
Abstract
Worldwide, ischaemic stroke is a major cause of death and the leading cause of acquired disability. To date, pharmacological thrombolysis with tissue-type plasminogen activator (alone or with mechanical thrombectomy) remains the gold standard acute treatment for ischaemic stroke. Many strategies of neuroprotection have been tested in the past, but none has ever succeeded in clinical trials. Accumulating evidence suggests that stroke activates the endoplasmic reticulum (ER) stress response, which likely deleteriously contribute to stroke damages. However, this remains to be clearly established. In the present chapter, we provide a snapshot on ER stress in ischaemic stroke, and intend to show that knowledge on ER stress in ischaemic context affecting other organs than the brain can provide interesting therapeutic avenues for neuroprotection.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 The ER Structure and Functions
1.1 General Information
The endoplasmic reticulum (ER ) is a major organelle in eukaryotic cells, consisting of a single membrane system with a continuous intraluminal space. The ER lumen occupies 10 % of the total cell volume while its membrane represents almost half of the total cell membranes [1]. It is thus a compartment completely separated from the cytosol, allowing the ER to maintain essential conditions (proteins for the molecular machinery, high concentration of calcium ions and oxidizing condition) for its various functions. The ER is involved in synthesis, folding and post-translational modification of proteins and also in protein transport and calcium homeostasis (the ER is one of the main calcium reserve in cells) [2, 3].
The ER is traditionally divided into three parts : the nuclear envelope (NE), the rough ER (RER) and the smooth ER (SER). By using electron microscopy, RER appears granular and tubular due to the attachment of ribosomes on its surface. It is found in all eukaryotic cells that synthesize and fold proteins [4]. Contrary to the RER, the SER does not contain ribosomes and has a more expanded and tortuous shape. The SER is the place of lipid synthesis, cell detoxification and calcium storage [4]. The NE surrounds the entire nucleus. It is an essential part of the ER as it defines the border between the nucleus and the cytoplasm. This physical limit is a fundamental characteristic of eukaryotic cells which can regulate gene expression at different levels [5]. The ER can also be divided into specialized regions depending on its morphology, its protein composition or its proximity to other organelles [1].
Even if all cytosolic proteins do not need to traffic through the ER, it is still an essential compartment for the maturation and the proper folding of the majority of proteins [1]. The oxidative environment of the ER lumen is optimal for the various mechanisms required for protein maturation, including the formation of disulphide bonds and their folding by chaperones proteins. Many cellular damages are linked with chaperone dysfunctions, representing then an essential target for neuroprotection (see part IV). Under physiological conditions, chaperones are involved in the synthesis, folding and degradation of newly synthesized proteins and can be divided into three main groups:
1.1.1 Chaperones of the Hsp70 (Heat Shock Protein 70) Family
They include Grp78 (Glucose Regulated Protein 78, also called Bip for Binding immunoglobulin protein) as its most abundant member, and Grp170 (Glucose Regulated Protein 170) [6]. Grp78 binds to proteins during their folding to prevent their aggregation and participates in the ERAD (Endoplasmic-reticulum-associated degradation) mechanism by directing misfolded proteins to the proteasome by retro-translocation via the translocon [7, 8]. Moreover, Grp78 is involved in calcium homeostasis and is a major player in the induction of the UPR (unfolded protein response; see part II) [2]. The roles of Grp170 are less known, but it seems to be involved in protein maturation during ER stress (such as hypoxia and glucose deprivation).
1.1.2 Grp94
It is the only known representative chaperone of the Hsp90 family. Grp94 roles overlap in many ways with those of Grp78 and both chaperones likely work in pairs [9]. While Grp78 binds to unfolded proteins since their entrance into the ER, Grp94 operates on partially folded proteins released by Grp78. Interestingly, Grp94 primarily promotes the biosynthesis of specific secreted or membrane proteins, such as immunoglobulins [7] or Toll-like receptors [10].
1.1.3 Calnexin/Calreticulin
They are lectin site chaperones that bind to proteins during the whole process of folding [11, 12]. Calnexin and calreticulin bind to all newly synthesized N-glycosylated proteins, allowing misfolded proteins to stay longer in the ER, leaving time to reach a correct folding [13]. Another important feature of these chaperones is the catching of calcium from the ER in very large quantities, hence their name.
The ER has a quality control machinery of newly synthesized proteins that discriminates correctly folded from misfolded proteins and allows them to reach a stable conformation or to be degraded. The main pathway for misfolded proteins destruction is called ERAD . ERAD includes two groups of proteins: chaperone proteins and glycan modifying enzymes (EDEM proteins (ER-Degradation-Enhancing alpha-Mannosidase-like protein)). Successive enzymatic reactions (digestion and addition of sugars) on the hydrocarbon chain allow the control of protein folding. Correctly folded glycoproteins are then transported to the Golgi apparatus. If the right protein structure cannot be achieved and maintained, EDEM proteins interact with these misfolded proteins and translocate them to the cytosol where they are poly-ubiquitinated and degraded by the proteasome [14, 15].
The ER is the main calcium storage site in many cells, playing then an important role in the regulation of calcium homeostasis. While the concentration of free calcium in the cytoplasm is 1 nM, the ER-calcium concentration is between 100 and 800 μM [16]. This difference of concentration is regulated by the presence of channel receptors and pumps on the ER surface. There are two types of channel receptors that allow calcium release into the cytosol:
1.1.4 IP3R (INOSITOL-1,4,5-Trisphosphate Receptor) Channels
They belong to a superfamily of ionic channels with 6 transmembrane domains. There are 3 isoforms in mammals which have different affinities for IP3 (inositol-1,4,5-trisphosphate) [17]. Even if IP3 is the main activator, calcium can also activate IP3R with a biphasic manner: activation at low concentrations of cytosolic calcium versus inhibition at high calcium concentrations.
1.1.5 RyR Channels (Ryanodine Receptor)
Three isoforms of RyR have been identified in mammals (RyR1, RyR2 and RyR3), sharing 65 % homology. Numerous agents including calcium and ATP modulate the activity of RyR. Calcium exerts a biphasic effect on its activity, like for IP3R [18].
To ensure a high concentration of calcium within the ER, the SERCA pump (Sarco endoplasmic reticulum Ca2+-transport ATPase), through an active ATP-dependent process, withdraws calcium from the cytosol to the ER [19].
1.2 The ER in Neurones
Neurones have a complex ER that extends all over the cell and formed by both RER and SER, depending on its localization. The RER is present from the nuclear outer membrane to the soma and is involved in protein synthesis. In dendritic spines and in the axon, the ER becomes SER and is involved in pre- and post-synaptic calcium signalling involved in synaptic plasticity [20].
In neurones, the nuclear envelope has a density of nuclear pores greater than in other cell types [21]. In the soma and at the beginning of dendrites, the ER forms sub-membrane tanks when close to the plasma membrane, whereas in axons, the RE is organized into interconnected tubules that run parallel to the axon axe. This ER axonal tubular network extends into the axon terminal, where it is closely linked to mitochondria. They form an interconnected dynamic network that collaborates with the genesis of the calcium signal, the RE extends into the dendrites down to the spines where it ends as “spine apparatus” [22]. This “spine apparatus” plays an important role in calcium regulation and in the delivery of components of the plasma membrane, as well as those of the axon terminal [23].
All situations leading to the impairment of ER functions prevent the correct protein folding. Unfolded proteins cannot move to the Golgi apparatus, then accumulate within the ER lumen and activate an adaptive programme named UPR (unfolded protein response). This adaptive cell response aims to reduce the amount of unfolded protein and to restore the ER homeostasis [24]. Many diseases are associated with UPR dysregulation or over activation: Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and also stroke [2, 3]. Modulation of the UPR activation appears then as an interesting and innovative therapeutic target for neuroprotection.
2 The ER Stress Response/The UPR
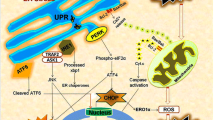
Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and ischaemia are known to activate ER stress and the UPR (Fig. 13.1). The UPR activates a set of cytoplasmic and nuclear signalling pathways characterized by three steps [25]: (1) a translational response to induce a quick reduction of the general protein synthesis by decreasing mRNA translation; (2) a transcriptional response to induce the expression of ER genes, like chaperone proteins, to increase the ER-folding capacity and (3) activation of the ERAD system to degrade unfolded proteins.

In healthy conditions (upper panel), eIF2β binds with eif2α to form the eIF2 complex to initiate protein translation. Newly synthesized proteins are translocated into the ER lumen and fold with the help of chaperones like Grp78 . Once correctly folded they traffic along the secretory pathway via the Golgi apparatus. ER stress sensors (PERK, ATF6 and IRE1) are inactivated by their association to Grp78. Under ER stress conditions (bottom panel), misfolded proteins accumulate within the ER lumen and sequester Grp78 from the three sensors. PERK phosphorylates eIF2α allowing the inhibition of translation but paradoxically it will induce the transcription of ATF4, CHOP and GADD34. CHOP activates pro-apoptotic genes such as Bim and caspase 12 to trigger apoptosis. CHOP also activates Ero1, leading to oxidative stress. GADD34 binds to PP1c to form an active eIF2α phosphatase, allowing a negative feedback of the PERK pathway to restore protein translation. IRE1 also contributes to the inhibition of translation by degraded mRNA associate with the ER. IRE1 splices one intron into the XBP1 mRNA sequence. XBP1s is an active transcription factor which activates the transcription of EDEM and IRE1. ATF6 migrates into the Golgi apparatus where it is cleaved by protease SP1 and SP2. ATF6 cytosolic portion acts as a transcription factors and activates the expression of XBP1 and IRE1
To control the accumulation of proteins in the ER, there are three anchored sensors at its membrane: PERK (protein kinase RNA-like endoplasmic reticulum kinase), IRE1 (Inositol-Requiring protein 1) and ATF6 (activating transcription factor 6) [25]. Under physiological conditions, Grp78 binds to the intraluminal domain of the three sensors and keeps them inactive. Grp78 regulates the signal by an exchange system of the balance between misfolded proteins and the sensors [26]. Following ER stress, the amount of misfolded proteins increases and the competitive binding of Grp78 to these proteins induces its dissociation from PERK, ATF6 and IRE1 allowing their respective activation [27].
ATF6, IRE1 and PERK control the regulation of the transcriptional and translational phases of the UPR [28]. The first phase is the attenuation of protein translation by PERK and aims to limit the addition of newly synthesized proteins within an already overloaded ER. During the second phase, IRE1 and ATF6 trigger a transcriptional activation of genes coding for chaperone and resident enzymes of the ER, as well as for components of the exporting and protein degradation pathways. The UPR not only helps cells to respond immediately to the presence of misfolded proteins but also prepare them to support further ER stress by changing its transcriptional programme in the long term [29]. If cells cannot restore a correct folding capacity, ER stress will eventually activate death signals [25, 30] (Fig. 13.1).
2.1 Origin of ER Stress Activation
Every situation that alters ER functions modifies the normal protein folding process. Unfolded proteins are no longer able to move towards the Golgi apparatus, then accumulate in the ER lumen and trigger the UPR. ER stress plays a particularly important physiological role in maintaining cellular homeostasis.
Activation of the UPR has many origins like glucose deficiencies, imbalance of redox status or hypoxia [25]. Protein folding within the ER is energetically costly, explaining the sensitivity of the ER to glucose deficient conditions. In addition, the absence of glucose alters post-translational modifications and more specifically glycosylation [31]. Furthermore, a decrease in extracellular glucose concentration results in the induction of ER stress genes through the PERK signalling pathway [32]. Hypoxia causes an alteration in protein conformations within the ER, especially due to the drop of ATP (Adenosine Tri-Phosphate) and leads to the activation of the UPR. Hypoxia affects various components of the protein folding machinery and induces an alteration of disulphide bridges formation [33], an increased expression of chaperones [34] and the inhibition of protein synthesis [35, 36].
The ER transiently produces ROS (reactive oxygen species) during disulphide bridges formation, which makes the ER sensitive to oxidative stress. Oxidative stress leads to protein aggregation that may be due to uncontrolled disulphide bridges formation or to the inactivation of proteins involved in ERAD [37]. These oxidative stresses induce UPR mainly by the activation of PERK. The PERK signalling pathway also engages the transcription factor Nrf2 to counter oxidative stress [38, 39].
2.2 PERK Pathway
2.2.1 Translational Response Mediated by PERK
The first event occurring after the activation of the UPR is the oligomerization and trans-autophosphorylation of PERK (by the detachment of Grp78), followed by the phosphorylation on the serine 51 of the α-subunit of eukaryotic initiation factor 2 (eIF2α), leading to the shutdown of translation [25, 40]. In eukaryotes, eIF2α is associated with eIF2β and eIF2γ to form the eIF2 complex, an essential element for the formation of the translation initiation complex [41]. In PERK-deficient cells, phosphorylation of eIF2α and inhibition of protein synthesis are indeed totally missing during ER stress [32] (Fig. 13.1).
2.2.2 Transcriptional Response Mediated by PERK
The global attenuation of protein synthesis paradoxically induces mRNA translation of UPR elements, including the activating transcription factor 4 (ATF4) [28]. Some studies show the presence of upstream open reading frames (uORFs) in the mRNA of eIF2α-phosphorylated target genes, like ATF4, CHOP (CAAT/enhancer binding protein homologous transcription factor; also known as GADD153 (growth arrest and DNA damage-inducible protein 153)) and GADD34 (growth arrest and DNA damage-inducible protein 34). These targets are more efficiently translated when eIF2α is phosphorylated. Indeed, under low phosphorylation of eIF2α, the presence of uORFs slows the reading of mRNA by the ribosome and is either poorly or not translated. In conditions of high phosphorylation of eIF2α, ribosomes are preferentially recruited at the uORFs and mRNAs containing them are more efficiently translated [42].
CHOP is an important protein activated by the PERK pathway and is crucial for the cell fate. Indeed, in high stress conditions (intensity and/or duration) CHOP activates, among others, the transcription of pro-apoptotic genes such as Bim and caspase-12 to trigger cell death and thus to eliminate damaged cells [43]. CHOP also activates the transcription of Ero1 (ER oxidoreductin I), which catalyses the exchange between proteins and the PDI to facilitate the formation of disulphide bridges [44]: taken together, CHOP will dramatically increase the oxidative stress (Fig. 13.1).
A feedback of the PERK pathway exists. ATF4, alone or in combination with CHOP, activates the transcription of GADD34, a binding factor of the catalytic subunit of the protein phosphatase 1 (PP1c) to form an active eIF2α phosphatase, allowing then a negative feedback of the PERK pathway [44, 45]. But, persistent or prolonged expression of GADD34 increases protein translation and potentially exacerbates protein misfolding. Thus, mammalian cells have developed complex mechanisms, including the GADD34 trafficking from the cytosol to the ER, and the poly-ubiquitination of the α-amino group at the N-terminus of GADD34, to actively degrade proteins via the proteasome [46, 47].
The CReP protein (Constitutive Repressor of eIF2α phosphorylation) can also bind to the PP1c to form an active phosphatase of eIF2α and induces its dephosphorylation. Whereas GADD34 expression is triggered by CHOP, CReP is constitutively expressed. Of note, the dephosphorylation of eIF2α by the CReP-PP1c complex is slower than the one induced by the GADD34-PP1c complex [48].
2.3 ATF6 Pathway
Under physiological conditions, Grp78 is associated with ATF6 at its GLS (Golgi localization signal) sequences and is sequestrated within the ER. ATF6 is also restraint in the ER by interaction between its glycosylated residues and calreticulin [49]. Under stress conditions, ATF6 is released from Grp78 and migrates to the Golgi apparatus where it is cleaved by proteases S1P (Site-1 protease) and S2P (Site-1 protease) [50]. S1P cleaves ATF6 in its luminal portion releasing its C-terminal part and leaving its N-terminal portion anchored in the membrane. S2P cleaves ATF6 in its transmembrane portion thus releasing the N-terminal cytoplasmic part. The cleaved cytosolic fragment acts as a transcription factor and also activates the expression of ER stress genes, such as Grp78 and other chaperones [25, 50]. The multiple steps involved in ATF6 trafficking, followed by the proteolytic processing and its entry into the nucleus, make the ATF6 pathway slower than the PERK pathway. Additionally, the half-life of Grp78 is longer than that of ATF4, CHOP or GADD34. Activation of the ATF6 pathway and the delayed increase of Grp78 expression allow cells to anticipate the arrival of other misfolded proteins, ensuring their survival [51] (Fig. 13.1).
Regarding feedbacks of the pathway, there are two isoforms of ATF6 : ATF6α and ATF6β. Both are induced by ER stress, but ATF6α is the predominant isoform in many cells. While studies in mice suggest that ATF6α and ATF6β have redundant functions [52], others show that the expression of ATF6β is slightly delayed compared to that of ATF6α and that the β isoform can reduce the α isoform’s functions [53]: the β isoform would be at the origin of a negative feedback.
2.4 IRE1 Pathway
Following ER stress, Grp78 dissociates from IRE1 allowing its homodimerization and activation by autophosphorylation [54]. IRE1 is an enzyme with both kinase and endonuclease activities. Its main function is the splicing of 26 nucleotides in the intron of the transcription factor XBP1 (X-box binding protein 1) mRNA, thereby generating a shift in the reading frame, and producing an active transcription factor [55, 56]. In mammals, this splicing seems to occur in the nucleus and IRE1 localizes at the inner nuclear membrane [57]. The spliced XBP1 mRNA (XBP1-s) encodes a transcription factor for UPR response genes. There is no difference in the regulation of translation of the XBP1-s mRNA and XBP1-u (unspliced form), but a competition between XBP1-s and XBP1-u for their dimerization with cofactors. Moreover, XBP1-u can inhibit the transcription induced by XBP1-s. Rapid degradation of XBP1-u by the proteasome is, however, observed via the activation of IRE1/XBP1-s pathway [58]. The XBP1 promoter also contains an unfolded protein response element (UPRE) sequence, suggesting a positive feedback loop [55]. Indeed, XBP1 can activate his own transcription, which keeps the IRE1 signalling pathway activated after the repression of the ATF6 and PERK pathways (Fig. 13.1).
IRE1 also degrades mRNA associated to the ER by a mechanism called IRE1-dependent decay of mRNA (RIDD) and can also contribute to the reduction of translation [59, 60].
2.5 UPR: Friend or Foe?
The origin of ER stress encountered by a cell dictates the nature of the UPR. Under physiological conditions, a secretory cell will experience considerable variations in the flow of newly synthesized proteins that will cross the ER according to its needs. When protein synthesis goes from a low flow to a high flow, the cell needs to increase its ability to fold proteins in the ER to avoid the overloading. In this case, the UPR regulates transcription factors which target genes of the ER machinery proteins. But this response includes a time delay, since new chaperones cannot be available instantly. To avoid further accumulation of misfolded proteins, the UPR must adjust downward the transcription of secreted proteins. The modulation of both transcription and translation by the UPR provides a coordinated response adapted to the cell situation. However, if the ER stress is too high and/or too long then this beneficial phenomenon becomes deleterious by inducing different cell death pathways [29].
3 ER Stress and Stroke
ER stress is a conserved mechanism which is thought to be beneficial if cells restore a correct cellular homeostasis; otherwise its overactivation becomes harmful and induces a cell death response. ER stress is activated in many neurodegenerative diseases [2] including stroke, for which restoring the blood flow is mandatory for the recovery of cellular homeostasis.
During cerebral ischaemia , the energetic depletion impairs calcium homeostasis within the ER. In neurones, the ER is the main calcium pool, and disturbance of calcium homeostasis is known to activate ER stress [61, 62]. Moreover, the energetic failure stops SERCA pumps. Therefore, the calcium initially stored in the ER translocates to the cytosol and contributes to the uncontrolled increase of its concentration causing chaperones dysfunction, and finally ER stress [63].
Activation of the UPR has been revealed in both in vitro and in vivo models of stroke [62, 64–67]. In these studies, protein synthesis is rapidly inhibited and accompanied by the phosphorylation of eIF2α [68, 69]. In parallel to the inhibition of protein translation, the UPR induces an increased expression of ER-response genes [25]. Many studies have clearly demonstrated the activation of ER stress in in vitro and in vivo models of stroke. ER stress markers such as PERK, IRE1, GADD34, ATF4, CHOP and Grp78 are shown to be clearly upregulated in various models, including in cultures of cortical neurones subjected to oxygen deprivation [70], cultures of rat astrocytes subjected to oxygen and glucose deprivation (OGD) [71], and primary cultures of mixed rat brain cortical cells under OGD [72]. ER-stress related genes and proteins are also increased in rodent models of middle cerebral artery occlusion [65, 67] and in transient models of common carotid arteries bilateral occlusion [62, 64, 73]. However, some discrepancies remain about the role of ER stress in stroke : some studies demonstrate a deleterious effect of the induction of ER stress after stroke [64, 71, 74], whereas few others show a beneficial effect [65, 70]. The consequences of ER stress likely depend on its level of activation. In the first phase of stroke, ER stress is moderate and promotes cell survival by decreasing general translation and promoting the degradation of misfolded proteins. But if ER stress is prolonged, it becomes deleterious by inducing cytoplasmic calcium overload, increasing toxic products and activating cell death pathways [75, 76]. All these studies use different models of stroke and the subsequent ER stress activation is then different between them , depending on the severity of the model.
4 ER Stress and Other Ischaemic Diseases
4.1 Cardiac Ischaemia
More recently, several studies have demonstrated the activation of ER stress in the myocardium after ischaemia or ischaemia/reperfusion (I/R). A microarray study shows that numerous ER stress response genes are induced 24 h after myocardial infarction in ischaemic mouse hearts in vivo [77]. ER stress markers are also increased in both mouse hearts subjected to I/R ex vivo, and in surviving cardiac myocytes bordering the infarct zone in a mouse model of myocardial infarction [78]. XBP1 splicing is also detected in cultured neonatal rat ventricular myocytes subjected to ischaemia [78, 79]. Dominant-negative XBP1 increases apoptosis in isolated cardiomyocytes in response to I/R, suggesting a cardioprotective effect of ER stress in cardiac ischaemia [77]. Moreover, the overexpression of activated ATF6 in transgenic mouse hearts decreases ischaemic damages and increases ventricular pump functions in an ex vivo I/R model [80].
Even if ER stress can be activated by cardiac ischaemia and I/R, it is not clear whether ER stress is protective or deleterious in this context [81]. Perhaps mild or brief episodes of ischaemia would favour the activation of pro-survival aspects of ER stress, whereas severe or long episodes would lead to an activation of pro-apoptotic factors. Further studies are required to delineate the circumstances under which ER stress is protective or deleterious in the myocardium and what ER stress-inducible genes and pathways are important contributors to these outcomes in the heart .
4.2 Renal Ischaemia
To date, only few studies have demonstrated the implication of ER stress in renal ischaemia . CHOP expression is increased after renal ischaemia and leads to apoptotic cell death [82], confirmed by another study showing an overexpression of both Grp78 and CHOP after I/R and confirming the link between CHOP and apoptotic cell death [83]. Moreover, these two studies demonstrate that deletion of CHOP prevents cell death in renal ischaemia. It is also suggested that ischaemic preconditioning attenuates oxidative and ER stresses leading to kidney protection [84]. All these studies demonstrate a deleterious effect of ER stress activation following renal ischaemia, consistent with studies showing a protective effect of ER stress inhibition [85–87].
5 ER Stress and Neuroprotection in Ischaemic Diseases
5.1 Through UPR Pathways
Modulation of ER stress holds a promise of therapeutic benefits in ischaemic conditions (Table 13.1). Stroke highly activates the PERK-eIF2α pathway [62, 88], leading to the deleterious activation of CHOP [64, 71]. Indeed, in an in vivo model of stroke, PERK deficient mice do not present eIF2α phosphorylation or protein aggregation [62]. Moreover, PERK activation participates to the loss of motor neurones of the upper airways in a model of obstructive apnoea sleep inducing recurrent cerebral hypoxia in mice [89]. Prevention of the activation of this pathway by salubrinal, an inhibitor of the eIF2α phosphatase GADD34 [90], decreases ER stress and kainate-induced neurotoxicity in vitro [68] and reduces infarct size in an in vivo stroke model [69].
Many studies consider CHOP as a pro-apoptotic factor. One of the first studies was performed by Tajiri and colleagues in 2004 who showed that hippocampal neurones from CHOP deficient mice were more resistant to hypoxia-reoxygenation than those from wild-type littermates [64], and this effect is also confirmed in an in vivo model of stroke [69, 74]. CHOP is also increased in astrocytes under OGD, leading to cell death [71]. So targeting CHOP by an inhibitor of p38MAPK which reduces its activity holds a possible strategy to inhibit UPR-induced cell death .
Other drugs have also demonstrated promising results in experimental models. Dantrolene, a ryanodine receptor antagonist used as muscle relaxant, significantly decreases the infarct volume and provides a neuroprotective effect in a rat stroke model, by reducing ER stress-mediated apoptosis in the ischaemic area [91]. Berberine, an alkaloid, also attenuates ischaemia/reperfusion (I/R) injury in kidney by inhibiting oxidative and ER stresses [85]. The flavonoid glycoside Baicalin, isolated from Scutellaria baicalensis, also protects kidney from I/R injury by reducing oxidative and ER stresses through the activation of the anti-oxidative Nrf2 signalling pathway [86]. Intermedin, a member of the calcitonin/calcitonin gene-related peptide family, down-regulates Grp78, CHOP and caspase 12 protein and prevents apoptosis after renal ischaemia [87] and the non-competitive inhibitor of nicotinic acetylcholine receptor Catestatin decreases ER stress markers after cardiac ischaemia [92]. Regarding cardiac ischaemia, elatoside C inhibits ER stress-associated markers (Grp78, CHOP, Caspase-12 and JNK) and provides a significant protection against ischaemia-induced cardiomyocytes death [93, 94].
5.2 Through Chaperones
Chaperone proteins also appear as good targets to counteract the deleterious events associated to ER stress, since they are implicated in both ER stress inhibition (by the binding of Grp78 to the ER stress sensors) and in protein folding (Table 13.1). For example, Grp78 expression increases up to 2 weeks after I/R with a maximum between 24 and 72 h [66, 95, 96] and protects neurones from death [67, 97]. Moreover, overexpression of Grp78 in astrocytes has a protective effect after OGD [72] and by opposition, deletion of Grp78 increases apoptotic mechanism in in vitro models of excitotoxicity or oxidative stress in primary culture of hippocampal neurones [98]. Grp170, another chaperon of the Hsp70 family, might have an anti-apoptotic role in cerebral [99, 100], renal [101], and cardiac [102] ischaemic injuries .
Developing chaperone-inducer compounds is also a strategy to protect cells from ischaemia. The injection of BIX (BiP protein Inducer X) before stroke induces Grp78 and reduces brain damages and the numbers of apoptotic neurones in the penumbra [97]. Sodium 4-phenylbutyrate (PBA), a chemical chaperone is also protective by decreasing the load of unfolded proteins within the ER during cellular stress, and preserves cells against ischaemic spinal cord damages [103].
5.3 Through Other Cell Death Pathways
As stated before, UPR activation often leads to cell death by activating several pathways , mainly initiated by CHOP and linked to apoptosis by the PI3K-Akt pathway [74]. The inhibition of CHOP effectors, like BIMEl (Bcl-2 interacting mediator of cell death), a pro-apoptotic BCl-2 family member, also leads to cellular protection and is a therapeutic target [43] (Table 13.1).
Oxidative stress is also induced after stroke, and as described previously, leads to protein aggregation due to uncontrolled disulphide bond formation or inactivation of proteins involved in ERAD [37]. Crosstalk between ER stress and oxidative stress potentiates each other [104] and so, activates endogenous anti-oxidant pathways: this represents another strategy for protecting cells after ischaemia. Compounds inducing the endogenous Nrf2-KEAP1 (Kelch-like ECH-associated protein 1) anti-oxidant pathway show neuroprotection after stroke [105, 106]. Moreover, others anti-oxidant drugs like edaravone and apocynin have shown promising results on the inhibition of ER stress in stroke models [107, 108].
6 Conclusion
The growing interest into the ER stress field opens new horizons for original therapeutic approaches. It is now clear that ER stress leads to UPR activation in cerebral, renal or cardiac ischaemia. Strategies to counteract the deleterious overactivation of ER stress have been developed, in particular in stroke. CHOP overexpression activates death pathways, but CHOP inhibitors are not yet fully developed or specific. For the moment, the use of chemical chaperones or chaperones inducer like BIX is a promising way for stroke: as chaperone induction is at the “end” of the UPR pathway, inducing their expression earlier during the acute disease seems to be protective. Other agents have demonstrated good results, but their mechanism of action is not fully understood or even not described at all: these agents decrease many ER markers without specificity. More work is necessary to decipher ER stress mechanisms and actions to propose specific inhibitors/strategies to counteract its deleterious actions in ischaemia, without affecting its beneficial effects.
Abbreviations
- ATF:
-
Activating transcription factor
- ATP:
-
Adenosine triphosphate
- Bip:
-
Binding immunoglobulin protein
- CHOP:
-
CAAT/enhancer binding protein homologous transcription factor
- CReP:
-
Constitutive repressor of eIF2α phosphorylation
- EDEM:
-
Endoplasmic reticulum degradation enhancing alpha mannosidase-like protein
- eIF2:
-
Eukaryotic initiation factor 2
- ER:
-
Endoplasmic reticulum
- ERAD:
-
Endoplasmic reticulum associated degradation
- Ero1:
-
ER oxidoreductin I
- GADD34:
-
Growth arrest and DNA damage-inducible protein 34
- GLS:
-
Golgi localization signal
- Grp:
-
Glucose related protein
- Hsp:
-
Heat shock protein
- I/R:
-
Ischaemia/reperfusion
- IP3R:
-
Inositol-1,4,5-trisphosphate receptor
- IRE1:
-
Inositol-requiring protein 1
- KEAP1:
-
Kelch-like ECH-associated protein 1
- NE:
-
Nuclear envelope
- OGD:
-
Oxygen and glucose deprivation
- PERK:
-
Protein kinase RNA-like endoplasmic reticulum kinase
- PP1:
-
Protein phosphatase 1
- RER:
-
Rough endoplasmic reticulum
- RIDD:
-
IRE1-dependent decay of mRNA
- ROS:
-
Reactive oxygen species
- RyR:
-
Ryanodine receptor
- S1P:
-
Site-1 protease
- S2P:
-
Site-2 protease
- SER:
-
Smooth endoplasmic reticulum
- SERCA:
-
Sarco endoplasmic reticulum Ca2+-transport ATPase
- tPA:
-
Tissue-type plasminogen activator
- UPR:
-
Unfolded protein response
- UPRE:
-
Unfolded protein response element
- XBP1:
-
X-box binding protein 1
References
Voeltz GK, Rolls MM, Rapoport TA (2002) Structural organization of the endoplasmic reticulum. EMBO Rep 3(10):944–950
Roussel BD, Kruppa AJ, Miranda E, Crowther DC, Lomas DA, Marciniak SJ (2013) Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol 12(1):105–118
Hetz C, Mollereau B (2014) Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci 15(4):233–249
Estrada de Martin P, Novick P, Ferro-Novick S (2005) The organization, structure, and inheritance of the ER in higher and lower eukaryotes. Biochem Cell Biol 83(6):752–761
Vertel BM, Walters LM, Mills D (1992) Subcompartments of the endoplasmic reticulum. Semin Cell Biol 3(5):325–341
Dudek J, Benedix J, Cappel S, Greiner M, Jalal C, Muller L et al (2009) Functions and pathologies of BiP and its interaction partners. Cell Mol Life Sci 66(9):1556–1569
Melnick J, Dul JL, Argon Y (1994) Sequential interaction of the chaperones BiP and GRP94 with immunoglobulin chains in the endoplasmic reticulum. Nature 370(6488):373–375
Brodsky JL, Werner ED, Dubas ME, Goeckeler JL, Kruse KB, McCracken AA (1999) The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct. J Biol Chem 274(6):3453–3460
Yang Y, Li Z (2005) Roles of heat shock protein gp96 in the ER quality control: redundant or unique function? Mol Cells 20(2):173–182
Randow F, Seed B (2001) Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nat Cell Biol 3(10):891–896
Ellgaard L, Frickel EM (2003) Calnexin, calreticulin, and ERp57: teammates in glycoprotein folding. Cell Biochem Biophys 39(3):223–247
Michalak M, Groenendyk J, Szabo E, Gold LI, Opas M (2009) Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem J 417(3):651–666
Helenius A, Aebi M (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem 73:1019–1049
Molinari M, Calanca V, Galli C, Lucca P, Paganetti P (2003) Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 299(5611):1397–1400
Oda Y, Hosokawa N, Wada I, Nagata K (2003) EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 299(5611):1394–1397
Orrenius S, Zhivotovsky B, Nicotera P (2003) Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 4(7):552–565
Mikoshiba K (2007) IP3 receptor/Ca2+ channel: from discovery to new signaling concepts. J Neurochem 102(5):1426–1446
Amador FJ, Stathopulos PB, Enomoto M, Ikura M (2013) Ryanodine receptor calcium release channels: lessons from structure-function studies. FEBS J 280(21):5456–5470
Brostrom MA, Brostrom CO (2003) Calcium dynamics and endoplasmic reticular function in the regulation of protein synthesis: implications for cell growth and adaptability. Cell Calcium 34(4-5):345–363
Berridge MJ (2002) The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32(5-6):235–249
Talamas JA, Capelson M (2015) Nuclear envelope and genome interactions in cell fate. Front Genet 6:95
Spacek J, Harris KM (1997) Three-dimensional organization of smooth endoplasmic reticulum in hippocampal CA1 dendrites and dendritic spines of the immature and mature rat. J Neurosci 17(1):190–203
Broadwell RD, Cataldo AM (1984) The neuronal endoplasmic reticulum: its cytochemistry and contribution to the endomembrane system. II. Axons and terminals. J Comp Neurol 230(2):231–248
Mollereau B, Rzechorzek NM, Roussel BD, Sedru M, Van den Brink DM, Bailly-Maitre B, et al. (2016) Adaptive preconditioning in neurological diseases - therapeutic insights from proteostatic perturbations. Brain Res. pii: S0006-8993(16)30092-0. doi:10.1016/j.brainres
Marciniak SJ, Ron D (2006) Endoplasmic reticulum stress signaling in disease. Physiol Rev 86(4):1133–1149
Schroder M, Kaufman RJ (2005) The mammalian unfolded protein response. Annu Rev Biochem 74:739–789
Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2(6):326–332
Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D (2000) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 5(5):897–904
Mollereau B, Manie S, Napoletano F (2014) Getting the better of ER stress. J Cell Commun Signal 8(4):311–321
Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8(7):519–529
Helenius A (1994) How N-linked oligosaccharides affect glycoprotein folding in the endoplasmic reticulum. Mol Biol Cell 5(3):253–265
Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P et al (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7(6):1165–1176
Gess B, Hofbauer KH, Wenger RH, Lohaus C, Meyer HE, Kurtz A (2003) The cellular oxygen tension regulates expression of the endoplasmic oxidoreductase ERO1-Lalpha. Eur J Biochem 270(10):2228–2235
Paris S, Denis H, Delaive E, Dieu M, Dumont V, Ninane N et al (2005) Up-regulation of 94-kDa glucose-regulated protein by hypoxia-inducible factor-1 in human endothelial cells in response to hypoxia. FEBS Lett 579(1):105–114
Blais JD, Filipenko V, Bi M, Harding HP, Ron D, Koumenis C et al (2004) Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol 24(17):7469–7482
Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N et al (2002) Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol 22(21):7405–7416
Stadtman ER, Levine RL (2003) Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 25(3-4):207–218
Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M et al (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11(3):619–633
Cullinan SB, Diehl JA (2004) PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem 279(19):20108–20117
Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397(6716):271–274
Roy AL, Chakrabarti D, Gupta NK (1987) Protein synthesis in rabbit reticulocytes: Mg2+-inhibition of ternary complex (Met-tRNA(f).eIF-2.GTP) formation by reticulocyte eIF-2. Biochem Biophys Res Commun 146(1):114–120
Vattem KM, Wek RC (2004) Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A 101(31):11269–11274
Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND et al (2007) ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129(7):1337–1349
Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R et al (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 18(24):3066–3077
Novoa I, Zeng H, Harding HP, Ron D (2001) Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol 153(5):1011–1022
Brush MH, Shenolikar S (2008) Control of cellular GADD34 levels by the 26S proteasome. Mol Cell Biol 28(23):6989–7000
Zhou W, Brush MH, Choy MS, Shenolikar S (2011) Association with endoplasmic reticulum promotes proteasomal degradation of GADD34 protein. J Biol Chem 286(24):21687–21696
Dalton LE, Healey E, Irving J, Marciniak SJ (2012) Phosphoproteins in stress-induced disease. Prog Mol Biol Transl Sci 106:189–221
Hong M, Li M, Mao C, Lee AS (2004) Endoplasmic reticulum stress triggers an acute proteasome-dependent degradation of ATF6. J Cell Biochem 92(4):723–732
Shen J, Chen X, Hendershot L, Prywes R (2002) ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell 3(1):99–111
Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM et al (2006) Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol 4(11), e374
Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H et al (2007) Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell 13(3):365–376
Thuerauf DJ, Morrison L, Glembotski CC (2004) Opposing roles for ATF6alpha and ATF6beta in endoplasmic reticulum stress response gene induction. J Biol Chem 279(20):21078–21084
Okamura K, Kimata Y, Higashio H, Tsuru A, Kohno K (2000) Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem Biophys Res Commun 279(2):445–450
Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107(7):881–891
Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP et al (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415(6867):92–96
Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T et al (2002) IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev 16(4):452–466
Lee AH, Iwakoshi NN, Glimcher LH (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23(21):7448–7459
Hollien J, Weissman JS (2006) Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313(5783):104–107
Maurel M, Chevet E, Tavernier J, Gerlo S (2014) Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem Sci 39(5):245–254
Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Chan PH (2005) Damage to the endoplasmic reticulum and activation of apoptotic machinery by oxidative stress in ischemic neurons. J Cereb Blood Flow Metab 25(1):41–53
Owen CR, Kumar R, Zhang P, McGrath BC, Cavener DR, Krause GS (2005) PERK is responsible for the increased phosphorylation of eIF2alpha and the severe inhibition of protein synthesis after transient global brain ischemia. J Neurochem 94(5):1235–1242
Szydlowska K, Tymianski M (2010) Calcium, ischemia and excitotoxicity. Cell Calcium 47(2):122–129
Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI et al (2004) Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ 11(4):403–415
McCaig D, Imai H, Gallagher L, Graham DI, Harland J, Moira Brown S et al (2005) Evolution of GADD34 expression after focal cerebral ischaemia. Brain Res 1034(1-2):51–61
Rissanen A, Sivenius J, Jolkkonen J (2006) Prolonged bihemispheric alterations in unfolded protein response related gene expression after experimental stroke. Brain Res 1087(1):60–66
Morimoto N, Oida Y, Shimazawa M, Miura M, Kudo T, Imaizumi K et al (2007) Involvement of endoplasmic reticulum stress after middle cerebral artery occlusion in mice. Neuroscience 147(4):957–967
Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L et al (2007) Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J Neurosci 27(4):901–908
Nakka VP, Gusain A, Raghubir R (2010) Endoplasmic reticulum stress plays critical role in brain damage after cerebral ischemia/reperfusion in rats. Neurotox Res 17(2):189–202
Halterman MW, Gill M, DeJesus C, Ogihara M, Schor NF, Federoff HJ (2010) The endoplasmic reticulum stress response factor CHOP-10 protects against hypoxia-induced neuronal death. J Biol Chem 285(28):21329–21340
Benavides A, Pastor D, Santos P, Tranque P, Calvo S (2005) CHOP plays a pivotal role in the astrocyte death induced by oxygen and glucose deprivation. Glia 52(4):261–275
Badiola N, Penas C, Minano-Molina A, Barneda-Zahonero B, Fado R, Sanchez-Opazo G et al (2011) Induction of ER stress in response to oxygen-glucose deprivation of cortical cultures involves the activation of the PERK and IRE-1 pathways and of caspase-12. Cell Death Dis 2, e149
Doutheil J, Althausen S, Gissel C, Paschen W (1999) Activation of MYD116 (gadd34) expression following transient forebrain ischemia of rat: implications for a role of disturbances of endoplasmic reticulum calcium homeostasis. Brain Res Mol Brain Res 63(2):225–232
Yuan Y, Guo Q, Ye Z, Pingping X, Wang N, Song Z (2011) Ischemic postconditioning protects brain from ischemia/reperfusion injury by attenuating endoplasmic reticulum stress-induced apoptosis through PI3K-Akt pathway. Brain Res 1367:85–93
Su Y, Li F (2015) Endoplasmic reticulum stress in brain ischemia. Int J Neurosci 126(8):681–91
DeGracia DJ, Montie HL (2004) Cerebral ischemia and the unfolded protein response. J Neurochem 91(1):1–8
Harpster MH, Bandyopadhyay S, Thomas DP, Ivanov PS, Keele JA, Pineguina N et al (2006) Earliest changes in the left ventricular transcriptome postmyocardial infarction. Mamm Genome 17(7):701–715
Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC (2006) Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res 99(3):275–282
Szegezdi E, Duffy A, O’Mahoney ME, Logue SE, Mylotte LA, O’Brien T et al (2006) ER stress contributes to ischemia-induced cardiomyocyte apoptosis. Biochem Biophys Res Commun 349(4):1406–1411
Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA et al (2006) Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res 98(9):1186–1193
Glembotski CC (2007) Endoplasmic reticulum stress in the heart. Circ Res 101(10):975–984
Dong B, Zhou H, Han C, Yao J, Xu L, Zhang M et al (2014) Ischemia/reperfusion-induced CHOP expression promotes apoptosis and impairs renal function recovery: the role of acidosis and GPR4. PLoS One 9(10), e110944
Noh MR, Kim JI, Han SJ, Lee TJ, Park KM (2015) C/EBP homologous protein (CHOP) gene deficiency attenuates renal ischemia/reperfusion injury in mice. Biochim Biophys Acta 1852(9):1895–1901
Mahfoudh-Boussaid A, Zaouali MA, Hadj-Ayed K, Miled AH, Saidane-Mosbahi D, Rosello-Catafau J et al (2012) Ischemic preconditioning reduces endoplasmic reticulum stress and upregulates hypoxia inducible factor-1alpha in ischemic kidney: the role of nitric oxide. J Biomed Sci 19:7
Yu W, Sheng M, Xu R, Yu J, Cui K, Tong J et al (2013) Berberine protects human renal proximal tubular cells from hypoxia/reoxygenation injury via inhibiting endoplasmic reticulum and mitochondrial stress pathways. J Transl Med 11:24
Lin M, Li L, Zhang Y, Zheng L, Xu M, Rong R et al (2014) Baicalin ameliorates H2O2 induced cytotoxicity in HK-2 cells through the inhibition of ER stress and the activation of Nrf2 signaling. Int J Mol Sci 15(7):12507–12522
Wang Y, Tian J, Qiao X, Su X, Mi Y, Zhang R et al (2015) Intermedin protects against renal ischemia-reperfusion injury by inhibiting endoplasmic reticulum stress. BMC Nephrol 16:169
Zhu Y, Fenik P, Zhan G, Sanfillipo-Cohn B, Naidoo N, Veasey SC (2008) Eif-2a protects brainstem motoneurons in a murine model of sleep apnea. J Neurosci 28(9):2168–2178
Zhu H, Fan X, Yu Z, Liu J, Murata Y, Lu J et al (2010) Annexin A2 combined with low-dose tPA improves thrombolytic therapy in a rat model of focal embolic stroke. J Cereb Blood Flow Metab 30(6):1137–1146
Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D et al (2005) A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307(5711):935–939
Li F, Hayashi T, Jin G, Deguchi K, Nagotani S, Nagano I et al (2005) The protective effect of dantrolene on ischemic neuronal cell death is associated with reduced expression of endoplasmic reticulum stress markers. Brain Res 1048(1-2):59–68
Liao F, Zheng Y, Cai J, Fan J, Wang J, Yang J et al (2015) Catestatin attenuates endoplasmic reticulum induced cell apoptosis by activation type 2 muscarinic acetylcholine receptor in cardiac ischemia/reperfusion. Sci Rep 5:16590
Wang M, Meng XB, Yu YL, Sun GB, Xu XD, Zhang XP et al (2014) Elatoside C protects against hypoxia/reoxygenation-induced apoptosis in H9c2 cardiomyocytes through the reduction of endoplasmic reticulum stress partially depending on STAT3 activation. Apoptosis 19(12):1727–1735
Wang M, Sun GB, Zhang JY, Luo Y, Yu YL, Xu XD et al (2015) Elatoside C protects the heart from ischaemia/reperfusion injury through the modulation of oxidative stress and intracellular Ca(2)(+) homeostasis. Int J Cardiol 185:167–176
Oida Y, Shimazawa M, Imaizumi K, Hara H (2008) Involvement of endoplasmic reticulum stress in the neuronal death induced by transient forebrain ischemia in gerbil. Neuroscience 151(1):111–119
Osada N, Kosuge Y, Ishige K, Ito Y (2010) Characterization of neuronal and astroglial responses to ER stress in the hippocampal CA1 area in mice following transient forebrain ischemia. Neurochem Int 57(1):1–7
Kudo T, Kanemoto S, Hara H, Morimoto N, Morihara T, Kimura R et al (2008) A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ 15(2):364–375
Yu Z, Luo H, Fu W, Mattson MP (1999) The endoplasmic reticulum stress-responsive protein GRP78 protects neurons against excitotoxicity and apoptosis: suppression of oxidative stress and stabilization of calcium homeostasis. Exp Neurol 155(2):302–314
Kuwabara K, Matsumoto M, Ikeda J, Hori O, Ogawa S, Maeda Y et al (1996) Purification and characterization of a novel stress protein, the 150-kDa oxygen-regulated protein (ORP150), from cultured rat astrocytes and its expression in ischemic mouse brain. J Biol Chem 271(9):5025–5032
Tamatani M, Matsuyama T, Yamaguchi A, Mitsuda N, Tsukamoto Y, Taniguchi M et al (2001) ORP150 protects against hypoxia/ischemia-induced neuronal death. Nat Med 7(3):317–323
Bando Y, Tsukamoto Y, Katayama T, Ozawa K, Kitao Y, Hori O et al (2004) ORP150/HSP12A protects renal tubular epithelium from ischemia-induced cell death. FASEB J 18(12):1401–1403
Aleshin AN, Sawa Y, Kitagawa-Sakakida S, Bando Y, Ono M, Memon IA et al (2005) 150-kDa oxygen-regulated protein attenuates myocardial ischemia-reperfusion injury in rat heart. J Mol Cell Cardiol 38(3):517–525
Mizukami T, Orihashi K, Herlambang B, Takahashi S, Hamaishi M, Okada K et al (2010) Sodium 4-phenylbutyrate protects against spinal cord ischemia by inhibition of endoplasmic reticulum stress. J Vasc Surg 52(6):1580–1586
Nakka VP, Prakash-Babu P, Vemuganti R (2016) Crosstalk Between Endoplasmic Reticulum Stress, Oxidative Stress, and Autophagy: Potential Therapeutic Targets for Acute CNS Injuries. Mol Neurobiol 53(1):532–544
Satoh T, Kosaka K, Itoh K, Kobayashi A, Yamamoto M, Shimojo Y et al (2008) Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J Neurochem 104(4):1116–1131
Shih AY, Li P, Murphy TH (2005) A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci 25(44):10321–10335
Qi X, Okuma Y, Hosoi T, Nomura Y (2004) Edaravone protects against hypoxia/ischemia-induced endoplasmic reticulum dysfunction. J Pharmacol Exp Ther 311(1):388–393
Chen H, Song YS, Chan PH (2009) Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J Cereb Blood Flow Metab 29(7):1262–1272
Lee MR, Dominguez C (2005) MAP kinase p38 inhibitors: clinical results and an intimate look at their interactions with p38alpha protein. Curr Med Chem 12(25):2979–2994
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Louessard, M., Lemarchand, E., Ali, C., Vivien, D., Roussel, B.D. (2017). Endoplasmic Reticulum Stress: An Opportunity for Neuroprotective Strategies After Stroke. In: Lapchak, P., Zhang, J. (eds) Neuroprotective Therapy for Stroke and Ischemic Disease. Springer Series in Translational Stroke Research. Springer, Cham. https://doi.org/10.1007/978-3-319-45345-3_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-45345-3_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-45344-6
Online ISBN: 978-3-319-45345-3
eBook Packages: MedicineMedicine (R0)