with EGFR TKI. It is unclear if the T790M mutation, which leads to treatment resistance, represents the development of a new mutation, or evolution of a resistant clone.
As T790M is not present in all resistant specimens, the search for alternative resistance mechanisms has also identified the MET pathway as a mechanism for resistance [29, 30]. Mutations in downstream effectors of the EGFR pathway, such as PI3-kinase, have also been shown to lead to resistance to EGFR TKI [32]. Blockade of these pathways in vitro has restored sensitivity to EGFR tyrosine kinase inhibition in pre-clinical models. Further development may prove that resistance can be thwarted by combination therapies.
Second Generation EGFR TKI
The acquired resistance to EGFR TKI’s has led to the development of second-generation compounds targeting EGFR. Several new compounds are in various phases of development. These compounds differ from the first generation in that they irreversibly block EGFR via covalent binding, and most of the drugs have selectivity for additional targets as well.
Neratinib, one of the first second generation TKI’s, is an oral inhibitor that targets EGFR and HER2. Preclinical data showed that covalently binding EGFR may overcome resistance mediated by T790M mutation. Neratinib, at the time HKI 272, showed efficacy in suppressing EGFR phosphorylation and phosphorylation of downstream effectors in gefitinib resistant clones [36]. A dose of 320 mg, determined in phase I testing, was used for the phase II study of neratinib in advanced NSCLC patients [37, 38]. The study involved three arms, patients with mutant EGFR, wild-type EGFR with at least 12 weeks or prior treatment with EGFR TKI, and TKI-naive patient with adenocarcinoma and light smoking histories. The study’s primary outcome was response rate. Grade 3 diarrhea in 50 % of the initial 39 patients led to dose reduction to 240 mg for all subsequent patients enrolled. Possibly related to the decreased dosage, the response rate in EGFR-mutated patients was only 3 %, and there were no responses in the other two groups. There were also no responses seen in the 12 patients (7 %) harboring T790M mutations. Of the total study population, 61 % had EGFR mutations, with the majority being exon 19 deletions or L858R mutations. There are no current studies investigating neratinib in patients with NSCLC.
Afatinib is also a dual inhibitor of EGFR and HER2. Afatinib (BIBW2992) is an orally available small molecule tyrosine kinase inhibitor that irreversibly binds the ATP pocket of EGFR as well as HER2. Preclinical activity was observed in cancer cells with activating EGFR mutations, and additionally has shown promise in the inhibition of tumors possessing T790M mutations [39]. Following preclinical and phase I work, the LUX-Lung program was initiated [40, 41]. The LUX-Lung program, which initially included two phase II studies and four phase III studies investigating afatinib in different clinical situations, now also has two additional trials.
LUX-Lung 1 was a phase IIb/III randomized, double blind, placebo-controlled study investigating the efficacy of afatinib in patients with prior treatment with chemotherapy and at least 12 weeks of either erlotinib or gefitinib [42•]. LUX-Lung 1 was an international study involving 86 centers that randomized 585 patients in a two-to-one fashion to afatinib plus best supportive care (BSC) versus placebo plus BSC. The feasibility of prospectively testing every patient for mutations was decreased by the size and scope of the study, so the investigators sought to enrich their population by enrolling patients who had been treated on EGFR TKI for at least 12 weeks. The two groups had similar response rates to prior EGFR-directed therapies, 46 % and 44 %, respectively, which is more consistent with EGFR mutated group than in previous EGFR TKI studies done on unselected populations. The primary endpoint of LUX-Lung 1 was overall survival. This endpoint was selected because of an expected short survival in patients who had progressed on multiple lines of therapy. Of the 585 patients assigned to treatment, EGFR mutation status was available on 141 patients, 93 in the afatanib group and 48 in the placebo group. Mutation rates were similar in both groups, 67 % and 71 %, respectively, with 79 % of the mutations being exon 19 deletion or L858R in exon 21. Eight patients were positive for T790M, four in each group, with testing most likely done pre-treatment. The study did not achieve its primary endpoint, with median OS in the afatinib group being 10.8 months, and 12.0 months in the placebo group (HR 1.08, 95 % CI 0.86–1.35; p = 0.74). Although there was no crossover to afatinib, the placebo group had a higher percentage of patients receiving subsequent systemic therapy. The percentage of patients in the placebo group that received additional therapy with EGFR TKI was double the percentage of patients in the afatinib group. The differences in subsequent therapy were not statistically significant. However, LUX-Lung 1 did show a significantly longer PFS in patients treated with afatinib compared to placebo, 3.3 months versus 1.1 months (HR 0.38, 95 % CI 0.31–0.48; p < 0.0001). The most common adverse events were diarrhea and rash, consistent with other EGFR TKIs. 38 % of patients required a dose reduction and 10 % of patients receiving afatinib experienced a serious adverse event. Despite the increase in side effects, higher percentages of patients receiving afatinib reported improvements in three pre-specified NSCLC-related, health-related quality of life (QoL) items. Although LUX-Lung 1 failed to achieve its primary endpoint, it was the first study to demonstrate a benefit in PFS, response rate (RR), and QoL in patients previously treated with chemotherapy and to have progressed on EGFR directed therapy [42•].
The LUX-Lung 2 trial was a phase II clinical trial single arm trial involving patients harboring activating EGFR mutations. [43]. Patients were allowed to have received no more than one line of chemotherapy, and be naive to EGFR-directed therapy. Overall, 61 % of 129 patients achieved an objective response, two complete and 77 partial. Those harboring the most common mutations, L858R or deletion 19, had a response rate of 66 % (70 of 106), whereas those with less common EGFR mutations had a 39 % response rate (nine of 23 patients). Two dose levels were studied in LUX-Lung 2, 50 mg in 99 patients, and 40 mg in 30 patients. The response rates of the two dosages were similar, although patients receiving the higher dose of 50 mg experienced more of the most common Grade 3 adverse events, as well as more possibly treatment-related serious adverse events. LUX-Lung 2 established the dosage of 40 mg to be used for future trials investigating afatinib.
Afatinib is being compared to front-line chemotherapy in NSCLC patients harboring EGFR activating mutations in the phase III LUX-Lung 3 trial (ClinicalTrials.gov identifier NCT00949650). The primary outcome of the study is PFS, with secondary outcomes looking at RR, disease control rate, OS, and QoL measures. The trial is currently closed to accrual, and estimated study completion date is December 2013. Results from LUX-Lung 3 were first presented at the ASCO 2012 Annual meeting [44]. A total of 345 patients were randomized 2:1 to afatinib or to cisplatin plus pemetrexed. Of the patients enrolled, 72 % were Asian, and 65 % were female. Forty-nine percent of the patients had deletion in exon 19; 40 % with L858R, and the other 11 % having other activating mutations. Overall, treatment with afatinib had a superior PFS compared to chemotherapy (11.1 months vs. 6.9 months; HR 0.58, 95 % CI 0.43–0.78; p = 0.0004). In the patients with the most common activating mutations, deletion in exon 19 and L858R, the difference was even more pronounced, with median PFS of 13.6 vs. 6.9 months (HR 0.47, 95 % CI 0.34–0.65; p = 0.0001). The finalized results of LUX-Lung 3 are eagerly anticipated as the response rates and PFS were very impressive, and likely warrant FDA approval in the front-line setting for patients with the most common mutations.
Additional studies in the LUX-Lung program, are currently accruing in different clinical scenarios. Studies are investigating afatinib in patients with clinical resistance to EGFR TKI (LUX-Lung 4) (ClinicalTrials.gov identifier NCT00711594); afatinib plus paclitaxel following progression on afatinib (LUX-Lung 5) (ClinicalTrials.gov identifier NCT01085136); Afatanib versus cisplatin plus gemcitabine in first line (LUX-Lung 6) (ClinicalTrials.gov identifier NCT01121393); afatinib versus gefitinib in the front line setting (LUX-Lung 7) (ClinicalTrials.gov identifier NCT01466660); and afatinib in patients with squamous cell histology (LUX-Lung 8) (ClinicalTrials.gov identifier NCT01523587).
Also, following preclinical data showing possible synergy between afatinib and other compounds in blocking cancer growth in cells with the T790M mutation, afatinib is also being studied in conjunction with cetuximab, a monoclonal antibody targeting EGFR (ClinicalTrials.gov identifierNCT01090011), and sirolimus, an mTOR inhibitor (ClinicalTrials.gov identifier NCT00993499). In T790M transgenic murine models, treatment with afatinib and cetuximab has induced near complete responses [45]. A phase I study of cetuximab plus afatinib in patients with clinically defined acquired resistance was commenced. Preliminary results presented at the American Society of Clinical Oncology (ASCO) 2011 Annual Meeting showed four of 13 (29 %) patients with T790M mutations treated with the recommended phase II dosing achieving a confirmed partial response [46]. These encouraging results have led to an expansion of this study.
Dacomitinib (PF-00299804) is an irreversible pan-HER inhibitor. It acts by covalently binding the tyrosine kinase domain of EGFR (HER1), HER2, and HER4. Dacomitinib also has demonstrated pre-clinical efficacy in inhibiting tumor cells with the T790M mutation [47]. The phase I study of dacomitinib showed activity in NSCLC patients, some of whom were previously treated with EGFR TKI, and led to the recommended phase II dose of 45 mg [48].
Following activity seen in the phase I study, as well as a phase II study in patients previously treated with chemotherapy followed by erlotinib, dacomitinib was compared to treatment with erlotinib in patients previously treated with chemotherapy [49•]. The primary endpoint of this study of an unenriched population was PFS. Dacomitinib had a longer median PFS compared to erlotinib, 2.86 months vs. 1.91 months (HR 0.66l, 95 % CI, 0.47–0.91; two-sided P = 0.12). Dacomitinib is now being compared to erlotinib in a phase III study, ARCHER 1009 (ClinicalTrials.gov identifier NCT01360554).
Dacomitinib is also being studied in an enriched population. A phase II, open-label trial is investigating dacomitinib in patients with former light or never smoking histories, patients with tumors positive for activating EGFR mutations, or patients with HER 2 mutation or amplification. Preliminary results of the patients with exon 19 or L858R mutations treated with dacomitinib were presented at the ASCO 2012 Annual Meeting [50]. Of the 92 patients enrolled, 47 were found to have EGFR mutations in exon 19 or 21. Patients with activating mutations treated with dacomitinib had a 74 % response rate (34/46 evaluable patients). The preliminary PFS in these patients was 17 months (95 % CI: 13-24). This study shows the promise of irreversible inhibition of EGFR in activating mutations, and will be compared to front-line gefitinib in a phase III study, ARCHER 1050, which is opening soon (ClinicalTrials.gov identifier NCT01774721). Table 1 lists the front line studies of erlotinib, gefitinib, afatinib, and dacomitinib.
Conclusions
The emergence of EGFR therapy for patients with activating mutations has changed the course of lung cancer management. Treatment with EGFR TKI in the frontline setting has led to greatly prolonged PFS, as well as improved QoL, as the therapy is less toxic in addition to being more efficacious. That none of the trials have demonstrated an OS benefit has left some daylight for a discussion of timing of therapy. A large determinant of first line treatment in some centers hinges on the turnaround time of EGFR testing. The IPASS study showed the detriment in attempting to treat patients based on clinical features, as those without activating mutations should be treated initially with chemotherapy. Studies of EGFR inhibitors in maintenance setting following chemotherapy suggest that patients with activating mutations will still benefit, even if they receive their personalized therapy following chemotherapy. The concern of many investigators is that in the patients who receive chemotherapy initially, a clinically significant number will not receive EGFR TKI-based therapy.
The development of resistance in all patients who receive first generation EGFR TKI also raises the issue of whether treatment with an irreversible EGFR TKI may lead to less resistance and improved duration without progression. The second generation of EGFR TKI’s, in particular afatinib and dacomitinib, look to further change the landscape of the treatment of patients with EGFR mutations. The niches of these exciting therapies remains to be seen. Afatinib is poised to be a potential alternative to chemotherapy in the first-line setting, following its early results in the pivotal phase III LUX-Lung 3 trial. Whether it is better than first generation TKI is currently in the phase II setting of testing. Afatinib also may potentially come to the clinic in a combination with other targeted therapies as a way to combat EGFR TKI resistance. To date, dacomitinib has shown its superiority over erlotinib in the second line setting in phase II testing, and hopes to further prove this benefit in phase III testing. Following encouraging phase II results in the activating mutation population, dacomitinib is now being compared to front-line gefitinib in the phase III setting. Like dacomitinib and afatinib, which inhibit other tyrosine kinases in addition to EGFR, many other dual inhibitors are in the pipeline in efforts to lead to better efficacy towards EGFR and in combating resistance. There is also a compound currently in early phase testing that specifically targets the T790M mutation. Results from the second generation EGFR TKI’s as well as these other promising compounds look to further improve the outlook for patients with non-small cell lung cancer.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30.
Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358(11):1160–74.
Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene. 2000;19(56):6550–65.
Hirsch FR, Varella-Garcia M, Bunn Jr PA, Di Maria MV, Veve R, Bremmes RM, et al. Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol. 2003;21(20):3798–807.
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–39.
Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–500.
Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101(36):13306–11. PMCID: 516528.
Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J Clin Oncol. 2003;21(12):2237–46.
Kris MG, Natale RB, Herbst RS, Lynch Jr TJ, Prager D, Belani CP, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA. 2003;290(16):2149–58.
Cohen MH, Williams GA, Sridhara R, Chen G, Pazdur R. FDA drug approval summary: gefitinib (ZD1839) (Iressa) tablets. Oncologist. 2003;8(4):303–6.
Thatcher N, Chang A, Parikh P, Rodrigues Pereira J, Ciuleanu T, von Pawel J, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet. 2005;366(9496):1527–37.
Maruyama R, Nishiwaki Y, Tamura T, Yamamoto N, Tsuboi M, Nakagawa K, et al. Phase III study, V-15-32, of gefitinib versus docetaxel in previously treated Japanese patients with non-small-cell lung cancer. J Clin Oncol. 2008;26(26):4244–52.
FDA Alert for Healthcare Professionals. 2005 [updated 2005; cited 4/1/2013]; Available from: http://www.fda.gov/downloads/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm126182.pdf.
Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123–32.
Inoue A, Kobayashi K, Usui K, Maemondo M, Okinaga S, Mikami I, et al. First-line gefitinib for patients with advanced non-small-cell lung cancer harboring epidermal growth factor receptor mutations without indication for chemotherapy. J Clin Oncol. 2009;27(9):1394–400.
Sequist LV. First-generation epidermal growth factor receptor tyrosine kinase inhibitors in EGFR mutation: positive non-small cell lung cancer patients. J Thorac Oncol. 2008;3(6 Suppl 2):S143–5.
Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361(10):958–67.
• Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–57. The IPASS study proved the critical importance of EGFR-targeted therapy based on genotype and not on phenotype. Treatment with gefitinib in patients harboring activating mutations leads to better PFS and quality of life when compared to chemotherapy.
Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380–8.
Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11(2):121–8.
Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735–42.
Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–46.
D'Angelo SP, Pietanza MC, Johnson ML, Riely GJ, Miller VA, Sima CS, et al. Incidence of EGFR exon 19 deletions and L858R in tumor specimens from men and cigarette smokers with lung adenocarcinomas. J Clin Oncol. 2011;29(15):2066–70. PMCID: 3296671.
Reinersman JM, Johnson ML, Riely GJ, Chitale DA, Nicastri AD, Soff GA, et al. Frequency of EGFR and KRAS mutations in lung adenocarcinomas in African Americans. J Thorac Oncol. 2011;6(1):28–31. PMCID: 3337520.
Cote ML, Haddad R, Edwards DJ, Atikukke G, Gadgeel S, Soubani AO, et al. Frequency and type of epidermal growth factor receptor mutations in African Americans with non-small cell lung cancer. J Thorac Oncol. 2011;6(3):627–30. PMCID: 3057407.
Ettinger DS, Akerley W, Borghaei H, Chang AC, Cheney RT, Chirieac LR, et al. Non-small cell lung cancer. J Natl Compr Canc Netw. 2012;10(10):1236–71.
Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–92.
Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012;2(10):922–33. PMCID: 3473100.
Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104(52):20932–7. PMCID: 2409244.
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–43.
Yu H, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of Mechanisms of Acquired Resistance to EGFR TKI therapy in 155 patients with EGFR-mutant Lung Cancers. Clin Cancer Res. 2013
Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. PMCID: 3132801.
Rosell R, Molina MA, Costa C, Simonetti S, Gimenez-Capitan A, Bertran-Alamillo J, et al. Pretreatment EGFR T790M mutation and BRCA1 mRNA expression in erlotinib-treated advanced non-small-cell lung cancer patients with EGFR mutations. Clin Cancer Res. 2011;17(5):1160–8.
Su KY, Chen HY, Li KC, Kuo ML, Yang JC, Chan WK, et al. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J Clin Oncol. 2012;30(4):433–40.
• Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. PMCID: 549606. Discovery of T790M mutation underlying development of resistance to gefitinib and erlotinib reported by Pao et al. This work demonstrated that resistance to EGFR directed therapy could be engendered by mutations in the tyrosine kinase domain, akin to the T315I mutation in imatinib resistant CML. It also showed the importance of re-biopsy at time of progression.
Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A. 2005;102(21):7665–70. PMCID: 1129023.
Wong KK, Fracasso PM, Bukowski RM, Lynch TJ, Munster PN, Shapiro GI, et al. A phase I study with neratinib (HKI-272), an irreversible pan ErbB receptor tyrosine kinase inhibitor, in patients with solid tumors. Clin Cancer Res. 2009;15(7):2552–8.
Sequist LV, Besse B, Lynch TJ, Miller VA, Wong KK, Gitlitz B, et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2010;28(18):3076–83.
Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27(34):4702–11. PMCID: 2748240.
Yap TA, Vidal L, Adam J, Stephens P, Spicer J, Shaw H, et al. Phase I trial of the irreversible EGFR and HER2 kinase inhibitor BIBW 2992 in patients with advanced solid tumors. J Clin Oncol. 2010;28(25):3965–72.
Metro G, Crino L. The LUX-Lung clinical trial program of afatinib for non-small-cell lung cancer. Expert Rev Anticancer Ther. 2011;11(5):673–82.
• Miller VA, Hirsh V, Cadranel J, Chen YM, Park K, Kim SW, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012;13(5):528–38. LUX-Lung 1 showed that treatment with afatinib in patients who progressed after at least 12 weeks of erlotinib or gefitinib could provide a PFS benefit (3.3 vs. 1.1 months) vs. placebo. No overall survival benefit was observed, although this was possibly related to subsequent treatments given to both groups.
Yang JC, Shih JY, Su WC, Hsia TC, Tsai CM, Ou SH, et al. Afatinib for patients with lung adenocarcinoma and epidermal growth factor receptor mutations (LUX-Lung 2): a phase 2 trial. Lancet Oncol. 2012;13(5):539–48.
Yang JC, Schuler M. H., Yamamoto N., et al. LUX-Lung 3: A randomized, open-label, phase III study of afatinib versus pemetrexed and cisplatin as first-line treatment for patients with advanced adenocarcinoma of the lung harboring EGFR-activating mutations. J Clin Oncol. 2012;30(No 18 Suppl).
Regales L, Gong Y, Shen R, de Stanchina E, Vivanco I, Goel A, et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest. 2009;119(10):3000–10. PMCID: 2752070.
Janjigian YY, Groen H.J., Horn L., et al. . Activity and tolerability of afatinib (BIBW 2992) and cetuximab in NSCLC patients with acquired resistance to erlotinib or gefitinib. J Clin Oncol. 2011;29(No 15 Suppl).
Engelman JA, Zejnullahu K, Gale CM, Lifshits E, Gonzales AJ, Shimamura T, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67(24):11924–32.
Janne PA, Boss DS, Camidge DR, Britten CD, Engelman JA, Garon EB, et al. Phase I dose-escalation study of the pan-HER inhibitor, PF299804, in patients with advanced malignant solid tumors. Clin Cancer Res. 2011;17(5):1131–9. PMCID: 3048920.
• Ramalingam SS, Blackhall F, Krzakowski M, Barrios CH, Park K, Bover I, et al. Randomized phase II study of dacomitinib (PF-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2012;30(27):3337–44. Dacomitinib showed a PFS benefit when compared to erlotinib in an unselected population of patients following first-line chemotherapy and naïve to previous EGFR-directed therapy.This result could have been skewed by a higher population of EGFR-mutant patients in the dacomitinib group. PFS for both drugs was 7.4 months in the mutant population.
Kris M, Mok T., Ou S.H., et al. . First-line dacomitinib (PF-00299804), an irreversible pan-HER tyrosine kinase inhibitor, ofr patients with EGFR-mutant lung cancers. J Clin Oncol. 2012;30(15 Suppl).
Compliance with Ethics Guidelines
Conflict of Interest
Kyle W. Robinson declares no potential conflict of interest.
Alan B. Sandler has been a consultant to Genentech/Roche, Lilly, Pfizer, Celgene, and Gilead, provided expert testimony for Genentech/Roche and Pfizer, and served on speakers’ bureaus for Genentech/Roche, Lilly, Celgene, and Pfizer.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Robinson, K.W., Sandler, A.B. EGFR Tyrosine Kinase Inhibitors: Difference in Efficacy and Resistance. Curr Oncol Rep 15, 396–404 (2013). https://doi.org/10.1007/s11912-013-0323-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11912-013-0323-7