
Abstract
Gonadotropin-releasing hormone (GnRH) mediates central control of reproductive function by activation of G-protein-coupled receptors on pituitary gonadotropes. These Gq-coupled receptors mediate acute effects of GnRH on the exocytotic secretion of luteinizing hormone and follicle-stimulating hormone and also chronic regulation of the synthesis of these gonadotropin hormones. GnRH is secreted in brief pulses and GnRH effects on its target cells are dependent upon the characteristics of these pulses. Here we provide an overview of GnRH receptors and their signaling network, emphasizing novel and atypical functional features of GnRH signaling, and mechanisms mediating pulsatile hormone signaling.
Access provided by CONRICYT-eBooks. Download reference work entry PDF
Similar content being viewed by others


Keywords
Gonadotropin-Releasing Hormone (GnRH) and Its Receptors
Gonadotropin-releasing hormone , (pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2), also known as luteinizing hormone-releasing hormone (LHRH) or GnRH-I, is a peptide that is synthesized in hypothalamic neurons. It is secreted into the hypothalamic-hypophyseal portal circulation in pulses each lasting for a few minutes (Belchetz et al. 1978; Clarke and Cummins 1985; Wildt et al. 1981), with the secretory activity of GnRH neurons being controlled largely by input from kisspeptin-containing neuronal circuits. After secretion GnRH exits the portal circulation and binds to its cognate receptors (GnRHRs) on the surface of anterior pituitary gonadotropes. It causes them to synthesize and secrete luteinizing hormone (LH) and follicle-stimulating hormone (FSH); these two gonadotropin hormones then control gametogenesis and steroidogenesis in the gonads (Cheng and Leung 2005; Ciccone and Kaiser 2009; McArdle and Roberson 2015; Millar et al. 2004; Naor 2009; Sealfon et al. 1997; Stojilkovic et al. 2010b; Wang et al. 2010). LH and FSH are heterodimeric glycoprotein hormones with distinct β-subunits (LHβ and FSHβ) and a common α-gonadotropin subunit (αGSU). The mature protein hormones are packaged into secretory vesicles for release from gonadotropes. Acutely GnRH regulates vesicle fusion with the plasma membrane whereas chronically, it increases synthesis of gonadotropins and thereby influences the hormone content of these vesicles. Together, these effects on synthesis and secretion underpin the central control of reproduction by GnRH.
The importance of this system is illustrated by the fact that GnRH and its receptors are both absolutely essential for mammalian reproduction (Cattanach et al. 1977; de Roux et al. 1997; Mason et al. 1986), but comparative studies have revealed multiple forms of GnRH and GnRHR. There are three distinct forms of the hormone: GnRH-I (often known simply as GnRH), GnRH-II, and GnRH-III. These have a common ancestral origin that predates vertebrates (Fernald and White 1999). Most classes of vertebrate have GnRH-I and GnRH-II, but GnRH-III is specific for teleosts (Cheng and Leung 2005; Millar et al. 2004; Schneider and Rissman 2008). GnRHRs are members of the rhodopsin-like G-protein-coupled receptor (GPCR) family and have a characteristic seven-transmembrane α-helical domain structure. They have been cloned from multiple species and can be classified into three groups based on sequence homology. All of the cloned mammalian GnRHRs are in groups I or II (Millar et al. 2004; Morgan and Millar 2004), and the type I GnRHRs of humans, rats, mice, pigs, sheep, and horses have >80% amino acid sequence identity. Some primates (notably rhesus and green monkeys and marmosets) express functional type II GnRHR (as well as type I GnRHR), but in humans there is a frameshift mutation and a premature stop codon in the GnRHR II (pseudo)gene so that a functional type II GnRHR is not expressed (Morgan and Millar 2004; Stewart et al. 2009; Wang et al. 2010). Accordingly, for humans, central control of reproduction is mediated by GnRH-I from the hypothalamus acting on type I GnRHR in gonadotropes. Further evidence of the importance of this system lies in the fact that GnRHRs are established therapeutic targets for manipulation with agonist and antagonist analogues of GnRH for assisted reproduction technology (Al-Inany et al. 2016; Siristatidis et al. 2015). In general, stimulatory effects of endogenous GnRH pulses can be mimicked with pulsatile agonists to induce ovulation or spermatogenesis. Alternatively, effects of endogenous GnRH can be blocked with GnRH antagonists to reduce circulating levels of gonadotropins and gonadal steroids and thereby treat sex hormone-dependent neoplasms such as those of the prostate, ovary, endometrium, or mammary glands (Chengalvala et al. 2003; Conn and Crowley 1994; Schally 1999). Paradoxically, sustained stimulation with GnRH agonists causes stimulation followed by desensitization of GnRHR-mediated gonadotropin secretion, and this is also exploited to treat sex steroid-dependent cancers (Cheng and Leung 2005; Ciccone and Kaiser 2009; McArdle and Roberson 2015; Millar et al. 2004; Naor 2009; Sealfon et al. 1997; Stojilkovic and Catt 1995; Wang et al. 2010).
GnRHR Signaling and Gonadotropin Secretion
In gonadotropes, GnRHR signaling (Fig. 1) is primarily mediated by activation of the heterotrimeric G-protein Gq which, in turn, activates the effector enzyme phospholipase C (PLC). PLC cleaves phosphatidylinositol (4,5)-bisphosphate to produce the second messengers inositol (1,4,5)-trisphosphate (IP3) and diacylglycerol (DAG). IP3 acts via its own intracellular receptors to increase Ca2+ release from intracellular stores, whereas DAG activates isozymes of protein kinase C (PKC). Ca2+ mobilization is followed by Ca2+ influx via L-type voltage-gated Ca2+ channels, and it is this Ca2+ entry across the plasma membrane that supports a more sustained increase in cytoplasmic Ca2+ concentration on continuous GnRH exposure (Ciccone and Kaiser 2009; Naor 2009; Stojilkovic and Catt 1995; Stojilkovic et al. 2010b). In some models, GnRH causes oscillations in cytoplasmic Ca2+, and the type of response depends on the model system and on GnRH concentration, with low concentrations having subthreshold effects, intermediate concentrations causing oscillatory responses, and high concentrations causing biphasic (spike-plateau) responses (Leong and Thorner 1991; Stojilkovic et al. 1991). For the latter, the initial spike phase is due to mobilization of Ca2+ from intracellular stores, whereas the plateau is dependent on Ca2+ entry through voltage-gated Ca2+ channels (Hansen et al. 1987; Izumi et al. 1989). For the oscillatory responses, a cytoplasmic oscillator model has been described, and with either response pattern, rapid effects of GnRH on gonadotropin secretion are driven by elevation of cytoplasmic Ca2+ (Hansen et al. 1987; Hille et al. 1994; Izumi et al. 1989; Stojilkovic et al. 1994).

A simplified GnRHR signaling network. GnRHR acts primarily via Gq, activating phospholipase C (PLC) to generate IP3 which drives IP3 receptor (IP3R)-mediated mobilization of Ca2+ from intracellular stores, and diacylglycerol (DAG) which, along with Ca2+, activates conventional PKC isozymes. Additional proteins involved in the control of the cytoplasmic Ca2+ ion concentration include Ca2+ sequestering sarcoplasmic and endoplasmic reticulum Ca2+ ATPase (SERCA), plasma membrane ATPase (PMCA), plasma membrane Na+/Ca2+ exchanger (NCX), Ca2+-sensitive K+ channels (K(Ca)), and voltage-gated Ca2+ channels (VGCC); GnRH increases cytoplasmic Ca2+ by coordinated effects on mobilization from intracellular stores and entry across the plasma membrane. Ca2+ is the primary driver for regulated release of gonadotropins that are contained in secretory vesicles and secreted largely by regulated exocytosis. Ca2+ also activates calmodulin (CaM), which activates CaM-dependent protein kinases (CaMK), which in turn phosphorylate and regulate effectors including CREB (cAMP response element binding protein). CaM also activates the protein phosphatase calcineurin (Cn), which activates a number of effectors including the Ca2+-dependent transcription factor NFAT (nuclear factor of activated T-cells). Furthermore, GnRH activates MAPK cascades, including the (largely PKC-mediated) activation of the Raf/MEK/ERK cascade shown. NFAT, CREB, and ERK-activated transcription factors are among the many inputs to the transcriptome mediating combinatorial control of gene expression. This includes the genes encoding the gonadotropin subunits, so GnRH has both acute effects on the rate of vesicle fusion with the plasma membrane and chronic effects on the gonadotropin synthesis to influence the content of these vesicles. This is a greatly simplified view of some of the network components and more detailed GnRH signaling models are described elsewhere (Bliss et al. 2010; Ciccone and Kaiser 2009; Fink et al. 2010; McArdle and Roberson 2015; Millar et al. 2004; Navratil et al. 2010; Stojilkovic and Catt 1995; Wang et al. 2010; Wurmbach et al. 2001)
The Ca2+ sensors mediating this regulated exocytosis have not been explored in detail, but early work implicated calmodulin as a mediator of GnRH-stimulated LH secretion (Conn et al. 1981) and also showed that PKC activation can mimic and modulate secretory effects of GnRH (McArdle et al. 1987; Stojilkovic et al. 1991; Zhu et al. 2002). Here, it is important to recognize that although Ca2+ drives regulated exocytosis, a proportion of gonadotropin secretion is via the constitutive pathway so that physiologically, pulses of GnRH-stimulated gonadotropin secretion overlay significant basal secretion (Pawson and McNeilly 2005). Indeed, the proportion of FSH secreted constitutively exceeds that for LH and when gonadotropins were measured in hypophyseal and peripheral blood, there was a high degree of synchrony between pulses of GnRH and LH, whereas FSH pulses are only associated with a small proportion of GnRH pulses (Clarke et al. 2002). LH and FSH are present in the same gonadotrope (Crawford and McNeilly 2002) so this requires sorting of the gonadotropins into distinct vesicles. Here it is noteworthy that only vesicles containing LH are associated with the storage protein secretogranin II and that the amount of LH stored in the pituitary can be 10–50 times higher than that of FSH (Pawson and McNeilly 2005). Thus, GnRH-stimulated Ca2+ transients drive regulated exocytotic release of storage vesicles containing LH, but have a less pronounced effect on FSH secretion, because FSH is less abundant in these vesicles and is directed largely for constitutive secretion (Pawson and McNeilly 2005).
GnRH Signaling and Gene Expression
Array studies have revealed that GnRH influences expression of many genes, several of which encode transcription factors, including c-Fos, Egr1, and ATF-3 (Ruf et al. 2003; Yuen et al. 2009; Yuen et al. 2002), but most work in this area has focused on transcriptional control of the gonadotrope signature genes for αGSU, LHβ, FSHβ, and GnRHR. GnRH increases transcription of each of these genes, and mechanistic studies have revealed regulatory roles for Ca2+-regulated proteins and also for mitogen-activated protein kinase (MAPK) cascades (McArdle and Roberson 2015). The most extensively studied MAPK is ERK , which phosphorylates and regulates numerous cytoplasmic and nuclear target proteins including Ets, ELK1, and SAP1 transcription factors. The ERK cascade is classically engaged by growth factors via tyrosine kinase receptors, but many other stimuli, including GPCR agonists, feed into the cascade (Caunt et al. 2006b). The mechanisms of GnRHR-mediated ERK activation differ between model systems, but it is largely mediated by PKC in αT3-1 and LβT2 gonadotropes (Naor 2009), and both PKC and ERK mediate transcriptional effects of GnRH on αGSU (Fowkes et al. 2002; Harris et al. 2003; Roberson et al. 1995; Weck et al. 1998), as well as LHβ (Call and Wolfe 1999; Harris et al. 2002; Kanasaki et al. 2005; Liu et al. 2002a) and FSHβ (Bonfil et al. 2004; Kanasaki et al. 2005; Vasilyev et al. 2002; Wang et al. 2008) subunits. However, other reports suggest roles for Ca2+ rather than ERK in GnRH-mediated LHβ (Weck et al. 1998) or αGSU expression (Ferris et al. 2007; Kowase et al. 2007). Moreover, in some models, GnRH engages the canonical ERK activation pathway by causing a PKC-dependent proteolytic release of membrane bound epidermal growth factor (EGF) receptor ligands, thereby activating EGF receptors (Cheng and Leung 2005; Kraus et al. 2001), whereas in others EGF receptor activation is not involved (Bonfil et al. 2004; Naor 2009). A particularly interesting feature here is that gene knockouts targeting the αGSU-expressing cells of the mouse pituitary revealed a requirement for ERK1 and/or ERK2 for ovulation and fertility in females but not for fertility in males (Bliss et al. 2009). These effects were attributed to LH insufficiency because LHβ transcript levels were reduced by knockout in females (but not in males) and levels of transcripts for other gonadotropin subunits and for the GnRHR were indistinguishable between control and knockout animals of either gender (Bliss et al. 2009). It is also important to recognize that GnRH can also activate other MAPKs. Thus, in rat pituitaries, αT3-1 and LβT2 cells, GnRH increases c-Jun N-terminal kinase (JNK) activity (Burger et al. 2004, 2009; Naor 2009), and JNK can mediate GnRH effects on αGSU expression as well as transcriptional activation of LHβ and FSHβ expression (Bonfil et al. 2004; Burger et al. 2009; Haisenleder et al. 2008). Similarly, GnRH activates p38 (also known as stress-activated protein kinase, SAPK) in these three model systems (Coss et al. 2007; Roberson et al. 1999), and this has been reported to have no role in LHβ, FSHβ, or αGSU subunit transcription (Haisenleder et al. 2008; Liu et al. 2002a; Roberson et al. 1999) or to mediate GnRH effects on FSHβ transcription in LβT2 cells (Bonfil et al. 2004; Coss et al. 2007). GnRH also activates ERK5, and this is thought to contribute to activation of FSHβ transcription in LβT2 cells (Lim et al. 2009).
Although Gq is the major mediator of GnRHR action, there is also evidence for regulation of the adenylyl cyclase/cyclic AMP (cAMP)/protein kinase A (PKA) pathway via Gs or Gi. GnRH was reported to increase cAMP production by pituitary cells (Borgeat et al. 1972), by gonadotrope-derived LβT2 cells (Lariviere et al. 2007; Liu et al. 2002b), and in heterologous GnRHR expression systems (Arora et al. 1998). GnRHR coupling to Gs has however remained controversial, because it was reported not to increase cAMP in some models (Conn et al. 1979; Horn et al. 1991), and where it does, some reports emphasize mediation by Gs (Liu et al. 2002b; Stanislaus et al. 1998), whereas others show GnRHR coupling exclusively to Gq (Grosse et al. 2000) or cAMP accumulation mediated by Ca2+/calmodulin-sensitive adenylyl cyclases (Lariviere et al. 2007). GnRHR apparently activate Gi in some cancer cell lines including JEG-3 human choriocarcinoma cells and BPH-1 human benign prostate hyperplasia cells (Maudsley et al. 2004), but perhaps the most compelling evidence for GnRHR coupling to multiple G-proteins comes from work with immortalized GnRH-expressing neurons where the endogenous mouse GnRHR of GT1-7 cells mediates the activation of Gs, Gi, and Gq as revealed by GnRH-stimulated release of G-protein subunits from membranes as well as associated functional responses (Krsmanovic et al. 2003). In LβT2 cells, a cAMP FRET sensor study (Tsutsumi et al. 2010) revealed that GnRH pulses cause pulsatile increases in cAMP and also that with constant stimulation, effects of GnRH on cAMP were more transient than its effects on Ca2+ or DAG (Tsutsumi et al. 2010). Furthermore, more recent work revealed that the GnRHRs interact directly with the proto-oncogene SET and that, in LβT2 cells, the SET protein facilitates cAMP production while inhibiting GnRHR-mediated elevation of cytoplasmic Ca2+ concentration (Avet et al. 2013).
Together, such studies highlight the possibility that GnRHR mediate effects on multiple heterotrimeric G-proteins and that the balance of signaling via these effectors varies with cell context and stimulation paradigm.
Regulation of cAMP levels by GnRH appears to have little or no acute effect on gonadotropin secretion but gonadotropin subunit promoters contain CREs (cAMP response elements), providing a direct mechanism for regulation by the cAMP/PKA pathway. In αT3-1 cells GnRH caused a 4–5-fold increase in phospho-CREB (CRE-binding protein) binding (Duan et al. 1999), and cAMP stimulates transcription of the mouse, rat, and human αGSU genes (Delegeane et al. 1987; Maurer et al. 1999). Moreover, a cAMP analogue increased αGSU mRNA in rat pituitary cells, although it did not alter mRNA levels for LHβ or FSHβ (Haisenleder et al. 1992). Nevertheless, it is possible that the ERK cascade mediates gonadotropin promoter CRE activation rather than the cAMP/PKA pathway (Brown and McNeilly 1999; Burger et al. 2004; Counis et al. 2005; Harris et al. 2002; Levi et al. 1998) as CREB integrates multiple inputs, being regulated not only by PKA but also by MAPKs, CaMKs, and PKC (Berridge 2012). Two known substrates of JNK (c-Jun and ATF-2) bind the CRE domain of the αGSU promoter (Haisenleder et al. 2005). Indeed, GnRH acts via p38 and JNK to phosphorylate ATF-2 and upon phosphorylation ATF-2 binds the CRE element within the c-Jun proximal promoter. Functional ATF-2 is needed for GnRH-mediated induction of both c-Jun and FSHβ (Fox et al. 2009). GnRH also increases ATF-3 expression, and ATF-3 is recruited along with c-Jun and c-Fos to CREs on the αGSU promoter that are essential for GnRH-stimulated αGSU gene expression (Chu et al. 2009).
In addition to the canonical Gq pathway that mediates GnRH effects on gonadotropin synthesis and secretion, it is important to recognize that GnRH activates additional signaling intermediates for which physiological roles are largely unknown. Thus, in addition to PLC, GnRH activates phospholipases D and A2 (Naor 2009), which hydrolyze phosphatidylcholine to produce phosphatidic acid (PA) and arachidonic acid (AA), respectively. PA and AA products (prostaglandins, thromboxanes, and leukotrienes) are thought to mediate GnRH signaling, and, conversely, GnRHR can activate a DAG kinase that phosphorylates DAG to produce PA (Davidson et al. 2004a). Similarly, although most work on cyclic nucleotide signaling has focused on cAMP, gonadotropes also express neuronal nitric oxide synthase (nNOS) which generates nitric oxide (NO), and thereby stimulates cyclic GMP production by NO activation of soluble guanylyl cyclase. Of particular interest here is that GnRH is not only able to increase expression of nNOS but would also be expected to cause a Ca2+/CaM-dependent activation of nNOS and that NO (which is membrane permeant and labile) has the potential to regulate guanylyl cyclase in gonadotropes and in neighboring cells (Garrel et al. 1997; Lozach et al. 1998). It has also been shown that GnRHRs mediate activation of the Wnt/β-catenin signaling pathway (Gardner et al. 2007), as well as proline-rich tyrosine kinase-2 (Davidson et al. 2004b) and AMP-activated protein kinase (AMPK), and the latter effect is implicated in control of LHβ gene transcription providing a potential common link between GnRH regulation and reproductive disorders due to metabolic dysregulation of gene expression (Andrade et al. 2013). Another particularly interesting feature here is that localization within the plasma membrane is crucial for GnRHR signaling. Indeed, GnRHRs are constitutively expressed in specialized plasma membrane micro-domains termed rafts, where they are co-localized with important effectors and GnRHR signaling (at least to ERK) is dependent on the integrity of such rafts (Bliss et al. 2007; Navratil et al. 2003; Navratil et al. 2010). A comprehensive overview of the signaling network in LβT2 cells (Fink et al. 2010) is available as a process diagram at http://tsb.mssm.edu/pathwayPublisher/GnRHR_Pathway/GnRHR_Pathway_ index.html.
Trafficking, Compartmentalization, and Desensitization of GnRHR
It has long been known that sustained agonist exposure causes activation followed by desensitization of GnRH-stimulated gonadotropin secretion that can be avoided with pulsatile stimulation. Indeed, early physiological studies revealed that GnRH pulses support circulating gonadotropin levels in ovariectomized primates, whereas sustained stimulation caused them to plummet, an effect that is reversed on return to pulsatile stimulation (Belchetz et al. 1978). GnRH causes GnRHR internalization, and this could certainly contribute to desensitization of GnRH-stimulated gonadotropin secretion. Sustained stimulation of GPCRs typically elicits a process known as rapid homologous receptor desensitization, in which G-protein receptor kinases phosphorylate Ser and Thr residues, most often within the receptor’s COOH-terminal tail, facilitating binding of nonvisual arrestins (arrestins 2 and 3). The arrestins prevent G-protein activation and also target the desensitized receptors for internalization, most often via clathrin-coated vesicles (CCVs) (Bockaert et al. 2003; Pierce and Lefkowitz 2001). Although GnRH was known to cause GnRHR internalization via CCVs (Hazum et al. 1980; Jennes et al. 1984), the cloning of mammalian type I GnRHR revealed most remarkably the complete absence of a COOH-terminal tail (Millar et al. 2004; Sealfon et al. 1997; Tsutsumi et al. 1992). Equally remarkable is the fact that all nonmammalian GnRHRs cloned to date have such tails, indicating that mammalian type I have undergone a period of rapidly accelerated molecular evolution with the advent of mammals being associated with the loss of COOH-terminal tails. In fact, it was established that type I mammalian GnRHR (where explored) do not rapidly desensitize or undergo agonist-induced phosphorylation or arrestin binding. Moreover, although they do show agonist-induced internalization, the process is relatively slow and is arrestin independent (Blomenrohr et al. 2002; Chen et al. 1995; Davidson et al. 1994; Finch et al. 2009; Heding et al. 1998; Hislop et al. 2000, 2001; McArdle et al. 1999; Pawson et al. 1998; Vrecl et al. 1998). Conversely, nonmammalian GnRHRs or type II mammalian GnRHRs (with COOH-terminal tails) do undergo agonist-induced phosphorylation, arrestin binding, and/or arrestin-dependent rapid homologous desensitization and are desensitized and internalized more rapidly than type I mammalian GnRHR. Furthermore, fusing the COOH-terminal of various nonmammalian GnRHRs to type I mammalian GnRHR can facilitate rapid desensitization, arrestin binding, and internalization (Finch et al. 2009; Hanyaloglu et al. 2001; Heding et al. 1998, 2000; Hislop et al. 2005). The fact that GnRH effects do undergo homologous desensitization seems initially at odds with the lack of desensitization of type I mammalian GnRH, but in reality just points to the importance of alternative downstream mechanisms as discussed in more detail below.
Arrestins are well known as terminators of GPCR signaling, but they can also act as scaffolds to mediate signaling (Pierce and Lefkowitz 2001). Notably, they bind MAPK cascade components so that some GPCRs can switch between two distinct modes of signaling with two waves of ERK activation, the first mediated by G-protein activation and the second reflecting G-protein-independent activation of arrestin-scaffolded ERK (Luttrell and Lefkowitz 2002; Shenoy and Lefkowitz 2003). This raised the possibility that the latter might be selectively engaged by GnRHR with COOH-terminal tails and consistent with this, it was shown that heterologously expressed mouse GnRHR mediate only G-protein-dependent ERK activation whereas a Xenopus laevis GnRHR (XGnRHR) provoked both G-protein- and arrestin-mediated ERK activation (Caunt et al. 2006a, c). A third area in which the absence or presence of GnRHR C-tails is important is for cell surface GnRHR expression. Here the key observation is that a large proportion of GnRHRs are actually intracellular (Brothers et al. 2006; Finch et al. 2009; Finch et al. 2008; Janovick and Conn 2010a, b; Janovick et al. 2012; Sedgley et al. 2006), as shown by work with human (h)GnRHR mutants that cause infertility and were found to be nonfunctional because of impaired trafficking rather than impaired signaling (Conn and Janovick 2009; Conn and Ulloa-Aguirre 2010; Janovick et al. 2009; Tao and Conn 2014; Ulloa-Aguirre and Conn 2009). Even wild-type hGnRHRs are relatively poorly expressed at the cell surface, and the presence of a primate specific Lys191, the absence of a second N-terminal glycosylation site, and the absence of a COOH-tail are all implicated in poor cell surface expression of hGnRHR (Conn and Janovick 2009; Conn and Ulloa-Aguirre 2010; Davidson et al. 1995; Janovick et al. 2009; Tao and Conn 2014; Ulloa-Aguirre and Conn 2009). Indeed, quantitative immunofluorescence revealed that <5% of HA-tagged GnRHRs are at the cell surface in several heterologous expression systems and that this value can be increased as much as 10–50-fold for GnRHR with COOH-terminal tails (Finch et al. 2008, 2010). Cell-permeant GnRHR ligands are currently being developed as potential orally active GnRHR antagonists (Betz et al. 2008) and the proportion of hGnRHRs at the cell surface can also be increased (10–20-fold) by non-peptide indole antagonists (Finch et al. 2008, 2010). Such compounds can rescue signaling by trafficking-impaired GnRHR mutants, acting as pharmacological chaperones (pharmacoperones) to aid the folding of endoplasmic reticulum (ER)-resident GnRHR into a suitable conformation to meet ER exit quality control criteria, thereby facilitating GnRHR trafficking to the cell surface (Conn and Janovick 2009; Conn and Ulloa-Aguirre 2010; Janovick and Conn 2010b; Janovick et al. 2009; Ulloa-Aguirre and Conn 2009). Perhaps the most exciting aspect of this work is the potential for such compounds to be used clinically to restore function of mutant receptors with impaired trafficking, and recent work provided proof of concept, using a knock-in mouse model with a recessive E90K mutation in the GnRHR (Stewart et al. 2012). This mutation impairs trafficking of GnRHR to the cell surface by causing ER retention. This causes hypogonadotropic hypogonadism in humans as well as in the mice, and pharmacoperone therapy restored testis function in this misfolded GnRHR model (Janovick et al. 2013).
Extrapituitary GnRHR, Context Dependence, and Ligand Bias
GnRHR expression is not restricted to the pituitary as they are found in many normal and neoplastic tissues. Thus, GnRHRs have been found in the brain, placenta, endometrium, ovary, breast, testes, and prostate, where they may be activated by locally produced GnRH (Harrison et al. 2004). Here, some of the earliest studies suggested a paracrine role with GnRHR expression in Leydig cells and GnRH production by Sertoli cells as well as well as effects of GnRH agonists on steroidogenesis in cultured testes (Bahk et al. 1995; Botte et al. 1998; Dufau et al. 1984; Harrison et al. 2004). Interestingly, rat testes have been shown to express high-affinity GnRHRs that mediate GnRH effects on steroidogenesis in vitro (Huhtaniemi et al. 1985), but blockade of testicular GnRHR did not alter Leydig cell function in vivo (Huhtaniemi et al. 1987) and early work showed that GnRHRs are not present in human gonadal tissues (Clayton and Huhtaniemi 1982). In general, physiological roles for extrapituitary GnRHR remain elusive, but interest in this field is fueled by the fact that GnRH analogs can stimulate apoptosis and can inhibit proliferation and migration, in cell lines derived from cancers of such tissues (Eidne et al. 1987). Thus, GnRH agonists can inhibit proliferation and/or migration of prostate cancer cell lines which together with evidence for GnRHR expression in reproductive duct cancers suggests a local role in tumor growth and metastasis (Cheng and Leung 2005; Cheung and Wong 2008; Franklin et al. 2003; Limonta et al. 2012; Limonta et al. 2003; Montagnani Marelli et al. 2009; Wang et al. 2010). There is also considerable interest in the possibility that such receptors may be targeted with cytotoxins conjugated to GnRH analogs. Notably a cytotoxin consisting of the agonist D-Lys6GnRH covalently coupled to doxorubicin (AEZS-108) is undergoing clinical trials for treatment of breast, endometrial, ovarian, and prostate cancers (Engel et al. 2012, 2016).
Interestingly, major functional differences have been reported between pituitary and extrapituitary GnRHR, most notable in early work suggesting that extrapituitary GnRHRs have lower affinity for peptide ligands than their pituitary counterparts, signal via Gi rather than via Gq, and are unable to distinguish agonists from antagonists in the same way as pituitary GnRHR do (Emons et al. 1998; Everest et al. 2001; Franklin et al. 2003; Grundker et al. 2001; Imai et al. 1997; Limonta et al. 2012). It was initially suspected that this reflected expression and activation of distinct receptors in different cell types, but this seems unlikely because, as noted above, the type II GnRHR pseudogene that does not encode functional GnRHR (Stewart et al. 2009), and in some hormone-dependent cancer cell line effects of GnRH (and, indeed, effects of GnRH-II), can be prevented by knockdown of type I GnRHR (Montagnani Marelli et al. 2009). The simplest alternative possibility is that GnRHRs are capable of activating multiple upstream effectors (i.e., G-proteins ), that the efficiency with which they do so is dependent on the relative amounts of such effectors in their immediate vicinity, and that this varies from one cell type to another. Although the endogenous GnRHR of breast cancer (MCF7) and prostate cancer (PC3) cells has been shown to mediate direct antiproliferative effects of GnRHR ligands, in our hands these cells did not express measurable GnRHR, as judged by binding and functional assays (Everest et al. 2001; Finch et al. 2004; Franklin et al. 2003). However, when recombinant adenovirus was used to express type I GnRHR in them, the heterologously expressed GnRHR had similar binding affinity, ligand specificity, and Gq coupling to the native GnRHR in gonadotropes. Moreover, activation of these receptors did reduce proliferation with effects apparently mediated by Gq rather than Gi. These experiments are consistent with a role for extrapituitary GnRHRs as regulators of cell fate in hormone-dependent cancer cells, but it remains unclear why the native type I GnRHR of GnRHR-positive breast and prostate cancer cells should mediate proliferation inhibition by a distinct mechanism to the type I GnRHR expressed heterologously in GnRHR-negative versions of the same cells. Cell context-dependent behavior was also seen when fluorescence microscopy was used to explore receptor compartmentalization, however (Finch et al. 2008; Sedgley et al. 2006). This revealed that <1% of HA-tagged hGnRHRs are at the cell surface in MCF7 and prostate cancer (DU145) cell lines and that this proportion is >5-fold higher in gonadotrope-lineage LβT2 cells.
Receptor dimerization may also be relevant to context-dependent GnRHR signaling as it is now well established that many GPCRs form dimers of higher-order oligomers and that such oligomerization can facilitate signaling and may be either constitutive or ligand induced. In some of the earliest work supporting this idea, Conn’s groups showed that GnRH antagonists could be converted to agonists by addition of bivalent antibodies to the ligand. No such effect was seen with monovalent antibodies so the simplest interpretation is that antibody-mediated GnRHR cross-linking is sufficient for activation (Conn et al. 1982), presumably because this cross-linking facilitates or mimics GnRHR dimerization. There is also now considerable evidence that agonists (but not antagonists) cause GnRHR oligomerization or at least bring GnRHR sufficiently close to one another to mediate FRET or BRET (Cornea et al. 2001; Horvat et al. 2001; Kroeger et al. 2003). However, the cellular compartments in which GnRHR oligomers form and the regulation of oligomer assembly remain poorly understood, and it has not been shown that oligomerization is required for GnRH signaling. It is also now well established that many GPCRs can form heterodimers (or higher-order oligomers) with other GPCRs. For example, type V somatostatin receptors (SSTR5) form heterodimers with type II dopamine receptors (D2R), both of which are Gi-coupled GPCRs (Rocheville et al. 2000). Some of the best evidence for this comes from early functional rescue studies showing, for example, that when signal dead (C-terminal truncated) SSTR5 are co-expressed with D2R, this rescues the ability of SST to activate Gi (Rocheville et al. 2000). To our knowledge dimerization of GnRHR with other GPCRs has not been explored, but if this were to occur, it could potentially facilitate GnRH signaling to G-proteins activated by the partner GPCR, and this could confer context-dependent signaling as the repertoire of partner GPCRs available would presumably also be dependent on cell type.
Finally, ligand bias (also known as biased signaling or pluridimensional efficacy) is another concept that may be important for cell context-dependent GnRHR signaling. Here, the fundamental idea is that GPCRs actually have multiple active conformations that may couple differentially to different effectors. They may also be differentially stabilized by different ligands, such that different ligands can bias signaling toward different effectors (Kenakin 2011; McArdle 2012). The simplest scenario is that there are two distinct active conformations, but in reality, for any given GPCR, there are thought to be many different tertiary structures in related groups of preferred conformations known as receptor ensembles (Kenakin 2011). If the effect of a given ligand on the distribution of receptors between possible conformations differs from one cell type to another (because other features of the receptors’ environments differ), ligand bias would itself be dependent on cell context. For cell context-dependent GnRHR effects, some experimental data cannot be easily explained without distinct active conformations of a single GnRHR type. Thus, the peptide “antagonist” cetrorelix is a pure antagonist of GnRH effects on inositol phosphate (IP) accumulation and gonadotropin secretion in pituitary cells, but it actually mimics antiproliferative effects of GnRH in some models (Grundker et al. 2004; Maudsley et al. 2004). Similarly, GnRH-I is more potent than GnRH-II at stimulation of IP accumulation by type I GnRHR in pituitary cells, which is the reverse of the situation for inhibition of proliferation in some models (Cheung and Wong 2008; Enomoto et al. 2004; Grundker et al. 2004; Hislop et al. 2000; Wang et al. 2010). Indeed, with only a single receptor target, ligand bias appears the most likely explanation for much data showing differences in ligand specificity when native GnRHR-mediated effects have been compared in different cell types (McArdle 2012). More direct evidence for ligand bias has been obtained in a number of models (Caunt et al. 2004; Davidson et al. 2004b; Lopez de Maturana et al. 2008; Maudsley et al. 2004) including a study comparing effects of GnRH analogues on different type I mammalian GnRHR-mediated responses. A series of GnRHR ligands all inhibited proliferation in JEG-3 cells and BPH-1 cells (both with native hGnRHR) and in SCL60 cells (which have exogenous rat GnRHR). They all apparently activated Gi and caused Gi-dependent inhibition of proliferation (Maudsley et al. 2004), and marked ligand bias was observed because GnRH-I stimulated IP accumulation; activated ERK, p38, and JNK; and inhibited proliferation, whereas a GnRH analogue (135–25) mimicked all other GnRH-I effects but failed to increase IP accumulation (Maudsley et al. 2004). Ligand bias at GnRHR is also evident in work on GnRHR localization and trafficking. As noted above, non-peptide pharmacoperones can increase GnRHR trafficking to the cell surface so work with these compounds provides a marked example of pluridimensional efficacy with non-peptide ligands acting as antagonists in terms of cell surface GnRHR signaling, but as agonists in terms of anterograde trafficking. This also indicates that the cell surface and intracellular GnRHRs have different conformations, which is not unexpected as most GnRHRs within the cell have apparently failed quality control criteria for ER exit, whereas those at the cell surface evidently have not. Experiments were performed with two peptide antagonists (antide and cetrorelix), which, being membrane-impermeant, did not have access to intracellular GnRHR and, as expected, had no effect on the proportion of hGnRHR at the cell surface (Finch et al. 2008). However, when the XGnRHR COOH-tail was added to the hGnRHR in order to increase cell surface expression, the peptide antagonists further increased cell surface expression of the chimeric receptor. Although the effect was modest, it raised the possibility that the peptides might act at the surface to increase GnRHR number by slowing internalization. Indeed, a pronounced synergism can occur when a non-peptide chaperone is used to increase GnRHR trafficking to the cell surface and a peptide antagonist is used to slow internalization from the cell surface (Finch et al. 2010). Thus, although the mechanisms are not known, this work clearly demonstrates that the cetrorelix-occupied hGnRHR is functionally distinct from the unoccupied receptor and that cetrorelix can be a pure antagonist for GnRH-I-stimulated IP accumulation and Ca2+ signaling and an inverse agonist for GnRHR internalization. Importantly, this form of ligand bias was seen with a compound that is used clinically and in gonadotropes, the only proven targets for GnRHR-directed therapy. Ligand bias has a number of implications for understanding and manipulating GnRHR signaling in pituitary and extrapituitary sites, but most importantly, it raises the exciting possibility of developing ligands that more selectively engage therapeutically beneficial responses. Here an obvious strategy would be to develop GnRHR ligands that are antagonists for Gq-mediated stimulation of gonadotropin secretion from the pituitary and agonists for direct Gi-mediated antiproliferative effects on hormone-dependent cancers.
Additional Hormonal and Local Regulators of Gonadotropes
In addition to GnRH, gonadotropes are targets for numerous other hormonal and local regulators. It is well established, for example, that gonadal steroids (estrogen, progesterone, and testosterone) mediate feedback within the hypothalamic-gonadal axis, acting centrally to influence GnRH secretion and at the pituitary to modulate GnRH effects on gonadotropes. In females estrogen exerts positive and negative feedback effects with positive feedback at the pituitary level being crucial for the preovulatory gonadotropin surge, whereas in males, testosterone exerts negative feedback effects both centrally and at the pituitary. At the pituitary level, testosterone influences expression of GnRHR, gonadotropin subunit expression, and GnRH signaling (Clayton and Catt 1981; Kaiser et al. 1993; Winters et al. 1992), and modulation of GnRH effects on cytoplasmic Ca2+ was shown to be dependent on local conversion of testosterone to dihydrotestosterone by 5α-reductase (Tobin and Canny 1998). Interestingly, a recent study in which the male reproductive axis of sheep was modeled mathematically incorporated regulation of GnRH pulsatility by central testosterone-mediated negative feedback (but not feedback at the pituitary) and illustrated the importance of a time delay that was attributed to conversion of testosterone to estrogen (Ferasyi et al. 2016). The proteins inhibin and activin also feedback from the gonads to inhibit and activate (respectively) FSH production but, in addition to this endocrine loop, are also synthesized in gonadotropes and act locally to regulate FSH synthesis. They are members of the TGF-β family and act via receptors with intrinsic serine/threonine kinase activity to exert effects that are modulated by locally produced follistatin (Bilezikjian et al. 2006).
Pituitary adenylyl cyclase-activating polypeptide (PACAP) is another ligand thought to mediate both endocrine and local regulation of gonadotropes. It was isolated from hypothalamic extracts based on its ability to stimulate cAMP production in pituitary cell cultures (Miyata et al. 1989) and has higher concentration in the portal circulation than in the periphery, supporting a hypothalamic-hypophysiotrophic hormone role (Counis et al. 2007; Rawlings and Hezareh 1996; Schomerus et al. 1994; Winters and Moore 2011). It has two major forms (PACAP27 and PACAP38) which act via three GPCRs: VPAC1 and VPAC2 that have similar affinity for PACAP and VIP (vasoactive polypeptide) and PAC1 that has higher affinity for PACAP than for VIP. PACAP causes a PAC1-mediated activation of both Gs and Gq in gonadotropes and gonadotrope-derived cell lines and influences gonadotropin secretion and synthesis both alone and by modulation of GnRH effects. It also targets PAC1 receptors on folliculo-stellate cells and evidence exists for its production by folliculo-stellate cells and gonadotropes, suggesting it to be an autocrine regulator of both (Denef 2008; Winters and Moore 2011). Interestingly, PACAP increases follistatin expression by folliculo-stellate cells and gonadotropes and may thereby modulate activin signaling in the pituitary (Winters and Moore 2011). Other ligands that act via GPCRs on gonadotropes include oxytocin, endothelin 1, galanin, β-endorphin, neuropeptide Y, and nucleotides (Denef 2008). The latter are of particular interest as ATP, ADP, uridine 5′ diphosphate, and uridine 5′ triphosphate (UDP and UTP) act via purinergic receptors that include both GPCRs and ligand-gated ion channels. P2X receptors (P2XRs) are ATP-activated ligand-gated ion channels that are permeable to Na+, K+, and Ca2+ so their activation characteristically increases Ca2+ entry across the plasma membrane either directly or as a consequence of membrane depolarization. P2Y receptors (P2YRs) and adenosine receptors (ARs) are GPCRs that are preferentially activated by ATP and adenosine (respectively), and since both classes include Gq-coupled receptors, their activation is also often associated with elevation of cytoplasmic Ca2+. Anterior pituitary cell expresses at least six types of P2XRs, two types of P2YR, and all four types of AR (Stojilkovic et al. 2010a; Stojilkovic and Koshimizu 2001). Early work revealed that ATP and UTP act via P2YRs in gonadotropes to drive a Gq-mediated increase in cytoplasmic Ca2+ (Chen et al. 1994, 1995), whereas later work revealed expression of P2XR in gonadotropes and, indeed, in all secretory cell types of the pituitary (Stojilkovic et al. 2010a, b; Stojilkovic and Koshimizu 2001). Pituitary cells store ATP in secretory vesicles and co-release it with hormones during agonist-stimulated exocytosis (Denef 2008), underlining the potential for a positive feedback loop in which GnRH stimulates ATP secretion and ATP stimulates LH secretion, either alone or by amplification of GnRHR-mediated LH secretion (Denef 2008). As noted above, GnRH increases nNOS expression and thereby increases cGMP production mediated by NO and soluble guanylyl cyclase, but pituitary cells are also responsive to natriuretic peptides that act via cell surface receptors with intrinsic guanylyl cyclase activity (Fowkes and McArdle 2000). Of particular interest here is C-type natriuretic peptide (CNP) that specifically activates NPR-2 (natriuretic peptide receptor 2, also known as guanylyl cyclase B) to increase cGMP levels in primary cultures of pituitary cells and in gonadotrope-derived cell lines (Fowkes and McArdle 2000; McArdle et al. 1994a; Thompson et al. 2009). CNP is highly expressed in the pituitary with particularly strong expression in gonadotropes where it is located largely in secretory vesicles (McArdle et al. 1996). Deletion of genes encoding both CNP and NPR-2 causes infertility (Chusho et al. 2001; Tamura et al. 2004), and although CNP does not stimulate LH secretion, it can stimulate the αGSU promoter (Thompson et al. 2009). Together these data suggest that autocrine and/or paracrine regulation of both particulate and soluble guanylyl cyclases influences gonadotrope function.
Gonadotropes (like all cells) sense multiple chemicals in their environment, and these different inputs act in combination. The importance of this combinatorial input is illustrated by cyclic nucleotide signaling; although GnRH increases cAMP and cGMP production in some models, its effects are much less pronounced than those of PACAP and CNP, and in the presence of PACAP, GnRH actually inhibits cAMP production (McArdle and Counis 1996; McArdle et al. 1994b), just as it actually inhibits cGMP production in the presence of CNP in gonadotrope cell lines (McArdle et al. 1994a). This raises the question of which effects predominate in normal gonadotropes and more generally, the issue that effects of GnRH seen in isolation and in vitro may differ from those seen in more complex and physiologically relevant extracellular environments. Furthermore, gonadotropes not only sense and respond to their environment but also influence it, as highlighted above for ATP, NO, CNP, PACAP, and inhibin, all of which are likely secreted in response to GnRH (Denef 2008). Recent work has shown how GnRH-stimulated secretion of inhibin and of growth differentiation factor 9 form incoherent feedforward loops controlling FSH production, highlighting the fact that the extracellular space can also mediate GnRH signaling in a concept that was termed “outside the box signaling” (Choi et al. 2012, 2014; Pincas et al. 2014).
Pulsatile GnRH Signaling
GnRH is secreted from hypothalamic neurons in pulses that drive pulses of gonadotropin release and are essential for normal reproduction (Clarke and Cummins 1982; Dierschke et al. 1970). Its effects are dependent on pulse frequency, as shown in early studies in which constant GnRH suppressed LH and FSH secretion, whereas restoration of GnRH pulses restored gonadotropin secretion (Belchetz et al. 1978; Knobil 1980; Wildt et al. 1981). In humans and other primates, GnRH pulses have a duration of a few minutes and intervals of 30 min to several hours, with pulse frequency differing under different physiological conditions. For example, changes in GnRH pulse frequency drive changes in reproductive status during development, with an increase in pulse frequency driving the increased gametogenesis and gonadal steroid production at puberty (Sisk and Foster 2004). Similarly, GnRH pulse frequency varies through the menstrual cycle, increasing before ovulation and contributing to generation of the preovulatory gonadotropin surge (Ferris and Shupnik 2006; Marshall et al. 1993). Moreover, stimulation paradigm is crucial for therapeutic intervention because agonist pulses can maintain or increase circulating gonadotropin levels whereas sustained agonist stimulation (after initial activation) reduces them, causing the chemical castration that is exploited in treatment of breast cancer, prostate cancer, and other sex steroid hormone-dependent conditions (Bliss et al. 2010; Ferris and Shupnik 2006; Marshall et al. 1993; Millar et al. 2004). Similar mechanisms mediate responses to sustained and pulsatile GnRH as for both, GnRH activates Gq and effectors including the Ca2+/calmodulin/calcineurin/NFAT module and ERK (Armstrong et al. 2009a, b; Bliss et al. 2009, 2010; Ciccone and Kaiser 2009; Ferris and Shupnik 2006; Millar et al. 2004). Moreover, pituitary ERK is essential for reproduction (Bliss et al. 2009) consistent with its role as an effector of pulsatile GnRHR in vivo.
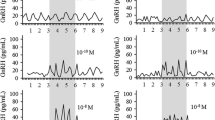
A fundamental question here is why GnRH is secreted in pulses, and we have explored this by monitoring effects of pulsatile GnRH on the nuclear translocation of ERK2-GFP as a readout for Raf/MEK/ERK activation and of NFAT-EFP as a readout for Ca2+/calmodulin activation (Fig. 2). We found that each 5 min pulse of GnRH elicits a rapid and transient ERK2-GFP translocation response and a somewhat slower NFAT-EFP translocation response (Armstrong et al. 2009a, 2010). With 30 min pulse intervals, there was insufficient time for the NFAT-EFP reporter to return to pre-stimulation values so that a cumulative or “saw-tooth” response was observed. Indeed, the NFAT-EFP translocation response to GnRH pulses was comparable to that seen with constant stimulation (Armstrong et al. 2009a), whereas the ERK2-GFP translocation response was not. This demonstrates two fundamental reasons why pulsatile signals are so prevalent in biological systems: first, the increase in efficiency (similar system output with pulsatile vs. constant stimulation) and, second, the possibility for selective effector activation (with 30 min pulses of GnRH causing maximal NFAT translocation and submaximal ERK activation). To explore this further, we developed an ordinary differential equation-based mathematical model of a GnRHR signaling network that was trained on experimental data (Perrett et al. 2014; Tsaneva-Atanasova et al. 2012) (Fig. 2). Model simulations were used to predict responses with varied GnRH concentration, pulse width and pulse frequency in order to explore system sensitivity to these distinct features of the dynamic input (Perrett et al. 2014; Tsaneva-Atanasova et al. 2012). These simulations revealed that a tenfold increase in GnRH concentration does not cause a tenfold increase in responses, primarily because it does not cause a tenfold increase in GnRHR occupancy. Moreover, increases in system outputs caused by a tenfold increase in GnRH pulse width are less than the increases caused by a tenfold increase in pulse frequency. Thus, the system is an integrative tracker (in that it is sensitive to pulse amplitude, frequency, and width, all of which influence the integral of the input), but there is certainly not a simple 1:1 relationship between integrated input and output. Instead, the kinetics of receptor occupancy and downstream effector activation create a system that is relatively robust to changes in pulse width and concentration but is highly sensitive to changes in pulse frequency, the input variable known to vary under different physiological conditions in vivo (Perrett et al. 2014).

Live cell monitoring and mathematical modelling of pulsatile GnRH signaling to ERK and NFAT. Panels A and B: Hela cells were transduced with recombinant adenovirus for expression of ERK2-GFP or NFAT-EFP (as indicated), both of which translocate to the nucleus on activation. Live cell imaging was used to capture responses during 5 min pulses of 10−7 M GnRH at intervals of 30 min (circles), 60 min (upright triangles) or 120 min (inverted triangles). Nuclear:cytoplasmic ratios were calculated for each reporter. These were normalized to the value at time 0 and are offset on the vertical axes for clarity (i.e., NFAT-EFP data are offset by 0, 1, or 2 and ERK2-GFP values are offset by 0, 0.5 or 1). Note that each GnRH pulse caused nuclear translocation of each reporter (although the ERK2-GFP translocation responses had more rapid onset and offset) and that with high pulse frequencies, there was insufficient time for the NFAT-EFP to return to the pre-stimulation value. Note also, that there was no obvious desensitization, in that amplitudes did not reduce over time. Panels C and D: An ordinary differential equation-based model for GnRH signaling was developed and trained against wet lab data for pulsatile GnRH signaling to ERK and NFAT. The data shown are simulations for ERK and NFAT translocation in cells receiving 5 min pulses of 10−7 M GnRH offset precisely as described for panels A and B to illustrate the close agreement between the wet lab data and the model predictions (Adapted from Tsaneva-Atanasova et al. 2012)
Another fundamentally important feature of the system is that responses can be maximal at submaximal pulse frequency (Bedecarrats and Kaiser 2003; Ciccone and Kaiser 2009; Ciccone et al. 2010; Dalkin et al. 1989; Ferris and Shupnik 2006; Haisenleder et al. 1991; Kaiser et al. 1993; Kanasaki et al. 2005; Shupnik 1990; Weiss et al. 1990). Moreover, the frequency eliciting maximal responses is dependent on the output measured, as seen in work with luciferase reporters for gonadotrope signature genes (Bedecarrats and Kaiser 2003), where optimal GnRH pulse frequencies for expression of LHβ, FSHβ, αGSU, and GnRHR reporters differ (maximal responses at pulse intervals of 2 h for LHβ and FSHβ, 0.5 h for αGSU, and 1 h for GnRHR, in LβT2 cells). In ovariectomized rhesus monkeys bearing hypothalamic lesions which reduced circulating LH and FSH to undetectable levels, hourly GnRH pulses favored LH secretion whereas pulses every 3 h favored FSH secretion (Wildt et al. 1981). Additional in vivo studies with GnRH-deficient men recapitulated this observation (Gross et al. 1987; Spratt et al. 1987), as do in vitro studies using pituitary cultures (Bedecarrats and Kaiser 2003; Dalkin et al. 1989; Ferris and Shupnik 2006; Haisenleder et al. 1991; Kaiser et al. 1993; Shupnik 1990; Weiss et al. 1990; Yasin et al. 1995). Moreover, in polycystic ovarian syndrome, the most common cause of infertility in women of reproductive age, there is an increase in GnRH activity and predominance of high-frequency GnRH pulses that are thought to drive the observed elevation of LH and suppression of FSH and the associated disruption of reproductive cycles (Ciccone et al. 2010; Hoffman and Ehrmann 2008).
In essence, the data above all illustrate the fact that for many GnRH effects, there is a non-monotonic (bell-shaped) pulse frequency-response curve. This could reflect the existence of feedback or feedforward loops (Krakauer et al. 2002), but the nature of these loops is unclear. Rapid homologous receptor desensitization can be excluded as a potential negative feedback loop because type I mammalian GnRHR do not show this behavior (as discussed earlier). However, GnRH does downregulate cell surface GnRHR, and a mathematical model of GnRH signaling predicts pulse frequency-dependent desensitization of upstream signals as a consequence of GnRHR downregulation (Washington et al. 2004). Alternative possible mechanisms for desensitization to GnRH have been described, including GnRHR-mediated induction of RGS-2 (regulator of G-protein signaling-2) which displays GTPase-activating protein activity and is known to inhibit Gα signaling (Karakoula et al. 2008; Wurmbach et al. 2001), direct interaction of GnRHR with SET protein which can inhibit Gα binding (Avet et al. 2013), induction of MAPK phosphatases (MKPs) which would modulate GnRHR-mediated ERK signaling (Lim et al. 2009), downregulation of IP3 receptors (Willars et al. 2001; Wojcikiewicz et al. 2003), induced expression of calmodulin-dependent small G-protein Kir/Gem (kinase-inducible Ras-like protein/GTP-binding protein overexpressed in skeletal muscle) (Ferris and Shupnik 2006), and ERK-mediated negative feedback (Armstrong et al. 2009b; Caunt et al. 2006a). However, such responses have been explored primarily with constant stimulation paradigms and may well have little or no effect with pulsatile stimulation. A thorough theoretical examination of pulse frequency decoding mechanisms also revealed how receptor dimerization can generate non-monotonic frequency-response relationships (Fletcher et al. 2014), and this is of particular interest in light of early studies suggesting that dimerization of GnRHR could elicit signaling (Conn et al. 1982, 1987), as well as work showing that agonists (but not antagonists) bring GnRHR closer to one another (Cornea et al. 2001; Navratil et al. 2006). However, as noted above, it is not established that dimerization of normal GnRHR is a prerequisite for signaling; the live cell imaging experiments described above also provide some insight here, as the ERK2-GFP and NFAT-EFP translocation responses were both reproducible with repeated GnRH pulses (Fig. 2) and the signals passing from the cytoplasm to the nucleus showed increasing monotonic frequency-response relationships. In support of this, Egr-1-responsive and NFAT-responsive luciferase reporters used as transcriptional readouts for ERK and NFAT activation both show maximal responses at maximal GnRH pulse frequency (Armstrong et al. 2009a, 2010).
If signaling inputs to the nucleus show increasing monotonic frequency-response relationships, the obvious possibility is that feedback and/or feedforward regulatory loops within the nucleus underlie the observed non-monotonic frequency-response relationships for gene expression. This has been explored most extensively for the FSHβ promoter, for which a number of incoherent feedforward loops have been described. These are signaling modules that fan out from an upstream node and reconverge at a downstream node and for which the two divergent branches have different overall signs (i.e., positive and negative effects). Thus, for example, stimulation of FSHβ gene expression by GnRH is, in part, mediated by its ability to phosphorylate and activate the transcription factor CREB, but GnRH can also increase expression of the inducible cAMP early repressor (ICER), which inhibits the effect of CREB, providing both positive and negative inputs to the promoter (Ciccone et al. 2010; Thompson et al. 2013). As noted above, pulsatile stimulation provides the potential for specificity in effector activation, and the inhibitory (ICER-mediated) loop is preferentially activated at high GnRH pulse frequency so that transcriptional activation is greatest at submaximal pulse frequency. Similarly, it was shown that expression of Fos and Jun (positive regulators of FSHβ expression) is increased at lower GnRH pulse frequencies than needed for expression of negative regulators (the co-repressors SKIL, CREM, and TGIF1) suggesting regulation by an alternative incoherent feedforward loop in which SKIL and/or TGIF1 inhibits activation by AP-1 factors Fos and Jun (Mistry et al. 2011). In addition to these nuclear mechanisms, incoherent feedforward loops have been described in which the inhibitory branch is due to GnRH-stimulated protein secretion. In the first, it is mediated by secretion of inhibin-α, which has long been known to suppress FSH expression, and in the second, it is mediated by inhibition of the secretion of growth differentiation factor 9, an autocrine inducer of FSHβ expression in LβT2 cells (Choi et al. 2012, 2014; Pincas et al. 2014). These studies are of particular interest as they effectively extend the GnRH signaling network to the extracellular space (as outlined above for autocrine regulation).
We have also used mathematical modeling to explore possible frequency decoding mechanisms, taking our model trained against NFAT-EFP and ERK2-GFP translocation data (Figs. 2 and 3), so that these could then be used as inputs to the transcriptome. In doing so, it was assumed that two transcription factors act at separate sites on a common gene promoter (using NFAT as the first transcription factor and an undefined ERK-dependent transcription factor as the second one). Three distinct logic gates were considered: an “and-gate,” an “or-gate” or a “cooperative gate.” This model predicted bell-shaped frequency-response relationships when two transcription factors act cooperatively. The characteristic feature of maximal response at submaximal frequency was never seen with the and-gate or with the or-gate, and this behavior was predicted without negative feedback (Tsaneva-Atanasova et al. 2012). A particularly interesting feature of these simulations is that they revealed GnRH pulse frequency-response relationship may be plastic, in that varying rate constants for transcription factor activation and inactivation, or varying balance of signaling via NFAT and ERK-dependent transcription factors, influenced the frequencies at which maximal response occurred (Tsaneva-Atanasova et al. 2012). This modelling clearly does not show that the bell-shaped frequency-response relationships seen for transcriptional effects of GnRH are mediated by convergence of NFAT and ERK-dependent transcription factors. In fact, multiple pathways converge to mediate GnRH effects on transcription (Nelson et al. 1998), and the relative importance and mechanisms of integration of these inputs are undoubtedly promoter/enhancer specific. Moreover, the bell-shaped frequency-response relationships seen in this model rely on a mathematical description of cooperative convergence for which biological substrates have not been identified, so it will be important to develop and test mathematical models for the biological pathways described above. Nevertheless, a common feature of much work in this field is that it highlights mechanisms for generation of non-monotonic frequency-response relationships in the absence of upstream negative feedback. Indeed, it seems likely that pulsatile GnRH secretion and the resistance of type I mammalian GnRHR to desensitization both serve to minimize negative feedback and thereby place increasing reliance on such alternative mechanisms.

Generation of bell-shaped pulse frequency-response relationships by convergent signaling. A previously described mathematical model for GnRH signaling (Tsaneva-Atanasova et al. 2012) was used to simulate transcriptional responses driven by ERK and NFAT assuming that they converge at a common promoter with one of three logic gates: an “and-gate,” an “or-gate,” or a “cooperative” gate. Predicted transcriptional responses (area under the curve for time-course data) are shown in panel A, as a function of pulse frequency (5 min pulses of 10−7 M GnRH) for the target gene (here given the generic term GSU, but not indicating any particular gonadotropin subunit gene). Such simulations always yielded increasing monotonic frequency-response relationships when a single pathway was considered or when convergence was modelled with an and-gate or an or-gate. Bell-shaped frequency-response relationships were only obtained with cooperative convergence of these two pathways at the transcriptome (panels A and B) (Adapted from Tsaneva-Atanasova et al. 2012)
An Information Theoretic Approach to GnRH Signaling
Most work on GnRH signaling has entailed measurement of average responses from populations of cells, and the mechanistic modelling outlined above effectively considers the behavior of a single cell, assuming it to be representative of the population. These approaches ignore cell-cell variation but such variation is in fact inevitable because cell signaling is inherently stochastic. It is also crucial for the behavior of cell populations (Bowsher and Swain 2014) because each individual cell has to sense its environment and make appropriate decisions (to express or suppress given genes, to survive or die, to proliferate or differentiate, etc.). Cell-cell variation in response to GnRH has been documented for many years, from early work on gonadotropin secretion and Ca2+ mobilization (Lewis et al. 1989; McArdle et al. 1992; Stojilkovic and Catt 1995) and more recent studies using transcriptional readouts and/or high content imaging (Armstrong et al. 2009a, b, 2010; Caunt et al. 2012; Garner et al. 2016; Ruf et al. 2006, 2007). Information theory was developed to analyze electronic communication but can also be applied to biological systems, where it provides tools with which the influence of cell-cell variation on the reliability of sensing can be determined (Bowsher and Swain 2014; Bowsher et al. 2013; Brennan et al. 2012; Cheong et al. 2011; Selimkhanov et al. 2014; Uda et al. 2013; Voliotis et al. 2014). In this context, information is defined as the uncertainty about the environment that is reduced by signaling and can be quantified as the mutual information (MI) between two stochastic variables (Bowsher and Swain 2014). MI measures the quality of the inference (or “prediction”) of the signal from the response. It is measured in bits with an MI of 1 bit indicating that the system can unambiguously distinguish between two equally probable states of the environment. Importantly, estimation of MI doesn’t require knowledge of the mechanism by which information is transferred, and MI values are unaffected by transformations of the signal or response (Bowsher and Swain 2014). Several groups have applied information theoretic approaches to analysis of cell signaling, treating signaling pathways as noisy communication channels and quantifying the information that they do (or could) carry. The value of this approach can be illustrated by considering a signaling pathway with multiple levels, such as a MAPK cascade. It is well established that signal amplification can occur from one tier to the next in the cascade, but it is less well recognized that information about the input cannot actually increase from one level in the cascade to the next. In fact there is normally loss (and never gain) of information through signaling cascades and any increase in numbers of activated molecules must therefore be associated with increased variability (noise) through the cascade. There is considerable interest in understanding how cells mitigate loss of information through signaling pathways, and here negative feedback loops are of particular interest because they can reduce information transfer (by reducing dynamic range of the output) or protect it (by reducing cell-cell variability).
In a recent study, ppERK and nuclear translocation of NFAT-EFP were measured as activation readouts, and Egr1- and NFAT response element-driven fluorophore expression were measured as transcription activation by ERK and NFAT. Responses were measured in large numbers of individual GnRH-stimulated cells (Garner et al. 2016) and used to calculate MI between GnRH concentration and ppERK (I(ppERK;GnRH)). This revealed information transfer between GnRHR and ERK to be <1 Bit in HeLa cells transduced with Ad-GnRHR (Fig. 4). This is comparable to values obtained for cytokine and growth factor signaling in other systems (Garner et al. 2016), but is still surprisingly low for two reasons. First, the cells were typically stimulated with eight GnRH concentrations so there was a 3 Bit input, of which <1 Bit of information was transferred. Second, population-averaged measures consistently show responses to GnRH being graded over a wide range of GnRH concentrations, yet an MI of <1 implies that single cells cannot unambiguously distinguish between just two inputs (i.e., with and without GnRH). This was not due to use of a heterologous expression system because information transfer values were similar in HeLa cells (with exogenous GnRHR) and LβT2 gonadotropes (with endogenous GnRHR). It was also not restricted to the ERK pathway because information transfer from GnRHR to NFAT was <0.5 Bits in both cell models (Garner et al. 2016). Another possible explanation for low information transfer is that single time-point measures underestimate information transfer. This would be expected where cells infer inputs (i.e., GnRH concentrations) from trajectories of outputs (i.e., ppERK levels) over time (Selimkhanov et al. 2014). For example, time-course experiments revealed that I(ppERK;GnRH) is higher at 5 than at 360 min (Fig. 4), but this clearly does not mean that a cell obtains less information over 360 min than it had over 5 min. Instead, it shows that the 360 min snapshot underestimates information transferred over the 360 min stimulation. Measuring MI for ERK-driven transcription is an alternative approach that could be sensitive to ppERK trajectory, and, consistent with this, work with imaging readouts for ERK-driven transcription revealed more reliable sensing of PDBu than of GnRH in HeLa cells (Fig. 4), presumably because PDBu has a more sustained effect than GnRH on ppERK and causes a more marked increase in Egr1-driven zsGREEN expression (Garner et al. 2016). Thus the system senses sustained stimulation more reliably and must therefore be sensitive to the dynamics of ERK activation. This information theoretic approach was also applied to consider possible effects of negative feedback, focusing on ERK-dependent feedback (i.e., rapid transcription-independent and slow transcription-dependent feedback) and on receptor desensitization (i.e., by comparison of type I mammalian GnRHRs that do not rapidly desensitize and XGnRHRs that do). The overriding observation from these first statistical measures of information transfer via GnRHR is that it is not measurably influenced by the occurrence or absence of rapid receptor desensitization, but is influenced by downstream adaptive processes (i.e., ERK-mediated feedback) with optimal GnRH sensing at intermediate feedback intensities.

MI as an information theoretic measure of GnRH sensing. Panels A and B show concentration and time-dependent effects of GnRH and phorbol 12,13 dibutyrate (PDBu) on ERK activity in LβT2 cells, with nuclear ppERK reported in arbitrary fluorescence units (AFU). Single cell measures underlying these plots were also used to calculate MI between ppERK and each of these stimuli and these values are plotted (MI in Bits on vertical axis) against time in panel C. The cells were also transduced with recombinant adenovirus for expression of an ERK-driven transcription reporter (Egr1-zsGREEN). Panel D shows the concentration-dependence of GnRH and PDBu on zsGREEN expression (in AFU) at 360 min and the MI between zsGREEN and each of these stimuli is also shown for this time (Adapted from Garner et al. 2016)
Summary
Since GnRH was isolated and sequenced in the 1970s, there have been immense advances in our understanding of GnRH signaling. This ranges from the early work identifying Ca2+ as a mediator of stimulus-secretion coupling through subsequent work mapping the GnRH signaling network as well as the extensive studies of gene expression focusing on gonadotrope signature genes or using omics approaches to elucidate regulatory networks. The ever-increasing complexity of GnRHR signaling networks highlights the necessity for mathematical and statistical analyses as illustrated by recent information theoretic work on GnRH signaling, where emphasis was on the amount of information transferred rather than identifying components of the paths through which it is conveyed. From the outset the system has provided remarkable surprises. Notably, the initial paradoxical observation that a peptide purified as a gonadotropin-releasing factor actually reduces circulating gonadotropins and causes chemical castration on sustained stimulation in vivo. With receptor cloning came the equally surprising observation that mammalian type I GnRHR lack COOH-terminal tails and do not rapidly desensitize, so alternative mechanisms must underlie the desensitization of GnRH-stimulated gonadotropin secretion. Compartmentalization has also emerged as a crucial determinant of GnRHR function, as highlighted by the discovery that most hGnRHRs are actually intracellular as well as the fact that GnRHR signaling is dependent upon its location within the plane of the plasma membrane. Similarly, the importance of dynamics cannot be overestimated because the CNS provides GnRH pulses as a frequency-encoded signal to be decoded by gonadotropes. We still do not have a detailed understanding of how they do so or how GnRHR compartmentalization is controlled, let alone how these systems may be modulated by other hormonal or local inputs. Accordingly, the authors believe that a major research challenge for future work is to overlay space and time on existing schema for GnRH action, whereas the clinical challenge lies in translating the large amount of mechanistic information into genuine therapeutic benefit.
References
Al-Inany HG, Youssef MA, Ayeleke RO, Brown J, Lam WS, Broekmans FJ. Gonadotrophin-releasing hormone antagonists for assisted reproductive technology. Cochrane Database Syst Rev. 2016;4:CD001750.
Andrade J, Quinn J, Becker RZ, Shupnik MA. AMP-activated protein kinase is a key intermediary in GnRH-stimulated LHbeta gene transcription. Mol Endocrinol. 2013;27:828–39.
Armstrong SP, Caunt CJ, Fowkes RC, Tsaneva-Atanasova K, McArdle CA. Pulsatile and sustained gonadotropin-releasing hormone (GnRH) receptor signaling: does the Ca2+/NFAT signaling pathway decode GnRH pulse frequency? J Biol Chem. 2009a;284:35746–57.
Armstrong SP, Caunt CJ, McArdle CA. Gonadotropin-releasing hormone and protein kinase C signaling to ERK: spatiotemporal regulation of ERK by docking domains and dual-specificity phosphatases. Mol Endocrinol. 2009b;23:510–9.
Armstrong SP, Caunt CJ, Fowkes RC, Tsaneva-Atanasova K, McArdle CA. Pulsatile and sustained gonadotropin-releasing hormone (GnRH) receptor signaling: does the ERK signaling pathway decode GnRH pulse frequency? J Biol Chem. 2010;285:24360–71.
Arora KK, Krsmanovic LZ, Mores N, O’Farrell H, Catt KJ. Mediation of cyclic AMP signaling by the first intracellular loop of the gonadotropin-releasing hormone receptor. J Biol Chem. 1998;273:25581–6.
Avet C, Garrel G, Denoyelle C, Laverriere JN, Counis R, Cohen-Tannoudji J, Simon V. SET protein interacts with intracellular domains of the gonadotropin-releasing hormone receptor and differentially regulates receptor signaling to cAMP and calcium in gonadotrope cells. J Biol Chem. 2013;288:2641–54.
Bahk JY, Hyun JS, Chung SH, Lee H, Kim MO, Lee BH, Choi WS. Stage specific identification of the expression of GnRH mRNA and localization of the GnRH receptor in mature rat and adult human testis. J Urol. 1995;154:1958–61.
Bedecarrats GY, Kaiser UB. Differential regulation of gonadotropin subunit gene promoter activity by pulsatile gonadotropin-releasing hormone (GnRH) in perifused L beta T2 cells: role of GnRH receptor concentration. Endocrinology. 2003;144:1802–11.
Belchetz PE, Plant TM, Nakai Y, Keogh EJ, Knobil E. Hypophyseal responses to continuous and intermittent delivery of hypothalamic gonadotropin-releasing hormone. Science. 1978;202:631–3.
Berridge MJ. Cell signalling biology. 2012.
Betz SF, Zhu YF, Chen C, Struthers RS. Non-peptide gonadotropin-releasing hormone receptor antagonists. J Med Chem. 2008;51:3331–48.
Bilezikjian LM, Blount AL, Donaldson CJ, Vale WW. Pituitary actions of ligands of the TGF-beta family: activins and inhibins. Reproduction. 2006;132:207–15.
Bliss SP, Navratil AM, Breed M, Skinner DC, Clay CM, Roberson MS. Signaling complexes associated with the type I gonadotropin-releasing hormone (GnRH) receptor: colocalization of extracellularly regulated kinase 2 and GnRH receptor within membrane rafts. Mol Endocrinol. 2007;21:538–49.
Bliss SP, Miller A, Navratil AM, Xie J, McDonough SP, Fisher PJ, Landreth GE, Roberson MS. ERK signaling in the pituitary is required for female but not male fertility. Mol Endocrinol. 2009;23:1092–101.
Bliss SP, Navratil AM, Xie J, Roberson MS. GnRH signaling, the gonadotrope and endocrine control of fertility. Front Neuroendocrinol. 2010;31:322–40.
Blomenrohr M, Bogerd J, Leurs R, Goos H. Differences in structure-function relations between nonmammalian and mammalian GnRH receptors: what we have learnt from the African catfish GnRH receptor. Prog Brain Res. 2002;141:87–93.
Bockaert J, Marin P, Dumuis A, Fagni L. The ‘magic tail’ of G protein-coupled receptors: an anchorage for functional protein networks. FEBS Lett. 2003;546:65–72.
Bonfil D, Chuderland D, Kraus S, Shahbazian D, Friedberg I, Seger R, Naor Z. Extracellular signal-regulated kinase, Jun N-terminal kinase, p38, and c-Src are involved in gonadotropin-releasing hormone-stimulated activity of the glycoprotein hormone follicle-stimulating hormone beta-subunit promoter. Endocrinology. 2004;145:2228–44.
Borgeat P, Chavancy G, Dupont A, Labrie F, Arimura A, Schally AV. Stimulation of adenosine 3′:5′-cyclic monophosphate accumulation in anterior pituitary gland in vitro by synthetic luteinizing hormone-releasing hormone. Proc Natl Acad Sci U S A. 1972;69:2677–81.
Botte MC, Chamagne AM, Carre MC, Counis R, Kottler ML. Fetal expression of GnRH and GnRH receptor genes in rat testis and ovary. J Endocrinol. 1998;159:179–89.
Bowsher CG, Swain PS. Environmental sensing, information transfer, and cellular decision-making. Curr Opin Biotechnol. 2014;28:149–55.
Bowsher CG, Voliotis M, Swain PS. The fidelity of dynamic signaling by noisy biomolecular networks. PLoS Comput Biol. 2013;9:e1002965.
Brennan MD, Cheong R, Levchenko A. Systems biology. How information theory handles cell signaling and uncertainty. Science. 2012;338:334–5.
Brothers SP, Janovick JA, Conn PM. Calnexin regulated gonadotropin-releasing hormone receptor plasma membrane expression. J Mol Endocrinol. 2006;37:479–88.
Brown P, McNeilly AS. Transcriptional regulation of pituitary gonadotrophin subunit genes. Rev Reprod. 1999;4:117–24.
Burger LL, Haisenleder DJ, Dalkin AC, Marshall JC. Regulation of gonadotropin subunit gene transcription. J Mol Endocrinol. 2004;33:559–84.
Burger LL, Haisenleder DJ, Aylor KW, Marshall JC. Regulation of Lhb and Egr1 gene expression by GNRH pulses in rat pituitaries is both c-Jun N-terminal kinase (JNK)- and extracellular signal-regulated kinase (ERK)-dependent. Biol Reprod. 2009;81:1206–15.
Call GB, Wolfe MW. Gonadotropin-releasing hormone activates the equine luteinizing hormone beta promoter through a protein kinase C/mitogen-activated protein kinase pathway. Biol Reprod. 1999;61:715–23.
Cattanach BM, Iddon CA, Charlton HM, Chiappa SA, Fink G. Gonadotrophin-releasing hormone deficiency in a mutant mouse with hypogonadism. Nature. 1977;269:338–40.
Caunt CJ, Hislop JN, Kelly E, Matharu AL, Green LD, Sedgley KR, Finch AR, McArdle CA. Regulation of gonadotropin-releasing hormone receptors by protein kinase C: inside out signalling and evidence for multiple active conformations. Endocrinology. 2004;145:3594–602.
Caunt CJ, Finch AR, Sedgley KR, McArdle CA. GnRH receptor signalling to ERK: kinetics and compartmentalization. Trends Endocrinol Metab. 2006a;17:308–13.
Caunt CJ, Finch AR, Sedgley KR, McArdle CA. Seven-transmembrane receptor signalling and ERK compartmentalization. Trends Endocrinol Metab. 2006b;17:276–83.
Caunt CJ, Finch AR, Sedgley KR, Oakley L, Luttrell LM, McArdle CA. Arrestin-mediated ERK activation by gonadotropin-releasing hormone receptors: receptor-specific activation mechanisms and compartmentalization. J Biol Chem. 2006c;281:2701–10.
Caunt CJ, Perett RM, Fowkes RC, McArdle CA. Mechanisms of GnRH-induced extracellular signal-regulated kinase nuclear localization. PLoS One. 2012;7:e40077.
Chen ZP, Levy A, McArdle CA, Lightman SL. Pituitary ATP receptors: characterization and functional localization to gonadotropes. Endocrinology. 1994;135:1280–3.
Chen ZP, Kratzmeier M, Levy A, McArdle CA, Poch A, Day A, Mukhopadhyay AK, Lightman SL. Evidence for a role of pituitary ATP receptors in the regulation of pituitary function. Proc Natl Acad Sci U S A. 1995;92:5219–23.
Cheng CK, Leung PC. Molecular biology of gonadotropin-releasing hormone (GnRH)-I, GnRH-II, and their receptors in humans. Endocr Rev. 2005;26:283–306.
Chengalvala MV, Pelletier JC, Kopf GS. GnRH agonists and antagonists in cancer therapy. Curr Med Chem Anticancer Agents. 2003;3:399–410.
Cheong R, Rhee A, Wang CJ, Nemenman I, Levchenko A. Information transduction capacity of noisy biochemical signaling networks. Science. 2011;334:354–8.
Cheung LW, Wong AS. Gonadotropin-releasing hormone: GnRH receptor signaling in extrapituitary tissues. FEBS J. 2008;275:5479–95.
Choi SG, Jia J, Pfeffer RL, Sealfon SC. G proteins and autocrine signaling differentially regulate gonadotropin subunit expression in pituitary gonadotrope. J Biol Chem. 2012;287:21550–60.
Choi SG, Wang Q, Jia J, Pincas H, Turgeon JL, Sealfon SC. Growth differentiation factor 9 (GDF9) forms an incoherent feed-forward loop modulating follicle-stimulating hormone beta-subunit (FSHbeta) gene expression. J Biol Chem. 2014;289:16164–75.
Chu Z, Andrade J, Shupnik MA, Moenter SM. Differential regulation of gonadotropin-releasing hormone neuron activity and membrane properties by acutely applied estradiol: dependence on dose and estrogen receptor subtype. J Neurosci. 2009;29:5616–27.
Chusho H, Tamura N, Ogawa Y, Yasoda A, Suda M, Miyazawa T, Nakamura K, Nakao K, Kurihara T, Komatsu Y, et al. Dwarfism and early death in mice lacking C-type natriuretic peptide. Proc Natl Acad Sci U S A. 2001;98:4016–21.
Ciccone NA, Kaiser UB. The biology of gonadotroph regulation. Curr Opin Endocrinol Diabetes Obes. 2009;16:321–7.
Ciccone NA, Xu S, Lacza CT, Carroll RS, Kaiser UB. Frequency-dependent regulation of follicle-stimulating hormone beta by pulsatile gonadotropin-releasing hormone is mediated by functional antagonism of bZIP transcription factors. Mol Cell Biol. 2010;30:1028–40.
Clarke IJ, Cummins JT. The temporal relationship between gonadotropin releasing hormone (GnRH) and luteinizing hormone (LH) secretion in ovariectomized ewes. Endocrinology. 1982;111:1737–9.
Clarke IJ, Cummins JT. Increased gonadotropin-releasing hormone pulse frequency associated with estrogen-induced luteinizing hormone surges in ovariectomized ewes. Endocrinology. 1985;116:2376–83.
Clarke I, Moore L, Veldhuis J. Intensive direct cavernous sinus sampling identifies high-frequency, nearly random patterns of FSH secretion in ovariectomized ewes: combined appraisal by RIA and bioassay. Endocrinology. 2002;143:117–29.
Clayton RN, Catt KJ. Regulation of pituitary gonadotropin-releasing hormone receptors by gonadal hormones. Endocrinology. 1981;108:887–95.
Clayton RN, Huhtaniemi IT. Absence of gonadotropin-releasing hormone receptors in human gonadal tissue. Nature. 1982;299:56–9.
Conn PM, Crowley Jr WF. Gonadotropin-releasing hormone and its analogs. Annu Rev Med. 1994;45:391–405.
Conn PM, Janovick JA. Trafficking and quality control of the gonadotropin releasing hormone receptor in health and disease. Mol Cell Endocrinol. 2009;299:137–45.
Conn PM, Ulloa-Aguirre A. Trafficking of G-protein-coupled receptors to the plasma membrane: insights for pharmacoperone drugs. Trends Endocrinol Metab. 2010;21:190–7.
Conn PM, Morrell DV, Dufau ML, Catt KJ. Gonadotropin-releasing hormone action in cultured pituicytes: independence of luteinizing hormone release and adenosine 3′,5′-monophosphate production. Endocrinology. 1979;104:448–53.
Conn PM, Chafouleas JG, Rogers D, Means AR. Gonadotropin releasing hormone stimulates calmodulin redistribution in rat pituitary. Nature. 1981;292:264–5.
Conn PM, Rogers DC, Stewart JM, Niedel J, Sheffield T. Conversion of a gonadotropin-releasing hormone antagonist to an agonist. Nature. 1982;296:653–5.
Conn PM, Huckle WR, Andrews WV, McArdle CA. The molecular mechanism of action of gonadotropin releasing hormone (GnRH) in the pituitary. Recent Prog Horm Res. 1987;43:29–68.
Cornea A, Janovick JA, Maya-Nunez G, Conn PM. Gonadotropin-releasing hormone receptor microaggregation. Rate monitored by fluorescence resonance energy transfer. J Biol Chem. 2001;276:2153–8.
Coss D, Hand CM, Yaphockun KK, Ely HA, Mellon PL. p38 mitogen-activated protein kinase is critical for synergistic induction of the FSH(beta) gene by gonadotropin-releasing hormone and activin through augmentation of c-Fos induction and Smad phosphorylation. Mol Endocrinol. 2007;21:3071–86.
Counis R, Laverriere JN, Garrel G, Bleux C, Cohen-Tannoudji J, Lerrant Y, Kottler ML, Magre S. Gonadotropin-releasing hormone and the control of gonadotrope function. Reprod Nutr Dev. 2005;45:243–54.
Counis R, Laverriere JN, Garrel-Lazayres G, Cohen-Tannoudji J, Lariviere S, Bleux C, Magre S. What is the role of PACAP in gonadotrope function? Peptides. 2007;28:1797–804.
Crawford JL, McNeilly AS. Co-localisation of gonadotrophins and granins in gonadotrophs at different stages of the oestrous cycle in sheep. J Endocrinol. 2002;174:179–94.
Dalkin AC, Haisenleder DJ, Ortolano GA, Ellis TR, Marshall JC. The frequency of gonadotropin-releasing-hormone stimulation differentially regulates gonadotropin subunit messenger ribonucleic acid expression. Endocrinology. 1989;125:917–24.
Davidson JS, Wakefield IK, Millar RP. Absence of rapid desensitization of the mouse gonadotropin-releasing hormone receptor. Biochem J. 1994;300(Pt 2):299–302.
Davidson JS, Flanagan CA, Zhou W, Becker II, Elario R, Emeran W, Sealfon SC, Millar RP. Identification of N-glycosylation sites in the gonadotropin-releasing hormone receptor: role in receptor expression but not ligand binding. Mol Cell Endocrinol. 1995;107:241–5.
Davidson L, Pawson AJ, Lopez de Maturana R, Freestone SH, Barran P, Millar RP, Maudsley S. Gonadotropin-releasing hormone-induced activation of diacylglycerol kinase-zeta and its association with active c-src. J Biol Chem. 2004a;279:11906–16.
Davidson L, Pawson AJ, Millar RP, Maudsley S. Cytoskeletal reorganization dependence of signaling by the gonadotropin-releasing hormone receptor. J Biol Chem. 2004b;279:1980–93.
de Roux N, Young J, Misrahi M, Genet R, Chanson P, Schaison G, Milgrom E. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N Engl J Med. 1997;337:1597–602.
Delegeane AM, Ferland LH, Mellon PL. Tissue-specific enhancer of the human glycoprotein hormone alpha-subunit gene: dependence on cyclic AMP-inducible elements. Mol Cell Biol. 1987;7:3994–4002.
Denef C. Paracrinicity: the story of 30 years of cellular pituitary crosstalk. J Neuroendocrinol. 2008;20:1–70.
Dierschke DJ, Bhattacharya AN, Atkinson LE, Knobil E. Circhoral oscillations of plasma LH levels in the ovariectomized rhesus monkey. Endocrinology. 1970;87:850–3.
Duan WR, Shin JL, Jameson JL. Estradiol suppresses phosphorylation of cyclic adenosine 3′,5′-monophosphate response element binding protein (CREB) in the pituitary: evidence for indirect action via gonadotropin-releasing hormone. Mol Endocrinol. 1999;13:1338–52.
Dufau ML, Warren DW, Knox GF, Loumaye E, Castellon ML, Luna S, Catt KJ. Receptors and inhibitory actions of gonadotropin-releasing hormone in the fetal Leydig cell. J Biol Chem. 1984;259:2896–9.
Eidne KA, Flanagan CA, Harris NS, Millar RP. Gonadotropin-releasing hormone (GnRH)-binding sites in human breast cancer cell lines and inhibitory effects of GnRH antagonists. J Clin Endocrinol Metab. 1987;64:425–32.
Emons G, Muller V, Ortmann O, Schulz KD. Effects of LHRH-analogues on mitogenic signal transduction in cancer cells. J Steroid Biochem Mol Biol. 1998;65:199–206.
Engel J, Emons G, Pinski J, Schally AV. AEZS-108: a targeted cytotoxic analog of LHRH for the treatment of cancers positive for LHRH receptors. Expert Opin Investig Drugs. 2012;21:891–9.
Engel JB, Tinneberg HR, Rick FG, Berkes E, Schally AV. Targeting of peptide cytotoxins to LHRH receptors for treatment of cancer. Curr Drug Targets. 2016;17:488–94.
Enomoto M, Endo D, Kawashima S, Park MK. Human type II GnRH receptor mediates effects of GnRH on cell proliferation. Zool Sci. 2004;21:763–70.
Everest HM, Hislop JN, Harding T, Uney JB, Flynn A, Millar RP, McArdle CA. Signaling and antiproliferative effects mediated by GnRH receptors after expression in breast cancer cells using recombinant adenovirus. Endocrinology. 2001;142:4663–72.
Ferasyi TR, Barrett PH, Blache D, Martin GB. Modeling the male reproductive endocrine axis: potential role for a delay mechanism in the inhibitory action of gonadal steroids on GnRH pulse frequency. Endocrinology. 2016;157:2080–92.
Fernald RD, White RB. Gonadotropin-releasing hormone genes: phylogeny, structure, and functions. Front Neuroendocrinol. 1999;20:224–40.
Ferris HA, Shupnik MA. Mechanisms for pulsatile regulation of the gonadotropin subunit genes by GNRH1. Biol Reprod. 2006;74:993–8.
Ferris HA, Walsh HE, Stevens J, Fallest PC, Shupnik MA. Luteinizing hormone beta promoter stimulation by adenylyl cyclase and cooperation with gonadotropin-releasing hormone 1 in transgenic mice and LBetaT2 Cells. Biol Reprod. 2007;77:1073–80.
Finch AR, Green L, Hislop JN, Kelly E, McArdle CA. Signaling and antiproliferative effects of type I and II gonadotropin-releasing hormone receptors in breast cancer cells. J Clin Endocrinol Metab. 2004;89:1823–32.
Finch AR, Sedgley KR, Caunt CJ, McArdle CA. Plasma membrane expression of GnRH receptors: regulation by antagonists in breast, prostate, and gonadotrope cell lines. J Endocrinol. 2008;196:353–67.
Finch AR, Caunt CJ, Armstrong SP, McArdle CA. Agonist-induced internalization and downregulation of gonadotropin-releasing hormone receptors. Am J Physiol Cell Physiol. 2009;297:C591–600.
Finch AR, Caunt CJ, Armstrong SP, McArdle CA. Plasma membrane expression of gonadotropin-releasing hormone receptors: regulation by peptide and nonpeptide antagonists. Mol Endocrinol. 2010;24:423–35.
Fink MY, Pincas H, Choi SG, Nudelman G, Sealfon SC. Research resource: gonadotropin-releasing hormone receptor-mediated signaling network in LbetaT2 cells: a pathway-based web-accessible knowledgebase. Mol Endocrinol. 2010;24:1863–71.
Fletcher PA, Clement F, Vidal A, Tabak J, Bertram R. Interpreting frequency responses to dose-conserved pulsatile input signals in simple cell signaling motifs. PLoS One. 2014;9:e95613.
Fowkes RC, McArdle CA. C-type natriuretic peptide: an important neuroendocrine regulator? Trends Endocrinol Metab. 2000;11:333–8.
Fowkes RC, King P, Burrin JM. Regulation of human glycoprotein hormone alpha-subunit gene transcription in LbetaT2 gonadotropes by protein kinase C and extracellular signal-regulated kinase 1/2. Biol Reprod. 2002;67:725–34.
Fox EM, Andrade J, Shupnik MA. Novel actions of estrogen to promote proliferation: integration of cytoplasmic and nuclear pathways. Steroids. 2009;74:622–7.
Franklin J, Hislop J, Flynn A, McArdle CA. Signalling and anti-proliferative effects mediated by gonadotrophin-releasing hormone receptors after expression in prostate cancer cells using recombinant adenovirus. J Endocrinol. 2003;176:275–84.
Gardner S, Maudsley S, Millar RP, Pawson AJ. Nuclear stabilization of beta-catenin and inactivation of glycogen synthase kinase-3beta by gonadotropin-releasing hormone: targeting Wnt signaling in the pituitary gonadotrope. Mol Endocrinol. 2007;21:3028–38.
Garner KL, Perrett RM, Voliotis M, Bowsher C, Pope GR, Pham T, Caunt CJ, Tsaneva-Atanasova K, McArdle CA. Information Transfer in Gonadotropin-releasing Hormone (GnRH) Signaling: extracellular signal-regulated kinase (ERK)-mediated feedback loops control hormone sensing. J Biol Chem. 2016;291:2246–59.
Garrel G, McArdle CA, Hemmings BA, Counis R. Gonadotropin-releasing hormone and pituitary adenylate cyclase-activating polypeptide affect levels of cyclic adenosine 3′,5′-monophosphate-dependent protein kinase A (PKA) subunits in the clonal gonadotrope alphaT3-1 cells: evidence for cross-talk between PKA and protein kinase C pathways. Endocrinology. 1997;138:2259–66.
Gross KM, Matsumoto AM, Bremner WJ. Differential control of luteinizing hormone and follicle-stimulating hormone secretion by luteinizing hormone-releasing hormone pulse frequency in man. J Clin Endocrinol Metab. 1987;64:675–80.
Grosse R, Schmid A, Schoneberg T, Herrlich A, Muhn P, Schultz G, Gudermann T. Gonadotropin-releasing hormone receptor initiates multiple signaling pathways by exclusively coupling to G(q/11) proteins. J Biol Chem. 2000;275:9193–200.
Grundker C, Volker P, Emons G. Antiproliferative signaling of luteinizing hormone-releasing hormone in human endometrial and ovarian cancer cells through G protein alpha(I)-mediated activation of phosphotyrosine phosphatase. Endocrinology. 2001;142:2369–80.
Grundker C, Schlotawa L, Viereck V, Eicke N, Horst A, Kairies B, Emons G. Antiproliferative effects of the GnRH antagonist cetrorelix and of GnRH-II on human endometrial and ovarian cancer cells are not mediated through the GnRH type I receptor. Eur J Endocrinol. 2004;151:141–9.
Haisenleder DJ, Dalkin AC, Ortolano GA, Marshall JC, Shupnik MA. A pulsatile gonadotropin-releasing hormone stimulus is required to increase transcription of the gonadotropin subunit genes: evidence for differential regulation of transcription by pulse frequency in vivo. Endocrinology. 1991;128:509–17.
Haisenleder DJ, Yasin M, Marshall JC. Enhanced effectiveness of pulsatile 3′,5′-cyclic adenosine monophosphate in stimulating prolactin and alpha-subunit gene expression. Endocrinology. 1992;131:3027–33.
Haisenleder DJ, Burger LL, Aylor KW, Dalkin AC, Walsh HE, Shupnik MA, Marshall JC. Testosterone stimulates follicle-stimulating hormone beta transcription via activation of extracellular signal-regulated kinase: evidence in rat pituitary cells. Biol Reprod. 2005;72:523–9.
Haisenleder DJ, Burger LL, Walsh HE, Stevens J, Aylor KW, Shupnik MA, Marshall JC. Pulsatile gonadotropin-releasing hormone stimulation of gonadotropin subunit transcription in rat pituitaries: evidence for the involvement of Jun N-terminal kinase but not p38. Endocrinology. 2008;149:139–45.
Hansen JR, McArdle CA, Conn PM. Relative roles of calcium derived from intra- and extracellular sources in dynamic luteinizing hormone release from perifused pituitary cells. Mol Endocrinol. 1987;1:808–15.
Hanyaloglu AC, Vrecl M, Kroeger KM, Miles LE, Qian H, Thomas WG, Eidne KA. Casein kinase II sites in the intracellular C-terminal domain of the thyrotropin-releasing hormone receptor and chimeric gonadotropin-releasing hormone receptors contribute to beta-arrestin-dependent internalization. J Biol Chem. 2001;276:18066–74.
Harris D, Bonfil D, Chuderland D, Kraus S, Seger R, Naor Z. Activation of MAPK cascades by GnRH: ERK and Jun N-terminal kinase are involved in basal and GnRH-stimulated activity of the glycoprotein hormone LHbeta-subunit promoter. Endocrinology. 2002;143:1018–25.
Harris D, Chuderland D, Bonfil D, Kraus S, Seger R, Naor Z. Extracellular signal-regulated kinase and c-Src, but not Jun N-terminal kinase, are involved in basal and gonadotropin-releasing hormone-stimulated activity of the glycoprotein hormone alpha-subunit promoter. Endocrinology. 2003;144:612–22.
Harrison GS, Wierman ME, Nett TM, Glode LM. Gonadotropin-releasing hormone and its receptor in normal and malignant cells. Endocr Relat Cancer. 2004;11:725–48.
Hazum E, Cuatrecasas P, Marian J, Conn PM. Receptor-mediated internalization of fluorescent gonadotropin-releasing hormone by pituitary gonadotropes. Proc Natl Acad Sci U S A. 1980;77:6692–5.
Heding A, Vrecl M, Bogerd J, McGregor A, Sellar R, Taylor PL, Eidne KA. Gonadotropin-releasing hormone receptors with intracellular carboxyl-terminal tails undergo acute desensitization of total inositol phosphate production and exhibit accelerated internalization kinetics. J Biol Chem. 1998;273:11472–7.
Heding A, Vrecl M, Hanyaloglu AC, Sellar R, Taylor PL, Eidne KA. The rat gonadotropin-releasing hormone receptor internalizes via a beta-arrestin-independent, but dynamin-dependent, pathway: addition of a carboxyl-terminal tail confers beta-arrestin dependency. Endocrinology. 2000;141:299–306.
Hille B, Tse A, Tse FW, Almers W. Calcium oscillations and exocytosis in pituitary gonadotropes. Ann N Y Acad Sci. 1994;710:261–70.
Hislop JN, Madziva MT, Everest HM, Harding T, Uney JB, Willars GB, Millar RP, Troskie BE, Davidson JS, McArdle CA. Desensitization and internalization of human and xenopus gonadotropin-releasing hormone receptors expressed in alphaT4 pituitary cells using recombinant adenovirus. Endocrinology. 2000;141:4564–75.
Hislop JN, Everest HM, Flynn A, Harding T, Uney JB, Troskie BE, Millar RP, McArdle CA. Differential internalization of mammalian and non-mammalian gonadotropin-releasing hormone receptors. Uncoupling of dynamin-dependent internalization from mitogen-activated protein kinase signaling. J Biol Chem. 2001;276:39685–94.
Hislop JN, Caunt CJ, Sedgley KR, Kelly E, Mundell S, Green LD, McArdle CA. Internalization of gonadotropin-releasing hormone receptors (GnRHRs): does arrestin binding to the C-terminal tail target GnRHRs for dynamin-dependent internalization? J Mol Endocrinol. 2005;35:177–89.
Hoffman LK, Ehrmann DA. Cardiometabolic features of polycystic ovary syndrome. Nat Clin Pract Endocrinol Metab. 2008;4:215–22.
Horn F, Bilezikjian LM, Perrin MH, Bosma MM, Windle JJ, Huber KS, Blount AL, Hille B, Vale W, Mellon PL. Intracellular responses to gonadotropin-releasing hormone in a clonal cell line of the gonadotrope lineage. Mol Endocrinol. 1991;5:347–55.
Horvat RD, Roess DA, Nelson SE, Barisas BG, Clay CM. Binding of agonist but not antagonist leads to fluorescence resonance energy transfer between intrinsically fluorescent gonadotropin-releasing hormone receptors. Mol Endocrinol. 2001;15:695–703.
Huhtaniemi IT, Catt KJ, Clayton RN. Newborn and immature rat testes contain gonadotropin-releasing hormone (GnRH) receptors, and their testosterone production is stimulated by a GnRH agonist in vitro. Mol Cell Endocrinol. 1985;40:41–4.
Huhtaniemi IT, Nikula H, Detta A, Stewart JM, Clayton RN. Blockade of rat testicular gonadotropin releasing hormone (GnRH) receptors by infusion of a GnRH antagonist has no major effects of Leydig cell function in vivo. Mol Cell Endocrinol. 1987;49:89–97.
Imai A, Horibe S, Takagi A, Tamaya T. Gi protein activation of gonadotropin-releasing hormone-mediated protein dephosphorylation in human endometrial carcinoma. Am J Obstet Gynecol. 1997;176:371–6.
Izumi S, Stojilkovic SS, Catt KJ. Calcium mobilization and influx during the biphasic cytosolic calcium and secretory responses in agonist-stimulated pituitary gonadotrophs. Arch Biochem Biophys. 1989;275:410–28.
Janovick JA, Conn PM. Salt bridge integrates GPCR activation with protein trafficking. Proc Natl Acad Sci U S A. 2010a;107:4454–8.
Janovick JA, Conn PM. Use of pharmacoperones to reveal GPCR structural changes associated with constitutive activation and trafficking. Methods Enzymol. 2010b;485:277–92.
Janovick JA, Patny A, Mosley R, Goulet MT, Altman MD, Rush 3rd TS, Cornea A, Conn PM. Molecular mechanism of action of pharmacoperone rescue of misrouted GPCR mutants: the GnRH receptor. Mol Endocrinol. 2009;23:157–68.
Janovick JA, Pogozheva ID, Mosberg HI, Cornea A, Conn PM. Rescue of misrouted GnRHR mutants reveals its constitutive activity. Mol Endocrinol. 2012;26:1179–88.
Janovick JA, Stewart MD, Jacob D, Martin LD, Deng JM, Stewart CA, Wang Y, Cornea A, Chavali L, Lopez S, et al. Restoration of testis function in hypogonadotropic hypogonadal mice harboring a misfolded GnRHR mutant by pharmacoperone drug therapy. Proc Natl Acad Sci U S A. 2013;110:21030–5.
Jennes L, Stumpf WE, Conn PM. Receptor-mediated binding and uptake of GnRH agonist and antagonist by pituitary cells. Peptides. 1984;5(Suppl 1):215–20.
Kaiser UB, Jakubowiak A, Steinberger A, Chin WW. Regulation of rat pituitary gonadotropin-releasing hormone receptor mRNA levels in vivo and in vitro. Endocrinology. 1993;133:931–4.
Kanasaki H, Bedecarrats GY, Kam KY, Xu S, Kaiser UB. Gonadotropin-releasing hormone pulse frequency-dependent activation of extracellular signal-regulated kinase pathways in perifused LbetaT2 cells. Endocrinology. 2005;146:5503–13.
Karakoula A, Tovey SC, Brighton PJ, Willars GB. Lack of receptor-selective effects of either RGS2, RGS3 or RGS4 on muscarinic M3- and gonadotropin-releasing hormone receptor-mediated signalling through G alpha q/11. Eur J Pharmacol. 2008;587:16–24.
Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther. 2011;336:296–302.
Knobil E. The neuroendocrine control of the menstrual cycle. Recent Prog Horm Res. 1980;36:53–88.
Kowase T, Walsh HE, Darling DS, Shupnik MA. Estrogen enhances gonadotropin-releasing hormone-stimulated transcription of the luteinizing hormone subunit promoters via altered expression of stimulatory and suppressive transcription factors. Endocrinology. 2007;148:6083–91.
Krakauer DC, Page KM, Sealfon S. Module dynamics of the GnRH signal transduction network. J Theor Biol. 2002;218:457–70.
Kraus S, Naor Z, Seger R. Intracellular signaling pathways mediated by the gonadotropin-releasing hormone (GnRH) receptor. Arch Med Res. 2001;32:499–509.
Kroeger KM, Pfleger KD, Eidne KA. G-protein coupled receptor oligomerization in neuroendocrine pathways. Front Neuroendocrinol. 2003;24:254–78.
Krsmanovic LZ, Mores N, Navarro CE, Arora KK, Catt KJ. An agonist-induced switch in G protein coupling of the gonadotropin-releasing hormone receptor regulates pulsatile neuropeptide secretion. Proc Natl Acad Sci U S A. 2003;100:2969–74.
Lariviere S, Garrel G, Simon V, Soh JW, Laverriere JN, Counis R, Cohen-Tannoudji J. Gonadotropin-RELEASING hormone couples to 3′,5′-cyclic adenosine-5′-monophosphate pathway through novel protein kinase Cdelta and -epsilon in LbetaT2 gonadotrope cells. Endocrinology. 2007;148:1099–107.
Leong DA, Thorner MO. A potential code of luteinizing hormone-releasing hormone-induced calcium ion responses in the regulation of luteinizing hormone secretion among individual gonadotropes. J Biol Chem. 1991;266:9016–22.
Levi NL, Hanoch T, Benard O, Rozenblat M, Harris D, Reiss N, Naor Z, Seger R. Stimulation of Jun N-terminal kinase (JNK) by gonadotropin-releasing hormone in pituitary alpha T3-1 cell line is mediated by protein kinase C, c-Src, and CDC42. Mol Endocrinol. 1998;12:815–24.
Lewis CE, Richards PS, Morris JF. Heterogeneity of responses to LH-releasing hormone and phorbol ester among rat gonadotrophs: a study using a reverse haemolytic plaque assay for LH. J Mol Endocrinol. 1989;2:55–63.
Lim S, Pnueli L, Tan JH, Naor Z, Rajagopal G, Melamed P. Negative feedback governs gonadotrope frequency-decoding of gonadotropin releasing hormone pulse-frequency. PLoS One. 2009;4:e7244.
Limonta P, Moretti RM, Montagnani Marelli M, Motta M. The biology of gonadotropin hormone-releasing hormone: role in the control of tumor growth and progression in humans. Front Neuroendocrinol. 2003;24:279–95.
Limonta P, Montagnani Marelli M, Mai S, Motta M, Martini L, Moretti RM. GnRH receptors in cancer: from cell biology to novel targeted therapeutic strategies. Endocr Rev. 2012. doi:10.1210/er.2012-1014.
Liu F, Austin DA, Mellon PL, Olefsky JM, Webster NJ. GnRH activates ERK1/2 leading to the induction of c-fos and LHbeta protein expression in LbetaT2 cells. Mol Endocrinol. 2002a;16:419–34.
Liu F, Usui I, Evans LG, Austin DA, Mellon PL, Olefsky JM, Webster NJ. Involvement of both G(q/11) and G(s) proteins in gonadotropin-releasing hormone receptor-mediated signaling in L beta T2 cells. J Biol Chem. 2002b;277:32099–108.
Lopez de Maturana R, Pawson AJ, Lu ZL, Davidson L, Maudsley S, Morgan K, Langdon SP, Millar RP. Gonadotropin-releasing hormone analog structural determinants of selectivity for inhibition of cell growth: support for the concept of ligand-induced selective signaling. Mol Endocrinol. 2008;22:1711–22.
Lozach A, Garrel G, Lerrant Y, Berault A, Counis R. GnRH-dependent up-regulation of nitric oxide synthase I level in pituitary gonadotrophs mediates cGMP elevation during rat proestrus. Mol Cell Endocrinol. 1998;143:43–51.
Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115:455–65.
Marshall JC, Dalkin AC, Haisenleder DJ, Griffin ML, Kelch RP. GnRH pulses – the regulators of human reproduction. Trans Am Clin Climatol Assoc. 1993;104:31–46.
Mason AJ, Hayflick JS, Zoeller RT, Young 3rd WS, Phillips HS, Nikolics K, Seeburg PH. A deletion truncating the gonadotropin-releasing hormone gene is responsible for hypogonadism in the hpg mouse. Science. 1986;234:1366–71.
Maudsley S, Davidson L, Pawson AJ, Chan R, Lopez de Maturana R, Millar RP. Gonadotropin-releasing hormone (GnRH) antagonists promote proapoptotic signaling in peripheral reproductive tumor cells by activating a Galphai-coupling state of the type I GnRH receptor. Cancer Res. 2004;64:7533–44.
Maurer RA, Kim KE, Schoderbek WE, Roberson MS, Glenn DJ. Regulation of glycoprotein hormone alpha-subunit gene expression. Recent Prog Horm Res. 1999;54:455–84; discussion 485.
McArdle CA. Gonadotropin-releasing hormone receptor signaling: biased and unbiased. Mini Rev Med Chem. 2012;12:841–50.
McArdle CA, Counis R. GnRH and PACAP action in gonadotropes: cross-talk between phosphoinositidase C and adenylyl cyclase mediated signaling pathways. Trends Endocrinol Metab. 1996;7:168–75.
McArdle CA, Roberson MS. Gonadotropes and gonadotropin-releasing hormone signaling. In: Plant TM, editor. Knobil and Neill’s physiology of reproduction. 4th ed. Amsterdam: Elsevier; 2015. p. 335–97.
McArdle CA, Huckle WR, Conn PM. Phorbol esters reduce gonadotrope responsiveness to protein kinase C activators but not to Ca2+−mobilizing secretagogues. Does protein kinase C mediate gonadotropin-releasing hormone action? J Biol Chem. 1987;262:5028–35.
McArdle CA, Bunting R, Mason WT. Dynamic video imaging of cystolic Ca(2+) in the alphaT3-1, gonadotrope-derived cell line. Mol Cell Neurosci. 1992;3:124–32.
McArdle CA, Olcese J, Schmidt C, Poch A, Kratzmeier M, Middendorff R. C-type natriuretic peptide (CNP) in the pituitary: is CNP an autocrine regulator of gonadotropes? Endocrinology. 1994a;135:2794–801.
McArdle CA, Poch A, Schomerus E, Kratzmeier M. Pituitary adenylate cyclase-activating polypeptide effects in pituitary cells: modulation by gonadotropin-releasing hormone in alpha T3-1 cells. Endocrinology. 1994b;134:2599–605.
McArdle CA, Willars GB, Fowkes RC, Nahorski SR, Davidson JS, Forrest-Owen W. Desensitization of gonadotropin-releasing hormone action in alphaT3-1 cells due to uncoupling of inositol 1,4,5-trisphosphate generation and Ca2+ mobilization. J Biol Chem. 1996;271:23711–7.
McArdle CA, Davidson JS, Willars GB. The tail of the gonadotrophin-releasing hormone receptor: desensitization at, and distal to, G protein-coupled receptors. Mol Cell Endocrinol. 1999;151:129–36.
Millar RP, Lu ZL, Pawson AJ, Flanagan CA, Morgan K, Maudsley SR. Gonadotropin-releasing hormone receptors. Endocr Rev. 2004;25:235–75.
Mistry DS, Tsutsumi R, Fernandez M, Sharma S, Cardenas SA, Lawson MA, Webster NJ. Gonadotropin-releasing hormone pulse sensitivity of follicle-stimulating hormone-beta gene is mediated by differential expression of positive regulatory activator protein 1 factors and corepressors SKIL and TGIF1. Mol Endocrinol. 2011;25:1387–403.
Miyata A, Arimura A, Dahl RR, Minamino N, Uehara A, Jiang L, Culler MD, Coy DH. Isolation of a novel 38 residue-hypothalamic polypeptide which stimulates adenylate cyclase in pituitary cells. Biochem Biophys Res Commun. 1989;164:567–74.
Montagnani Marelli M, Moretti RM, Mai S, Januszkiewicz-Caulier J, Motta M, Limonta P. Type I gonadotropin-releasing hormone receptor mediates the antiproliferative effects of GnRH-II on prostate cancer cells. J Clin Endocrinol Metab. 2009;94:1761–7.
Morgan K, Millar RP. Evolution of GnRH ligand precursors and GnRH receptors in protochordate and vertebrate species. Gen Comp Endocrinol. 2004;139:191–7.
Naor Z. Signaling by G-protein-coupled receptor (GPCR): studies on the GnRH receptor. Front Neuroendocrinol. 2009;30:10–29.
Navratil AM, Bliss SP, Berghorn KA, Haughian JM, Farmerie TA, Graham JK, Clay CM, Roberson MS. Constitutive localization of the gonadotropin-releasing hormone (GnRH) receptor to low density membrane microdomains is necessary for GnRH signaling to ERK. J Biol Chem. 2003;278:31593–602.
Navratil AM, Farmerie TA, Bogerd J, Nett TM, Clay CM. Differential impact of intracellular carboxyl terminal domains on lipid raft localization of the murine gonadotropin-releasing hormone receptor. Biol Reprod. 2006;74:788–97.
Navratil AM, Bliss SP, Roberson MS. Membrane rafts and GnRH receptor signaling. Brain Res. 2010;1364:53–61.
Nelson SB, Eraly SA, Mellon PL. The GnRH promoter: target of transcription factors, hormones, and signaling pathways. Mol Cell Endocrinol. 1998;140:151–5.
Pawson AJ, McNeilly AS. The pituitary effects of GnRH. Anim Reprod Sci. 2005;88:75–94.
Pawson AJ, Katz A, Sun YM, Lopes J, Illing N, Millar RP, Davidson JS. Contrasting internalization kinetics of human and chicken gonadotropin-releasing hormone receptors mediated by C-terminal tail. J Endocrinol. 1998;156:R9–12.
Perrett RM, Voliotis M, Armstrong SP, Fowkes RC, Pope GR, Tsaneva-Atanasova K, McArdle CA. Pulsatile hormonal signaling to extracellular signal-regulated kinase: exploring system sensitivity to gonadotropin-releasing hormone pulse frequency and width. J Biol Chem. 2014;289:7873–83.
Pierce KL, Lefkowitz RJ. Classical and new roles of beta-arrestins in the regulation of G-protein-coupled receptors. Nat Rev Neurosci. 2001;2:727–33.
Pincas H, Choi SG, Wang Q, Jia J, Turgeon JL, Sealfon SC. Outside the box signaling: secreted factors modulate GnRH receptor-mediated gonadotropin regulation. Mol Cell Endocrinol. 2014;385:56–61.
Rawlings SR, Hezareh M. Pituitary adenylate cyclase-activating polypeptide (PACAP) and PACAP/vasoactive intestinal polypeptide receptors: actions on the anterior pituitary gland. Endocr Rev. 1996;17:4–29.
Roberson MS, Misra-Press A, Laurance ME, Stork PJ, Maurer RA. A role for mitogen-activated protein kinase in mediating activation of the glycoprotein hormone alpha-subunit promoter by gonadotropin-releasing hormone. Mol Cell Biol. 1995;15:3531–9.
Roberson MS, Zhang T, Li HL, Mulvaney JM. Activation of the p38 mitogen-activated protein kinase pathway by gonadotropin-releasing hormone. Endocrinology. 1999;140:1310–8.
Rocheville M, Lange DC, Kumar U, Patel SC, Patel RC, Patel YC. Receptors for dopamine and somatostatin: formation of hetero-oligomers with enhanced functional activity. Science. 2000;288:154–7.
Ruf F, Fink MY, Sealfon SC. Structure of the GnRH receptor-stimulated signaling network: insights from genomics. Front Neuroendocrinol. 2003;24:181–99.
Ruf F, Park MJ, Hayot F, Lin G, Roysam B, Ge Y, Sealfon SC. Mixed analog/digital gonadotrope biosynthetic response to gonadotropin-releasing hormone. J Biol Chem. 2006;281:30967–78.
Ruf F, Hayot F, Park MJ, Ge Y, Lin G, Roysam B, Sealfon SC. Noise propagation and scaling in regulation of gonadotrope biosynthesis. Biophys J. 2007;93:4474–80.
Schally AV. LH-RH analogues: I. Their impact on reproductive medicine. Gynecol Endocrinol. 1999;13:401–9.
Schneider JS, Rissman EF. Gonadotropin-releasing hormone II: a multi-purpose neuropeptide. Integr Comp Biol. 2008;48:588–95.
Schomerus E, Poch A, Bunting R, Mason WT, McArdle CA. Effects of pituitary adenylate cyclase-activating polypeptide in the pituitary: activation of two signal transduction pathways in the gonadotrope-derived alpha T3-1 cell line. Endocrinology. 1994;134:315–23.
Sealfon SC, Weinstein H, Millar RP. Molecular mechanisms of ligand interaction with the gonadotropin-releasing hormone receptor. Endocr Rev. 1997;18:180–205.
Sedgley KR, Finch AR, Caunt CJ, McArdle CA. Intracellular gonadotropin-releasing hormone receptors in breast cancer and gonadotrope lineage cells. J Endocrinol. 2006;191:625–36.
Selimkhanov J, Taylor B, Yao J, Pilko A, Albeck J, Hoffmann A, Tsimring L, Wollman R. Systems biology. Accurate information transmission through dynamic biochemical signaling networks. Science. 2014;346:1370–3.
Shenoy SK, Lefkowitz RJ. Multifaceted roles of beta-arrestins in the regulation of seven-membrane-spanning receptor trafficking and signalling. Biochem J. 2003;375:503–15.
Shupnik MA. Effects of gonadotropin-releasing hormone on rat gonadotropin gene transcription in vitro: requirement for pulsatile administration for luteinizing hormone-beta gene stimulation. Mol Endocrinol. 1990;4:1444–50.
Siristatidis CS, Gibreel A, Basios G, Maheshwari A & Bhattacharya S. Gonadotrophin-releasing hormone agonist protocols for pituitary suppression in assisted reproduction. Cochrane Database Syst Rev. 2015; CD006919.
Sisk CL, Foster DL. The neural basis of puberty and adolescence. Nat Neurosci. 2004;7:1040–7.
Spratt DI, Finkelstein JS, Butler JP, Badger TM, Crowley Jr WF. Effects of increasing the frequency of low doses of gonadotropin-releasing hormone (GnRH) on gonadotropin secretion in GnRH-deficient men. J Clin Endocrinol Metab. 1987;64:1179–86.
Stanislaus D, Ponder S, Ji TH, Conn PM. Gonadotropin-releasing hormone receptor couples to multiple G proteins in rat gonadotrophs and in GGH3 cells: evidence from palmitoylation and overexpression of G proteins. Biol Reprod. 1998;59:579–86.
Stewart AJ, Katz AA, Millar RP, Morgan K. Retention and silencing of prepro-GnRH-II and type II GnRH receptor genes in mammals. Neuroendocrinology. 2009;90:416–32.
Stewart MD, Deng JM, Stewart CA, Mullen RD, Wang Y, Lopez S, Serna MK, Huang CC, Janovick JA, Pask AJ, et al. Mice harboring Gnrhr E90K, a mutation that causes protein misfolding and hypogonadotropic hypogonadism in humans, exhibit testis size reduction and ovulation failure. Mol Endocrinol. 2012;26:1847–56.
Stojilkovic SS, Catt KJ. Novel aspects of GnRH-induced intracellular signaling and secretion in pituitary gonadotrophs. J Neuroendocrinol. 1995;7:739–57.
Stojilkovic SS, Koshimizu T. Signaling by extracellular nucleotides in anterior pituitary cells. Trends Endocrinol Metab. 2001;12:218–25.
Stojilkovic SS, Iida T, Merelli F, Torsello A, Krsmanovic LZ, Catt KJ. Interactions between calcium and protein kinase C in the control of signaling and secretion in pituitary gonadotrophs. J Biol Chem. 1991;266:10377–84.
Stojilkovic SS, Tomic M, Kukuljan M, Catt KJ. Control of calcium spiking frequency in pituitary gonadotrophs by a single-pool cytoplasmic oscillator. Mol Pharmacol. 1994;45:1013–21.
Stojilkovic SS, He ML, Koshimizu TA, Balik A, Zemkova H. Signaling by purinergic receptors and channels in the pituitary gland. Mol Cell Endocrinol. 2010a;314:184–91.
Stojilkovic SS, Tabak J, Bertram R. Ion channels and signaling in the pituitary gland. Endocr Rev. 2010b;31:845–915.
Tamura N, Doolittle LK, Hammer RE, Shelton JM, Richardson JA, Garbers DL. Critical roles of the guanylyl cyclase B receptor in endochondral ossification and development of female reproductive organs. Proc Natl Acad Sci U S A. 2004;101:17300–5.
Tao YX, Conn PM. Chaperoning G protein-coupled receptors: from cell biology to therapeutics. Endocr Rev. 2014;35:602–47.
Thompson IR, Chand AN, Jonas KC, Burrin JM, Steinhelper ME, Wheeler-Jones CP, McArdle CA, Fowkes RC. Molecular characterisation and functional interrogation of a local natriuretic peptide system in rodent pituitaries, alphaT3-1 and LbetaT2 gonadotroph cells. J Endocrinol. 2009;203:215–29.
Thompson IR, Ciccone NA, Xu S, Zaytseva S, Carroll RS, Kaiser UB. GnRH pulse frequency-dependent stimulation of FSHbeta transcription is mediated via activation of PKA and CREB. Mol Endocrinol. 2013;27:606–18.
Tobin VA, Canny BJ. The regulation of gonadotropin-releasing hormone-induced calcium signals in male rat gonadotrophs by testosterone is mediated by dihydrotestosterone. Endocrinology. 1998;139:1038–45.
Tsaneva-Atanasova K, Mina P, Caunt CJ, Armstrong SP, McArdle CA. Decoding GnRH neurohormone pulse frequency by convergent signalling modules. J R Soc Interface. 2012;9:170–82.
Tsutsumi M, Zhou W, Millar RP, Mellon PL, Roberts JL, Flanagan CA, Dong K, Gillo B, Sealfon SC. Cloning and functional expression of a mouse gonadotropin-releasing hormone receptor. Mol Endocrinol. 1992;6:1163–9.
Tsutsumi R, Mistry D, Webster NJ. Signaling responses to pulsatile gonadotropin-releasing hormone in LbetaT2 gonadotrope cells. J Biol Chem. 2010;285:20262–72.
Uda S, Saito TH, Kudo T, Kokaji T, Tsuchiya T, Kubota H, Komori Y, Ozaki Y, Kuroda S. Robustness and compensation of information transmission of signaling pathways. Science. 2013;341:558–61.
Ulloa-Aguirre A, Conn PM. Targeting of G protein-coupled receptors to the plasma membrane in health and disease. Front Biosci. 2009;14:973–94.
Vasilyev VV, Pernasetti F, Rosenberg SB, Barsoum MJ, Austin DA, Webster NJ, Mellon PL. Transcriptional activation of the ovine follicle-stimulating hormone-beta gene by gonadotropin-releasing hormone involves multiple signal transduction pathways. Endocrinology. 2002;143:1651–9.
Voliotis M, Perrett RM, McWilliams C, McArdle CA, Bowsher CG. Information transfer by leaky, heterogeneous, protein kinase signaling systems. Proc Natl Acad Sci U S A. 2014;111:E326–33.
Vrecl M, Anderson L, Hanyaloglu A, McGregor AM, Groarke AD, Milligan G, Taylor PL, Eidne KA. Agonist-induced endocytosis and recycling of the gonadotropin-releasing hormone receptor: effect of beta-arrestin on internalization kinetics. Mol Endocrinol. 1998;12:1818–29.
Wang Y, Fortin J, Lamba P, Bonomi M, Persani L, Roberson MS, Bernard DJ. Activator protein-1 and smad proteins synergistically regulate human follicle-stimulating hormone beta-promoter activity. Endocrinology. 2008;149:5577–91.
Wang L, Chadwick W, Park SS, Zhou Y, Silver N, Martin B, Maudsley S. Gonadotropin-releasing hormone receptor system: modulatory role in aging and neurodegeneration. CNS Neurol Disord Drug Targets. 2010;9:651–60.
Washington TM, Blum JJ, Reed MC, Conn PM. A mathematical model for LH release in response to continuous and pulsatile exposure of gonadotrophs to GnRH. Theor Biol Med Model. 2004;1:9.
Weck J, Fallest PC, Pitt LK, Shupnik MA. Differential gonadotropin-releasing hormone stimulation of rat luteinizing hormone subunit gene transcription by calcium influx and mitogen-activated protein kinase-signaling pathways. Mol Endocrinol. 1998;12:451–7.
Weiss J, Jameson JL, Burrin JM, Crowley Jr WF. Divergent responses of gonadotropin subunit messenger RNAs to continuous versus pulsatile gonadotropin-releasing hormone in vitro. Mol Endocrinol. 1990;4:557–64.
Wildt L, Hausler A, Marshall G, Hutchison JS, Plant TM, Belchetz PE, Knobil E. Frequency and amplitude of gonadotropin-releasing hormone stimulation and gonadotropin secretion in the rhesus monkey. Endocrinology. 1981;109:376–85.
Willars GB, Royall JE, Nahorski SR, El-Gehani F, Everest H, McArdle CA. Rapid down-regulation of the type I inositol 1,4,5-trisphosphate receptor and desensitization of gonadotropin-releasing hormone-mediated Ca2+ responses in alpha T3-1 gonadotropes. J Biol Chem. 2001;276:3123–9.
Winters SJ, Moore Jr JP. PACAP, an autocrine/paracrine regulator of gonadotrophs. Biol Reprod. 2011;84:844–50.
Winters SJ, Ishizaka K, Kitahara S, Troen P, Attardi B. Effects of testosterone on gonadotropin subunit messenger ribonucleic acids in the presence or absence of gonadotropin-releasing hormone. Endocrinology. 1992;130:726–34.
Wojcikiewicz RJ, Xu Q, Webster JM, Alzayady K, Gao C. Ubiquitination and proteasomal degradation of endogenous and exogenous inositol 1,4,5-trisphosphate receptors in alpha T3-1 anterior pituitary cells. J Biol Chem. 2003;278:940–7.
Wurmbach E, Yuen T, Ebersole BJ, Sealfon SC. Gonadotropin-releasing hormone receptor-coupled gene network organization. J Biol Chem. 2001;276:47195–201.
Yasin M, Dalkin AC, Haisenleder DJ, Kerrigan JR, Marshall JC. Gonadotropin-releasing hormone (GnRH) pulse pattern regulates GnRH receptor gene expression: augmentation by estradiol. Endocrinology. 1995;136:1559–64.
Yuen T, Wurmbach E, Ebersole BJ, Ruf F, Pfeffer RL, Sealfon SC. Coupling of GnRH concentration and the GnRH receptor-activated gene program. Mol Endocrinol. 2002;16:1145–53.
Yuen T, Ruf F, Chu T, Sealfon SC. Microtranscriptome regulation by gonadotropin-releasing hormone. Mol Cell Endocrinol. 2009;302:12–7.
Zhu H, Hille B, Xu T. Sensitization of regulated exocytosis by protein kinase C. Proc Natl Acad Sci U S A. 2002;99:17055–9.
Acknowledgments
This work was funded Project Grants from MRC (93447) and the BBSRC (J014699). KTA gratefully acknowledges the financial support of the EPSRC via grant EP/N014391/1.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this entry
Cite this entry
Garner, K.L., Tsaneva-Atanasova, K., McArdle, C.A. (2017). GnRH Action. In: Simoni, M., Huhtaniemi, I. (eds) Endocrinology of the Testis and Male Reproduction. Endocrinology. Springer, Cham. https://doi.org/10.1007/978-3-319-44441-3_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-44441-3_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-44440-6
Online ISBN: 978-3-319-44441-3
eBook Packages: MedicineReference Module Medicine