
Abstract
The endoplasmic reticulum(ER) stress plays a vital role in mediating ischemic neuronal cell death. However, very little is known about the role of ER stress in mediating pathophysiological reactions to acute brain injuries. An attempt was therefore made to assess the role of cerebral ischemia/reperfusion (I/R) induced ER stress and its modulation on outcome of ischemic insult. Focal cerebral ischemia was induced in rats by middle cerebral artery occlusion (MCAO) for 2 h followed by varying time points of reperfusion. The brain loci specific and time-dependent alterations were seen in the expression pattern of molecular markers, i.e., heat-shock protein 70 (HSP70) for cytoplasmic dysfunction, glucose-regulated protein 78 (GRP78), Caspase-12, C/EBP homologous protein/growth arrest and DNA damage-inducible gene 153 (CHOP/GADD153), activating transcription factor 4 (ATF-4), and Processed X-box protein 1 (xbp1) mRNA for ER dysfunction. Further, histological examinations indicated pronounced brain damage, massive neuronal loss, and DNA fragmentation predominantly in the striatum and cortex. The enhanced expression of GRP78, Caspase-12, CHOP/GADD153, ATF4 and processing of xbp1 mRNA in the affected brain regions clearly indicate the critical involvement of ER-mediated cell death/survival mechanisms and also collectively demonstrated the activation of unfolded protein response (UPR). Moreover, Salubrinal, a selective inhibitor of eIF2α dephosphorylation was used to counteract ER stress, which significantly increased the phosphorylation of eukaryotic translation initiation factor 2 subunit α (eIF2α), leading to reduced brain damage after I/R injury. Therefore, inhibition of ER stress following I/R injury may be used as key therapeutic target for neuroprotection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Stroke is referred as rapidly developing loss of brain functions in humans due to interruption in the vessels supplying blood to the brain. Estimates indicate that stroke is responsible for nearly half of all the patients hospitalized for acute neurological disorders (Dirnagl et al. 2003). It is the number two cause of death and leading cause of disability world-wide (Feigin 2005). According to the WHO estimation over 50 million healthy life-years across the globe will be lost by 2015 as a result of stroke, and 90% of this burden will be borne in low-income and middle-income countries (Strong et al. 2007). Although there are available drugs that can dissolve a clot, for acute stroke patients requiring thrombolysis, currently no strategies exist to prevent or limit ischemic damage resulting from stroke. Because, I/R injury triggers multiple and distinct but overlapping cell signaling pathways, which may lead to cell survival or cell damage (Nakka et al. 2008).
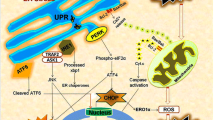
ER is an important subcellular organelle that is responsible for the proper folding and sorting of proteins. Cellular stress conditions, such as glucose deprivation, depletion of ER Ca2+ stores, exposure to free radicals, and accumulation of unfolded/misfolded proteins, disrupt the proper function of ER and initiates cell stress response, the unfolded protein response (UPR) to cope with this adverse situation (Paschen and Doutheil 1999; Ma and Hendershot 2001; DeGracia et al. 2002; Hayashi et al. 2005; Boyce and Yuan 2006). UPR is characterized by the activation of three ER transmembrane effector proteins: PKR-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1), and the activating transcription factor-6 (ATF-6) (DeGracia and Montie 2004). Activated PERK phosphorylates eukaryotic translation initiation factor 2 subunit α (eIf2α), which leads to inhibition of global protein synthesis (Prostko et al. 1993; Ron 2002). Moreover, phosphorylated eIF2α also leads to the paradoxical increased translation of ATF-4 and CHOP/GADD153 (Harding et al. 2000a; Lu et al. 2004a). Activated IRE1 on the other hand specifically cuts out a 26-nucleotide intron from the xbp1 mRNA, which leads to a shift of the open reading frame of xbp1 mRNA and the transfer RNA ligase-gated transcript that leads to enhanced translation of XBP-1 protein, a transcription factor for ER-resident enzymes and chaperones (Calfon et al. 2002). However, prolonged ER stress with the failure of cellular protective mechanisms by UPR eventually results in cell death. Accumulation of unfolded proteins in the ER lumen (Hu et al. 2000), phosphorylation of eIF2α (Althausen et al. 2001; Kumar et al. 2001), inhibition of protein synthesis (DeGracia et al. 2002), depletion of Ca2+ from ER stores (Paschen and Doutheil 1999), activation of CHOP/GADD153 expression (Paschen et al. 1998), and activation of caspase-12 (Mouw et al. 2003; Shibata et al. 2003), all these events suggest the crucial role of ER in neuronal cell death signaling after cerebral ischemia.
However, the precise molecular mechanisms by which ER stress leads to cell survival/death remains enigmatic, with multiple potential participants described above but without or very little clarity about which specific death effectors dominate particularly in the context of I/R injury. This has prompted us to study the impact of ER stress in a regional and time-dependent manner following I/R injury. Further, a specific ER stress inhibitor Sal would be used for neuroprotection against I/R injury. These findings may certainly help in understanding the basic events involved in ER stress-mediated cell survival/death signaling pathways during I/R injury.
Materials and Methods
Induction of Focal Cerebral Ischemia in Rats
Adult male Sprague–Dawley (SD) rats weighing 260 ± 20 g were procured from National Animal Laboratory Centre (NALC) of Central Drug Research Institute (CDRI), Lucknow, India and used throughout the study. Animal experiments were conducted after approval and in accordance with the strict guidelines of the Institutional Animal Ethics Committee (IAEC). Focal cerebral ischemia was induced in the rats by occlusion of the middle cerebral artery (MCA) using a modification of the intraluminal technique (Longa et al. 1989; Belayev et al. 1999). Briefly, the left MCA was occluded with a 3-0 nylon monofilament (Ethicon, Johnsons & Johnsons Ltd., Mumbai, India). Recirculation/reperfusion of cerebral blood flow was allowed by gently removing the monofilament carefully after 2 h ischemia followed by different time intervals of reperfusion. In sham-operated animals, all the procedure except for the insertion of the nylon filament was carried out.
Histological Analysis
Rats subjected to MCAO were transcardially perfused under deep anesthesia with chilled normal saline, and then with 4% paraformaldehyde prepared in PBS (pH—7.4) for internal fixation. Brains were removed carefully and then frozen immediately in embedding matrix. Cryostat sections of about 6–8 μ thickness were collected on poly-l-lysine coated slides for further processing. Briefly, for NeuN staining, cell permeabilization was done by using Triton X-100; sections were blocked in 1% BSA or serum for 1 h. After blocking, sections were incubated with the biotinylated NeuN antibody overnight at 4°C. After three or four times washing in 1× PBS, sections were incubated with the FITC conjugated avidin (Santa Cruz) for 2 h at 4°C to detect the biotinylated NeuN antibody binding. Sections were counterstained for nucleus with 4′,6′-diamino-2-phenylendolehydrochloride (DAPI) (Santa Cruz, USA) and then visualized under fluorescence microscope (Leica DM5000 B, Leica Microsystems). The signal for both FITC (green) and DAPI (blue) were superimposed by using IM50 software (Leica, Germany).
For detection of DNA fragmentation, terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) assay was performed according to the manufacturer’s protocol (DeadEnd™ TUNEL system, Promega) with slight modifications. Briefly, the brain sections were incorporated with biotinylated nucleotides at the 3′-OH DNA ends using the enzyme TdT. Further, sections were incubated with the FITC conjugated avidin and counterstained for nucleus with DAPI and then visualized under fluorescence microscope as mentioned above.
2,3,5-Triphenyltetrazolium chloride (TTC) staining was done on coronal brain sections of about 2 mm thickness in order to map out the cerebral infract of the affected brain tissue (Bederson et al. 1986). Briefly, the brains were rapidly dissected out and sectioned coronally at 2 mm intervals from the rostrum to caudal end after designated time points of I/R injury. All slices were incubated for 20 min in a 1% solution of TTC at 37°C. The sections were scanned and analyzed by using computerized image analysis system (Biovis Image Plus). The infarct area of all brain sections of each rat of a particular group were analyzed and multiplied by section thickness to obtain the infarct volume.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated from the cortex and striatum brain tissue subjected to I/R injury and sham using TRI reagent (Sigma) according to manufacturer’s protocol. Briefly, after quantification 2 μg RNA was reverse transcribed into complementary DNA (cDNA) using Omniscript-RT kit (Qiagen) and oligodT23 anchored primers (Sigma). PCR was performed using Taq PCR core kit (Qiagen) according to manufacturer’s protocol. PCR was performed in thermal cycler PTC-200 (M.J. Research, USA). Briefly, each PCR was performed using 0.6 μm primer concentration under conditions: denaturation (94°C for 1 min), annealing (47–59.6°C for 30 s), and extension (72°C for 1 min) for 30–35 cycles. The amplified RT-PCR products were separated by 1.2% agarose gel electrophoresis and visualized by ethidium bromide (0.5 μg/μl) staining under UV light. The relative band intensity was measured by using gel documentation system (Alphamager TM 2200) and the results were expressed as relative changes in mRNA levels following spot densitometry analysis. Primers used for gene expression studies were either designed using online free primer designing software or used the reported primer sequences. All primers were procured from Sigma-Genosys. For designing of the primers, nucleotide sequences of selected genes were downloaded from reference sequence (Ref Seq) and which served as input data files. Primer sequences were checked for their self complementary properties and avoided if they did not fulfill default parameters. The set of primers were used: ATF4: forward 5′-CTACTAGGTACCGCCAGAAG-3′, reverse 5′-GCCTTACGGACCTCTTCTAT-3′; caspase-12: forward 5′-GGCCGTCCAGAGCACCAGT-3′, reverse 5′-CAGTGGCTATCCCTTTGCTTGTG-3′; processed xbp1: forward 5′-CTGAGTCCGCAGCAG-3′, reverse 5′-GGATCTCTAAAACTAGAGGCT-3′; GRP78: forward; 5′-GTTCTGCTTGATGTGTGTCC-3′, reverse 5′-TTTGGTCATTGGTGATGGTG-3′; CHOP/GADD153: forward 5′-TCAGATGAAATTGGGGGCAC-3′, reverse 5′-TTTCCTCGTTGAGCCGCTCG-3′; HSP70: forward 5′-TATTCATCCACTCCATCGCCTCAT-3′, reverse 5′-ACGCTTGGCCTGGTGGACA-3′; β-Actin: forward 5′-GGCTGTGTTGTCCCTGTAT-3′, reverse 5′-CCGCTCATTGCCGATAGTG-3′.
Western Blot Analysis
Rats were killed by an overdose of anesthetic ether and the major affected brain loci viz., cortex and striatum were quickly excised in the chilled medium. The tissue was homogenized in five volumes of ice cold lysis buffer containing 200 mmol/l HEPES (pH 7.4), 250 mmol/l sucrose, 1 mmol/l dithiothreitol, 1.5 mmol/l MgCl2, 10 mmol/l KCl, 1 mmol/l EDTA, 1 mmol/l EGTA, 0.1 mmol/l PMSF, and 1% protease and phosphatase inhibitor cocktails (Sigma) using a teflon homogenizer. For whole tissue/cell lysates homogenates were spun at 10,000g for 10 min and the supernatant was used. The protein concentration was determined by Lowry method (Lowry et al. 1951). For western blot analysis an equal amount of protein (60–100 μg) was loaded in each well and subjected to 8–15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The separated proteins were then transferred onto nitrocellulose membrane and blocked in 5% non-fat dry milk prepared in 1× PBS. The membranes were incubated with the primary antibodies for 3 h at RT or overnight at 4°C (dilutions 1:500–1:1,000). The following primary antibodies were used: phospho-eIF2α (Ser51) and eIF2α (both from Cell signaling), HSP70 and actin (both from Santacruz). After washing the membranes were incubated with secondary antibodies (1:2,000–1:4,000) for 1 h at RT. The secondary antibodies used were horseradish peroxidase (HRP) conjugated anti-IgG corresponding to the primary antibodies. The blots were developed by using ECL Plus detection system (GE Healthcare). The relative band intensity was measured by spot densitometry analysis using AlphamagerTM 2200 software.
Salubrinal Treatment
Salubrinal (Sal; Tocris) was dissolved in dimethylsulfoxide (DMSO) and was administered at the dose of 1 mg/kg intraperitoneally 30 min before (Sokka et al. 2007) induction of cerebral ischemia. An equal volume of DMSO was injected in vehicle control animals. Rats were killed after 2 h ischemia followed by 24 h reperfusion under deep anesthesia. The brains were quickly removed and the affected brain regions were dissected out and frozen in liquid nitrogen and stored at −80°C until processed for western blot analysis.
Statistical Analysis
Significant differences between sham and MCAO groups were made using ANOVA followed by Newman–Keuls multiple comparison test. The comparison between identical time points of I/R was made using Student’s t-test and represented as mean ± SEM. * P < 0.05, ** P < 0.01, and *** P < 0.001 were considered statistically significant.
Results
Temporal Progression of Brain Damage After I/R Injury
TTC staining of rat brain sections subjected to 2 h ischemia followed by different time intervals of reperfusion has shown varying degree of infarction, predominantly in the striatum and cortex. Although, the damage at early time points of reperfusion (i.e., 0, 3, and 6 h) was confined to the striatum only, however, it extended to the cortical region with increasing time points of reperfusion with the maximum infarction observed after 24 h of reperfusion (Supplementary Figure). Moreover, NeuN immunoreactivity after 24 h of reperfusion revealed that I/R injury caused dramatic reduction of NeuN staining both in the cortex and striatum, which indicates neuronal loss after 24 h of reperfusion (Fig. 1). These changes were more or less similar both in the core of infract and surrounding striatal area. Interestingly, DAPI staining of the same sections revealed that these nuclei were preserved and yet some of the nuclei were morphologically intact. Further, TUNEL staining was performed to evaluate I/R injury induced apoptotic DNA fragmentation in the cortex and striatal regions to specify the mode of cell death. TUNEL positive cells were not observed in the non-ischemic contralateral side. In contrast, the ischemic side (ipsilateral) exhibited intense TUNEL positive staining both in the striatum and cortex. The TUNEL positive cells was counted by considering at least four non-overlapping microscopic fields and divided by total number of DAPI stained cells. Because, the necrotic cells may also be TUNEL positive, albeit with diffuse staining, therefore, the cells with intense signals were only considered for counting. In striatum the number of TUNEL positive cells was about 33% ± 5.3 and in cortex it was about 25% ± 3.7 (Fig. 2d). Moreover, the morphology of some TUNEL positive cells in both the regions have shown features of classical apoptosis like nuclear condensation and formation of apoptotic bodies (Fig. 2a–c). TUNEL positive staining was confirmed within the nucleus by overlapping with the nuclear stain DAPI. The number of TUNEL positive cells appeared at maximum in the striatum as compared to cortex.

A representative photomicrographs showing the neurons stained with neuronal specific marker NeuN in the cortex (upper panel) and in the striatum (lower panel) of rats subjected to 2 h/24 h of I/R injury. Note the reduced NeuN staining or neuronal loss in the ipsilateral cortex and striatum. Nuclei were counterstained with DAPI. The non-ischemic contralateral hemisphere was used as control. Bar = 100 μm

Representative photomicrographs showing the TUNEL positive staining in the striatal brain region following 2 h/24 h of I/R injury in rats. Nuclei were counterstained with DAPI. a Comparison was made between ipsilateral (ischemic) and contralateral (non-ischemic) brain areas. Note the intense TUNEL positive staining in the ischemic damaged area (ipsilateral), bar = 100 μm. b Shows the morphological features of TUNEL positive cells in the striatum of ipsilateral brain area. Cells with classical features of apoptosis are highlighted in close up. Original magnification ×400. c Shows the morphological features of TUNEL positive cells in the ipsilateral cortical brain area. Morphological features of cells are highlighted in close up. Original magnification ×400. d Bar represents the percent of TUNEL positive cells in striatum (2/24S) and in cortex (2/24C) following I/R injury. The non-ischemic contralateral hemisphere considered as control (n = 3, at least four non-overlapping microscopic fields were considered for quantitation). Statistically significant differences in comparison to the control are indicated by *** P < 0.001
Induction of HSP70 Expression After I/R Injury
Enhanced expression of HSP70 indicates the cytoplasmic dysfunction and often used as potentially useful marker of cytoplasmic stress response. Therefore, the expression of HSP70 was used to confirm, whether ER stress is a consequence of intra-cellular dysfunction in the affected brain regions after I/R injury at various time points of reperfusion injury. Western blot analysis revealed that I/R injury induced robust increase in the HSP70 protein levels both in the striatum (Fig. 3a) and cortex (Fig. 3b) at various reperfusion time points of 6, 12, and 24 h, respectively. Moreover, the temporal profile of HSP70 mRNA levels was also in correlation to the protein levels as observed in the striatum (Fig. 3c). Thus, the temporal profile of HSP70 expression is indicative of the severity of I/R injury induced cellular stress, which seems to have direct impact on subcellular organelles.

Representative western blot shows the banding pattern of HSP70 protein, a in striatum, b in cortex following various time intervals of I/R. Note the relative increase in HSP70 protein levels after 6 h reperfusion. β-Actin was used as loading control. c Represents relative HSP70 mRNA expression in striatum following various time intervals of I/R (n = 3 per group). Semi quantitative changes of HSP70 m-RNA are represented as mean ± SEM. Statistically significant differences to the sham are indicated by * P < 0.05, ** P < 0.01, and *** P < 0.001 vs. sham
Induction of GRP78 Expression After I/R Injury
The GRP78 expression serves as a good marker of ER stress as this gene is being specifically activated under the conditions of ER dysfunction. Therefore, in the present study GRP78 mRNA expression was assessed by RT-PCR in the affected brain regions at different time points of I/R injury. In striatum the expression of GRP78 was significantly increased in a graded fashion from 3 h post reperfusion reaching at peak at 12 h and which were maintained until 24 h (Fig. 4a). While, in the cortex the expression of GRP78 had a tendency to increase significantly from 0 h, but increased at 3 and 24 h post reperfusion (Fig. 4b). These results noticeably indicate the similarities in the upregulation of mRNA levels following I/R injury and also demonstrate the effect of severity of injury in both brain regions on the GRP78 mRNA expression.

Represents relative GRP78 mRNA expression a in striatum and b in cortex following various time intervals of I/R. Semi quantitative changes of GRP78 mRNA expression are represented as mean ± SEM (n = 3 per group). Statistically significant differences to the sham are indicated by * P < 0.05 and ** P < 0.01 vs. sham. β-Actin was used as loading control
Induction of xbp1 mRNA Processing After I/R Injury
The processing of xbp1 mRNA is a reaction, where by a sequence of 26 bases are cut out from xbp1 mRNA by the activated form of IRE1. Processed xbp1 mRNA is an indicative of activation of unfolded protein response. It is further translated into a new protein of 54 kDa, which translocates to the nucleus and functions as an active transcription factor for inducing expression of ER stress genes including grp78, grp94, etc. Therefore, we have evaluated I/R injury induced changes in xbp1 mRNA processing by semi quantitative RT-PCR using specific primers. The brain regional and time-dependent changes of processed xbp1 mRNA levels were quantified following I/R injury. In the striatum, a significant increase in processed xbp1 mRNA levels was observed at 6 and 24 h (Fig. 5a). While, in cortex the mRNA levels were significantly observed at 0 and 6 h (Fig. 5b). The processed xbp1 mRNA levels seem to be more pronounced at 6 h post reperfusion both in the striatum and cortex. It is noteworthy that merely all the given time points of I/R induced the processing of xbp1 mRNA, though may not be significant at all places. The temporal expression profile of processed xbp1 mRNA after cerebral injury, therefore may implies the activation of UPR.

Represents I/R injury induced processing of xbp1 mRNA a in striatum and b in cortex following various time intervals of I/R. Data represented as mean ± SEM (n = 3 per group). Statistically significant differences to the sham are indicated by * P < 0.05, ** P < 0.01, and *** P < 0.001 vs. sham. β-Actin was used as loading control
Induction of Caspase-12 Expression After I/R Injury
Consistent with the upregulation of GRP78 and HSP70, it became imperative to analyze the involvement of caspase-12, which plays an important role especially in ER stress-mediated cell death. Therefore, an attempt was made to evaluate the altered gene expression of caspase-12 during cerebral injury, using semi-quantitative RT-PCR. In the striatum the levels of mRNA showed significant increase after 6 h of reperfusion, which continued till 24 h of reperfusion (Fig. 6a). Similarly, in the cortex significant caspase-12 mRNA expression was detected from 6 h onwards peaking at 24 h of reperfusion (Fig. 6b). Thus, this data is in conformity with that of GRP78 mRNA expression pattern.

Represents relative caspase-12 mRNA expression a in striatum and b in the cortex following various time intervals of I/R. Semi quantitative changes of caspase-12 m-RNA are represented as mean ± SEM (n = 3 per group). Statistically significant differences to the sham are indicated by * P < 0.05 and ** P < 0.01 vs. sham. β-Actin was used as loading control
Induction of CHOP/GADD153 and ATF4 Expression After I/R Injury
The expression of the gene encoding the CHOP/GADD153 has been shown to be specifically activated under conditions that disturb the functioning of the ER. In the striatum significant increase in the mRNA levels were observed at 12 and 24 h after I/R injury (Fig. 7a). In the cortex the significant increase in mRNA levels as observed predominantly at 3 and 12 h after I/R injury (Fig. 7b). Therefore, the results suggested that activation of CHOP/GADD153, which is an indicative of activation of UPR following I/R injury. ATF-4 is a downstream molecule of PERK pathway, which may play a significant role in cell survival/death mechanisms after ER stress. In the striatum as well as cortex the mRNA levels of ATF-4 were increased significantly from 3 h onwards and peaked at 24 h of reperfusion (Fig. 8a, b). Thus, the above results indicated the increased expression of ATF-4 mRNA levels in affected brain regions which correlates with that of major ER stress marker GRP78 expression.

Represents relative CHOP/GADD153 mRNA expression a striatum b cortex following various time intervals of I/R. Data represented as mean ± SEM (n = 3 per group). β-Actin was used as loading control. Statistical significance * P < 0.05, ** P < 0.01, and *** P < 0.001 vs. sham

Represents relative mRNA expression of ATF-4 a in striatum and b in cortex following various time intervals of I/R. Semi quantitative changes of ATF-4 m-RNA are represented as mean ± SEM (n = 3 per group). Statistically significant differences are indicated by * P < 0.05, ** P < 0.01, and *** P < 0.001 vs. sham
Effect of Salubrinal After I/R Injury
The primary response of the mammalian cells to ER stress response is transient global attenuation of protein translation. Phosphorylation of serine-51 residue of eIF2α subunit is an indicative of global protein synthesis inhibition. In the present study, it was found that I/R injury induced robust phopshorylation of eIF2α at very early time points, i.e., 0 and 3 h after ischemia indicating suppression of protein synthesis. Of note, the phosphorylation was relatively reduced after 6 h of reperfusion and which remained unchanged till 24 h (Fig. 9). It is suggested that Salubrinal; a small molecule counteracts ER stress-induced cell degeneration by selectively inhibiting eIF2α dephosphorylation both in vitro and in vivo (Boyce et al. 2005; Sokka et al. 2007). It is also suggested that Sal is able to penetrate into the brain tissue in vivo and thereby affords protection against excitotoxicity (Sokka et al. 2007). Western blot analysis was revealed that Sal induced the phosphorylation of eIF2α significantly after 2/24 h of I/R injury, indicating Sal in line with eIF2α for its action (Fig. 10a). Further, Sal reduced infarct volume significantly after 2/24 h of I/R injury as revealed by TTC staining (Fig. 10b). The expression of HSP70 protein was also analyzed to know whether Sal has any effect on cytoplasmic stress and there was no significant change observed in the expression of HSP70 (Fig. 11). Thus, it indicates that Sal does not cause any cellular stress.

Representative western blot shows the phosphorylation of eIF2α after I/R injury in the striatum. Note the relative decrease of eIF2α phosphorylation after 6 h reperfusion. Total eIF2α was used as loading control

a Representative western blot shows the phosphorylation of eIF2α in Sal administered as compared to vehicle control rats following I/R injury. Note the relative increase of eIF2α phosphorylation after 24 h reperfusion in the striatum. Statistically significant difference to the vehicle control is indicated by * P < 0.05. β-Actin was used as loading control (n = 4 per group). b Effect of Sal on infarct volume of rats subjected to 2/24 h of I/R injury (n = 5 per group). Infarct volume in the brain sections was significantly reduced by Sal treatment. Statistically significant differences to the vehicle control are indicated by ** P < 0.01

Representative western blot shows the expression of HSP70 in Sal administered as compared to vehicle control rats following I/R injury. a in the striatum b in the cortex after 24 h reperfusion. β-Actin was used as loading control (n = 4 per group)
Discussion
Cerebral ischemia, particularly reperfusion injury triggers multiple and distinct but overlapping cell signaling pathways in the brain, which may lead to cell survival or cell damage (Mehta et al. 2007; Nakka et al. 2008). However, the degree of ischemia and the period of reperfusion determine the outcome for individual cells. It has been suggested that ER stress-induced neuronal cell death plays an important role in stroke pathophysiology (DeGracia and Montie 2004; Tajiri et al. 2004; Paschen and Mengesdorf 2005). The mechanisms by which ER stress leads to cell death following I/R injury is not elusive with multiple potential participants described but with little clarity about which specific death effectors dominate in particular cellular contexts. In this study, we demonstrated the critical involvement of ER stress after I/R injury in rats with the activation of multiple cell death regulators. Further, we also demonstrated that inhibition of ER stress leads to reduced brain damage after I/R injury.
The primary ischemic injury observed predominantly in the striatum during early reperfusion after 2 h ischemia expanded over a time period towards cortex and resulted in a larger infarct after 24 h of reperfusion. These results are consistent with the previous report that 2 h of ischemia followed by reperfusion typically involves maximal infarct expansion with increasing reperfusion (Zhang et al. 1994). Further, the NeuN staining confirmed massive neuronal loss within the ischemic lesion mainly in the striatum and then in the cortex. Of particular note, the nuclei of cells, which are not stained by NeuN, are still preserved and some of the nuclei are morphologically intact after staining with the DAPI (Fig. 1). This also supports the previous report that labeling of neurons with the caspase-3 antibodies that had lost NeuN positivity, revealed that these neurons still preserved their integrity (Unal-Cevik et al. 2004). Furthermore, the number of TUNEL positive cells appeared was also more in striatum than in the cortex (Fig. 2). Thus, it seems that a quite simplistic view implies poor prospects regarding cell survival in the core of the cerebral infarction and therapeutic expectations to control cell death and cell survival in the penumbra.
Increased expression of HSP70 not only indicates the cytoplasmic dysfunction but often used as potential molecular marker for the identification of ischemic penumbra. Ample evidence suggests that the protective effects of HSP70 occurs mainly in the ischemic penumbra but not in the core (Kinouchi et al. 1993; Sharp et al. 1993; Hossmann 1994; Yenari et al. 1999; Rajdev et al. 2000; Weinstein et al. 2004). In the present study, we have observed increased expression of HSP70 both in the striatum and cortex after 6 h and maintained till 24 h following I/R injury (Fig. 3). Therefore, the increased expression of HSP70 at different time points of I/R injury further strengthened our view regarding events observed in the striatum and cortex. Because, cell death in the penumbra is considered as an active process largely dependent on the activation of cell death programmes (Ferrer 2006).
Under conditions associated with ER stress, unfolded or misfolded proteins accumulate in the ER lumen, a pathologic process resulting in the activation of the UPR pathway to combat the harmful effects of ER stress. Therefore, the present study examined the expression of wide range of molecules that are associated with ER stress in specific brain region and time-dependent manner. Increased expression of GRP78 serves as a good marker for ER stress. A marked increase in the expression of GRP78 mRNA levels were found both in the striatum and cortex indicated I/R injury induced ER stress in affected brain regions (Fig. 4). In physiological state, GRP78 binds to the effectors of UPR, i.e., PERK, IRE1 to suppress their activity (Bertolotti et al. 2000). Under conditions of ER stress, when misfolded proteins accumulate in the ER lumen, GRP78 dissociates from these effectors allowing their activation. Upon activation, the endonuclease activity of IRE1 specifically cuts out a 26-nucleotide intron from the xbp1 mRNA, which leads to a shift of the open reading frame of xbp1 mRNA (Calfon et al. 2002). Therefore, processing of xbp1 mRNA is indicative of activation UPR branch IRE1 pathway and ER dysfunction. In the present study, we did indeed observe a marked increase in the processed xbp1 mRNA levels after I/R injury in the affected brain regions. A marked increase in processed xbp1 mRNA levels was observed during reperfusion being most pronounced after 6 and 24 h in the striatum and at 0 and 6 h of reperfusion in the cortex, respectively (Fig. 5). Processed xbp1 mRNA is translated into a new protein of 54 kDa that functions as a transcription factor and has diverse targets specific for ER stress genes including GRP78, GRP94, etc. (Lee et al. 2003). Thus, IRE1–xbp1 pathway seems to be pro-survival by activating the transcription of ER chaperones. The temporal profile of the processed xbp1 and GRP78 mRNA expression after I/R injury might fit into the above mentioned view. However, prolonged ER stress accompanied by failure of adaptive response may eventually results in apoptotic cell death.
Activated PERK phosphorylates eIF2α to avoid further accumulation of proteins by suppressing protein synthesis but also leads to the paradoxical increased translation of transcription factors ATF-4 and CHOP/GADD153 (DeGracia and Montie 2004). It has been reported that ER stress induced by thapsigargin is associated with increased expression of ATF-4 (Luo et al. 2003). Indeed, mice lacking ATF-4 have shown significantly smaller infarcts and improved behavioral outcome as compared with wild-type mice subjected to ischemic stroke (Lange et al. 2008). Thus, the increased expression of ATF-4 in the present scenario seems to play an important pro-death role after I/R injury (Fig. 8). There is ample evidence that CHOP/GADD153 participates in apoptosis signaling pathways and the expression of CHOP/GADD153 mRNA serves as a hallmark of ER stress (DeGracia and Montie 2004; Oyadomari and Mori 2004). The increased mRNA levels of CHOP/GADD153 observed in the present study after I/R injury (Fig. 7) are also in agreement with the previous observation after bilateral common carotid artery occlusion in mice (Tajiri et al. 2004). Indeed, mice lacking CHOP/GADD153 exhibits decreased neuronal apoptosis (Tajiri et al. 2004). Thus, it indicates that CHOP/GADD153 might be crucial in mediating ER stress-induced apoptotic cell death pathways after ischemic injury. The temporal profile of ATF-4 and CHOP/GADD153, GRP78 and processed xbp1 mRNA collectively demonstrated the activation of UPR signaling pathways, i.e., PERK and IRE1 after I/R injury. Recent studies also suggested the role of ER stress response in brain injury after global ischemia/reperfusion in rats (Urban et al. 2009). Moreover, the neuroprotective effect of ischemic preconditioning has been also attributed to the attenuation of ER stress response after ischemic insults (Hayashi et al. 2003; Lehotský et al. 2009).
Caspase-12 (Van de Craen et al. 1997), which is mainly located on cytoplasmic side of the ER, regarded as a representative lead molecule implicated in cell death executing mechanisms related to ER stress (Nakagawa et al. 2000). It has been suggested that activated caspase-12 lacking its prodomain may directly process downstream caspases either caspase-3 or -9 in the cytosol, or possibly target some other unidentified substrate that influence the progression of apoptosis (Rao et al. 2001; Morishima et al. 2002). We have observed that I/R injury caused significant increase in the caspase-12 mRNA levels both in the striatum and cortex peaking at 24 h after reperfusion (Fig. 5). Of note, the increase in caspase-12 mRNA levels appeared to correlate with that of GRP78, ATF-4, and CHOP/GADD153 mRNA expression profile observed in the present study.
Phosphorylation of eIF2α on ser51 by PERK is one of the downstream processes of UPR. Phosphorylated eIF2α mediates a both transient decrease in global translation of proteins and translational upregulation of selected stress induced mRNAs (Prostko et al. 1993; Ron 2002). It was found that I/R injury-induced robust phosphorylation of eIF2α on ser51 immediate after ischemia and early at 3 h reperfusion. After 6 h reperfusion the phosphorylation was relatively reduced and remained till 24 h as observed in the affected striatum (Fig. 9). Phosphorylation of eIF2α suggested to be cytoprotective during ER stress; indeed, cells become susceptible, when this pathway is genetically ablated (Harding et al. 2000b; Scheuner et al. 2001) and protected when it is ectopically enforced (Jousse et al. 2003; Lu et al. 2004b). Therefore, the increased eIF2α phosphorylation early after I/R injury may be correlated to the protective mechanisms initiated by the UPR, whereas, the relative reduction of phospho-eIF2α indicates its dephosphorylation. Activated PERK phosphorylates eIF2α to avoid further accumulation of proteins by suppressing protein synthesis but also leads to the paradoxical increased expression of transcription factors ATF-4 and CHOP/GADD153 (DeGracia and Montie 2004). The temporal profile of both the ATF-4 and CHOP/GADD153 after I/R injury supports this phenomenon at least in part. Nevertheless, further studies at translational level may provide more optimistic picture regarding the molecular mechanisms of ER stress response after I/R injury.
Attenuation of translation is thought to be an adaptive response that helps cells to survive from ER stress. Indeed, mice lacking PERK and eIF2α cannot have translational control in their cells via PERK/eIF2α (Harding et al. 2001; Zhang et al. 2002). Inhibiting translational recovery by selectively inhibiting eIF2α dephosphorylation protects some cells from ER stress-induced apoptosis (Boyce et al. 2005). It has been reported that selective inhibition of eIF2α dephosphorylation by a small molecule inhibitor, Salubrinal (Sal) protects cells from ER stress in a dose-dependent manner (Boyce et al. 2005). We have observed that intraperitoneal administration of Sal significantly induced the phosphorylation of eIF2α after 2/24 h of I/R injury (Fig. 10a). Therefore, it appeared that Sal may act via eIF2α for its neuroprotective effect. Interestingly, Sal reduced infarct volume significantly after 2/24 h of I/R injury as revealed by TTC staining (Fig. 10b). Moreover, Sal did not show any significant effect on the expression of HSP70, indicating that Sal does not cause any cellular stress (Fig. 11).Our results were also supported by the previous study that intraperitoneal administration of Sal decreases neuronal death after kainic acid injury induced seizures and excitiotoxic cell death in the rat brain (Sokka et al. 2007). Therefore, the treatment with salubrinal tends to inhibit the ER stress and also seems to be neuroprotective after I/R injury, the inhibition of ER stress thus may be used as therapeutic target for neuroprotection in neurodegenerative diseases including cerebral stroke.
References
Althausen S, Mengesdorf T, Mies G, Olah L, Nairn AC, Proud CG, Paschen W (2001) Changes in the phosphorylation of initiation factor eIF-2alpha, elongation factor eEF-2 and p70 S6 kinase after transient focal cerebral ischaemia in mice. J Neurochem 78:779–787
Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H (1986) Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke 17:472–476
Belayev L, Busto R, Zhao W, Fernandez G, Ginsberg MD (1999) Middle cerebral artery occlusion in the mouse by intraluminal suture coated with poly-l-lysine: neurological and histological validation. Brain Res 833:181–190
Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2:326–332
Boyce M, Yuan J (2006) Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ 13:363–373
Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J (2005) A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science 307:935–939
Calfon M, Zeng H, Urano F, Till JH, Hubbart SR, Harding HP, Clark SG, Ron D (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing XBP-1 mRNA. Nature 415:92–96
DeGracia DJ, Montie HL (2004) Cerebral ischemia and the unfolded protein response. J Neurochem 91:1–8
DeGracia DJ, Kumar R, Owen CR, Krause GS, White BC (2002) Molecular pathways of protein synthesis inhibition during brain reperfusion: implications for neuronal survival or death. J Cereb Blood Flow Metab 22:127–141
Dirnagl U, Simon RP, Hallenbeck JM (2003) Ischemic tolerance and endogenous neuroprotection. Trends Neurosci 26:248–254
Feigin VL (2005) Stroke epidemiology in the developing world. Lancet 365(9478):2160–2161
Ferrer I (2006) Apoptosis: future targets for neuroprotective strategies. Cerebrovasc Dis 2:9–20
Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D (2000a) Regulated translation initiation controls stress induced gene expression in mammalian cells. Mol Cell 6:1099–1108
Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D (2000b) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mole Cell 5:897–904
Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D (2001) Diabetes Mellitus and exocrine pancreatic dysfunction in Perk−/−mice reveals a role for translational control in secretory cell survival. Mol Cell 7:1153–1163
Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Chan PH (2003) Induction of GRP78 by ischemic preconditioning reduces endoplasmic reticulum stress and prevents delayed neuronal cell death. J Cereb Blood Flow Metab 23:949–961
Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Chan PH (2005) Damage to the endoplasmic reticulum and activation of apoptotic machinery by oxidative stress in ischemic neurons. J Cereb Blood Flow Metab 25:41–53
Hossmann KA (1994) Viability thresholds and the penumbra of focal ischemia. Ann Neurol 36:557–565
Hu BR, Martone ME, Jones YZ, Liu CL (2000) Protein aggregation after transient cerebral ischemia. J Neurosci 20:3191–3199
Jousse C, Oyadomari S, Novoa I, Lu PD, Zhang Y, Harding HP, Ron D (2003) Inhibition of a constitutive translation initiation factor 2a phosphatase, CReP, promotes survival of stressed cells. J Cell Biol 163:767–775
Kinouchi H, Sharp FR, Koistinaho J, Hicks K, Kamii H, Chan PH (1993) Induction of heat shock hsp70 mRNA and HSP70 kDa protein in neurons in the “penumbra” following focal cerebral ischemia in the rat. Brain Res 619:334–338
Kumar R, Azam S, Sullivan JM, Owen C, Cavener DR, Zhang P, Ron D, Harding HP, Chen JJ, Han A, White BC, Krause GS, DeGracia DJ (2001) Brain ischemia and reperfusion activates the eukaryotic initiation factor 2alpha kinase, PERK. J Neurochem 77:1418–1421
Lange PS, Chavez JC, Pinto JT, Coppola G, Sun CW, Townes TM, Geschwind DH, Ratan RR (2008) ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J Exp Med 205:1227–1242
Lee AH, Iwakoshi NN, Glimcher LH (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23:7448–7459
Lehotský J, Urban P, Pavlíková M, Tatarková Z, Kaminska B, Kaplán P (2009) Molecular mechanisms leading to neuroprotection/ischemic tolerance: effect of preconditioning on the stress reaction of endoplasmic reticulum. Cell Mol Neurobiol 29:917–925
Longa EZ, Weinstein PR, Carlson S, Cummins R (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20:84–91
Lowry OH, Rosebrouge NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin Phenol reagent. J Biol Chem 193:265–275
Lu PD, Harding HP, Ron D (2004a) Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. Mol Cell Biol 167:27–33
Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, Kaufman RJ, Ron D, Harding HP (2004b) Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J 23:169–179
Luo S, Baumeister P, Yang S, Abcouwer SF, Lee AS (2003) Induction of Grp78/BiP by translational block: activation of the Grp78 promoter by ATF4 through and upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J Biol Chem 278:37375–37385
Ma Y, Hendershot LM (2001) The unfolding tale of the unfolded protein response. Cell 107:827–830
Mehta SL, Manhas N, Raghubir R (2007) Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev 54:34–66
Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y (2002) An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem 277:34287–34294
Mouw G, Zechel JL, Gamboa J, Lust WD, Selman WR, Ratcheson RA (2003) Activation of caspase-12, an endoplasmic reticulum resident caspase, after permanent focal ischemia in rat. Neuroreport 14:183–186
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum specific apoptosis and cytotoxicity by amyloid-beta. Nature 403:98–103
Nakka VP, Gusain A, Mehta SL, Raghubir R (2008) Molecular mechanisms of apoptosis in cerebral ischemia: multiple neuroprotective opportunities. Mol neurobiol 37:7–38
Oyadomari S, Mori M (2004) Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11:381–389
Paschen W, Doutheil J (1999) Disturbances of the functioning of endoplasmic reticulum: a key mechanism underlying neuronal cell injury? J Cereb Blood Flow Metab 19:1–18
Paschen W, Mengesdorf T (2005) Cellular abnormalities linked to endoplasmic reticulum dysfunction in cerebrovascular disease—therapeutic potential. Pharmacol Ther 108:362–375
Paschen W, Gissel C, Linden T, Althausen S, Doutheil J (1998) Activation of gadd153 expression through transient cerebral ischemia: evidence that ischemia causes endoplasmic reticulum dysfunction. Brain Res Mol Brain Res 60:115–122
Prostko CR, Brostrom MA, Brostrom CO (1993) Reversible phosphorylation of eukaryotic initiation factor 2 alpha in response to endoplasmic reticular signaling. Mol Cell Biochem 128:255–265
Rajdev S, Hara K, Kokubo Y, Mestril R, Dillmann W, Weinstein PR, Sharp FR (2000) Mice overexpressing rat heat shock protein 70 are protected against cerebral infarction. Ann Neurol 47:782–791
Rao RV, Hermel E, Castro-Obregon S, del Rio G, Ellerby LM, Ellerby HM, Bredesen DE (2001) Coupling endoplasmic reticulum stress to the cell death program: mechanism of caspase activation. J Biol Chem 276:33869–33874
Ron D (2002) Translational control in the endoplasmic reticulum stress response. J Clin Invest 110:1383–1388
Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7:1165–1176
Sharp FR, Kinouchi H, Koistinaho J, Chan PH, Sagar SM (1993) HSP70 heat shock gene regulation during ischemia. Stroke 24:I72–I75
Shibata M, Hattori H, Sasaki T, Gotoh J, Hamada J, Fukuuchi Y (2003) Activation of caspase-12 by endoplasmic reticulum stress induced by transient middle cerebral artery occlusion in mice. Neuroscience 118:491–499
Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L, Belluardo N, Lindholm D, Korhonen L (2007) Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J Neurosci 27:901–908
Strong K, Mathers C, Bonita R (2007) Preventing stroke: saving lives around the world. Lancet Neurol 6:182–187
Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI, Ushio Y, Mori M (2004) Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ 11:403–415
Unal-Cevik I, Kılınc M, Gursoy-Ozdemir Y, Gurer G, Dalkara T (2004) Loss of NeuN immunoreactivity after cerebral ischemia does not indicate neuronal cell loss: a cautionary note. Brain Res 1015:169–174
Urban P, Pavlíková M, Sivonová M, Kaplán P, Tatarková Z, Kaminska B, Lehotský J (2009) Molecular analysis of endoplasmic reticulum stress response after global forebrain ischemia/reperfusion in rats: effect of neuroprotectant simvastatin. Cell Mol Neurobiol 29:181–192
Van de Craen M, Vandenabeele P, Declercq W, Van den Brande I, Van Loo G, Molemans F, Schotte P, Van Criekinge W, Beyaert R, Fiers W (1997) Characterization of seven murine caspase family members. FEBS Lett 403:61–69
Weinstein PR, Hong S, Sharp FR (2004) Molecular identification of the ischemic penumbra. Stroke 35:2666–2670
Yenari MA, Giffard RG, Sapolsky RM, Steinberg GK (1999) The neuroprotective potential of heat shock protein 70 (HSP70). Mol Med Today 5:525–531
Zhang RL, Chopp M, Chen H, Garcia JH (1994) Temporal profile of ischemic tissue damage, neutrophil response, and vascular plugging following permanent and transient (2H) middle cerebral artery occlusion in the rat. J Neurol Sci 125:3–10
Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR (2002) The PERK eukaryotic initiation factor 2alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol 22:3864–3874
Acknowledgment
Mr. Venkata Prasuja Nakka and Ms. Anchal Gusain received Senior Research Fellowship from the Council of Scientific and Industrial Research, New Delhi, India.
Author information
Authors and Affiliations
Corresponding author
Additional information
+CDRI Communication No. 7749.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Supplementary Figure
Coronal brain sections of TTC staining showing the ipsilateral brain damage after 2 h ischemia followed by different time intervals of reperfusion (TIFF 536 kb)
Rights and permissions
About this article
Cite this article
Nakka, V.P., Gusain, A. & Raghubir, R. Endoplasmic Reticulum Stress Plays Critical Role in Brain Damage After Cerebral Ischemia/Reperfusion in Rats. Neurotox Res 17, 189–202 (2010). https://doi.org/10.1007/s12640-009-9110-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-009-9110-5