
Abstract
Inhibitors of cancer-specific pathways can selectively kill off tumor cells. However, heterogeneity of neoplastic tissue often allows other cancer cells to repopulate the tissue area, leading to regeneration of resistant disease. β-Lapachone is a novel bioactivatable drug that relies specifically on tumor-directed upregulated levels of NAD(P)H:quinone oxidoreductase 1 (NQO1) to kill most solid cancers, such as 90 % of pancreatic and non-small cell lung, 60 % of breast, colon, and prostate, as well as 50 % of head and neck cancers. Once β-lapachone is bioactivated by the NQO1 enzyme, massive levels of hydrogen peroxide are produced that, in turn, damage the DNA of cancer cells, while associated normal tissues, which lack NQO1, are protected by high levels of catalase. If tumors are irradiated prior to applying β-lapachone, the drug (clinical form, ARQ761) can work in combination with the vast spectrum of DNA lesions created by ionizing radiation, particularly DNA base lesions, single and double strand breaks (SSBs and DSBs), in addition to the massive hydrogen peroxide-based lesions created by β-lapachone, to cause tumor-dependent poly(ADP-ribose) polymerase 1 (PARP1) hyperactivation. Once tumor-selective PARP hyperactivation is induced in cancer cells, they die due to low concomitant catalase levels. In contrast, associated normal tissue, as well as other normal tissue, lack elevated levels of NQO1 and have high catalase levels. Cancer cell death ultimately occurs by NAD+-depletion, where resistance to NQO1 bioactivatable drugs has not been noted to date. Current studies are focused on pancreatic and non-small cell lung cancers, as NQO1 is elevated in nearly all of these cancers.
*Author contributed equally with all other contributors.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
10.1 Introduction
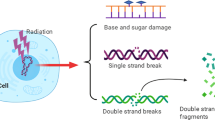
Developing effective agents that can selectively increase the sensitivity of cancer cells to ionizing radiation (IR), so called ‘radiosensitizers,’ while not affecting normal cells or tissues, has been difficult. IR therapy is effective due to the spectrum of DNA lesions produced and its ability to generate complex DNA double strand breaks (DSBs), where only one non-repaired lesion is required for cancer cell lethality. Nevertheless, IR therapy is subject to the classical four Rs of radiobiology [1, 2]: (i) Re-oxygenation and resistance by hypoxia-efficient sensitization of cells (see Chap. 2); (ii) Repair where tumor cells often have heightened DNA repair mechanisms; (iii) Redistribution of tumor cells into more radioresistant phases of the cell cycle; (iv) Repopulation in which resistant tumor cells (e.g. cancer stem cells [CSCs]) rapidly expand and often have increased capacity for metastasis. In recent years, another R has been added, (v) Radioresistance, to indicate ‘inherent radiation resistance’ mechanisms that develop during carcinogenesis. Strategies for developing radiosensitizers (briefly summarized below and specifically addressed in this review), have attempted to overcome one of these five Rs. Unfortunately, many of these strategies have focused on differential cell division and not on underlying non-dividing tumor cells (that may include CSCs) that remain unperturbed by nearly all radiosensitizing agents. In this review, we will present data on NQO1 bioactivatable drugs from the past 20 years, and suggest that these agents should be explored as radiosensitizing agents which, when used properly, can kill independent of cell cycle status, or presence of tumor suppressor (e.g., p53, pRb) or oncogene (e.g., KRAS, MYC) status. Additionally, NQO1 bioactivatable agents can efficiently alter tumor-specific metabolism and inhibit a spectrum of DNA repair pathways by a unique poly(ADP-ribose) polymerase (PARP) hyperactivation mechanism. This occurs via the X-ray-inducible transcript leading to protein (xip3) gene [3], now known as NAD(P)H:quinone oxidoreductase 1 (NQO1) which is overexpressed 5- to 100-fold in most solid cancers, including extremely recalcitrant cancers, such as non-small cell lung, pancreas, as well as breast, prostate, colon, bladder, and head and neck cancers [4–8]. We briefly review current and past radiosensitizer strategies, and then describe the mechanism of action of NQO1 bioactivatable drugs alone [7, 9–11], and how these agents exploit a novel NQO1-dependent, tumor-selective PARP hyperactivation mechanism when combined with IR therapy.
10.2 Prior and Current Strategies for Radiosensitizing Cancers (Fig. 10.1)

Cellular map of radiosensitization targets
1. Halogenated Pyrimidines. Halogenated pyrimidines (HPs), such as chloro-, bromo- and iodo-deoxyuridine (CldU, BrdU, IdU), are well-known radiosensitizers and have been reviewed in detail elsewhere [12–17]. In an effort to develop additional tumor-selectivity and to exploit elevated deoxycytidine deaminase (dCD)-deoxycytidine kinase (dCK) levels, halogenated deoxycytidine derivatives (particularly CldC and FdC) were developed by Dr. Sheldon Greer [9, 18–20].
Since the 1960s, HPs were investigated for their potential to replace thymidine and incorporate into the DNA of replicating tumor cells to increase cell sensitivity to IR [21–28]. Bromine, chlorine and iodine have van der Waals radii that are larger than hydrogen and similar to the 5-methyl group of thymine. They mimic thymidine and become better substrates for thymidine kinase [29]. Several mechanisms are simultaneously at work to explain how HPs cause radiosensitization. First, IR exposure of cells that have unifilar or bifilar incorporation of HPs causes de-halogenation and subsequent formation of HP● radicals that cause complex DSBs due to the formation of multiple damaged sites, making these lesions difficult to repair [30]. Accordingly, bifilar incorporation is more effective than unifilar incorporation [31]. Multiple clinical trials and applications have been developed for HPs over the years with varying degrees of success, with the main challenge being the incorporation of sufficient HPs into DNA to achieve a therapeutic result [32–38]. Clinical trials have not been definitive about the use of HPs in cancer treatments, but there may be a subset of patients that benefit [39, 40]. Toxicities, including increased sensitization of the eyes to light and blindness, have greatly impeded the use of HPs as radiosensitizers [41].
2. Oxygenation mimics: The use of oxygen to increase IR effects . The aggressive nature of tumors relies on their ability to adapt to environmental stress factors. Regions of the tumor contain necrotic areas where oxygenation levels remain low (i.e., are hypoxic, with oxygen (O2) levels less than 10 mmHg). This hypoxic environment in tumors induces the activation of certain compensatory pathways to protect the cell, including the hypoxia-inducible factor (HIF) pathway [42, 43]. Activation of these pathways in response to low O2 levels allows for a selective advantage against apoptosis and can lead to resistance to chemo- and radiotherapies [44, 45]. When the level of O2 is enhanced in the tumors, radiosensitization is increased up to threefold [46]. This increased radiobiological effect (RBE) led to the development of oxygenation methods to enhance the response to IR therapy.
One technique to sensitize tumors to radiotherapy is the use of oxygenation mimetics. These mimics utilize the chemical properties of oxygen without being rapidly metabolized by cells undergoing cellular respiration, which allows for increased distribution into hypoxic areas of tumors [44]. The most common class of oxygenation mimetics is the nitroimidazoles. These agents are able to “fix” and prolong DNA radical lesions produced by IR. Nitroimidazoles take advantage of nitroreductase enzymes that are upregulated in hypoxic conditions found in tumors to generate anion radicals [47, 48]. The radical anions created are highly reactive and undergo irreversible fragmentation that promote cross-linking in DNA, rendering irradiated cells unable to divide, eventually leading to apoptosis [49].
3. Signal transduction inhibition strategies. Aberrant activation of various signal transduction pathways is frequent in neoplastic growth. In response to IR, cancer cells critically depend on a cascade of multiple signal transduction responses, such as the stimulation of (1) plasma membrane receptors [50, 51]; (2) cytoplasmic protein kinases [52, 53]; (3) specific transcriptional activation [54–56]; and (4) altered cell cycle regulation [53, 57] to ultimately evade the toxic effects of IR-induced damage. In cancer, for example, overexpression or mutational activation of RAS and PTEN can regulate the PI3K pathway needed for the eventual repair of IR-induced DSBs [58]. This inherent “addiction”, however, opens new avenues for innovative therapeutic strategies toward the development of novel anticancer drugs that inhibit key signal transduction cascade steps to potentiate the effects of low-dose IR [59–63]. The PI3K/mTOR dual inhibitor, NVP-BEZ235, is an excellent example of a radiosensitizer [61–63]. This inhibitor targets both ATM and DNA-PKcs that are apical kinases involved in repair of IR-induced DSB [62]. Simultaneous inhibition of these two kinases blocks both homologous recombination (HR) and non-homologous end joining (NHEJ) pathways that are critical for repair of IR-induced DNA damage [62]. In combination with IR, this inhibitor potently induces apoptosis both in vitro and in vivo using models of mutant KRAS-driven non-small cell lung cancer (NSCLC) and glioblastoma [61–63]. This combination therapy could be broadly applicable to other cancers that exhibit aberrant activation of PI3K. Pharmacological inhibition of specific signal transduction pathways is often quite effective initially, but over time, cancer cells upregulate compensatory pathways to overcome their dependence on the targeted pathways. In such scenarios, combination therapies simultaneously targeting multiple pathways become a necessity to eliminate cancer.
4. Hormone Regulation. The most well-studied growth factor pathways modulating radiosensitivity have been targetable EGFR and insulin receptors. The mechanism of radiosensitization by inhibiting EGFR are under intense investigation, but are likely related to both regulation of the cell cycle [64–71] and modulation of DSB repair [72, 73]. Kriegs et al., demonstrated that in NSCLC, radiosensitivity increases by promoting G1/S-arrest in tumor cells [64]. In clinical trials, the most substantial benefit of EGFR inhibitors appears to be in a subset of head and neck cancers, with significant enhanced survival in patients receiving combination therapy with IR plus Cetuximab [74]. The main difficulty in improving clinical outcomes has been identifying the cohort of patients who would best respond to therapy. Less well investigated are other hormonal pathways, such as insulin and insulin-like growth factor 1 (IGF-1) regulation. Wang et al. demonstrated radiosensitization in pancreatic cancer via metformin’s effect on the G2-checkpoint and increased rates of mitotic catastrophe [75] and Fasih showed similar results via the AMPK pathway [76]. Another strategy being developed is to alter the TGFβ1-IGF-1 extracellular expression axis, which leads to the pro-survival protein and extracellular protein chaperone, secretory clusterin (sCLU) [77–79]. Suppressing the TGFβ1-IGF-1-sCLU expression pathway is likely to decrease resistance and suppress glycolytic and TCA metabolic reprogramming that can occur post-IR by suppression of fatty acid synthase (FASN) and lipogenesis [79].
5. Hyperthermia. Modifying the tumor microenvironment has long been a therapeutic target in cancer therapy. Multiple studies in vitro demonstrated the dramatic radiosensitization effects of hyperthermia [12, 80]. Heating tissue over 43 °C impairs the cell’s ability to effectively perform DNA repair, which is potentiated by its combination with IR [81–83]. Among a host of other responses, hyperthermia can cause enhanced ATM kinase activity, inactivation of HSP70, and increased telomerase activity [82]. Heat also diminishes chromatin condensation and leads to nucleolar disintegration as a marker for impaired DNA repair [84]. Though many of these effects were demonstrated in various cell lines, clinical application of hyperthermia continues to be problematic. Strategies are now being employed to target heat shock proteins instead. As technology has improved, there is now considerable interest in the use of magnetic resonance high intensity focused ultrasound (MR-HIFU) for delivery of chemotherapy and palliative pain control [85–91].
6. Metals. Various metals, with a predominant emphasis on gold nanoparticles, have been investigated as radiosensitizers [92–94]. Gold particles absorb energy from IR, thereby potentiating SSBs and DSBs in vitro, as well as by generation of toxic free radicals [95–98]. Studies in vivo have also demonstrated antitumor effects in mice [99–103]. The main difficulty has been enhancing the delivery of gold nanoparticles to tumor sites. Other metals with radiosensitization properties such as copper, iron, nickel, and gadolinium are in early phases of exploration for potential clinical use [104–110]. Most metals, even gold, however, have major toxicity issues that must be dealt with for future therapies [111].
7. Dietary supplements, vitamins, and complementary and alternative medicine. Over the years, a large effort has been devoted to developing complementary and alternative medicines for the treatment of cancers, and many of these agents have been investigated as potential radiosensitizers. Caffeine has been described in several papers to induce radiation sensitivity via elimination of the radiation-induced G2 checkpoint and inhibition of both ATM and ATR [112–114]. Indeed, examination of the literature on caffeine [115] led to a search for DNA repair inhibitors, and ultimately, the identification of β-lapachone, the first NQO1 bioactivatable drug [10]. Neem leaf extract was also implied in radiosensitization by modulation of apoptotic pathways in neuroblastoma, and via NF-kB in pancreatic cancer cell lines, where curcuma and black raspberry extract were also indicated to be effective [116–120]. Likewise, caffeic acid phenethyl ester (CAPE), the active ingredient in honeybee resin, demonstrated growth inhibition in human lung cancer cell lines via decreased glutathione and elevated H2O2 levels by unknown mechanisms. [121, 122] Soy products, likely via antioxidant effects, demonstrate synergistic anticancer effects in combination with radiation therapy in prostate cancer [123]. Finally, vitamins, such as riboflavin, were suggested as a radiosensitizer in mouse thymocyte models and human hepatoma cells [124]. Though none of these compounds have become adjuncts to radiation therapy, the widespread use of complementary and alternative medicines warrants clinical awareness of the effects of these particular xenobiotics, and specifically inquiring about their use.
10.3 Targeting NQO1 Expression for Cancer Therapy
1. NQO1: A Phase II detoxification enzyme. The cellular detoxification of foreign chemicals occurs in a step-wise manner facilitated by enzymes that carry out specific metabolic processes, including biotransformations (Phase I), conjugations (Phase II) and transportation (Phase III) [125]. Phase II detoxification involves glucuronidation, acetylation, and sulfonation conjugation reactions. These reactions add polar moieties to very reactive and toxic xenobiotic molecules, rendering them more water-soluble. Following conjugation reactions, the newly created quinone-metabolites are less reactive, and thus, are less harmful to cells. For most quinones, NQO1 is a phase II detoxification enzyme, and as such, interacts with various xenobiotic quinone substrates. NQO1 converts these reactive quinones to more stable hydroquinones, allowing more efficient phase II conjugation reactions, and subsequent rapid excretion [126]. The classic example of NQO1 metabolism of a quinone resulting in its detoxification is the interaction between NQO1 and menadione. In the early 1980s, studies involving menadione metabolism in hepatocytes discovered that in the presence of the NQO1 inhibitor dicoumarol, increased free radical formation and elevated oxygen consumption [127]. The relevance of NQO1’s detoxification of menadione was further confirmed in the late 1990s by Jaiswal’s characterization of NQO1-deficient mice who were hypersensitive to quinoid compounds, such as menadione [128].
2. NQO1 expression in cancers. In normal tissue, particularly normal lung, expression of NQO1 has been shown to be inducible, and the induction of endogenous NQO1 levels in tissues is primarily a response to increased levels of oxidative stress [129]. NQO1’s inducible expression is tightly regulated by the transcription factor, Nrf2. Nrf2 is held in abeyance in the cytosol by Keap1, an E3 ubiquitin ligase. In the presence of Keap1, Nrf2 undergoes rapid proteasomal degradation. The Keap1-Nrf2 pathway controls expression of NQO1 as well as many other oxidative stress regulatory genes, resulting in their expression only when Nrf2 is released from Keap1. The dissociation of Nrf2 from Keap1 occurs when cells are confronted with oxidative stress. The severance of Nrf2 from Keap1 permits Nrf2 to translocate to the nucleus where it interacts with the chaperone, small Maf-1 protein. Together, the Nrf2/Maf-1 complex regulates the transcription of various antioxidant genes whose common thread is the presence of an antioxidant response element (ARE) within specific promoters of certain genes. These Nrf2-activated genes, including NQO1, are activated and tightly controlled in normal cells to protect the genome from various deleterious forms of oxidant stress. In contrast to the tightly regulated low levels of NQO1 in normal cells, expression of NQO1 in cancer cells was constitutively elevated well above those observed in normal tissues [130, 131]. In retrospective analyses, NQO1 expression in most solid cancerous tissue is elevated 5- to 100-fold more than levels noted in associated normal tissue for the same patient [130, 131]. Increased NQO1 expression in cancerous tissues is typically a disruption in the Keap1-Nrf2 association [132]. In fact, studies have reported finding mutations in Keap1 in various cancers, where NQO1 levels were elevated [133, 134]. These studies showed that when Keap1 was mutated, expression levels of Nrf2 were increased in the nucleus and an elevated expression of NQO1 and other Nrf2-regulated genes were observed. Thus, elevated NQO1 expression in many cancers has led to an increase in studies investigating the plausibility of developing quinones whose bioactivation could potentially be harnessed for development of NQO1-directed antitumor chemotherapeutics.
3. Previous studies on initial NQO1 bioactivated drugs: Mitomycin C, Geldanamycin, EO9 and Streptonigrin. NQO1 plays a significant role in ‘bioactivating’ a select few quinone substrates. These NQO1 activatable drugs fall into two classes: (i) compounds, such as mitomycin C (MMC) and geldanamycin that are converted to their hydroquinone forms in one-step reactions, and become either DNA alkylating agents (MMC) or inhibitors of specific pathways, such as HSP90 or HSP70; or (ii) compounds that undergo futile redox cycling (e.g., EO9 and streptonigrin), potentially generating tremendous oxidative stress. Streptonigrin is unique in that it causes both elevated reactive oxygen species (ROS) levels and induces DNA alkylation.
Mitomycin C is a naturally occurring compound originally derived from the Gram-positive bacterium, Streptomyces. MMC is used clinically to treat a variety of tumor types, including stomach, pancreas, breast, and lung [135]. However, its use as an NQO1-directed killing compound has been limited by its dependence on a narrow pH range [136]. Outside this pH range, its antitumor activity is not NQO1-specific. In fact, MMC, in most cases, acts as an inhibitor of NQO1 activity. Thus, this compound, although used to treat a wide assortment of cancers with limited success, does not truly define the potential of selective NQO1-directed chemotherapy.
Geldanamycin is a 1,4-benzoquinone originally isolated from Streptomyces in 1970 [137]. Its antitumor activity has been correlated with three main factors, including free radical formation, tyrosine kinase inhibition, and binding and interfering with heat shock proteins. 17-AAG is a geldanamycin derivative produced to avoid dose-limiting toxicities that included hemolytic anemia and hepatotoxicity [138]. In clinical studies with patients with NQO1 homozygous *2 polymorphisms, and thus no NQO1 expression, no correlation was observed between responses to 17-AAG therapy and NQO1 status, suggesting that although NQO1 bioactivates 17-AAG [139], the activity of the drug was not related to NQO1 expression, but to its off-target effects.
EO9 (3-hydroxy-5-aziridinyl-1-methyl-2(1H-indole-4,7-dione)prop-β-en-α-ol), also known as apazoquinone, is another example of an NQO1-bioactivated compound that has undergone clinical trials [140]. Studies in vitro showed that EO9 bioreduction by NQO1 caused DNA damage in the form of SSBs, which was suppressed by catalase, implicating hydrogen peroxide formation during EO9 reduction by NQO1 [141]. In clinical studies with EO9, its low water solubility led to extremely poor systemic pharmacokinetics, actually due to metabolism by peripheral red blood cells themselves [142]. Thus, poor responses in patients with solid cancers were noted [143, 144]. However, when used in trials to treat local regional bladder tumors, the compound faired much better. Although EO9 had limited success in clinical trials, an interest still exists in its utilization in treating local regional tumors [140].
Streptonigrin is another quinone antibiotic discovered in the early 1960s to have antitumor activity. However, the drug was extremely difficult to synthesize and subsequently failed in initial clinical trials due to poor water solubility [145]. As a result, limited studies are available to prove its entire mechanism of action. In more recent studies, streptonigrin was found to be active in an NQO1-dependent manner in human colon cancer cells [146]. As noted above, streptonigrin represents a potent potentially NQO1-dependent agent that undergoes NQO1-dependent redox cycling, but also alkylates DNA [147]. Further studies on streptonigrin, including improved and more efficient synthetic procedures, may be warranted.
4. NQO1 bioactivatable drug mechanisms of action and cellular consequences. β-Lapachone (β-lap) is a unique NQO1 bioactivatable drug that exploits the NQO1:catalase therapeutic window due to its elevated expression in many solid tumors [5]. β-Lapachone undergoes a futile redox cycle, which attempts to detoxify the drug, thereby forming its unstable hydroquinone (Fig. 10.2). The hydroquinone form of β-lapachone undergoes a spontaneous back-reaction, creating two superoxides. The reaction is robust, using 60 mol of NAD(P)H and creating 120 mol of superoxide in just 2 min in NQO1 positive cancer cells. Reactions are prevented by dicoumarol (DIC), a specific inhibitor of NQO1 and do not occur in NQO1 negative cells. Impressively, a 1 h exposure of NQO1+ pancreatic ductal adenocarcinoma (PDA) cells to 4 μM β-lapachone is equivalent to 300–500 μM H2O2 [8, 148]. A minimum of 35–120 min of exposure to NQO1 bioactivatable drugs is required to kill all NQO1+ cancer cells in vitro, strongly suggesting that key lethal responses occur in this time frame [149, 150]. Loss of NAD(P)H reducing equivalents, accumulation of NAD+ pools, DNA base damage, and Ca2+ release from the endoplasmic reticulum (ER) result in ‘PARP1 hyperactivation’ which degrades NAD+ pools and causes tumor-selective NQO1-mediated cell death (Fig. 10.2). Major advantages of using β-lapachone include its unique NQO1-dependent mechanism of action and lack of major exposure-related resistance pathways. A small population of NQO1- polymorphic individuals (~5 %) are predicted not to respond. Prior work [4, 8, 131, 149–154] demonstrates that β-lapachone-induced cell death is: (i) not dependent on p53 status; (ii) not dependent on cell cycle; (iii) not affected by bak/bax loss; (iv) not affected by known oncogenic driver or carrier mutations [5]; and (v) not affected by caspase (e.g., bcl2 expression or caspase loss in MCF-7 cells) loss [7, 153, 155–157]. β-Lapachone also causes a potent bystander effect, wherein NQO1low (<1 Unit) cancer cells in a mixed NQO1+/NQO1− tumor are killed by apoptosis, while normal tissues are unaffected due to high catalase levels [5, 6, 8, 158]. Cell death is mediated by μ-calpain/AIF activation [131, 148, 151, 152]. β-Lapachone causes extensive DNA base damage and SSBs, even at sublethal doses (≤2.5 μM) for NQO1+ cancers [5].

Tumor-selective, NQO1-mediated futile redox cycle of β-lapachone (β-lap) triggers a novel PARP hyperactivation-dependent pathway of programmed necrosis, referred to as NAD+-Keresis. Using β-lapachone, this pathway is being exploited for cancer therapy, but also as a general treatment against metabolic syndromes to correct NAD(P)H:NAD(P)+ ratios
5. Metabolic consequences of NQO1 bioactivatable drugs. The loss of NAD+ can be seen in several examples of NQO1 bioactivatable drugs. The mechanism of NQO1 bioactivable drugs (Fig. 10.2) shows how NQO1 catalyzes the rapid conversion of NADH to NAD+. The buildup of NAD+ in the cell is quickly depleted upon PARP1 hyperactivation. A lethal dose of the drug deoxynyboquinone (DNQ) revealed NAD+ is depleted within 30–60 min, as well as complete loss of ATP within the cell [154]. A similar phenomenon is observed with a lethal dose of β-lapachone [159]. β-Lap treatment also caused a persistent reduction in the activity of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in glycolysis, noted by the accumulation of glyceraldehyde 3-phosphate (GA3P) in the cells. This observation was also complemented by a decrease in glucose utilization and subsequent lactate production [160].
The exploitation of metabolic pathways that utilize NAD+/NADH and ATP can be advantageous in enhancing the effects of NQO1 bioactivatable drugs. In theory, the rapid depletion of these essential cofactors suggests downstream metabolic consequences might occur. One pathway to target would be the nicotinamide recycling pathway which is primarily responsible for NAD+ synthesis. The cell naturally tries to recover from the dramatic NAD+ loss caused by NQO1 bioactivatable drugs by regenerating this essential nucleotide. The rate-limiting step of the reaction is catalyzed by nicotinamide phosphoribosyltransferase (NAMPT) and the most well-known inhibitor is FK866 [161, 162]. Pre-treatment with FK866 reduced overall NAD+/NADH pool sizes prior to β-lap treatment, which leads to an accelerated decrease in overall NAD+/NADH and shifted lethality of β-lap to smaller doses of the drug [160]. Overall, treatment with FK866 attenuated the effects of β-lap, without causing excess PAR formation (due to lower NAD+ levels) and renders the NAMPT inhibitor tumor-selective.
Alternate pathways to target for combination therapy are those that feed directly into glycolysis and/or the TCA cycle. One approach targets cancer cells whose metabolism is driven by mutant KRAS. KRAS-driven metabolism utilizes glutamine as an anaplerotic carbon source. The first step in this pathway is catalyzed by mitochondrial glutaminase (GLS1) and is responsible for converting glutamine to glutamate within the mitochondria [163]. Current inhibitors against GLS1 include BPTES (bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide) and CB-839 [164, 165]. Recent studies have shown that treatment with BPTES sensitized pancreatic cells to β-lap in a mutant KRAS-dependent manner and caused a significant decrease in NAD+ at lower, sublethal β-lap doses. In vivo studies with CB-839 reveals an increase in overall PAR formation and an increase in a survival advantage when treated in combination with β-lapachone [6, 166].
A more recent NQO1 bioactivatable drug, KP-372, shows many downstream metabolic effects. A KP-372 treatment results in an increase in the cytosolic NAD+/NADH redox state in a dose-dependent manner. The most significant observation is the increase (seven to eightfold) of several intermediate metabolites within the pentose phosphate pathway (PPP), which most likely occurs to regenerate depleted NAD(P)H. Additionally, the glycolytic and TCA cycle metabolites also showed a perturbation upon treatment. However, the lactate production after treatment showed no significant change, indicating KP-372 had no measurable effect on glycolysis [167].
6. Pathways of sensitizing cells to NQO1 bioactivatable drugs. Early on, in searching for a DNA repair inhibitor, β-lapachone was discovered for its abilities to inhibit recovery of irradiated cells, only if added during or immediately after exposure to IR, affording efficacy as a radiosensitizing agent in vitro [9–11]. In those early studies, NQO1low radioresistant malignant melanoma (U1Mel) cells were used in an effort to counter potentially lethal DNA damage repair (PLDR) processes [10]. Simultaneously, it was discovered that IR-treated U1Mel cells significantly induced NQO1 expression, then identified as X-ray-inducible transcript leading to protein 3 (xip3) [3]. Therefore, IR treatment predisposed U1Mel cells to β-lap, which were relatively resistant to this NQO1 bioactivatable drug in its basal state. Dose enhancement ratios (DERs) of 1.8- to 2.5-fold, with >3.5-fold when halogenated pyrimidines (HPs) were incorporated, were found [9]. Further analyses revealed that numerous cancers have constitutively elevated levels of NQO1 [131, 148], and that NQO1 expression appears to be a pro-survival gene for CSCs [168]. Additionally, cancer cells with constitutively elevated levels of NQO1 could still be radiosensitized to the same extent as NQO1low cells [4], dramatically broadening use of β-lap and other NQO1 bioactivatable drugs as radiosensitizers. Mechanistically, low doses of IR, which do not hyperactivate PARP, combine with sublethal doses of β-lap (not capable of hyperactivating PARP) synergistically create enough DNA base damage, SSBs, and DSBs to hyperactivate PARP. In this case, synergy is the culmination of a number of events in the following sequence: (a) treatment with IR, causing a spectrum of DNA lesions, including DNA base alkylation, as well as single- and double strand breaks, with DSBs being the most lethal; (b) simultaneous treatment with β-lapachone, resulting in significant H2O2 production and specific DNA base damage and SSBs. Co-treatment reaches the threshold level of DNA base, SSBs and DSBs, resulting in PARP hyperactivation, where significant decreases in NAD(P)+ result creating conditions where DNA repair is prevented (Fig. 10.2). Most significantly, DSB repair is prevented, causing a synergistic killing effect which triggers a form of programmed necrosis (PN), known as NAD+-Keresis. Due to the spectrum of DNA lesions, specifically DSBs, and triggering of PARP hyperactivation-dependent NAD+/ATP loss, the combination shows significant synergistic lethality against many NQO1+ human cancers, including neoplasms of the prostate, breast, non-small cell lung, and head and neck carcinomas.
7. Other synergistic combinations with β-lapachone. Elucidating the mechanism of action of β-lap allows the prediction of various synergistic combinations with other pathways, and the development of specific inhibitors of these pathways for improved efficacy of treating NQO1+ human cancers (Table 10.1). Note that IR exposure is clearly the most efficacious, but also the only combination therapy where DNA lesions are initiated by both agents. In all cases, tumor selectivity arises from β-lap, where exposures exploit the elevated NQO1 levels present in most solid cancers, with concomitant lower levels of catalase [5]. After IR + β-lapachone, the most efficacious combinations with β-lap are indicated in decreasing order of efficacy, and include combination with (Table 10.1):
-
(a)
Gemcitabine, the DNA base analog incorporates into DNA and creates DNA lesions with or without DNA repair mechanisms. The lesions synergize with β-lapachone-induced DNA damage to decrease survival. The exact mechanism of synergy is believed to result from PARP hyperactivation-dependent NAD+ loss. However, studies confirming this have yet to be performed.
-
(b)
Methoxyamine (MeOX), the base excision repair (BER) inhibitor and abasic-modifying agent. MeOX prolongs abasic sites allowing enhanced PARP binding and hyperactivation. DNA base damage by β-lapachone is the essential DNA lesion forming mechanism.
-
(c)
CB-839, glutamine transaminase 1 (GLS1) inhibitor. Pancreatic cancers with activated KRAS concomitantly elevate NQO1, as well as GLS1 and other glutamine anaplerotic pathways, in order to move electrons within the cell for the ultimate synthesis of NAD(P)H. Depleting cells at the first steps of this pathway with GLS1 inhibitors, BPTES or CB-839, depletes NAD(P)H making β-lapachone-induced PARP hyperactivation-dependent NAD+ loss more efficient.
-
(d)
FK866, NAMPT inhibitor. NAMPT is the sole de novo enzyme responsible for the major pool of NAD+ in the cells. Since most cancer cells, particularly pancreatic cancer cells overexpress the enzyme, inhibiting this salvage NAD+ synthetic pathway lowers the pool and makes β-lapachone-induced PARP hyperactivation more efficient, causing NQO1+-dependent efficacy. Studies in vivo are warranted.
A common mode of synergy between the agents listed in Table 10.1 and β-lapachone is the formation of threshold levels of DNA lesions leading to PARP hyperactivation, NAD+/ATP loss, repair inhibition and programmed necrosis-medicated cell death. Accordingly, agents that do not induce DNA lesions that PARP (specifically PARP1) typically binds, such as ultraviolet radiation (UV), DNA alkylators or cross-linking agents, do not synergize with β-lapachone (Table 10.1).
10.4 NQO1 Bioactivatable Drugs as Tumor-Selective Radiosensitizers
1. Prostate Cancer. In general, therapy for advanced prostate cancer therapy, particularly androgen-independent castration-resistant prostate cancer, highlights well the five Rs of radiobiology and the difficulties in treating these diseases. Its slow growing, metastatic nature makes it difficult to treat with agents developed for therapy against actively replicating cancer cells. The ability of β-lap to kill cells, regardless of cell cycle position, is a major advantage and its increased efficacy in combination with very low doses of IR makes such therapy using NQO1 bioactivatable drugs very attractive [4]. β-Lap kills prostate cancer cells by NQO1 metabolic bioactivation, triggering a massive induction of ROS, irreversible SSB and DNA base damage that hyperactivate PARP1, resulting in NAD+/ATP depletion and μ-calpain–induced programmed necrosis [151, 152]. In combination with IR, β-lap radiosensitizes NQO1+ prostate cancer cells under conditions where nontoxic doses of either agent alone achieved threshold levels of DNA base damage and SSBs required for hyperactivation of PARP. Combination therapy significantly elevates DNA base and SSB lesions, γH2AX foci formation, and poly(ADP-ribosylation) of PARP1, which are associated with NAD+/ATP losses and induction of μ-calpain–induced programmed necrosis [4]. Radiosensitization by β-lap was blocked by the NQO1 inhibitor dicoumarol, or temporarily by PARP inhibitors. β-Lap synergized with IR to promote antitumor efficacy in a mouse xenograft model of prostate cancer. NQO1 levels were elevated in 60 % of human prostate tumors evaluated relative to adjacent normal tissue, where β-lap might be efficacious alone or in combination with ionizing radiation [4]. These data warrant a clinical trial to use β-lap as a radiosensitizer against prostate cancers that overexpress NQO1, offering a potentially synergistic targeting strategy to exploit PARP hyperactivation. Completion of the ongoing first-in-man clinical trial of ARQ761 against solid cancers should pave the way for future β-lap radiosensitization trials.
2. Head and neck cancer (HNC). This aggressive cancer accounts for ~3–5 % of all cancers in the United States with over 45,000 new cases and 8000 deaths estimated as of 2015 [173]. The majority of these cancers are squamous cell carcinomas and are highly curable with surgery in combination with radiotherapy and/or chemotherapy if detected early. Although there has been a progressive improvement in therapy, current treatment approaches still result in an overall survival (OS) rate of only ~50 % for locally advanced HNCs.
NQO1 was overexpressed in ~50 % HNC tissues compared to adjacent normal tissues [191]. Expression of catalase is also examined since NQO1-mediated β-lap lethality kills cancer cells through an NQO1-dependent futile redox cycling to generate massive amounts of H2O2, which is degraded by catalase [8, 148]. Interestingly, catalase was significantly elevated in adjacent normal tissues compared to HNC tissues. This inverse expression pattern of NQO1 and catalase suggests an ideal therapeutic strategy, since the NQO1-dependent anticancer mechanism of β-lapachone will selectively kill HNC cancer cells. In contrast, catalase will efficiently protect adjacent normal cells by detoxifying the H2O2 generated by NQO1-mediated β-lapachone futile redox cycle. In addition to NQO1/Catalase IHC staining in HNC tissue microarrays (TMA), Western blotting was used to examine NQO1 and catalase expression in 41 HNC cell lines, including several pairs of primary and lymph node metastasis-originated cell lines, as well as one normal human fibroblast cell line. A corresponding inverse relationship was found between NQO1 and catalase expression compared to normal human IMR90 fibroblasts.
Since many HNC cell lines overexpress NQO1, cell survival was determined after treating HNC lines with β-lapachone. A total of 41 HNC cell lines were selected and cells with elevated levels of NQO1 expression respond very well with β-lapachone exposure, while NQO1-deficient cell lines, which carry *2 or *3 single nucleotide polymorphisms (SNPs), did not. HNC cell death was dramatically increased with increasing concentrations of β-lapachone through a very sharp inflection point in dose–response studies, and completely abrogated by co-treatment with dicoumarol, and significantly decreased by an shRNA specific knockdown of NQO1 [191]. Furthermore, NQO1 activity (in terms of units of enzyme) were determined for a panel of 41 HNC cell lines and one primary normal fibroblast, where the data further confirmed that ~100 NQO1 enzymatic units were required for lethality due to NQO1 futile redox recycling of β-lapachone. In cells with higher NQO1 enzymatic activity, NAD(P)H (electron donor) most likely became rate-limiting, in which case higher NQO1 levels did not confer enhanced lethality or increased β-lapachone lethality (i.e., lowered LD50 values) for β-lapachone treatments in HNC cells. In addition, NQO1-mediated, β-lapachone-induced cell death was partially blocked in the presence of exogenous catalase in HNC cell lines, as previously noted [131, 148]. These data further confirm that β-lap selectively kills HNC cancer cells in an NQO1-dependent manner, regardless of clinic pathological status, while sparing adjacent normal tissue. Finally, β-lapachone killed NQO1+ HNC cells through massive formation of ROS/H2O2, dramatic increases in Ca2+ efflux, creation of SSBs and DSBs, PARP1 hyperactivation, NAD+/ATP depletion, and programmed necrosis.
3. β-Lapachone radiosensitizes NQO1 overexpressing HNC cell lines. Given the massive amount of ROS generation and SSBs and DSBs, enhanced anticancer lethality with a combination of IR + β-lap could be used for the treatment of HNC. Since radiation therapy is used to treat a majority of HNCs, radiosensitization using sublethal doses of β-lapachone was explored using a combination of relative cell survival assays and colony formation assays. In agreement with previous data, HNC cell lines that express NQO1 were all radiosensitized by β-lap at low (otherwise nontoxic) concentrations, while there was no radiosensitization of NQO1-deficient cells. To further assess the role of β-lap in radiosensitization, DNA damage status was assessed with γH2AX foci formation 2 h after a 2 Gy exposure in the absence or presence of sublethal doses of β-lapachone. Damage was assessed using immunofluorescence in vitro assays, as well as in vivo mouse models. Data clearly demonstrated that γH2AX formation was significantly increased in the presence of sublethal doses of β-lapachone (doses that cause significant stress, but no lethality) with 2 Gy compared to β-lapachone or IR alone [191]. As in prostate cancer, significant NAD+/ATP losses was confirmed, and programmed necrosis played an essential role in NQO1-mediated radiosensitization of HNC cells with β-lapachone.
IR is a central therapeutic modality for the treatment of locally advanced and locally regional recurrent HNC [174]. Currently, chemotherapy is used as the standard of care to radiosensitize HNC tumors, but the use of chemotherapy also suffers from non-specific normal tissue cytotoxicity. Since the current standard of care, which includes radiation therapy with concomitant chemotherapy only cures ~50 % of patients with locally advanced disease, there is a need to identify better tumor radiosensitizers to increase the effectiveness of IR. The inverse expression of the NQO1:catalase ratio provides a favorable microenvironment to exploit the therapeutic window of NQO1 bioactivatable drugs, such as β-lapachone, for the treatment of HNCs, since catalase in normal tissue protects against NQO1-dependent β-lapachone lethality by neutralizing the bystander effect (H2O2-generated) of the β-lapachone futile redox cycle [8]. Since a similar inverse expression pattern was seen in HNC cell lines and patient samples, these data support further evaluation of radiation therapy + β-lapachone as a treatment strategy for HNC. This therapeutic strategy will leverage tumor-selective cytotoxicity, as well as the radiosensitizing capacity of β-lapachone [9, 10], while greatly reducing exposure to β-lapachone-HP-β-CD-induced side-effects, which are restricted to a non-NQO1-induced methemoglobinemia. These data warrant a clinical trial of IR + β-lapachone for the treatment of HNCs, where methemoglobinemia would not be an issue with far lower doses of β-lapachone needed for therapy.
10.5 Conclusions
Use of NQO1 bioactivatable drugs, for example β-lapachone (ARQ761), in combination with ionizing radiation (IR) is applicable to many of the most non-treatable forms of cancer (e.g., pancreatic and non-small cell lung cancers). Mechanistically, IR + β-lapachone synergistically kills cancer cells by combinational DNA lesion formation, with DNA base, SSB and DSB lesions leading to PARP hyperactivation, as PARPs bind all of these lesions with differential hypersensitivity. However, the dramatic down-regulation of glucose utilization via glycolysis (GAPDH) and TCA cycle suppression, along with tremendous NAD+/ATP losses likely plays havoc on DSB repair. Studies on DSB repair activities in cells exposed to IR + β-lap, compared to control, low doses of β-lap and IR are ongoing. The IR + β-lap synergistic responses in cells likely causes interesting DNA metabolic alterations in cells leading to atypical routes of attempted recovery. The massive production of H2O2 may also cause alterations in the tumor microvasculature that could have impacts beyond just what is happening within cancer tissue. Limited experiments demonstrate that hypoxia plays no role in cancer cell metabolism to β-lap alone, or contribute to IR + β-lap responses.
10.6 Future Directions
1. Improved delivery is key to enhancing therapy with NQO1 bioactivatable drugs. Although β-lapachone shows significant synergy with many drugs in NQO1-specific tumors, its poor water solubility (0.038 mg/mL) limits its systematic administration and clinical applications in vivo [175]. To improve solubility, Nasongkla et al., formulated β-lapachone with cyclodextrins (CD), finding that hydroxypropyl-β-cyclodextrin (HP-β-CD) can improve solubility to 16.0 mg/mL and the complex offers a major improvement in bioavailability [175].
However, non-specific distribution raises the risk of methemoglobinemia, and quick clearance significantly impedes drug effectiveness. To improve tumor specific distribution and exposure, biocompatible polymers that can adjust drug release and drug distribution become the first choice. Blanco et al. [176, 177] developed β-lapachone-containing poly(ethylene glycol)-co-poly(D,L-lactic acid) (PEG-PLA) polymer micelles for the treatment of NQO1-overexpressing tumors. Compared with β-lapachone-HP-β-CD, β-lapachone-PEG5k-PLA5k in mice bearing subcutaneous A549 lung tumors showed prolonged circulation (t1/2, ~28 h) of the drug and increased accumulation in tumors. In addition, antitumor efficacy analyses in mice bearing subcutaneous A549 lung tumors and orthotopic Lewis lung carcinoma models showed significant tumor growth delay and increased survival relative to HP-β-CD [177]. Wang et al. [178] designed poly(D,L-lactide-co-glycolide) (PLGA) millirods through cyclodextrin complexation and Díaz-Rodríguez et al. [179] designed Pluronic F127® (PF127) gel that forms a complex of β-lap and cyclodextrin. Dong et al. [180] intratumorally delivered β-lap via polymer implants for prostate cancer therapy showing that inclusion complexes of β-lap-HP-β-CD in PLGA millirods released constant β-lap (~0.4 mg/kg/day) after a burst of 0.5 mg in 12 h and improved antitumor efficacy. Zhang et al. [181] encapsulated β-lap with paclitaxel into the PEG-PLA micelles with significantly (>10 fold) improved drug encapsulation efficiency, although only additive effects resulted. Ma et al. [182–184] developed an esterase-activatable prodrug of β-lap formulated into PEG-PLA micelles. They synthesized diester derivatives of β-lap and the resulting micelles yielded fairly high β-lap solubility (>7 mg/mL), physical stability, and an ability to reconstitute after lyophilization. Moreover, β-lapachone-dC3 prodrug micelles significantly improved antitumor efficacy against orthotopic A549 mouse models versus β-lapachone-HP-β-CD and provide a promising strategy for NQO1-targeted therapy of lung cancer with improved safety [182].
Besides polymers, liposomes have also been used to formulate β-lapachone. Cavalcanti et al. [185, 186] encapsulated β-lapachone-HP-β-CD into liposomes and evaluated their antimicrobial activity. Release kinetics in vitro of β-lap from liposomes showed initial faster drug release (almost 40 % in the first 4 h). In sum, formulation of β-lap has significantly improved over previous studies. Nevertheless, to overcome blood toxicity seen in the clinic caused by non-specific distribution and fast release from capsules, tumor-specific accumulation, and sustainable effective release at the tumor site is an important future direction for β-lap delivery to improve the clinical efficacy and safety.
2. Metabolic consequences and anaplerotic recovery. The one phenotype that is consistent among all the NQO1 bioactivatable drugs is the drastic change in the metabolic state of the cell due to the depletion of NAD+ and/or NADH. The cell is expected to rescue this phenotype through anaplerotic pathways that produce NAD+/NADH in order to recover a metabolic steady state. There are multiple pathways in cancer cells that are upregulated, therefore targeting those highly expressed pathways, which also generate NAD+/NADH, provide several targets that could synergize with β-lapachone treatment. Several of these pathways are highlighted in Fig. 10.3. These pathways can regenerate the energy necessary in both the cytosol and mitochondria for proper metabolism to occur while also providing substrates needed to feedback into glycolysis and/or TCA cycle (Fig. 10.3).

Multiple pathways are upregulated in cancer cells, with some known for generating NAD+/NADH. (1) Pentose phosphate pathway (PPP): The enzymes glucose 6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase both produce NADPH from NADP+; (2) Tryptophan Biosynthesis: The intermediate metabolite 2-amino-3-carboxymuconate semialdehyde is a precursor for de novo NAD+ and NADP+ biosynthesis while 2-aminomuconate semialdehyde dehydrogenase generates NADH from NAD+ and 2-aminomuconate reductase produces NAD+ from NADH; (3) Pyruvate-malate cycling: The cytosolic isozyme of malate dehydrogenase is responsible for producing NAD+ from NADH while malic enzyme further generates NADPH from NADP+; (4) Serine/glycine metabolism: The enzyme 3-hydroxypyruvate dehydrogenase can convert NADH to NAD+ from mitochondrial serine; (5) β-oxidation: The breakdown of fatty acids within the mitochondria afford an NADH molecule from NAD+ through 3-hydroxyacyl dehydrogenase; (6) Glutamine/glutamate metabolism: Uptake of glutamine into the mitochondria can convert to glutamate which will then produce NADH from NAD+ through glutamate dehydrogenase
3. NQO1 in Cancer Stem Cells and reducing recurrence and metastatic spread with NQO1 directed therapies. Developing novel chemotherapeutics that target tumor associated genes that are overexpressed in tumor tissues versus associated normal tissues, such as the KRAS protein, EGFR, and hepatocyte growth factor receptor (cMet), has become a staple in the anticancer drug discovery field [187]. Over the past few years, there has been an increasing interest in genes that regulate oxidant stress, such as catalase, hemoxygenease-1, and glutathione transferase, as these genes have been found to be critical factors in tumor associated oxidative stress regulation [188]. As such, these genes may also be credible targets for the development of anticancer therapies [189]. Interestingly, NQO1 overexpression in tumors is partly due to its critical role in quelling oxidative stress associated with tumor development. Depleting NQO1 in lung cancer cells causes an increase in oxidative stress, inhibits anchorage independent growth, increases sensitization to anoikis, inhibits tumor invasion, blocks cell proliferation, and reduces the growth of tumors in vivo [168]. NQO1’s role in tumorigenesis suggest that the regulation of oxidative stress plays a critical role in tumorigenesis and developing therapeutics targeting oxidative stress regulatory genes, such as NQO1, is critically needed as an anticancer strategy [189].
One common goal for the development of novel anticancer drugs is discovering therapeutics that can kill bulk tumors, as well as chemo-resistant tumor initiating/cancer stem cells that are purportedly the cause of tumor recurrence [190]. Interestingly, our findings on NQO1’s role in tumorigenesis also revealed that depletion of tumor associated NQO1 levels decreased the population of ALDHhigh cancer cells, suggesting that the reason we observed far less tumor growth in our in vivo NQO1 knockdown studies was due to the depletion of the ALDHhigh cancer stem cell population within the bulk tumor [168]. Thus, further studies on NQO1’s role in tumorigenesis may lead to the development of novel therapeutics targeting NQO1 expression levels directly in patient tumors.
References
Hall EJ. Radiobiology for the Radiologist. 4th ed. Philadelphia: J.B.Lippincott Company; 1994. p. 219–21.
Hall EJ. Radiation, the two-edged sword: cancer risks at high and low doses. Cancer J. 2000;6:343–50.
Boothman DA, Meyers M, Fukunaga N, Lee SW. Isolation of x-ray-inducible transcripts from radioresistant human melanoma cells. Proc Natl Acad Sci U S A. 1993;90:7200–4.
Dong Y, et al. Prostate cancer radiosensitization through poly(ADP-Ribose) polymerase-1 hyperactivation. Cancer Res. 2010;70:8088–96.
Chakrabarti G, et al. Tumor-selective use of DNA base excision repair inhibition in pancreatic cancer using the NQO1 bioactivatable drug, beta-lapachone. Sci Rep. 2015;5:17066.
Chakrabarti G, et al. Targeting glutamine metabolism sensitizes pancreatic cancer to PARP-driven metabolic catastrophe induced by ß-lapachone. Cancer Metabol. 2015;3:12.
Pink JJ, et al. NAD(P)H:Quinone oxidoreductase activity is the principal determinant of beta-lapachone cytotoxicity. J Biol Chem. 2000;275:5416–24.
Cao L, et al. Tumor-selective, futile redox cycle-induced bystander effects elicited by NQO1 bioactivatable radiosensitizing drugs in triple-negative breast cancers. Antioxid Redox Signal. 2014;21:237–50.
Boothman DA, Greer S, Pardee AB. Potentiation of halogenated pyrimidine radiosensitizers in human carcinoma cells by beta-lapachone (3,4-dihydro-2,2-dimethyl-2H-naphtho[1,2-b]pyran- 5,6-dione), a novel DNA repair inhibitor. Cancer Res. 1987;47:5361–6.
Boothman DA, Trask DK, Pardee AB. Inhibition of potentially lethal DNA damage repair in human tumor cells by beta-lapachone, an activator of topoisomerase I. Cancer Res. 1989;49:605–12.
Boothman DA, Pardee AB. Inhibition of radiation-induced neoplastic transformation by beta-lapachone. Proc Natl Acad Sci U S A. 1989;86:4963–7.
Franken NA, Barendsen GW. Enhancement of radiation effectiveness by hyperthermia and incorporation of halogenated pyrimidines at low radiation doses as compared with high doses: implications for mechanisms. Int J Radiat Biol. 2014;90:313–7.
Watanabe R, Nikjoo H. Modelling the effect of incorporated halogenated pyrimidine on radiation-induced DNA strand breaks. Int J Radiat Biol. 2002;78:953–66.
Miller EM, Fowler JF, Kinsella TJ. Linear-quadratic analysis of radiosensitization by halogenated pyrimidines. II Radiosensitization of human colon cancer cells by bromodeoxyuridine. Radiat Res. 1992;131:90–7.
Miller EM, Fowler JF, Kinsella TJ. Linear-quadratic analysis of radiosensitization by halogenated pyrimidines. I Radiosensitization of human colon cancer cells by iododeoxyuridine. Radiat Res. 1992;131:81–9.
Iliakis G, Pantelias G, Okayasu R, Seaner R. Comparative studies on repair inhibition by araA, araC and aphidicolin of radiation induced DNA and chromosome damage in rodent cells: comparison with fixation of PLD. Int J Radiat Oncol Biol Phys. 1989;16:1261–5.
Franken NA, Van Bree CV, Kipp JB, Barendsen GW. Modification of potentially lethal damage in irradiated Chinese hamster V79 cells after incorporation of halogenated pyrimidines. Int J Radiat Biol. 1997;72:101–9.
Perez LM, Greer S. Sensitization to X ray by 5-chloro-2′-deoxycytidine co-administered with tetrahydrouridine in several mammalian cell lines and studies of 2′-chloro derivatives. Int J Radiat Oncol Biol Phys. 1986;12:1523–7.
Perez LM, Mekras JA, Briggle TV, Greer S. Marked radiosensitization of cells in culture to X ray by 5-chlorodeoxycytidine coadministered with tetrahydrouridine, and inhibitors of pyrimidine biosynthesis. Int J Radiat Oncol Biol Phys. 1984;10:1453–8.
Santos O, Perez LM, Briggle TV, Boothman DA, Greer SB. Radiation, pool size and incorporation studies in mice with 5-chloro-2′-deoxycytidine. Int J Radiat Oncol Biol Phys. 1990;19:357–65.
Djordjevic B, Szybalski W. Genetics of human cell lines. III. Incorporation of 5-bromo- and 5-iododeoxyuridine into the deoxyribonucleic acid of human cells and its effect on radiation sensitivity. J Exp Med. 1960;112:509–31.
Lett JT, Parkins G, Alexander P, Ormerod MG. Mechanisms of Sensitization to X-Rays of Mammalian Cells by 5-Bromodeoxyuridine. Nature. 1964;203:593–6.
Delihas N, Rich MA, Eidinoff ML. Radiosensitization of a mammalian cell line with 5-bromodeoxyuridine. Radiat Res. 1962;17:479–91.
Rich MA. Mechanism of radiosensitization of mammalian cells by exposure to halogenated pyrimidines. J Albert Einstein Med Cent (Phila). 1963;11:10–2.
Dewey WC, Sedita BA, Humphrey RM. Radiosensitization of X chromosome of Chinese hamster cells related to incorporation of 5-bromodeoxyuridine. Science. 1966;152:519–21.
Hotz G. On the mechanism of radiosensitization by 5-bromouracil. Studies on 60-Co-gamma irradiated phage phi-X-174 and its single-stranded infectious DNA. Mol Gen Genet. 1968;102:44–9.
Hoshino T, Sano K. Radiosensitization of malignant brain tumours with bromouridine (thymidine analogue). Acta Radiol Ther Phys Biol. 1969;8:15–26.
Sano K, Hoshino T, Nagai M. Radiosensitization of brain tumor cells with a thymidine analogue (bromouridine). J Neurosurg. 1968;28:530–8.
Mekras JA, Boothman DA, Greer SB. Use of 5-trifluoromethyldeoxycytidine and tetrahydrouridine to circumvent catabolism and exploit high levels of cytidine deaminase in tumors to achieve DNA- and target-directed therapies. Cancer Res. 1985;45:5270–80.
Taverna P, et al. Inhibition of base excision repair potentiates iododeoxyuridine-induced cytotoxicity and radiosensitization. Cancer Res. 2003;63:838–46.
Fornace Jr AJ, Dobson PP, Kinsella TJ. Enhancement of radiation damage in cellular DNA following unifilar substitution with iododeoxyuridine. Int J Radiat Oncol Biol Phys. 1990;18:873–8.
Urtasun RC, et al. Survival improvement in anaplastic astrocytoma, combining external radiation with halogenated pyrimidines: final report of RTOG 86-12. Phase I-II study. Int J Radiat Oncol Biol Phys. 1996;36:1163–7.
Epstein AH, et al. Treatment of locally advanced cancer of the head and neck with 5′-iododeoxyuridine and hyperfractionated radiation therapy: measurement of cell labeling and thymidine replacement. J Natl Cancer Inst. 1994;86:1775–80.
Chang AE, et al. A phase I study of intraarterial iododeoxyuridine in patients with colorectal liver metastases. J Clin Oncol Off J Am Soc Clin Oncol. 1989;7:662–8.
Young MM, et al. Treatment of sarcomas of the chest wall using intensive combined modality therapy. Int J Radiat Oncol Biol Phys. 1989;16:49–57.
Sondak VK, et al. Preoperative idoxuridine and radiation for large soft tissue sarcomas: clinical results with five-year follow-up. Ann Surg Oncol. 1998;5:106–12.
Kinsella TJ, et al. Pharmacology and phase I/II study of continuous intravenous infusions of iododeoxyuridine and hyperfractionated radiotherapy in patients with glioblastoma multiforme. J Clin Oncol Off J Am Soc Clin Oncol. 1988;6:871–9.
Lawrence TS, Davis MA, Maybaum J, Stetson PL, Ensminger WD. The dependence of halogenated pyrimidine incorporation and radiosensitization on the duration of drug exposure. Int J Radiat Oncol Biol Phys. 1990;18:1393–8.
Prados MD, et al. Influence of bromodeoxyuridine radiosensitization on malignant glioma patient survival: a retrospective comparison of survival data from the Northern California Oncology Group (NCOG) and Radiation Therapy Oncology Group trials (RTOG) for glioblastoma multiforme and anaplastic astrocytoma. Int J Radiat Oncol Biol Phys. 1998;40:653–9.
Hegarty TJ, et al. Intra-arterial bromodeoxyuridine radiosensitization of malignant gliomas. Int J Radiat Oncol Biol Phys. 1990;19:421–8.
Phillips TL, Scott CB, Leibel SA, Rotman M, Weigensberg IJ. Results of a randomized comparison of radiotherapy and bromodeoxyuridine with radiotherapy alone for brain metastases: report of RTOG trial 89-05. Int J Radiat Oncol Biol Phys. 1995;33:339–48.
Smith TG, Robbins PA, Ratcliffe PJ. The human side of hypoxia-inducible factor. Br J Haematol. 2008;141:325–34.
Miyata T, Takizawa S, van Ypersele de Strihou C. Hypoxia. 1. Intracellular sensors for oxygen and oxidative stress: novel therapeutic targets. Am J Physiol Cell Physiol. 2011;300:C226–31.
Oronsky BT, Knox SJ, Scicinski J. Six degrees of separation: the oxygen effect in the development of radiosensitizers. Transl Oncol. 2011;4:189–98.
Williams KJ, Cowen RL, Stratford IJ. Hypoxia and oxidative stress. Tumour hypoxia—therapeutic considerations. Breast Cancer Res. 2001;3:328–31.
Dasu A, Denekamp J. New insights into factors influencing the clinically relevant oxygen enhancement ratio. Radiother Oncol. 1998;46:269–77.
Brown J. Selective radiosensitization of the hypoxic cells of mouse tumors with the nitroimidazoles metronidazole and Ro 7-0582. Radiat Res. 1975;64(3):633–47.
Brown JM. Clinical trials of radiosensitizers: what should we expect? Int J Radiat Oncol Biol Phys. 1984;10:425–9.
Weiss GJ, et al. Phase 1 study of the safety, tolerability, and pharmacokinetics of TH-302, a hypoxia-activated prodrug, in patients with advanced solid malignancies. Clin Cancer Res. 2011;17:2997–3004.
Corre I, Niaudet C, Paris F. Plasma membrane signaling induced by ionizing radiation. Mutat Res. 2010;704:61–7.
Galabova-Kovacs G, et al. ERK and beyond: insights from B-Raf and Raf-1 conditional knockouts. Cell Cycle. 2006;5:1514–8.
Caron RW, et al. Activated forms of H-RAS and K-RAS differentially regulate membrane association of PI3K, PDK-1, and AKT and the effect of therapeutic kinase inhibitors on cell survival. Mol Cancer Ther. 2005;4:257–70.
Caron RW, et al. Radiation-stimulated ERK1/2 and JNK1/2 signaling can promote cell cycle progression in human colon cancer cells. Cell Cycle. 2005;4:456–64.
Dent P, et al. Radiation-induced release of transforming growth factor alpha activates the epidermal growth factor receptor and mitogen-activated protein kinase pathway in carcinoma cells, leading to increased proliferation and protection from radiation-induced cell death. Mol Biol Cell. 1999;10:2493–506.
Shvartsman SY, et al. Autocrine loops with positive feedback enable context-dependent cell signaling. Am J Physiol Cell Physiol. 2002;282:C545–59.
Amorino GP, et al. Epidermal growth factor receptor dependence of radiation-induced transcription factor activation in human breast carcinoma cells. Mol Biol Cell. 2002;13:2233–44.
Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys. 2004;59:928–42.
McKenna WG, Muschel RJ, Gupta AK, Hahn SM, Bernhard EJ. The RAS signal transduction pathway and its role in radiation sensitivity. Oncogene. 2003;22:5866–75.
Jones HA, Hahn SM, Bernhard E, McKenna WG. Ras inhibitors and radiation therapy. Semin Radiat Oncol. 2001;11:328–37.
Brunner TB, et al. Farnesyltransferase inhibitors as radiation sensitizers. Int J Radiat Biol. 2003;79:569–76.
Konstantinidou G, et al. Dual phosphoinositide 3-kinase/mammalian target of rapamycin blockade is an effective radiosensitizing strategy for the treatment of non-small cell lung cancer harboring K-RAS mutations. Cancer Res. 2009;69:7644–52.
Mukherjee B, et al. The dual PI3K/mTOR inhibitor NVP-BEZ235 is a potent inhibitor of ATM- and DNA-PKCs-mediated DNA damage responses. Neoplasia. 2012;14:34–43.
Gil del Alcazar CR, et al. Inhibition of DNA double-strand break repair by the dual PI3K/mTOR inhibitor NVP-BEZ235 as a strategy for radiosensitization of glioblastoma. Clin Cancer Res. 2014;20:1235–48.
Kriegs M, et al. Radiosensitization of NSCLC cells by EGFR inhibition is the result of an enhanced p53-dependent G1 arrest. Radiother Oncol. 2015;115:120–7.
Kim K, Wu HG, Jeon SR. Epidermal growth factor-induced cell death and radiosensitization in epidermal growth factor receptor-overexpressing cancer cell lines. Anticancer Res. 2015;35:245–53.
Tsai YC, et al. Targeting epidermal growth factor receptor/human epidermal growth factor receptor 2 signalling pathway by a dual receptor tyrosine kinase inhibitor afatinib for radiosensitisation in murine bladder carcinoma. Eur J Cancer. 2013;49:1458–66.
Diaz Miqueli A, et al. Radiosensitisation of U87MG brain tumours by anti-epidermal growth factor receptor monoclonal antibodies. Br J Cancer. 2009;100:950–8.
Maddineni SB, Sangar VK, Hendry JH, Margison GP, Clarke NW. Differential radiosensitisation by ZD1839 (Iressa), a highly selective epidermal growth factor receptor tyrosine kinase inhibitor in two related bladder cancer cell lines. Br J Cancer. 2005;92:125–30.
Sartor CI. Mechanisms of disease: radiosensitization by epidermal growth factor receptor inhibitors. Nat Clin Pract Oncol. 2004;1:80–7.
Bonner JA, et al. Anti-EGFR-mediated radiosensitization as a result of augmented EGFR expression. Int J Radiat Oncol Biol Phys. 2004;59:2–10.
Stea B, et al. Time and dose-dependent radiosensitization of the glioblastoma multiforme U251 cells by the EGF receptor tyrosine kinase inhibitor ZD1839 (‘Iressa’). Cancer Lett. 2003;202:43–51.
Feng FY, de Bono JS, Rubin MA, Knudsen KE. Chromatin to clinic: the molecular rationale for PARP1 inhibitor function. Mol Cell. 2015;58:925–34.
Mateo J, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–708.
Rosenthal DI, et al. Association of human papillomavirus and p16 status with outcomes in the IMCL-9815 phase Iii registration trial for patients with locoregionally advanced oropharyngeal squamous cell carcinoma of the head and neck treated with radiotherapy with or without cetuximab. J Clin Oncol Off J Am Soc Clin Oncol. 2015;34(12):1300–8.
Wang Z, et al. Radiosensitization of metformin in pancreatic cancer cells via abrogating the G2 checkpoint and inhibiting DNA damage repair. Cancer Lett. 2015;369:192–201.
Fasih A, Elbaz HA, Huttemann M, Konski AA, Zielske SP. Radiosensitization of pancreatic cancer cells by metformin through the AMPK pathway. Radiat Res. 2014;182:50–9.
Yang CR, et al. Nuclear clusterin/XIP8, an x-ray-induced Ku70-binding protein that signals cell death. Proc Natl Acad Sci U S A. 2000;97:5907–12.
Sutton D, et al. Efficient suppression of secretory clusterin levels by polymer-siRNA nanocomplexes enhances ionizing radiation lethality in human MCF-7 breast cancer cells in vitro. Int J Nanomedicine. 2006;1:155–62.
Jiang L, et al. Metabolic reprogramming during TGFbeta1-induced epithelial-to-mesenchymal transition. Oncogene. 2015;34:3908–16.
Raaphorst GP, Feeley MM. Hyperthermia radiosensitization in human glioma cells comparison of recovery of polymerase activity, survival, and potentially lethal damage repair. Int J Radiat Oncol Biol Phys. 1994;29:133–9.
Kampinga HH, Dynlacht JR, Dikomey E. Mechanism of radiosensitization by hyperthermia (> or = 43 degrees C) as derived from studies with DNA repair defective mutant cell lines. Int J Hyperthermia. 2004;20:131–9.
Pandita TK, Pandita S, Bhaumik SR. Molecular parameters of hyperthermia for radiosensitization. Crit Rev Eukaryot Gene Expr. 2009;19:235–51.
Xu M, et al. The effects of 41 degrees C hyperthermia on the DNA repair protein, MRE11, correlate with radiosensitization in four human tumor cell lines. Int J Hyperthermia. 2007;23:343–51.
Iliakis GE, Pantelias GE. Effects of hyperthermia on chromatin condensation and nucleoli disintegration as visualized by induction of premature chromosome condensation in interphase mammalian cells. Cancer Res. 1989;49:1254–60.
Camphausen K, Tofilon PJ. Inhibition of Hsp90: a multitarget approach to radiosensitization. Clin Cancer Res. 2007;13:4326–30.
Pruitt R, Sasi N, Freeman ML, Sekhar KR. Radiosensitization of cancer cells by hydroxychalcones. Bioorg Med Chem Lett. 2010;20:5997–6000.
Sekhar KR, et al. Novel chemical enhancers of heat shock increase thermal radiosensitization through a mitotic catastrophe pathway. Cancer Res. 2007;67:695–701.
Hijnen N, Langereis S, Grull H. Magnetic resonance guided high-intensity focused ultrasound for image-guided temperature-induced drug delivery. Adv Drug Deliv Rev. 2014;72:65–81.
Negussie AH, et al. Formulation and characterisation of magnetic resonance imageable thermally sensitive liposomes for use with magnetic resonance-guided high intensity focused ultrasound. Int J Hyperthermia. 2011;27:140–55.
Staruch RM, Hynynen K, Chopra R. Hyperthermia-mediated doxorubicin release from thermosensitive liposomes using MR-HIFU: therapeutic effect in rabbit Vx2 tumours. Int J Hyperthermia. 2015;31:118–33.
Yeo SY, et al. Bone metastasis treatment using magnetic resonance-guided high intensity focused ultrasound. Bone. 2015;81:513–23.
Butterworth KT, McMahon SJ, Currell FJ, Prise KM. Physical basis and biological mechanisms of gold nanoparticle radiosensitization. Nanoscale. 2012;4:4830–8.
Dorsey JF, et al. Gold nanoparticles in radiation research: potential applications for imaging and radiosensitization. Transl Cancer Res. 2013;2:280–91.
Mesbahi A. A review on gold nanoparticles radiosensitization effect in radiation therapy of cancer. Rep Pract Oncol Radiother. 2010;15:176–80.
Zheng Y, Hunting DJ, Ayotte P, Sanche L. Radiosensitization of DNA by gold nanoparticles irradiated with high-energy electrons. Radiat Res. 2008;169:19–27.
Xu W, et al. RGD-conjugated gold nanorods induce radiosensitization in melanoma cancer cells by downregulating alpha(v)beta(3) expression. Int J Nanomedicine. 2012;7:915–24.
Jeynes JC, Merchant MJ, Spindler A, Wera AC, Kirkby KJ. Investigation of gold nanoparticle radiosensitization mechanisms using a free radical scavenger and protons of different energies. Phys Med Biol. 2014;59:6431–43.
Brun E, Sanche L, Sicard-Roselli C. Parameters governing gold nanoparticle X-ray radiosensitization of DNA in solution. Colloids Surf B Biointerfaces. 2009;72:128–34.
Jain S, et al. Cell-specific radiosensitization by gold nanoparticles at megavoltage radiation energies. Int J Radiat Oncol Biol Phys. 2011;79:531–9.
Masood R, et al. Gold nanorod-sphingosine kinase siRNA nanocomplexes: a novel therapeutic tool for potent radiosensitization of head and neck cancer. Integr Biol (Camb). 2012;4:132–41.
Joh DY, et al. Selective targeting of brain tumors with gold nanoparticle-induced radiosensitization. PLoS One. 2013;8, e62425.
Al Zaki A, et al. Gold-loaded polymeric micelles for computed tomography-guided radiation therapy treatment and radiosensitization. ACS Nano. 2014;8:104–12.
Hebert EM, Debouttiere PJ, Lepage M, Sanche L, Hunting DJ. Preferential tumour accumulation of gold nanoparticles, visualised by Magnetic Resonance Imaging: radiosensitisation studies in vivo and in vitro. Int J Radiat Biol. 2010;86:692–700.
Bhattacharyya SN, Mandal PC, Chakraborty S. Copper (II) induced radiosensitization of thymine. Anticancer Res. 1989;9:1181–4.
Roy MB, Mandal PC, Bhattacharyya SN. Radiosensitization of thymine by copper(II) and nickel(II) complexes of metronidazole. Int J Radiat Biol. 1996;69:471–80.
Savoye C, Sabattier R, Charlier M, Spotheim-Maurizot M. Sequence-modulated radiosensitization of DNA by copper ions. Int J Radiat Biol. 1996;70:189–98.
Rae C, et al. The role of copper in disulfiram-induced toxicity and radiosensitization of cancer cells. J Nucl Med. 2013;54:953–60.
Khoei S, Mahdavi SR, Fakhimikabir H, Shakeri-Zadeh A, Hashemian A. The role of iron oxide nanoparticles in the radiosensitization of human prostate carcinoma cell line DU145 at megavoltage radiation energies. Int J Radiat Biol. 2014;90:351–6.
Skov KA, Adomat H, Farrell NP. Radiosensitization by nickel lapachol. Int J Radiat Biol. 1993;64:707–13.
Dufort S, et al. Nebulized gadolinium-based nanoparticles: a theranostic approach for lung tumor imaging and radiosensitization. Small. 2015;11:215–21.
Zhao J, Lee P, Wallace MJ, Melancon MP. Gold nanoparticles in cancer therapy: efficacy, biodistribution, and toxicity. Curr Pharm Des. 2015;21:4240–51.
Wang TJ, et al. Caffeine enhances radiosensitization to orthotopic transplant LM3 hepatocellular carcinoma in vivo. Cancer Sci. 2010;101:1440–6.
Wang X, Wang H, Iliakis G, Wang Y. Caffeine-induced radiosensitization is independent of nonhomologous end joining of DNA double-strand breaks. Radiat Res. 2003;159:426–32.
Sinn B, et al. Caffeine confers radiosensitization of PTEN-deficient malignant glioma cells by enhancing ionizing radiation-induced G1 arrest and negatively regulating Akt phosphorylation. Mol Cancer Ther. 2010;9:480–8.
Boothman DA, Schlegel R, Pardee AB. Anticarcinogenic potential of DNA-repair modulators. Mutat Res. 1988;202:393–411.
Veeraraghavan J, et al. Neem leaf extract induces radiosensitization in human neuroblastoma xenograft through modulation of apoptotic pathway. Anticancer Res. 2011;31:161–70.
Veeraraghavan J, et al. Impact of curcumin, raspberry extract, and neem leaf extract on rel protein-regulated cell death/radiosensitization in pancreatic cancer cells. Pancreas. 2011;40:1107–19.
Li M, Zhang Z, Hill DL, Wang H, Zhang R. Curcumin, a dietary component, has anticancer, chemosensitization, and radiosensitization effects by down-regulating the MDM2 oncogene through the PI3K/mTOR/ETS2 pathway. Cancer Res. 2007;67:1988–96.
Jagetia GC. Radioprotection and radiosensitization by curcumin. Adv Exp Med Biol. 2007;595:301–20.
Garg AK, Buchholz TA, Aggarwal BB. Chemosensitization and radiosensitization of tumors by plant polyphenols. Antioxid Redox Signal. 2005;7:1630–47.
Chen MF, Wu CT, Chen YJ, Keng PC, Chen WC. Cell killing and radiosensitization by caffeic acid phenethyl ester (CAPE) in lung cancer cells. J Radiat Res. 2004;45:253–60.
Lin YH, et al. Antiproliferation and radiosensitization of caffeic acid phenethyl ester on human medulloblastoma cells. Cancer Chemother Pharmacol. 2006;57:525–32.
Raffoul JJ, Sarkar FH, Hillman GG. Radiosensitization of prostate cancer by soy isoflavones. Curr Cancer Drug Targets. 2007;7:759–65.
Liu G, et al. Radiosensitization mechanism of riboflavin in vitro. Sci China C Life Sci. 2002;45:344–52.
Xu C, Li CY, Kong AN. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch Pharm Res. 2005;28:249–68.
Jancova P, Anzenbacher P, Anzenbacherova E. Phase II drug metabolizing enzymes. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2010;154:103–16.
Thor H, et al. The metabolism of menadione (2-methyl-1,4-naphthoquinone) by isolated hepatocytes. A study of the implications of oxidative stress in intact cells. J Biol Chem. 1982;257:12419–25.
Radjendirane V, et al. Disruption of the DT diaphorase (NQO1) gene in mice leads to increased menadione toxicity. J Biol Chem. 1998;273:7382–9.
Begleiter A, Fourie J. Induction of NQO1 in cancer cells. Methods Enzymol. 2004;382:320–51.
Marin A, et al. DT-diaphorase and cytochrome B5 reductase in human lung and breast tumours. Br J Cancer. 1997;76:923–9.
Bey EA, et al. An NQO1- and PARP-1-mediated cell death pathway induced in non-small-cell lung cancer cells by beta-lapachone. Proc Natl Acad Sci U S A. 2007;104:11832–7.
Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45–9.
Singh A, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3, e420.
Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179–91.
Siegel D, Yan C, Ross D. NAD(P)H:quinone oxidoreductase 1 (NQO1) in the sensitivity and resistance to antitumor quinones. Biochem Pharmacol. 2012;83:1033–40.
Siegel D, Gibson NW, Preusch PC, Ross D. Metabolism of mitomycin C by DT-diaphorase: role in mitomycin C-induced DNA damage and cytotoxicity in human colon carcinoma cells. Cancer Res. 1990;50:7483–9.
DeBoer C, Meulman PA, Wnuk RJ, Peterson DH. Geldanamycin, a new antibiotic. J Antibiot. 1970;23:442–7.
Jilani K, Qadri SM, Lang F. Geldanamycin-induced phosphatidylserine translocation in the erythrocyte membrane. Cell Physiol Biochem. 2013;32:1600–9.
Goetz MP, et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2005;23:1078–87.
Phillips RM, Hendriks HR, Peters GJ, EORTC-Pharmacology and Molecular Mechanism Group. EO9 (Apaziquone): from the clinic to the laboratory and back again. Br J Pharmacol. 2013;168:11–8.
Phillips RM, et al. Bioreductive activation of a series of indolequinones by human DT-diaphorase: structure-activity relationships. J Med Chem. 1999;42:4071–80.
Loadman PM, Bibby MC, Phillips RM. Pharmacological approach towards the development of indolequinone bioreductive drugs based on the clinically inactive agent EO9. Br J Pharmacol. 2002;137:701–9.
Dirix LY, et al. EO9 phase II study in advanced breast, gastric, pancreatic and colorectal carcinoma by the EORTC Early Clinical Studies Group. Eur J Cancer. 1996;32A:2019–22.
Pavlidis N, et al. A randomized phase II study with two schedules of the novel indoloquinone EO9 in non-small-cell lung cancer: a study of the EORTC Early Clinical Studies Group (ECSG). Ann Oncol. 1996;7:529–31.
Smith GM, Gordon JA, Sewell IA, Ellis H. A trial of streptonigrin in the treatment of advanced malignant disease. Br J Cancer. 1967;21:295–301.
Lewis AM, et al. Targeting NAD(P)H:quinone oxidoreductase (NQO1) in pancreatic cancer. Mol Carcinog. 2005;43:215–24.
Testoni MI, Bianchi NO, Bianchi MS. The kinetics of chromosome and DNA damage by streptonigrin in CHO cells. Mutat Res. 1995;334:23–31.
Bey EA, et al. Catalase abrogates beta-lapachone-induced PARP1 hyperactivation-directed programmed necrosis in NQO1-positive breast cancers. Mol Cancer Ther. 2013;12:2110–20.
Bentle MS, Reinicke KE, Dong Y, Bey EA, Boothman DA. Nonhomologous end joining is essential for cellular resistance to the novel antitumor agent, beta-lapachone. Cancer Res. 2007;67:6936–45.
Bentle MS, Reinicke KE, Bey EA, Spitz DR, Boothman DA. Calcium-dependent modulation of poly(ADP-ribose) polymerase-1 alters cellular metabolism and DNA repair. J Biol Chem. 2006;281:33684–96.
Tagliarino C, Pink JJ, Dubyak GR, Nieminen AL, Boothman DA. Calcium is a key signaling molecule in beta-lapachone-mediated cell death. J Biol Chem. 2001;276:19150–9.
Tagliarino C, et al. Mu-calpain activation in beta-lapachone-mediated apoptosis. Cancer Biol Ther. 2003;2:141–52.
Wuerzberger SM, et al. Induction of apoptosis in MCF-7:WS8 breast cancer cells by beta-lapachone. Cancer Res. 1998;58:1876–85.
Huang X, et al. An NQO1 substrate with potent antitumor activity that selectively kills by PARP1-induced programmed necrosis. Cancer Res. 2013;72:3038–47.
Pink JJ, et al. Activation of a cysteine protease in MCF-7 and T47D breast cancer cells during beta-lapachone-mediated apoptosis. Exp Cell Res. 2000;255:144–55.
Planchon SM, et al. beta-Lapachone-induced apoptosis in human prostate cancer cells: involvement of NQO1/xip3. Exp Cell Res. 2001;267:95–106.
Planchon SM, et al. Beta-lapachone-mediated apoptosis in human promyelocytic leukemia (HL-60) and human prostate cancer cells: a p53-independent response. Cancer Res. 1995;55:3706–11.
Gerber DE et al. Phase 1 correlative study of ARQ761, a β-lapachone analogue that promotes NQ01-mediated programmed cancer cell necrosis. In: 26th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics. Barcelona, Spain, (Poster presentation by DE Gerber); 2015
Li LS, et al. Modulating endogenous NQO1 levels identifies key regulatory mechanisms of action of beta-lapachone for pancreatic cancer therapy. Clin Cancer Res. 2011;17:275–85.
Moore Z, et al. NAMPT inhibition sensitizes pancreatic adenocarcinoma cells to tumor-selective, PAR-independent metabolic catastrophe and cell death induced by beta-lapachone. Cell Death Dis. 2015;6, e1599.
Chini CC, et al. Targeting of NAD metabolism in pancreatic cancer cells: potential novel therapy for pancreatic tumors. Clin Cancer Res. 2014;20:120–30.
Nahimana A, et al. The NAD biosynthesis inhibitor APO866 has potent antitumor activity against hematologic malignancies. Blood. 2009;113:3276–86.
Son J, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–5.
Shukla K, et al. Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J Med Chem. 2012;55:10551–63.
Gross MI, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. 2014;13:890–901.
Chakrabarti G, Gerber DE, Boothman DA. Expanding antitumor therapeutic windows by targeting cancer-specific nicotinamide adenine dinucleotide phosphate-biogenesis pathways. Clin Pharmacol. 2015;7:57–68.
Zhao Y, et al. SoNar, a highly responsive NAD+/NADH sensor. Allows high-throughput metabolic screening of anti-tumor agents. Cell Metab. 2015;21:777–89.
Madajewski B, Boatman MA, Chakrabarti G, Boothman DA, Bey EA. Depleting tumor-NQO1 potentiates anoikis and inhibits growth of NSCLC. Mol Cancer Res. 2016;14:14–25.
Choi EK, et al. Upregulation of NAD(P)H:quinone oxidoreductase by radiation potentiates the effect of bioreductive beta-lapachone on cancer cells. Neoplasia. 2007;9:634–42.
Park HJ, et al. Susceptibility of cancer cells to beta-lapachone is enhanced by ionizing radiation. Int J Radiat Oncol Biol Phys. 2005;61:212–9.
Kim H, et al. Antagonistic effects of anti-EMMPRIN antibody when combined with chemotherapy against hypovascular pancreatic cancers. Mol Imaging Biol. 2014;16:85–94.
Breton CS, et al. Combinative effects of beta-Lapachone and APO866 on pancreatic cancer cell death through reactive oxygen species production and PARP-1 activation. Biochimie. 2015;116:141–53.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29.
Bonner JA, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–78.
Nasongkla N, et al. Enhancement of solubility and bioavailability of β-lapachone using cyclodextrin inclusion complexes. Pharm Res. 2003;20:1626–33.
Blanco E, et al. β-Lapachone-containing PEG-PLA polymer micelles as novel nanotherapeutics against NQO1-overexpressing tumor cells. J Control Release. 2007;122:365–74.
Blanco E, et al. β-lapachone micellar nanotherapeutics for non-small cell lung cancer therapy. Cancer Res. 2010;70:3896–904.
Wang F, Blanco E, Ai H, Boothman DA, Gao J. Modulating β-lapachone release from polymer millirods through cyclodextrin complexation. J Pharm Sci. 2006;95:2309–19.
Díaz-Rodríguez P, Landin M. Smart design of intratumoral thermosensitive β-lapachone hydrogels by Artificial Neural Networks. Int J Pharm. 2012;433:112–8.
Dong Y, et al. Intratumoral delivery of β-lapachone via polymer implants for prostate cancer therapy. Clin Cancer Res. 2009;15:131–9.
Zhang L, et al. β-lapachone and paclitaxel combination micelles with improved drug encapsulation and therapeutic synergy as novel nanotherapeutics for NQO1-targeted cancer therapy. Mol Pharm. 2015;12:3999–4010.
Ma X, et al. Prodrug strategy to achieve lyophilizable, high drug loading Micelle formulations through diester derivatives of β-lapachone. Adv Healthc Mater. 2014;3:1210–6.
Ma X, et al. Esterase-activatable β-lapachone prodrug micelles for NQO1-targeted lung cancer therapy. J Control Release. 2015;200:201–11.
Ma X, et al. Nanotechnology-enabled delivery of NQO1 bioactivatable drugs. J Drug Target. 2015;23:672–80.
Cavalcanti IMF, et al. The encapsulation of β-lapachone in 2-hydroxypropyl-β- cyclodextrin inclusion complex into liposomes: A physicochemical evaluation and molecular modeling approach. Eur J Pharm Sci. 2011;44:332–40.
Cavalcanti IMF, et al. Antimicrobial activity of β-lapachone encapsulated into liposomes against meticillin-resistant Staphylococcus aureus and Cryptococcus neoformans clinical strains. J Glob Antimicrob Resist. 2015;3:103–8.
Huang M, Shen A, Ding J, Geng M. Molecularly targeted cancer therapy: some lessons from the past decade. Trends Pharmacol Sci. 2014;35:41–50.
DeNicola GM, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–9.
Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–47.
Rich JN, Bao S. Chemotherapy and cancer stem cells. Cell Stem Cell. 2007;1:353–5.
Li LS, et al. NQO1-mediated tumor-selective lethality and radiosensitization for head and neck cancer. Mol Cancer Ther. 2016;15(7):1757–67.
Acknowledgements
Work is supported by the National Institutes of Health, CA102972 to DAB.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Bey, E.A. et al. (2016). NQO1 Bioactivatable Drugs Enhance Radiation Responses. In: Anscher, M., Valerie, K. (eds) Strategies to Enhance the Therapeutic Ratio of Radiation as a Cancer Treatment. Springer, Cham. https://doi.org/10.1007/978-3-319-45594-5_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-45594-5_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-45592-1
Online ISBN: 978-3-319-45594-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)