
Abstract
Thermal analysis is a series of laboratory techniques that measure physical and chemical properties of materials as a function of temperature and time.Thermal analysis results provide insight into the structure and quality of both starting materials and finished products. The physical structure (amorphous, crystalline, semi-crystalline) of a material creates a set of physical properties, which in turn define end-use properties, such as texture and storage stability. Areas of application include quality assurance, product development, and research into new materials, formulations, and processing conditions.This chapter covers the principles, experimental conditions, common measurements, and applications of the two most frequently used thermal analysis techniques, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC).Also covered is modulated temperature DSC (MDSC®).This chapter includes illustrated application of thermal analysis to a variety of food materials.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Thermal analysis is a term used to describe a broad range of analytical techniques that measure physical and chemical properties as a function of temperature, time, and atmosphere (inert or oxidizing gas, pressure, and relative humidity). Depending on the technique, test temperatures can range from −180 to 1,000 °C or more, allowing investigation into a range of applications, including low-temperature stability and processing (e.g., freezing and freeze-drying) to high-temperature processing and cooking (e.g., extrusion, spray drying, and frying).
Thermal analysis results provide insight into the structure and quality of starting materials, as well as finished products. The physical structure (amorphous, crystalline, semicrystalline) of a material creates a set of physical properties, which in turn define end-use properties, such as texture and storage stability. Areas of application include quality assurance, product development, and research into new materials, formulations, and processing conditions [1–4].
Specific instruments are typically used for characterization of a particular property. Instrumentation includes a transducer, used to measure the property of interest, and a temperature-measuring device, such as a thermocouple, thermopile, or platinum resistance thermometer, used to record the sample’s temperature. Experiments are performed while heating, cooling, or at a constant temperature (isothermal), and measured signals are stored for analysis.
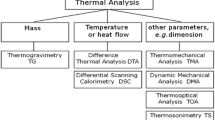
Thermal analysis techniques of major interest to the food researcher and the properties they measure are listed in Table 30.1, with the first three widely used techniques discussed in detail in Sect. 30.2.
Thermogravimetric analysis (TGA) is typically the first thermal analysis measurement done when characterizing a new material. TGA data can detect and quantify the presence of bulk water and/or associated water and identify the temperature at which molecular decomposition (chemical change) begins. The change in weight measured by TGA is quantitative; however, no information on the chemistry of evolved gases is obtained. If chemical knowledge of evolved gases is desired, TGA can be coupled to a mass spectrometer (MS) (see Chap. 11) and/or Fourier transform infrared (FTIR) spectrometer (see Chap. 8) [5].
Once composition and thermal stability are obtained from TGA results, the physical structure or “form” of the material is typically determined using differential scanning calorimetry (DSC) and/or modulated temperature DSC (MDSC®). Structure and the temperature(s) at which the structure changes [transition(s)] significantly influence physical and chemical properties of a material. By understanding structure and related physical properties, formulations can be developed that provide desired end-use properties, such as crispness, fast dissolution rate, and extended shelf life.
2 Materials Science
Since the pioneering work of Slade and Levine [e.g., 6, 7], food scientists have been actively applying the principles of materials science to the study of food materials [e.g., 8, 9]. One of the main driving forces underlying the application of materials science principles to foods is that the end-use properties (functionality) of a material at a specific temperature are dependent on the structure of the components at that temperature. Therefore, it is necessary to measure structure as a function of temperature. The primary use of thermal analysis is to determine structure by measuring the physical properties (e.g., heat capacity, flow, expansion, rigidity) associated with that structure.
2.1 Amorphous Structure
Many food products are amorphous or have a high amorphous content (semicrystalline). Examples include extruded snacks and breakfast cereals, low-moisture cookies and crackers, sugar-based hard candies, and spray-dried powdered drink mixes. Amorphous structure has no regular or systematic molecular order, which means that it has the highest energy and entropy content, the highest molecular mobility, and the fastest rate of dissolution, compared to crystalline structure. A potential problem with amorphous structure is that physical properties can change by orders of magnitude at a specific temperature, termed the glass transition temperature (Tg).
At temperatures below Tg, an amorphous material acts like a glass. It is rigid, has low molecular mobility, and has very high viscosity (low ability to flow). Above Tg, these materials act like a rubbery material, viscous liquid, or gel with greater free volume and much higher molecular mobility. Some amorphous materials (e.g., fats, oils, and water) have the ability to crystallize when cooled to lower temperatures. However, these materials may associate (e.g., hydrogen bond) with another material in the formulation and not crystallize, even at temperatures well below their freezing point. In the case of water, it is well known that not all the water in a food material freezes, even at very low storage temperatures [10].
Since properties change so significantly at Tg, it is important to be able to measure and control Tg by selecting appropriate ingredients in the correct weight ratio to other ingredients in a formulation or recipe. In foods, Tg can change by 50 °C or more via changes in moisture content. Crisp snack foods, such as crackers and potato chips, are classic examples of foods whose Tg and resulting physical properties are affected by changes in water content. For example, the Tg of freshly processed potato chips is much higher than room temperature; thus, the chips are crisp and have a pleasant texture. If the chips are exposed to ambient temperature and high humidity conditions for several hours, they become soft and pliable, which are textural characteristics typical of a low-moisture food held above Tg. Once the package of chips is opened, the low-moisture chips begin to absorb moisture from the air, which lowers Tg and creates a different set of physical properties at the consumption temperature.
Tg can be measured using most thermal analysis techniques due to significant changes in physical properties, such as heat capacity (for DSC), coefficient of thermal expansion (for thermomechanical analysis, TMA), and stiffness (for dynamic mechanical analysis, DMA), that accompany Tg [8]. However, DSC is usually the technique of choice because of easy sample preparation, short test times, straightforward data interpretation, and the ability to use sealed pans (hermetic), which prevent loss of moisture as the sample is heated. Because of the significant increase in molecular mobility and heat capacity that occurs as the sample is heated to a temperature above Tg, there is a corresponding increase in the heat flow rate measured by DSC. Data analysis software measures the temperature and magnitude of the change in heat flow that occurs, which is proportional to the amount of amorphous material in the sample (Fig. 30.1).

The glass transition of amorphous structure can be measured by differential scanning calorimetry (DSC) due to the significant increase in heat capacity that occurs as the material is heated to a temperature above the glass transition temperature (Tg). Typical Tg analysis includes the extrapolated onset, midpoint (temperature of one-half of the heat capacity change), and end point temperatures, as well as the difference in heat capacity (∆Cp, J/g°C)
2.2 Crystalline Structure
Crystalline structure is different from amorphous structure in many ways. Molecules have long-range order, lower energy (heat content) and entropy, higher density, and a different set of physical properties. Molecular mobility is low, which means that heat capacity is low. Melting of the crystalline material creates an amorphous liquid. Upon cooling of the amorphous liquid, some materials (e.g., fats) will crystallize, while others (e.g., sucrose and fructose) transition into an amorphous glass below their Tg.
Because of the increased density and molecular order within the crystal, there is a reduction in the ability of crystalline material to form hydrogen bonds and thus a reduced tendency to “absorb” moisture into their bulk structure from the atmosphere. However, crystalline materials “adsorb” water onto their surface quite quickly, mirroring the % relative humidity of the surrounding environment [11]. The melting of crystalline material occurs at a higher temperature than the glass transition associated with amorphous material, which makes crystalline material more stable and much more rigid (often gritty like table sugar and salt) than amorphous material over a wide temperature range. Because the crystalline structure is more stable, physical properties change less with time.
Since crystalline material has lower heat content than amorphous material, crystalline material must absorb heat (endothermic process) to become amorphous. The absorption of heat during a DSC experiment is seen as an endothermic peak, termed the melting peak. Data analysis software can measure the onset, peak, and end temperatures of the endothermic melting peak and calculate the heat (J/g) required to melt the sample by determining the area under the peak. The area of the melting peak (J/g) increases as the percent crystallinity of the material increases. Figure 30.2 shows the DSC data for melting of the crystalline sugar alcohol (polyol) mannitol, which is commonly used in confectioneries, such as “breath-freshening” mints and gums.

DSC data showing melting of the crystalline sugar alcohol (polyol) mannitol. Analysis of the data shows the extrapolated onset and peak melting temperatures and heat required to melt the crystalline structure (heat of fusion, joules/gram, J/g)
Normally, crystalline structure is converted to amorphous structure by heating the sample to a temperature (an energy level) that is high enough to overcome the energy associated with the crystalline lattice (termed thermodynamic melting). However, crystalline structure can also be lost due to processes such as dissolving of crystals in a solvent (dissolution), dehydration of a hydrated crystalline form, chemical interaction of functional groups between two materials in a mixture, and breaking of chemical bonds (decomposition) at temperatures below the true melting point of the material. Since these are time-dependent (kinetic) processes, the endothermic peak observed in DSC data shifts to a higher temperature as heating rate increases. A material that illustrates this behavior is the monosaccharide sugar fructose (Fig. 30.3). Figure 30.3 contains multiple Y-axes, as do other figures in this chapter. Since heat flow rate (W/g) is proportional to heating rate, it is necessary to scale the heat flow curves to different sensitivities to visually compare curves run at different heating rates, resulting in the need for more than one Y-axis.

The endothermic peak associated with loss of crystalline structure in fructose shifts to higher temperatures at higher heating rates, indicating the influence of a time-dependent (kinetic) process
2.3 Semicrystalline Structure
Many foods contain both amorphous and crystalline structures. In some cases, such as a lipid that melts over a temperature range, one ingredient can exist in both phases. A term that is sometimes used to describe a mixture of phases in a lipid is solid-fat content (SFC) (see Chap. 23, Sect. 23.3.11). At a particular temperature (usually room temperature, 72 °F or 22 °C), a certain fraction or percentage of the lipid material is solid (crystalline), and the remainder is liquid (melted). One lipid that illustrates this property at room temperature is cocoa butter, a common ingredient in chocolate. Cocoa butter can crystallize into six known forms (termed polymorphs), with the lowest stability form (form I) melting at approximately 17 °C and the highest stability form (form IV) melting at approximately 35 °C. Form V is the most desirable crystalline form of cocoa butter in chocolate, yielding a dark brown, glossy appearance, a satisfying snap when broken, and a melting temperature near mouth temperature.
Figure 30.4 shows the broad melt of a sugar-coated chocolate candy. There are several small overlapping melting peaks below 22 °C that are due to melting of the less stable lipid crystal forms in the sample. By measuring the percentage of melting below and above 22 °C, the ratio of liquid to solid phases can be determined for room temperature. A feature of the DSC data analysis software, termed the “running integral,” can plot percent melted versus temperature. Results show that 22.9 % of the cocoa butter is liquid (melted) at room temperature in this particular sample. The presence of the liquid lipid provides a creamy texture to the chocolate.

DSC data for sugar-coated chocolate candy, containing cocoa butter, shows several small overlapping melting peaks below 22 °C, due to melting of the less stable crystal lipid forms in the sample. In this particular sample, results show that 22.9 % of the cocoa butter is liquid (melted) at 22 °C; the remainder is crystalline (solid)
2.4 Thermodynamic and Kinetic Properties
As discussed in Sect. 30.3.2.1, a natural limitation of DSC, and other thermal analysis techniques used to measure structure, is that heating of the sample is typically required. As temperature increases, molecular mobility increases, which permits the structure to change in ways that are not always obvious in the data. Since the purpose of the experiment is typically to measure the existing structure in the material, users of thermal analysis instrumentation must be able to recognize whether observations are due to changes in structure (e.g., phase transitions) or composition (e.g., solvent evaporation).
Thermodynamic properties (e.g., heat capacity, enthalpy, and density) have absolute values as a function of temperature, while kinetic properties are always a function of time and temperature. Some common examples of kinetic processes observed in foods are freezing of water during cold storage, crystallization of fats, adsorption/desorption of water, staling of bread, and decomposition/oxidation during processing, such as deep-frying.
The easiest way to determine if an observed event in the data is due to thermodynamics or kinetics is to change the heating rate. Since heating rate has the units of °C/min, the reciprocal is min/°C. The higher the heating rate, the less time the sample experiences at each temperature. Therefore, high heating rates decrease the probability of structural change, while low heating rates increase the probability of structural change. If the onset temperature of the observed event remains relatively constant (changes <1 °C) with a tenfold change in heating rate, it is typically a thermodynamic event, while an increase in event temperature with heating rate indicates the influence of a kinetic process. An example of substantial heating rate dependence is shown in Fig. 30.4 for fructose, for which the onset temperature of the endothermic DSC peak shifts from 112.7 °C at a heating rate of 5 °C/min to 120.8 °C at a heating rate of 20 °C/min. As discussed previously, the kinetic event associated with the observed heating rate dependence in fructose is thermal decomposition [12].
3 Principles and Methods
This section describes the working principles of the most frequently used thermal techniques and makes recommendations on optimum experimental conditions for characterizing common materials.
3.1 Thermogravimetric Analysis
3.1.1 Overview
Thermogravimetric analysis (TGA) should be the first thermal analysis technique used to characterize a new material. TGA provides information about the composition (number of components) of the material and its thermal or oxidative stability (decomposition in inert and oxidizing atmospheres, respectively). TGA instruments use a specially designed and very sensitive analytical balance to measure weight changes, since samples are typically heated from room temperature to 1,000 °C or more. A thermocouple is located close to the sample to continuously record the temperature as weight changes occur. The heated sample chamber is typically purged with an inert gas, such as nitrogen or helium; however, air or oxygen can be used when measuring oxidative stability. Most weight changes are weight losses due to volatilization or decomposition, but weight gain may be observed during early stages of oxidation. A specialized version of TGA is designed with humidity control so that the rate of moisture sorption (both absorption and desorption) can be measured as a function of time, temperature, and relative humidity.
Figure 30.5 shows a schematic of possible designs for both conventional TGA and a humidity-controlled sorption analysis TGA. Both contain sample and reference pans attached to the balance. The reference pan is empty, since its purpose is to offset the weight of the sample pan. With conventional TGA, the reference pan is typically not heated. The reference pan for sorption analysis is exposed to the same temperature and relative humidity as the sample. This greatly improves the stability and baseline performance of the measurement. Most modern instruments have autosamplers so that many samples can be run sequentially without need of operator presence.

Thermogravimetric instrumentation measures weight change using a sensitive analytical balance. The sample container is typically suspended into a temperature- and atmosphere-controlled chamber (Adapted from figure by TA Instruments, New Castle, DE)
Most TGA instruments have several natural limitations. Although TGA provides a quantitative measurement of weight change, it is often difficult to quantify the weight of a specific component because weight losses typically overlap in temperature. Improved temperature resolution of these weight losses can usually be obtained by slowing the heating rate and reducing the sample weight. However, the lower heating rate increases test time (reduced productivity), and the smaller sample size reduces accuracy of small weight changes. Another limitation of TGA is that it cannot identify the chemistry of gases evolved from the sample. Knowledge of the gas composition helps to distinguish between water loss and loss of low molecular weight additives, such as flavors and fragrances, and helps to determine the chemical mechanisms involved in cooking and decomposition. Most manufacturers of mass spectrometers and Fourier transform infrared instruments offer interfaces between their products and the TGA instrument so that off-gases can be chemically identified.
3.1.2 Experimental Conditions
Although TGA experiments can be performed over a wide range of conditions, a good starting point for most materials is as follows:
-
Sample weight: 10–20 mg (larger samples improve sensitivity for detection of minor components)
-
Pan type: Platinum
-
Purge gas: Nitrogen
-
Start temperature: Room temperature, typically 20 °C
-
Heating rate: 10 °C/min (lower rates improve resolution of overlapping weight losses)
-
Final temperature: 300 °C
3.1.3 Common Measurements
TGA experiments are primarily heating experiments; however, isothermal (constant temperature) conditions can be used to determine drying rates or follow weight changes at processing/cooking temperatures. The most common measurements include the following:
-
Temperature of weight change
-
Free (or bulk) moisture content
-
“Bound” or associated water content (part of the structure)
-
Composition (multiple components)
-
Decomposition temperature
It should be noted that in reality, there is no such thing as a decomposition temperature. Decomposition is a kinetic process, which means that it is a function of both time and temperature. Therefore, the temperature of weight loss due to decomposition increases if the heating rate is increased.
Figure 30.6 shows TGA data for sugar-frosted cornflakes. Because it contains multiple minor components, a relatively large sample size of 58.6 mg was used to improve sensitivity to detect those components. The Y1-axis is expanded to focus on only the weight loss from 90 % to 100 %. In addition, the Y2- and Y3-axes show the derivative signal, which is the rate of weight loss, at two sensitivities in order to better detect and measure the first two components that contribute to sample weight loss. The first weight loss starts immediately and is typical of unassociated or free water within the sample. The peak in the derivative signal occurs well above 100 °C (boiling point of water) because time is required for diffusion of the water through the sample. Loss of weight due to the second component starts near 178 °C. At this temperature, the sample has lost about 2.7 % of its weight. Since decomposition of some component starts near 178 °C, the chemistry of the sample is changing, making it difficult to measure any meaningful structure by other techniques, such as those described in Sect. 30.3.2.

Thermogravimetric analysis (TGA) data for a 58 mg sample of sugar-coated cornflakes. The derivative signals show rate of weight loss and are plotted at two sensitivities to better illustrate loss of moisture and the first stage of thermal decomposition. At the start of decomposition (178 °C), the weight % curve shows that the sample contains at least 2.7 % water
3.2 Differential Scanning Calorimetry
3.2.1 Overview
Differential scanning calorimetry (DSC) is the most frequently used thermal analysis technique and probably accounts for 70 % of all thermal analysis measurements. Since every change in structure (transition) either absorbs or releases heat, DSC is the universal detector for measuring structure. The only major limitation of the technique is the sensitivity of the instrument, which is its ability to detect small transitions or very slow kinetic processes, where the rate of heat flow is similar to or less than the signal noise of the instrument.
The ability of DSC to measure very small rates (microwatts, μJ/s) of heat flow is greatly enhanced because it uses a differential signal. An empty reference pan is subjected to the same thermal environment as the sample pan, and the measured signal from the DSC is the difference in heat flow rate between the sample and reference. This effectively eliminates signal noise and drift caused by heat exchanges with the environment or atmosphere around the pans.
There are two general approaches to making the differential measurement. One approach uses a common furnace for both the sample and reference (heat flux design), while the other uses individual furnaces (power compensation design). Each design has theoretical advantages and limitations, but performance is mostly based on the manufacturer’s ability to build a completely symmetric system so that instrumental effects on the measurement are minimized. This provides the highest sensitivity (ability to detect weak transitions), resolution (ability to separate transitions close in temperature), accuracy, and precision. Heat flux is the most common approach to making DSC measurements and is discussed below.
Figure 30.7 is a cross-sectional view of a heat flux DSC. A common furnace is used for the sample and reference pans, and is typically purged with high purity, ultra-dry nitrogen. The furnace and cooling accessory provide a wide temperature range from −180 % to 725 °C. The sample and reference sensors are temperature sensors, such as thermocouples, thermopiles, or platinum resistance thermometers. They provide the basis of the differential heat flow measurement and directly measure temperature.

Cross-sectional view of a heat flux DSC. The sample and reference pans are located in a common chamber that is temperature controlled over the range of −180 to 725 °C. The chamber is typically purged with high purity, ultra-dry nitrogen gas (Courtesy of TA Instruments, New Castle, DE)
Even though DSC is the most useful of the thermal techniques for measuring structure and changes in structure, it has a number of natural limitations, including the following:
-
DSC measures the sum of all heat flows within the calorimeter. It is sometimes difficult to interpret data because of overlapping events (multiple transitions occurring at the same time and temperature).
-
Most measurements involve heating the sample to higher temperatures. As temperature increases, mobility increases, permitting the structure to change in ways that are not always obvious. The measured structure may not be the original structure at the start of the experiment.
-
DSC uses a single heating rate. However, higher heating rates provide better sensitivity, while lower heating rates provide better resolution. Therefore, it is not possible to optimize both sensitivity and resolution in a single DSC experiment.
-
DSC cannot measure heat capacity under isothermal conditions. Therefore, DSC cannot use heat capacity as a way to follow changes in structure at constant temperature.
3.2.2 Experimental Conditions
Significantly different conditions are used depending on the measurement. However, larger sample weights and higher heating rates always improve sensitivity (ability to detect an event), while smaller sample weights and slower heating rates improve resolution. The conditions listed below provide a good starting point, but require optimization for the best results. The first decision in selecting conditions is the type of pan that will be used. If the sample is dry (less than 0.5 % volatile components at 100 °C in TGA), then standard aluminum crimped pans will provide the best results. Crimped pans have lower mass and typically provide better heat transfer between the sample and sensor. However, they are not sealed, which permits evaporation of volatile components as the sample is heated. Hermetic pans are sealed and recommended for samples with volatile components. However, they are heavier than crimped pans and generally provide poorer contact between the sample and sensor, which can lower sensitivity and resolution. Recommended starting conditions are as follows:
-
Sample weight: 8–12 mg
-
Pan type: Aluminum hermetic
-
Start temperature: At least 25 °C below the first transition of interest. This gives the baseline time to stabilize before the temperature of the event is reached, and permits better quantification of the change in heat content (enthalpy) resulting from the change in structure.
-
Purge gas: Dry nitrogen
-
Heating rate: 10 °C/min
-
Final temperature: Temperature of 5 % weight loss due to decomposition in TGA data should be the maximum temperature for a DSC experiment. In general, it is bad to decompose samples in the DSC cell because decomposition products can condense and affect the quality of future data.
3.2.3 Common Measurements
DSC can be used to measure the properties and structure of most ingredients used in the food industry. Typically, heating experiments are performed; however, measurements also are made while cooling or under constant temperature (isothermal). Examples of common measurements include the following:
-
Glass transition temperature of amorphous structure
-
Melting temperature of crystalline structure
-
Percent crystallinity of semicrystalline material
-
Crystallization of amorphous material
-
Denaturation of proteins
-
Gelatinization of starch
-
Analysis of frozen solutions used for freeze-drying
-
Oxidative stability of fats and oils
Changes in structure (transitions) are either endothermic, i.e., the sample absorbs additional energy (heat), or exothermic, i.e., heat is released. Figure 30.8 shows endothermic transitions associated with denaturation of albumin from chicken egg at concentrations of 1 and 10 % (w/w) solution with water. Unfolding of the protein results in an increase in enthalpy, free volume, and molecular mobility, and this requires heat to occur. Denaturation and gelatinization are low-energy processes and therefore require large sample sizes to provide sufficient sensitivity. In the case of Fig. 30.8, sample weights of approximately 80 mg were used in high-volume stainless steel pans.

DSC data showing protein denaturation for two concentrations (1 and 10 % w/w in water) of albumin from chicken egg. Large samples (>50 mg) in high-volume pans are recommended to improve sensitivity of the weak transitions associated with protein denaturation and starch gelatinization
3.3 Modulated DSC®
3.3.1 Overview
Modulated DSC® (MDSC®) is a special type of DSC that applies two simultaneous heating rates to the sample [13]. A linear rate is used to obtain the same information as provided by conventional DSC, while a sinusoidal rate superimposed on the linear rate permits measurement of the heat capacity component of the total heat flow signal. As described above, one of the natural limitations of the DSC technique is that it can only measure the sum of all heat flows, and this often makes interpretation of the data difficult. This is illustrated by a brief review of Eq. 30.1, which is often used to describe the heat flow signal from DSC:
where:
-
dH/dt = measured heat flow rate (mW = mJ/s)
-
Cp = heat capacity (J/°C), product of specific heat (J/g°C) × sample weight (g)
-
dT/dt = heating rate (°C/min)
-
f (T,t) = heat flow rate due to time-dependent, kinetic processes (mW)
As indicated in Eq. 30.1, the heat flow signal measured by conventional DSC has two components: one associated with heat capacity and the other with kinetic processes that are a function of both time and temperature. DSC only measures the sum of the two components. By applying two simultaneous heating rates, MDSC® can separate the total signal into its individual components. Figure 30.9 shows temperature versus time and heating rate versus time for an MDSC® experiment. The MDSC® average temperature and linear heating rate would be typical of a DSC experiment, while the modulated temperature and sinusoidal heating rate only occur with MDSC®.

Modulated temperature DSC (MDSC®) applies two simultaneous heating rates (linear and sinusoidal) to separate the total heat flow (equivalent to conventional DSC) signal into the heat capacity and kinetic components
As with every analytical technique, MDSC® also has limitations, including:
-
Slow average heating rates (typically 1–5 °C/min) must be used to obtain good separation of overlapping events. This decreases the productivity (number of samples per day) of MDSC® as compared to DSC.
-
MDSC® is more complex because it requires additional experimental parameters and creates more signals than DSC.
-
Separation of overlapping events requires the ability to modulate the sample’s temperature during the events. This is not possible during melting of relatively pure materials that melt over just a few degrees.
-
The probability of structural change is increased in the sample while heating due to the slower heating rates of MDSC®.
3.3.2 Experimental Conditions
As seen in Fig. 30.9, the applied temperature of MDSC® has both linear and sinusoidal components. Therefore, it is necessary to specify conditions for both. Recommended starting conditions that will work for most samples include the following:
-
Average linear heating rate = 2 °C/min
-
Temperature modulation period = 60 s (use longer periods for larger samples)
-
Temperature modulation amplitude = ±1.0 °C
-
Other conditions: same as DSC
3.3.3 Common Measurements
MDSC® is used to make the same measurements as DSC, but has the significant advantage of being able to separate the heat flow signal into the heat capacity and kinetic components. The benefit of this can be seen in Fig. 30.10, which is an analysis of the structure of sugar-frosted cornflakes. Many cereal products exhibit a broad endothermic peak (most likely a relaxation process, also termed physical aging) in DSC data at temperatures above room temperature; however, the expected and very important glass transition is not visible. The total heat flow signal of the MDSC® data in Fig. 30.10 (equivalent to conventional DSC) shows such a peak. However, MDSC® also shows the kinetic and heat capacity components of the total signal, allowing for straightforward analysis of the glass transition seen in the reversing heat flow signal. In general, the reversing heat flow signal includes heat capacity, changes in heat capacity, and most melting. All kinetic events such as crystallization, decomposition, evaporation, and physical aging appear in the nonreversing heat flow signal.

Modulated temperature DSC (MDSC®) data for a 20 mg sample of sugar-coated cornflakes. A large sample was used to improve sensitivity, and a hermetically sealed pan was used to prevent moisture loss during heating. The total signal shows the broad endothermic peak typically observed in DSC data, while the reversing signal (heat capacity component) shows the underlying glass transition. Since the glass transition temperature (Tg) is above room temperature, the cornflakes will be crispy at room temperature
4 Applications
This section will examine some of the difficulties associated with applying thermal analysis to food products, as well as illustrate some additional uses of the thermal techniques discussed previously to determine food system composition and structure.
4.1 Sample Preparation Challenges
-
Poor Sample Contact with Pan. It is necessary to contain the sample in a DSC pan (typically aluminum hermetic sealed) to perform a DSC experiment. When preparing the sample pan, keep in mind that heat transfer between the sample and DSC sensor takes place through the bottom of the pan. Therefore, it is important to provide good contact between the sample and the bottom of the DSC pan to avoid artifacts in the data due to sample movement during the experiment. This can be done by lightly compacting the sample in the pan or using an inner crimping lid to force the solid sample to the bottom of the pan prior to sealing with the hermetic lid.
-
Lack of Homogeneity. Most food products are heterogeneous, having large variations in composition and structure in their cross sections. Since thermal analysis techniques use relatively small sample sizes, users must look for ways to obtain representative samples. Two simple techniques to address this problem are to use the largest sample possible (that still fits in pan) and do all initial measurements in triplicate to detect compositional inconsistencies. Note that to better detect the weak glass transition in the sugar-frosted cornflakes of Fig. 30.10, a sample weight of 20 mg was used. Although large samples negatively affect resolution, the first objective should be to detect the transition of interest.
-
Controlling Moisture Content. Food products typically have high surface area and amorphous content. This can result in moisture being absorbed or lost during sample storage and/or preparation. Moisture exchange with the atmosphere needs to be kept to a minimum, since just a few percent change in water content can change the glass transition temperature by tens of degrees. In addition, hermetic DSC pans should be used to prevent moisture loss during heating, and open samples should not be left in a TGA autosampler tray.
-
Previous Thermal History. It is often difficult to analyze a glass transition obtained during the first heat scan of a DSC experiment. This is due to numerous kinetic processes caused by previous thermal history (time and temperature) that can occur in the temperature region of the glass transition, including enthalpic relaxation and recovery, stress relaxation, and flow. To eliminate interference from these kinetic processes, it is often useful to heat the sample to a temperature of about 25 °C above the end of the glass transition and then cool it back to the desired starting temperature. This is typically called a heat-cool-heat experiment. An initial experiment will be required to determine this temperature, but it is important to not change the crystalline structure or cause decomposition during the first heat.
4.2 Additional Applications
Figure 30.11 is a comparison of the TGA results from the center (crumb) and crust of a loaf of bread. As expected, both samples show a similar decomposition temperature starting just above 200 °C, which can be seen in the derivative signal. Clear differences can be observed in the DSC data in regard to moisture content, the first peak in the derivative signal, and minor components (second minor peak). These differences result in the substantially different physical properties between the center and the crust of the loaf of bread.

A comparison of TGA data on the center (crumb) and crust of a loaf of bread baked at 190 °C shows substantial differences at temperatures below 200 °C. This is because the center of the loaf remains much cooler than the crust during baking due to the evaporation of water
Figure 30.12 is a comparison of DSC results from a sugar-coated chocolate candy and the sugar coating alone. Note that the melting of the sugar is different in the presence of the melted chocolate. This is due to the sugar starting to dissolve in the liquid chocolate.

Comparison of DSC data on sugar-coated chocolate candy and the sugar coating alone. Melting of the sugar changes in the presence of the liquid chocolate due to its solubility in the chocolate
Many materials can exist in either an amorphous or crystalline form depending on how the material was processed. Since the structure or form of the material is a function of its previous thermal history (e.g., time, temperature, relative humidity, pressure), the structure can change as it is heated during the DSC experiment. This can be seen in Fig. 30.13, a DSC experiment on amorphous sucrose that was prepared by freeze-drying. The first observed transition is the glass transition near 53 °C. The Tg of dry (<0.1 % moisture content) sucrose is approximately 68 °C, which decreases as moisture content increases. A Tg of 53 °C indicates that this sample has moisture content of approximately 1.5–2.0 % (dry basis) [14]. The second useful piece of information is the size of the glass transition, which is the magnitude of the step change in heat capacity and is typically expressed in heat capacity units of J/g°C. A value of 0.75 J/g°C for this sample indicates that it is very close to 100 % amorphous, since the value for a 100 % amorphous melt-quenched sample, given in the literature, is 0.78 J/g°C [15]. An endothermic peak is seen at the end of the glass transition. The presence of this peak is common to materials with a glass transition slightly above room temperature and is due to the process of physical aging. The material relaxes over time to a lower energy state and must reabsorb this energy (termed enthalpic recovery) to achieve an equilibrium state above Tg, thus producing the observed endothermic peak [16].

DSC data obtained using a 10 °C/min heating rate shows that a sample of freeze-dried sucrose is 100 % amorphous based on the size of the glass transition and a comparison of the energies (J/g) of crystallization and melting
The second transition near 100 °C is an exothermic peak caused by crystallization (commonly termed thermally induced crystallization or cold crystallization) of the amorphous material. The size of the peak (J/g) provides information on the amount of amorphous material that was able to crystallize. By dividing the measured area of the peak (88.57 J/g) by the heat of crystallization for 100 % conversion (approximately 131 J/g obtained from a separate DSC experiment on 100 % crystalline sucrose at 10 °C/min, reference [15]), it appears that only about 68 % of the amorphous sucrose crystallized during heating.
The last observed transition is an endothermic peak between approximately 150 and 180 °C caused by conversion of the crystalline structure to an amorphous form, typically termed melting. The shape of the peak is not very symmetric, because sucrose begins to decompose (chemical transformation) at the same temperature [12]. Thus, the overlapping of the two processes creates the nonsymmetric peak. As with thermally induced crystallization, the size of the peak (J/g) is a quantitative measure of the amount of structural change. In this case, the size of the melting peak (87.89 J/g) is within experimental error (typically ±2 %) of the size of the cold crystallization peak (88.57 J/g), indicating that all observed melting is the result of the crystallization that occurred during heating and that the original structure was 100 % amorphous.
For the interested reader, additional applications of thermal analysis techniques to food materials, such as gelatinization of starch, oxidation of vegetable oils, and denaturation of meat protein, can be found in the Thermal Analysis Food Application Handbook [17].
5 Summary
Thermal analysis is a series of laboratory techniques that measures physical and chemical properties of materials as a function of temperature and time. In a thermal analysis experiment, temperature is either held constant (isothermal) or programmed to increase or decrease at a linear rate. Since temperature and time are controlled in all food preparation processes, thermal analysis instruments can simulate these processes on a very small scale (milligrams) and measure the response of the material. The most frequently used techniques include TGA and DSC.
The utility of thermal analysis to the food scientist is due to the fact that end-use properties (functionality) at a specific temperature are dependent on the structure of the components at that temperature. Therefore, it is necessary to measure structure as a function of temperature. Structure can be defined as amorphous (no molecular order), crystalline, or semicrystalline, with most food products having both amorphous and crystalline components.
Amorphous structure is characterized by analysis of the glass transition, which involves a significant change in the material’s heat capacity and molecular mobility. Crystalline structure melts as temperature increases, creating an endothermic peak that provides information on the quantity of crystalline structure and its melting temperature. This chapter has illustrated application of thermal analysis to a variety of food materials.
6 Study Questions
-
1.
You have two brands of caramels, one that is hard and one that is chewy.
-
(a)
What structural transition would you expect to detect in the DSC data for each type of caramel?
-
(b)
How would the DSC data be useful in understanding the differences in texture of the two caramel types?
-
(a)
-
2.
If amorphous material crystallizes during heating (called cold crystallization), would you expect it to crystallize below or above its glass transition temperature? Explain your choice.
-
3.
Explain in your own words why a cold crystallization peak and a melting peak show opposite DSC heat flow signals. See Fig. 30.13 for an example of this behavior.
-
4.
In Sect. 30.3.3.3, the glass transition is referred to as “very important” – why?
-
5.
Assume that you have been asked to develop a recipe for a crisp cookie based on your knowledge of material properties. To develop the recipe, you need to answer the following questions:
-
(a)
Should the finished cookie be in the glassy or rubbery state at room temperature? Explain your choice.
-
(b)
In selecting TGA experimental conditions, would you use a sealed or unsealed sample pan and why?
-
(c)
In selecting DSC experimental conditions, would you typically use a sealed or unsealed pan and why?
-
(d)
If you were to run a MDSC® experiment, which MDSC® signal would contain the glass transition and why?
-
(e)
You find out that high-fructose corn syrup (HFCS) is less expensive than sucrose. Can you substitute the sucrose in your crisp cookie formula for HFCS? Be sure to give reasons for your response.
-
(f)
While performing a DSC or MDSC® experiment, an amorphous component in the sample begins to crystallize. Would this cause an increase or decrease in the measured heat capacity?
-
(g)
As stated in Sect. 30.3.2, a limitation of DSC is that it cannot measure heat capacity under isothermal conditions. However, MDSC® can measure heat capacity during an isothermal experiment. Explain this difference between DSC and MDSC® using Eq. 30.1 in Sect. 30.3.3.
-
(a)
References
Farkas J, Mohácsi-Farkas C (1996) Application of differential scanning calorimetry in food research and food quality assurance. J Therm Anal 47: 1787–1803
Raemy A (2003) Behavior of foods studied by thermal analysis. J Therm Anal Calorim 71: 273–278
Ievolella J, Wang M, Slade L, Levine H (2003) Application of thermal analysis to cookie, cracker, and pretzel manufacturing, Ch. 2. In: Kaletunc G, Breslauer KJ (eds) Characterization of cereals and flours: properties, analysis, and applications. CRC Press, Boca Raton, FL, pp 37–63
Sahin S, Sumnu SG (2006) Physical properties of foods. Springer, New York, p 257
Kamruddin M, Ajikumar PK, Dash S, Tyagi K, Baldev RAJ (2003) Thermogravimetry-evolved gas analysis–mass spectrometry system for materials research. Bull Mater Sci 26(4): 449–460
Schmidt SJ (2004) Water and solids mobility in foods. Advances in Food and Nutrition Research, vol 48. Academic Press, London, UK, pp 1–101
Slade L, Levine H (1988) Non-equilibrium behavior of small carbohydrate-water systems. Pure Appl Chem 60(12):1841–1864
Slade L, Levine H (1991) Beyond water activity: recent advances based on an alternative approach to the assessment of food quality and safety. Crit Rev Food Sci Nutr 30(2–3): 115–360
Aguilera JM, Lillford PJ (2007) Food materials science: principles and practice. Springer, New York, p 622
Sun D-W (2005) Handbook of frozen food processing and packaging. CRC Press, Raton, FL, p 760
Schmidt SJ (2012) Exploring the sucrose-water state diagram: Applications to hard candy cooking and confection quality and stability. Manufacturing Confectioner, January: 79–89
Lee JW, Thomas LC and Schmidt SJ (2011) Investigation of the heating rate dependency associated with the loss of crystalline structure in sucrose, glucose, and fructose using a thermal analysis approach (Part I). J Ag Food Chem (59): 684–701
Thomas L (2006) Modulated DSC technology manual. TA Instruments, New Castle, DE
Yu X, Kappes SM, Bello-Perez LA, Schmidt SJ (2008) Investigating the moisture sorption behavior of amorphous sucrose using a dynamic humidity generating instrument. J Food Sci 73(1): E25–E35
Magoń A, Wurm A, Schick C, Pangloli P, Zivanovic S, Skotnicki M, Pyda M (2014) Heat capacity and transition behavior of sucrose by standard, fast scanning and temperature-modulated calorimetry. Thermochim Acta 589: 183–196
Wungtanagorn R and Schmidt SJ (2001) Thermodynamic properties and kinetics of the physical aging of amorphous glucose, fructose, and their mixture. J Therm Anal Calorim 65: 9–35
Widmann G and Oberholzer T (2014) Thermal Analysis Application Handbook, Food Collected Applications. Mettler Toledo, Columbus, OH, p. 66
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing
About this chapter
Cite this chapter
Thomas, L.C., Schmidt, S.J. (2017). Thermal Analysis. In: Nielsen, S.S. (eds) Food Analysis. Food Science Text Series. Springer, Cham. https://doi.org/10.1007/978-3-319-45776-5_30
Download citation
DOI: https://doi.org/10.1007/978-3-319-45776-5_30
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-45774-1
Online ISBN: 978-3-319-45776-5
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)