
Abstract
The evolutionary conserved control of behaviour by glucocorticoids translates into a key role for glucocorticoids in the control of neuropsychological functioning. In accordance, both animal and human models of uncontrolled exposure to glucocorticoids show impaired stress responsiveness, cognitive dysfunction, and a broad spectrum of neuropsychiatric disorders, ranging from severe depression and anxiety disorders to acute psychosis and delirium. Importantly, exogenous glucocorticoid administration can induce the same phenotype, proving the causal role of glucocorticoids per se on neurocognitive and neuropsychiatric functioning. Recent findings now indicate that these effects may be long-lasting and even may not be completely reversible because cognitive dysfunction and maladaptive personality traits persist in patients long-term after successful correction of glucocorticoid excess in the presence of altered coping strategies and affected illness perceptions. This implies that long-term care for both patients with pituitary and adrenal disorders and patients using glucocorticoids should incorporate self-management interventions that help to improve quality of life
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Glucocorticoids
- Brain
- Cortisol
- Adrenal Insufficiency
- Cushing’s syndrome
- Animal models
- Neurocognitive function
- Neuropsychiatric function
- Coping strategies
- Illness perceptions
- Quality of life
Introduction: Regulation of the Stress Response (From an Evolutionary Perspective)
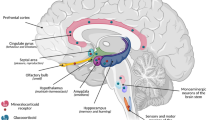
Evolution has provided us with powerful tools to ensure survival, and an adequate response to a stressor in this respect is fundamental. A normal stress response is a prerequisite for a normal behavioural and metabolic adaptation to the stressor. When an individual is exposed to a stressor, the response is characterized by stimulation of the sympathetic nervous system (leading to catecholamine release) and activation of the hypothalamus–pituitary–adrenal (HPA) axis. Cortisol, or corticosterone in the rodent, is the main mediator of the adrenocortical stress response that ultimately serves only one purpose: to induce the required behavioural and metabolic adaptations enabling the individual to adequately cope with the stressor. Thus, activation of the HPA axis, and consequently, increased cortisol secretion is fundamental for modelling the stress response [1]. Corticotrophin releasing hormone (CRH ), secreted from parvocellular neurons of the paraventricular nucleus (PVN) in the hypothalamus, stimulates the pituitary to release adrenocorticotropin (ACTH) after cleavage from the pro-opiomelanocortin precursor. Subsequently, activation of ACTH receptors in the adrenal cortex leads to the synthesis and secretion of glucocorticoids.
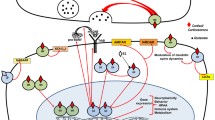
The regulation of stress-induced HPA activation occurs by so-called negative glucocorticoid feedback at the level of the anterior pituitary and hypothalamus. In clinical endocrinology, this negative feedback action exerted at the pituitary by synthetic glucocorticoids is exploited in the diagnostic workup and subsequent treatment of primary and secondary adrenal insufficiency. However, this clinical model of the HPA axis actually is a truncated model from a biological perspective, because higher centres, including brain stem catecholamines, modulate CRH production by the hypothalamus and limbic brain structures such as the amygdala [2]. This activation is of paramount importance in the responses to psychological stressors, which trigger emotional arousal and require cognitive operations for coping and storing the experience in the memory for future use. Glucocorticoids exert a strong feedback and feedforward action on these limbic forebrain areas [3]. Two nuclear receptor types mediate this action exerted by these steroids: the mineralocorticoid (MR ) and the glucocorticoid receptor (GR) [1].
In addition to the well-known genomic actions of glucocorticoids, recent evidence suggests that rapid, non-genomic effects of glucocorticoids are mediated via lower affinity MR and GR variants localized in the cell membrane [4, 5]. This so-called fast negative-feedback control of glucocorticoid action appears to be mediated by another pleiotropic physiological system: the endocannabinoid system. Endocannabinoids play a pivotal role in the control of glucocorticoid action, via modulation of the excitatory action of glutamate on CRH neurons in the PVN [6]. Glutamate activation is a crucial step in the activation of the HPA axis and the inhibition of glutamate release appears to be specifically mediated by cannabinoids in the hypothalamic PVN.
Dysregulation of the activity of the HPA axis occurs when the glucocorticoid response is either inadequate, or too extreme and prolonged. This aberrant glucocorticoid response to stressors can have deleterious consequences for the organism. The inability to effectively terminate the stress response may lead to continued hypersecretion of glucocorticoids, which eventually leads to wear and tear of tissues and organs with an increased risk for metabolic and cardiovascular diseases, compromised immune responses, and psychopathology. Alternatively, an inadequate cortisol response is unable to restrain the initial stress reactions, as is the case for instance in inflammatory disorders and autoimmune diseases.
The Regulation of Emotion and Cognition by the HPA Axis (For Coping and Storing Experience in the Memory for Future use)
As stated in the introduction, the action of cortisol in the central nervous system is mediated by two steroid receptors, the mineralocorticoid (MR) and glucocorticoid receptor ( GR). An appropriate balance of MR and GR activation is key for optimal control of emotion and cognition that is regulated by the limbic system. In accordance, MR and GR expression is high, especially in the hippocampus, amygdala, and prefrontal cortex [7, 8]. Basal levels of cortisol via MR stimulate neuronal excitation and determine the initial defence against the stressor, a finding that translates to vulnerability and resilience to psychiatric disease [9]. In contrast, stress-induced activation of GR coordinates the recovery, processing of information, and storage of the experience in the memory through reduction of neuronal excitation . In a general sense, these effects on excitability affect the overall activity of brain regions and circuits in ways that bias emotional and behavioural responses towards survival (e.g. by increasing likelihood of habitual rather than goal-directed responses [10]).
MR and GR activation depends foremost on binding of cortisol. High-affinity MRs are already occupied by low, basal levels of hormone, whereas GR affinity is such that substantial activation takes place during the circadian peak and after stress. Thus, mildly elevated trough levels may bias receptor activation towards the MR [11]. Intracellular prereceptor metabolism and differential tissue access are two other factors that determine cortisol levels ‘seen’ by the receptors [12, 13].
Next to hormone levels, absolute and relative MR/GR activation depends on expression and posttranslational modifications. Expression can vary as a consequence of genetic variation [14], early life programming effects [15], and regulation during adult life (see below). Because MRs can be considered tonically activated even at relatively low levels of cortisol, it has been argued that regulation of receptor amount is an important level of regulation. However, receptor regulation of expression is also a relevant variable for GR, for example, in view of its homologous down-regulation upon chronic hormone exposure [16].
The MR- and GR-dependent effects are not autonomous, but occur in conjunction with central stress-responsive transmitters such as noradrenalin, corticotrophin-releasing hormone (CRH), and urocortins. A prime example is the interaction between noradrenalin and glucocorticoid hormones in the amygdala and hippocampus that underlies stress-induced facilitation of memory consolidation [17]. The effects of cortisol interact with those of other neurotransmitters in two ways.
First, because cortisol affects neuronal excitability rather than neuronal firing per se [18], the effects are permissive : they bias how the brain responds depends on the current state of activity and demands on the system. For example, neuropsychiatric symptoms that can be induced by cortisol and its synthetic homologues include psychosis [19]. It can be expected that this particular vulnerability is highest in subjects that—in absence of any psychopathology—have high basal activity of dopaminergic signalling, or other pathways that can be causal to psychotic states. Permissive effects imply that ‘moving parts’ of the circuit are affected most strongly. A hypothesis that is testable is that this vulnerability becomes manifest in an interaction between high cortisol and variation in psychosis-related genes.
A second context-dependence lies in effects of neurotransmitters on functionality of the MR and GR. Animal studies have shown that activation of brain-derived neurotropic factor (BDNF) increases the phosphorylation of the GR in the hypothalamus. This in turn potentiates many effects of GR on gene expression [20]. Likewise, a prior history of stressful circumstances led to a dramatic change in the genes that were regulated in the rat hippocampus upon treatment with a single dose of corticosterone. Genome-wide analysis revealed that corticosterone could regulate the expression of around 600 genes in the hippocampus both in naïve and in chronically stressed rats. Strikingly, only 50 % of these genes were common to both groups. This implies that previous, recent history substantially remodels—via unknown mechanisms—the way in which the neuronal circuits respond to glucocorticoid exposure [21].
Animal Models of HPA Axis Disturbances
Animal studies have been indispensable to gain insight in the many effects of corticosteroids and their underlying mechanisms [22]. Of note, the sole glucocorticoid in rodents is corticosterone, which does differ from cortisol in some aspects, most notably in relation to transport into tissues [12]. Such species differences become even more pronounced when studying cortisol in the context of stress-related brain circuitry , as readouts of psychological state are necessarily indirect in rodents. A prime example has been the Porsolt forced swim test, in which active swimming/struggling is compared to passive floating. This behaviour is surely strongly dependent on glucocorticoids, but the interpretation of these effects has been given very differently, either as inducing a depression-like state or rather as adaptive memory processes [23].
Nevertheless, animal models do give insights on the brain effects of glucocorticoids per se and on their roles as mediators of the consequences of physical and psychological stress . Classic models of glucocorticoid exposure include treatment via implanted pellets and drinking water. Such studies—in absence of stressors—have revealed many principles of feedback regulation [24] and genomic targets predominantly in the hippocampus. Many of these targets are evolutionary conserved [25]. Such studies have also shown the consequences of chronic hypercortisolemia for the morphology of neurons and size of brain areas. Earlier studies revealed the vulnerability of the hippocampus to glucocorticoid exposure, including shrinking of dendrites of the principal cells in the CA3 area and effects on adult neurogenesis in the dentate gyrus.
Of note, it is not only the overall amount of cortisol that is important, but also the pattern of exposure over the day—as is clear from the imperfections of current replacement therapies. An elegant approach to studying the importance of circadian variation has been to treat animals with low, constant levels of corticosterone, which leads to suppression of the endogenous secretion at the time of circadian peak. This regimen ensures flattened diurnal rhythms in absence of hypercorticism [26]. It has been useful to study both negative feedback and corticosterone effects on hippocampal gene expression [11, 16]. Also, the importance of the ultradian rhythm of glucocorticoid rhythms was revealed in rats, showing marked effects on behavioural and endocrine stress responsiveness that correlated with changes in neuronal activation in the amygdala . Twelve hours of constant low, rather than absence of a corticosterone rhythm led to a blunted neuronal response to an acute stressor stressor, in conjunction with a blunted ACTH response to the stressor. In this setting, also the timing of the stressor relative to the phase of ultradian peaks was of consequence, suggesting rapid feedback effects from these one-hour corticosterone peaks [27].
A last approach to study the effects of glucocorticoids on the brain makes use of the fact that dexamethasone strongly suppresses ACTH secretion at the level of the pituitary, but at low doses do not penetrate into the brain [28, 29]. In this way, a state of selective central hypocorticism can be created [30]. This approach was used to demonstrate the importance of glucocorticoid rhythmicity for the plasticity of dendritic spines—the contact points for synaptic contacts that form the structural basis for plasticity of the brain. Circadian glucocorticoid peaks allowed the formation of dendritic spine, while troughs were required for stabilizing newly formed spines, which are important for long-term memory retention [31].
The role of MR and GR in individual cell types of the brain has also been approached using transgenic methodologies , using either advanced transgenic mice [32] or local manipulation of expression in adult mice [33, 34].
There is a plethora of models for glucocorticoids as mediators of the effects of stress. Steroids in general can have either long-term programming effects, or more adaptive activational effects. In line, there are models that focus on early life stressors, stressors during adolescence, and stressors during adult life. The latter have a logical extension to any animal model for disease that is available.
Early life experience—even in utero—can have major consequences for the development of emotional reactivity in later life [35]. Consequences of early life stress often include the development of anxiety and reprogramming of the HPA axis [36, 37]. This type of programming was recognized in animal studies as early as the 1950s [38]. Many types of early life stressors have been used, ranging from 24 h separation between mother and pup to creating ‘disorganized mothers’ by limiting the amount of bedding material that is available to the dam [39]. The direct contribution of glucocorticoids in the development of later life changes has mainly been studied in the prenatal models, also in relationship to the barrier function of the placenta for maternal cortisol [40].
The effects of stress-induced corticosterone have also been extensively studied using rodent models. The different types of stressors differ in physical and psychological components, intensity, duration, predictability, and controllability. Much is known on the role of glucocorticoids in models for single traumatic events , based on fear-conditioning paradigms [41]. However, many clinical issues involve more chronic exposure to stress and cortisol. The often-used stressor of repeated restraint can lead to substantial habituation of at least the HPA-axis response [42], and while this is accompanied by strong changes in the brain reactivity [21], it does not model chronically elevated cortisol. Therefore, many recent studies have taken to the non-habituating models of chronic unpredictable stress [43]. Certainly, many effects observed in these models depend on elevation of glucocorticoid levels [44].
However, even if stress and glucocorticoids predispose to disease, a stress-model per se may not suffice to study particular pathologies. In this respect, there is more direct information in combining existing disease models and treatment with MR and GR agonists or antagonists. A case in point is a recent impressive study where the GR antagonist mifepristone was efficacious both in a rat model of alcohol abuse and in a group of addicted human subjects [45]. In particular, such studies using receptor antagonists (or cortisol-lowering agents [46]) point to involvement of cortisol in pathogenic processes, even in situations without an obvious or dominant stress-related component.
Human Models for the Effects of Glucocorticoids on Neuropsychological Function
Cushing’s Syndrome
Cushing’s syndrome is a rare endocrine disorder characterized by long-term exposure to elevated endogenous glucocorticoid levels . Cushing’s syndrome is caused by either an ACTH secreting pituitary adenoma (70 % of cases), ectopic ACTH secretion (mostly bronchial carcinoids), or by autonomous cortisol hyper-secretion secondary to an adrenal adenoma/carcinoma, or adrenal hyperplasia. Cushing’s syndrome can also be induced by long-term administration of supraphysiological doses of synthetic corticosteroids, as is prescribed in clinical practice for a variety of inflammatory conditions and autoimmune diseases. This so-called exogenous Cushing’s syndrome is highly prevalent and insufficiently recognized in routine clinical practice, especially in the milder cases.
In accordance with the earlier described biological effects of glucocorticoids, the vast majority of patients with Cushing’s syndrome have both physical and psychological morbidity [47]. In patients with active or uncontrolled disease, neurocognitive function (that includes cognition, mood, and personality) is affected, and psychopathology is also often observed. In active Cushing’s syndrome, the frequency of psychiatric symptoms was reported starting in the early 1980s, demonstrating that symptoms like irritability, depressed mood, and anxiety were present in the majority of the patients [48]. In accordance, depression was present in more than 50 % of patients in a large cohort of patients with Cushing’s disease reported by Sonino and colleagues, and was significantly associated with older age, female sex, higher pretreatment urinary cortisol levels, a more severe clinical condition, and no pituitary adenoma on pituitary imaging [49]. Intriguingly, an increased overall psychiatric disability score was associated with increased cortisol secretion. In addition, patients with active Cushing’s syndrome report cognitive impairments , like memory problems and lack of concentration [50, 51]. Thus, the most common comorbid disorder is major depression, and a severe clinical presentation of Cushing’s often also includes depression (though to a lesser extent mania and anxiety disorders have also been reported). These observations are in line with the pivotal evolutionary role ascribed to cortisol in the control of mood and behaviour. Because limbic structures like the hippocampus and the prefrontal cortex are rich in glucocorticoid-receptors, these clinical observations suggest that these structures are particularly vulnerable to the cortisol excess as is present in Cushing’s syndrome.
The limited numbers of patients who have been reported after treatment indicate that a significant improvement occurs within the first year after treatment [52, 53]. In addition, reduction of glucocorticoid synthesis or action, either with metyrapone, ketoconazole, or mifepristone, rather than treatment with antidepressant drugs, is generally successful in relieving depressive symptoms, as well as other disabling symptoms [54, 55]. Thus, following successful correction of hypercortisolism, both physical and psychiatric signs and symptoms improve substantially. In the long-term, however, it now becomes evident from an accumulating number of studies that patients do not completely return to their premorbid level of functioning. These studies demonstrated residual physical and psychopathological morbidity despite long-term biochemical remission [56–59]. In addition, patients with long-term remission of CD reported persistent impairments in cognitive functioning [58, 60] and a reduced quality of life [61]. To which extent psychopathology still affects general well-being after long-term cure of CS is still, however, not clear.
An emerging topic of interest in this respect is the relation between glucocorticoid excess and changes in brain structure and function, and consequently, its relation with neuropsychological dysfunction .
The first observations in the human indicating that long-term exposure to elevated glucocorticoids may affect the brain were reported by Lupien and colleagues [62]. In that particular study, exposure to prolonged elevated cortisol levels in aged humans led to reduced hippocampal volumes as well as memory deficits (when compared to controls with normal cortisol levels). In later studies, however (in healthy young men), a larger hippocampal volume got associated with a greater cortisol response both in a social stress test (Trier social stress test ) and in the cortisol awakening response, questioning the relevance of the former finding in aged individuals for younger individuals [63]. Many psychiatric diseases, like major depressive and bipolar disorder, have been linked to alterations in the HPA axis [64, 65], and GC receptor polymorphisms that alter glucocorticoid sensitivity have been associated with depression (reviewed in [66]). In addition, other studies in patients with psychiatric diseases indicate that limbic structure volumes , like the hippocampus and the amygdala, are smaller [67, 68], though these changes may also be associated with brain aging and interact with the progression of the disorder [69].
The effects of Cushing’s syndrome on the brain, reflecting long-term excessive overexposure to endogenous cortisol, were recently reported in a systematic review [52]. This review systematically evaluated all studies in patients with active and remitted Cushing’s disease or syndrome using MRI (n = 19). These studies demonstrated that structural abnormalities in the grey matter were present in patients with active disease, which were characterized by smaller hippocampal volumes, enlarged ventricles, and cerebral atrophy (see also: [70]). In addition, functional changes occurred, characterized by alterations in neurochemical concentrations and functional activity . Intriguingly, the reversibility of structural and neurochemical alterations after correction of cortisol excess was incomplete, even when patients were evaluated after long-term remission. The structural alterations after long-term remission included smaller grey matter volumes of the anterior cingulate cortex, greater grey matter volume of the left posterior lobe of the cerebellum [71], and widespread reductions in white matter integrity [72, 73]. Long-lasting functional alterations included increased resting state functional connectivity between the limbic network and the subgenual subregion of the anterior cingulate cortex [74] and altered neural processing of emotional faces [75]. Some findings as obtained using MRI were related to the severity of the cortisol excess, and others also to neuropsychological functioning (as reflected by mood, cognition, and emotional functioning) and quality of life. This points towards persistent changes in brain function after previous exposure to hypercortisolism.
Adrenal Insufficiency
Adrenal insufficiency per se, by definition, will result in impaired stress responsiveness. In the human, this is best exemplified by the clinical application of the insulin tolerance test that is considered the golden standard for the diagnosis of adrenal insufficiency. The test is based upon induction of the stress response by insulin-induced hypoglycaemia, which from an evolutionary perspective is one of the most potent physiological stressors because it is potentially lethal. In accordance, the response to severe hypoglycaemia is characterized both by a sympathetic noradrenergic response (tachycardia, agitation, sweating, etc.) and stimulation of cortisol secretion through activation of the HPA axis. Patients with adrenal insufficiency (regardless the cause) are not able to secrete sufficient cortisol after hypoglycaemia (and fail this test). The subsequent metabolic and behavioural adaptations orchestrated by cortisol via the mineralo- and glucocorticoid receptor are not or insufficiently induced. Thus, by definition, these patients exhibit impaired stress responsiveness, and in accordance, even patients with adrenal insufficiency that were on stable hydrocortisone replacement reported impairments in quality of life [76–78].
Cognitive function in patients with adrenal insufficiency on hydrocortisone replacement has been reported only in seven studies involving a total of 195 patients [79–85]. These studies indicate that mild cognitive deficits may persist, especially in memory and executive functioning tasks. Intriguingly, patients performed better on concentration and attentional tasks when compared with controls [83], and cognitive function was neither affected by the dose used (high vs. low daily dose) [85], nor by postponement of the first daily dose by a few hours [83].
Besides cognition, neurocognitive functioning also includes mood and personality. Adrenal insufficiency may present solely with psychiatric manifestations [86, 87] and epidemiological studies indicate that patients with adrenal insufficiency may be at increased risk of developing severe affective disorders. When hospitalized patients with Addison’s disease were compared to hospitalized patients with osteoarthritis, the former had a more than two times greater rate of affective disorders and 1.7 times greater rate of depressive disorders [88]. In the Leiden cohort, we observed more psychosocial morbidity (irritability and somatic arousal) in the presence of impairments in quality of life when patients with adrenal insufficiency were compared with controls. Patients and controls did not differ regarding maladaptive personality traits; however, the daily hydrocortisone dose proved to be strongly associated both with the prevalence of maladaptive personality traits and with depression [78].
Patients Using Glucocorticoids
Glucocorticoids are frequently prescribed for various conditions like chronic obstructive pulmonary diseases and autoimmune diseases to inhibit the inflammatory response. Soon after their introduction in the 1950s, the first cases were reported on severe neuropsychiatric manifestations after the initiation of glucocorticoid therapy [89, 90]. In agreement with the studies in endogenous CS reported by Sonino and colleagues, more than 50 % of patients exposed to glucocorticoids for more than 3 months developed neuropsychiatric symptoms/manifestations [91]. A recent review beautifully summarized the topic of the adverse neuropsychological consequences of glucocorticoid therapy [19]. The acute and long-term effects on both mood and cognition have been studied in prospective studies, and the severe neuropsychiatric effects in case studies and with the use of epidemiological databases [92]. The observed rates and spectrum of manifestations of depression, anxiety disorders, and cognitive dysfunction are similar to those as observed in endogenous Cushing’ syndrome and exemplifies that glucocorticoids can induce the same neuropsychological phenotype (in pre-disposed individuals). The most prominent risk factors identified were gender (male patients being more prone to develop mania and delirium, and female patients being more prone for depression), a past history for psychiatric disorders, and the initial daily glucocorticoid dose (in general above 40 mg of prednisone daily equivalent). Finally, withdrawal from long-term glucocorticoid therapy also increases the risk for severe psychiatric manifestations. Again, a past history of psychiatric disease and also the use of long-acting glucocorticoids (especially dexamethasone) increased the risk for depression and delirium following discontinuation of glucocorticoid therapy [93].
Summary and Conclusions
Glucocorticoids play a key role in the control of neuropsychological functioning, which is exemplified by the evolutionary conserved control of behaviour in the ‘fight or flight response’. In accordance, both animal and human models of uncontrolled (and therefore abnormal) exposure to glucocorticoids show impaired stress responsiveness, cognitive dysfunction, and a broad spectrum of neuropsychiatric disorders, ranging from severe depression and anxiety disorders to acute psychosis and delirium (for a summary, see Table 1). The fact that the same phenotype can be induced by exogenous glucocorticoid administration proves the causal role of glucocorticoids per se on neurocognitive and neuropsychiatric functioning. Finally, it now becomes clear that these effects may be long-lasting and even may not be completely reversible because cognitive dysfunction and maladaptive personality traits persist in the presence of altered coping strategies and affected illness perception s despite long-term optimal treatment. This implies that long-term care for both patients with pituitary and adrenal disorders and patients using glucocorticoids should incorporate self-management interventions that help to improve quality of life.
References
de Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6(6):463–75.
McCall JG, et al. CRH engagement of the locus coeruleus noradrenergic system mediates stress-induced anxiety. Neuron. 2015;87(3):605–20.
Laugero KD, et al. Corticosterone infused intracerebroventricularly inhibits energy storage and stimulates the hypothalamo-pituitary axis in adrenalectomized rats drinking sucrose. Endocrinology. 2002;143(12):4552–62.
Jiang CL, Liu L, Tasker JG. Why do we need nongenomic glucocorticoid mechanisms? Front Neuroendocrinol. 2014;35(1):72–5.
Karst H, et al. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci U S A. 2005;102(52):19204–7.
Evanson NK, et al. Fast feedback inhibition of the HPA axis by glucocorticoids is mediated by endocannabinoid signaling. Endocrinology. 2010;151(10):4811–9.
Datson NA, et al. Identification of corticosteroid-responsive genes in rat hippocampus using serial analysis of gene expression. Eur J Neurosci. 2001;14(4):675–89.
de Kloet ER, et al. Brain mineralocorticoid receptors and centrally regulated functions. Kidney Int. 2000;57(4):1329–36.
Klok MD, et al. A common and functional mineralocorticoid receptor haplotype enhances optimism and protects against depression in females. Transl Psychiatry. 2011;1(12), e62.
Sousa N, Almeida OF. Disconnection and reconnection: the morphological basis of (mal)adaptation to stress. Trends Neurosci. 2012;35(12):742–51.
Meijer OC, Van Oosten RV, De Kloet ER. Elevated basal trough levels of corticosterone suppress hippocampal 5-hydroxytryptamine(1A) receptor expression in adrenally intact rats: implication for the pathogenesis of depression. Neuroscience. 1997;80(2):419–26.
Karssen AM, et al. Multidrug resistance P-glycoprotein hampers the access of cortisol but not of corticosterone to mouse and human brain. Endocrinology. 2001;142(6):2686–94.
Wyrwoll CS, Holmes MC, Seckl JR. 11β-hydroxysteroid dehydrogenases and the brain: from zero to hero, a decade of progress. Front Neuroendocrinol. 2011;32(3):265–86.
van Leeuwen N, et al. Human mineralocorticoid receptor (MR) gene haplotypes modulate MR expression and transactivation: implication for the stress response. Psychoneuroendocrinology. 2011;36(5):699–709.
Turecki G, Meaney MJ. Effects of the social environment and stress on glucocorticoid receptor gene methylation: a systematic review. Biol Psychiatry. 2016;79(2):87–96.
Sarabdjitsingh RA, et al. Disrupted corticosterone pulsatile patterns attenuate responsiveness to glucocorticoid signaling in rat brain. Endocrinology. 2010;151(3):1177–86.
Roozendaal B, McGaugh JL. Memory modulation. Behav Neurosci. 2011;125(6):797–824.
Joels M, Sarabdjitsingh RA, Karst H. Unraveling the time domains of corticosteroid hormone influences on brain activity: rapid, slow, and chronic modes. Pharmacol Rev. 2012;64(4):901–38.
Judd LL, et al. Adverse consequences of glucocorticoid medication: psychological, cognitive, and behavioral effects. Am J Psychiatry. 2014;171(10):1045–51.
Lambert WM, et al. Brain-derived neurotrophic factor signaling rewrites the glucocorticoid transcriptome via glucocorticoid receptor phosphorylation. Mol Cell Biol. 2013;33(18):3700–14.
Datson NA, et al. Previous history of chronic stress changes the transcriptional response to glucocorticoid challenge in the dentate gyrus region of the male rat hippocampus. Endocrinology. 2013;154(9):3261–72.
de Kloet ER, et al. Glucocorticoid signaling and stress-related limbic susceptibility pathway: about receptors, transcription machinery and microRNA. Brain Res. 2009;1293:129–41.
Molendijk ML, de Kloet ER. Immobility in the forced swim test is adaptive and does not reflect depression. Psychoneuroendocrinology. 2015;623:89–91.
Dallman MF, et al. Regulation of ACTH secretion: variations on a theme of B. Recent Prog Horm Res. 1987;43:113–73.
Datson NA, et al. Specific regulatory motifs predict glucocorticoid responsiveness of hippocampal gene expression. Endocrinology. 2011;152(10):3749–57.
Akana SF, et al. Feedback sensitivity of the rat hypothalamo-pituitary-adrenal axis and its capacity to adjust to exogenous corticosterone. Endocrinology. 1992;131(2):585–94.
Sarabdjitsingh RA, et al. Stress responsiveness varies over the ultradian glucocorticoid cycle in a brain-region-specific manner. Endocrinology. 2010;151(11):5369–79.
De Kloet R, Wallach G, McEwen BS. Differences in corticosterone and dexamethasone binding to rat brain and pituitary. Endocrinology. 1975;96(3):598–609.
Meijer OC, et al. Penetration of dexamethasone into brain glucocorticoid targets is enhanced in mdr1A P-glycoprotein knockout mice. Endocrinology. 1998;139(4):1789–93.
Karssen AM, et al. Low doses of dexamethasone can produce a hypocorticosteroid state in the brain. Endocrinology. 2005;146(12):5587–95.
Liston C, et al. Circadian glucocorticoid oscillations promote learning-dependent synapse formation and maintenance. Nat Neurosci. 2013;16(6):698–705.
Ambroggi F, et al. Stress and addiction: glucocorticoid receptor in dopaminoceptive neurons facilitates cocaine seeking. Nat Neurosci. 2009;12(3):247–9.
Fitzsimons CP, et al. Knockdown of the glucocorticoid receptor alters functional integration of newborn neurons in the adult hippocampus and impairs fear-motivated behavior. Mol Psychiatry. 2013;18(9):993–1005.
Kolber BJ, et al. Central amygdala glucocorticoid receptor action promotes fear-associated CRH activation and conditioning. Proc Natl Acad Sci U S A. 2008;105(33):12004–9.
Bock J, et al. Stress in utero: prenatal programming of brain plasticity and cognition. Biol Psychiatry. 2015;78(5):315–26.
Klengel T, Binder EB. Epigenetics of stress-related psychiatric disorders and gene × environment interactions. Neuron. 2015;86(6):1343–57.
Schmidt MV, Wang XD, Meijer OC. Early life stress paradigms in rodents: potential animal models of depression? Psychopharmacology (Berl). 2011;214(1):131–40.
Levine S. Infantile experience and resistance to physiological stress. Science. 1957;126(3270):405.
Molet J, et al. Naturalistic rodent models of chronic early-life stress. Dev Psychobiol. 2014;56(8):1675–88.
Chapman K, Holmes M, Seckl J. 11β-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev. 2013;93(3):1139–206.
Kaouane N, et al. Glucocorticoids can induce PTSD-like memory impairments in mice. Science. 2012;335(6075):1510–3.
Grissom N, Bhatnagar S. Habituation to repeated stress: get used to it. Neurobiol Learn Mem. 2009;92(2):215–24.
Willner P, et al. Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology (Berl). 1987;93(3):358–64.
Joels M, et al. Chronic stress: implications for neuronal morphology, function and neurogenesis. Front Neuroendocrinol. 2007;28(2-3):72–96.
Vendruscolo LF, et al. Glucocorticoid receptor antagonism decreases alcohol seeking in alcohol-dependent individuals. J Clin Invest. 2015;125(8):3193–7.
Sooy K, et al. Cognitive and disease-modifying effects of 11β-hydroxysteroid dehydrogenase type 1 inhibition in male Tg2576 mice, a model of Alzheimer’s disease. Endocrinology. 2015;156(12):4592–603.
Newell-Price J, et al. Cushing’s syndrome. Lancet. 2006;367(9522):1605–17.
Starkman MN, Schteingart DE. Neuropsychiatric manifestations of patients with Cushing’s syndrome. Relationship to cortisol and adrenocorticotropic hormone levels. Arch Intern Med. 1981;141(2):215–9.
Sonino N, et al. Clinical correlates of major depression in Cushing’s disease. Psychopathology. 1998;31(6):302–6.
Starkman MN, et al. Elevated cortisol levels in Cushing’s disease are associated with cognitive decrements. Psychosom Med. 2001;63(6):985–93.
Webb SM, et al. Evaluation of health-related quality of life in patients with Cushing’s syndrome with a new questionnaire. Eur J Endocrinol. 2008;158(5):623–30.
Andela CD, et al. Mechanisms in endocrinology: Cushing’s syndrome causes irreversible effects on the human brain: a systematic review of structural and functional magnetic resonance imaging studies. Eur J Endocrinol. 2015;173(1):R1–14.
Hook JN, et al. Patterns of cognitive change over time and relationship to age following successful treatment of Cushing’s disease. J Int Neuropsychol Soc. 2007;13(1):21–9.
Jeffcoate WJ, et al. Psychiatric manifestations of Cushing’s syndrome: response to lowering of plasma cortisol. Q J Med. 1979;48(191):465–72.
Sonino N, Fava GA. Psychiatric disorders associated with Cushing’s syndrome. Epidemiology, pathophysiology and treatment. CNS Drugs. 2001;15(5):361–73.
Dorn LD, et al. The longitudinal course of psychopathology in Cushing’s syndrome after correction of hypercortisolism. J Clin Endocrinol Metab. 1997;82(3):912–9.
Milian M, et al. Similar psychopathological profiles in female and male Cushing’s disease patients after treatment but differences in the pathogenesis of symptoms. Neuroendocrinology. 2014;100(1):9–16.
Resmini E, et al. Verbal and visual memory performance and hippocampal volumes, measured by 3-Tesla magnetic resonance imaging, in patients with Cushing’s syndrome. J Clin Endocrinol Metab. 2012;97(2):663–71.
Tiemensma J, et al. Increased prevalence of psychopathology and maladaptive personality traits after long-term cure of Cushing’s disease. J Clin Endocrinol Metab. 2010;95(10):E129–41.
Tiemensma J, et al. Subtle cognitive impairments in patients with long-term cure of Cushing’s disease. J Clin Endocrinol Metab. 2010;95(6):2699–714.
van Aken MO, et al. Quality of life in patients after long-term biochemical cure of Cushing’s disease. J Clin Endocrinol Metab. 2005;90(6):3279–86.
Lupien SJ, et al. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat Neurosci. 1998;1(1):69–73.
Pruessner M, et al. The associations among hippocampal volume, cortisol reactivity, and memory performance in healthy young men. Psychiatry Res. 2007;155(1):1–10.
Antonijevic IA. Depressive disorders—is it time to endorse different pathophysiologies? Psychoneuroendocrinology. 2006;31(1):1–15.
Belvederi Murri M, et al. The HPA axis in bipolar disorder: systematic review and meta-analysis. Psychoneuroendocrinology. 2016;63:327–42.
Spijker AT, van Rossum EF. Glucocorticoid receptor polymorphisms in major depression. Focus on glucocorticoid sensitivity and neurocognitive functioning. Ann N Y Acad Sci. 2009;1179:199–215.
Harrisberger F, et al. BDNF Val66Met polymorphism and hippocampal volume in neuropsychiatric disorders: a systematic review and meta-analysis. Neurosci Biobehav Rev. 2015;55:107–18.
Malykhin NV, Coupland NJ. Hippocampal neuroplasticity in major depressive disorder. Neuroscience. 2015;309:200–13.
Alves GS, et al. Structural neuroimaging findings in major depressive disorder throughout aging: a critical systematic review of prospective studies. CNS Neurol Disord Drug Targets. 2014;13(10):1846–59.
Burkhardt T, et al. Hippocampal and cerebellar atrophy in patients with Cushing’s disease. Neurosurg Focus. 2015;39(5), E5.
Andela CD, et al. Smaller grey matter volumes in the anterior cingulate cortex and greater cerebellar volumes in patients with long-term remission of Cushing’s disease: a case-control study. Eur J Endocrinol. 2013;169(6):811–9.
Pires P, et al. White matter alterations in the brains of patients with active, remitted, and cured cushing syndrome: a DTI study. AJNR Am J Neuroradiol. 2015;36(6):1043–8.
van der Werff SJ, et al. Widespread reductions of white matter integrity in patients with long-term remission of Cushing’s disease. Neuroimage Clin. 2014;46:59–67.
van der Werff SJ, et al. Resting-state functional connectivity in patients with long-term remission of Cushing’s disease. Neuropsychopharmacology. 2015;40(8):1888–98.
Bas-Hoogendam JM, et al. Altered neural processing of emotional faces in remitted Cushing’s disease. Psychoneuroendocrinology. 2015;59:134–46.
Aulinas A, Webb SM. Health-related quality of life in primary and secondary adrenal insufficiency. Expert Rev Pharmacoecon Outcomes Res. 2014;14(6):873–88.
Bancos I, et al. Diagnosis and management of adrenal insufficiency. Lancet Diabetes Endocrinol. 2015;3(3):216–26.
Tiemensma J, et al. Psychological morbidity and impaired quality of life in patients with stable treatment for primary adrenal insufficiency: cross-sectional study and review of the literature. Eur J Endocrinol. 2014;171(2):171–82.
Harbeck B, Kropp P, Monig H. Effects of short-term nocturnal cortisol replacement on cognitive function and quality of life in patients with primary or secondary adrenal insufficiency: a pilot study. Appl Psychophysiol Biofeedback. 2009;34(2):113–9.
Henry M, Thomas KG, Ross IL. Episodic memory impairment in Addison’s disease: results from a telephonic cognitive assessment. Metab Brain Dis. 2014;29(2):421–30.
Klement J, et al. Effects of glucose infusion on neuroendocrine and cognitive parameters in Addison disease. Metabolism. 2009;58(12):1825–31.
Schultebraucks K, et al. Cognitive function in patients with primary adrenal insufficiency (Addison’s disease) and the role of mineralocorticoid receptors. Psychoneuroendocrinology. 2015;55:1–7.
Tiemensma J, et al. Mild cognitive deficits in patients on stable treatment for primary adrenal insufficiency. Psychoneuroendocrinology. 2015;61:46.
Tytherleigh MY, Vedhara K, Lightman SL. Mineralocorticoid and glucocorticoid receptors and their differential effects on memory performance in people with Addison’s disease. Psychoneuroendocrinology. 2004;29(6):712–23.
Werumeus Buning J, et al. The effects of two different doses of hydrocortisone on cognition in patients with secondary adrenal insufficiency—results from a randomized controlled trial. Psychoneuroendocrinology. 2015;55:36–47.
Anglin RE, Rosebush PI, Mazurek MF. The neuropsychiatric profile of Addison’s disease: revisiting a forgotten phenomenon. J Neuropsychiatry Clin Neurosci. 2006;18(4):450–9.
Pavlovic A. Sivakumar V. Hypoadrenalism presenting as a range of mental disorders. BMJ Case Rep. 2011. pii: bcr0920103305. doi:10.1136/bcr.09.2010.3305.
Thomsen AF, et al. The risk of affective disorders in patients with adrenocortical insufficiency. Psychoneuroendocrinology. 2006;31(5):614–22.
Manzini B. Psychotic reactions during prednisone therapy. Riv Sper Freniatr Med Leg Alien Ment. 1958;82(2):417–29.
Piguet B. Study of attacks of tetany and psychological disorders appearing during adrenal cortex hormone therapy: attacks of tetany and grave psychoses initiated by substitution of delta-cortisone for hydrocortisone and subsequently by ACTH. Rev Rhum Mal Osteoartic. 1958;25(12):814–28.
Fardet L, et al. Corticosteroid-induced clinical adverse events: frequency, risk factors and patient’s opinion. Br J Dermatol. 2007;157(1):142–8.
Fardet L, Petersen I, Nazareth I. Suicidal behavior and severe neuropsychiatric disorders following glucocorticoid therapy in primary care. Am J Psychiatry. 2012;169(5):491–7.
Fardet L, et al. Severe neuropsychiatric outcomes following discontinuation of long-term glucocorticoid therapy: a cohort study. J Clin Psychiatry. 2013;74(4):e281–6.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Pereira, A.M., Meijer, O.C. (2017). Glucocorticoid Regulation of Neurocognitive and Neuropsychiatric Function. In: Geer, E. (eds) The Hypothalamic-Pituitary-Adrenal Axis in Health and Disease. Springer, Cham. https://doi.org/10.1007/978-3-319-45950-9_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-45950-9_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-45948-6
Online ISBN: 978-3-319-45950-9
eBook Packages: MedicineMedicine (R0)