
Abstract
The obtaining of nutrients is the most important task in our lives. Energy is central to life’s evolutions; this was one of the aspect that induced the selection of the more adaptable and more energetically profitable species. Nowadays things have changed in our modern society. A high proportion of people has access to plenty amount of food and the obesity appear as one of the pathological characteristics of our society. Energy is obtained essentially in the mitochondria with the transfer of protons across the inner membrane that produce ATP. The exactly regulation of the synthesis and degradation of ATP (ATP ↔ ADP + phosphate) is essential to all form of life. This task is performed by the 5' adenosine monophosphate-activated protein kinase (AMPK). mtDNA is highly exposed to oxidative damage and could play a central role in human health and disease. This high potential rate of abnormalities is controlled by one of the most complex mechanism: the autophagy. AMPK appears to be the key cellular energy sensor involved in multiple cellular mechanisms and is essential to have a good metabolic homeostasis to face all the aggression and start the inflammatory reaction. Therefore its disturbances have been related with multiple diseases. Recent findings support the role of AMPK in inflammation and immunity such as Metabolic Syndrome, Obesity and Diabetes. All these Metabolic Disorders are considered pandemics and they need an adequate control and prevention. One important way to achieve it is deepen in the pathogenic mechanisms. Mitochondria and AMPK are the key elements through which it happen, their knowledge and research allow us to a better management. The discovery and use of drugs that can modulate them is imperative to improve our way of manage the metabolic disorders.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- AMPK
- Mitochondria
- mtDNA
- Metabolic Diseases
- Metabolic Syndrome
- Obesity
- Diabetes
- Inflammation
- Inflammation treatment
1 Introduction
All forms of life need energy to survive. Energy is central to life’s evolutions; we can only understand the properties of life if we bring energy to equation. The way the energy is produced gives advantage to one organism against the other one. During the evolution of all the form of lives in the earth, this was one of the aspects that induced the selection of the more adaptable and more energetically profitable species. The Homo sapiens as an animal species has suffered famine period like other animal species, but we could survive. The obtaining of nutrients is the most important task in our lives. This was essential in the past because large periods of nutrient shortage were very common. But nowadays things have changed in our modern society. A high proportion of people have access to plenty amount of food. The way to deal with this time of plenty is to consume the calories with exercise or accumulate the nutrients in adipose tissue. In a sedentary attitude of the population, the obesity appears as one of the pathological characteristics of our society. Our entire metabolism, controlled by genes, needs to be adapted to this abundance of nutrient but usually it cannot be achieved. The breaks of the homeostasis and equilibrium in the metabolism involve most of the times to the development of metabolic disorder (Ezzati et al. 2002).
Essentially, all living cells power themselves through the flow of protons (positively charged hydrogen atoms), in what amounts to a kind of electricity with protons in place of electrons. The energy we gain from burning food in respiration is used to pump protons across a membrane, forming a reservoir on one side of the membrane. The flow of protons back from this reservoir can be used to power work in the same way as a turbine in a hydroelectric dam. This transfer of protons across a membrane is known as chemiosmotic hypothesis (Mitchell 1961). This use of proton gradients is universal across life on earth; proton power is as much an integral part of all life as the universal genetic code. This mechanism produces energy, but the requirements of energy to live is extremely high. The energy currency used by all living cells is a molecule called ATP (adenosine triphosphate). A single cell consumes around ten million molecules of ATP every second. If we calculate 40 trillion cells contained in the human body, we used about 60–100 kg per day, roughly our own body weight. ATP is usually split into two unequal pieces, ADP (adenosine diphosphate) and inorganic phosphate (PO4 3−). The energy of respiration (the energy released from the reaction of food with oxygen) is used to make ATP from ADP and Pi. This energy comes from just one particular type of chemical reaction known as a redox reaction in which electrons are transferred from one molecule to another. This takes place in the inner membrane of the mitochondria in the electron transport chain that comprise five protein membrane-bound complexes. The electrons pass through this respiratory complexes, protons are ferried across the membrane, the enzyme ATP synthase is activated, and ATP is produced (Rich 2003).
An appreciation of the rising global burden of chronic, noncommunicable diseases and its influence in mortality and global health cost is a main concern in our society. It has been calculated that 35 million people died in 2005 from heart disease, stroke, cancer, and other chronic diseases (Strong et al. 2005). Physicians and health managers have applied effective measures, including behavioral interventions and pharmaceutical treatment, in the prevention and management of chronic diseases, but these are neither widely used nor equitably distributed. Most of them are related to modern life style, mainly with sedentary way of living and obesity. Therefore, the most effective treatment is the prevention of changing these parameters. But the results are not adequate, for instance, overweight and obesity continue to grow its prevalence, even in children and adolescent (Brown et al. 2015). The only way to improve our knowledge of the mechanism of metabolic diseases is to deepen the pathogenic mechanism involved in the energy production and its control. Energy production inside a cell is a very complex and accurate process that needs the proper function of the electron transport chain placed in the internal mitochondrial membrane. It is essential to attend all the environmental requirements as soon as possible, with the exact amount that does not permit to waste energy.
2 Mitochondria
Mitochondria are ubiquitous in eukaryotes and are essential for survival. Their primary function is to support aerobic respiration and to provide energy substrates (such as ATP) for intracellular metabolic pathways. Mitochondria have also been shown to play an important role in iron and calcium homeostasis, amino acid, fatty acid and steroid hormone metabolism, and in cell signaling, particularly in signaling for apoptotic cell death. Mitochondria host several metabolic pathways, including the Krebs cycle, β-oxidation, and lipid and cholesterol synthesis (Schapira 2006). Mitochondria are intracellular double membrane-bound structures. Although traditionally considered as small isolated organelles within the cell, it is more likely that mitochondria form a complex branching network. They are derived from prokaryotic cells (bacteria) that were assimilated by another cell (archaea) through a process called endosymbiosis. Although these organelles usually retain some DNA, most of their original genome has moved to the host nucleus through endosymbiotic gene transfer (Archibald 2015). Human mtDNA is a circular double-stranded molecule about 16 · 6 Kb long. It is much smaller than most nuclear genes. MtDNA codes for 22 transfer and 2 ribosomal RNAs and for 13 proteins. Human mtDNA is extremely compact and contains virtually non-intronic (noncoding) regions. By encoding proteins of the respiratory chain, mtDNA allows individual mitochondria to respond, by gene expression, to changes in membrane potential. Although human mtDNA encodes the basic machinery for protein synthesis, it remains entirely dependent upon the nucleus for the provision of enzymes for replication, repair, transcription, and translation. This dependency lies at the heart of several newly recognized human diseases that are characterized by secondary abnormalities of mtDNA.
There are good reasons to believe that genes affecting the mitochondria could play a central role in human health and disease. Most of the genes that have remained in the mitochondrion have been linked to a series of devastating diseases, indicating the importance of fully functional mitochondria to human health. Genes residing in the mitochondria pose a particular problem, in part because they are unusually prone to damage. Unlike nuclear genes, which are wrapped in protective proteins and stored safely away in the nucleus, mitochondrial genes are vulnerable to attack from highly reactive molecules called free radicals; these are generated during energy production. In mammals, the mutation rate of mitochondrial genes is 10–20 times higher than that of the nuclear genes. The idea that mutations in mitochondrial DNA could cause metabolic diseases, or even aging, is widely accepted (Lane 2006). These alterations range from changes to single DNA bases to deletions of large sections of the genome. Mitochondria that lose their genome (hydrogenosomes and mitosomes) lose the ability to synthesize ATP by chemiosmotic coupling (Lane and Martin 2010). Respiration rates do correlate with the amount of mtDNA in the cell (Williams 1986), and mutations that deplete mtDNA usually cause mitochondrial diseases (Schapira 2006). Oxidative phosphorylation is under tight control by the amount of mtDNA in the cell, and the full complement of mtDNA is necessary to maintain a normal energy production level (Rocher et al. 2008). Mitochondria, along with their tiny genomes, are normally inherited only from the mother—they are present in huge numbers in the egg, whereas the handful in sperm is marked up for destruction in the fertilized egg. This gives at least some mitochondrial diseases a maternal-inheritance pattern. Even so, trying to spot mitochondrial diseases by looking to the mother can be grossly misleading and has own played the importance of these organelles in disease. More than 80 % of diseases known to be linked to faulty mitochondria do not follow a maternal-inheritance pattern at all. Why not? At least partly because some mitochondrial diseases may be caused by mutations in the nuclear genes encoding mitochondrial proteins. So far, mutations in more than 30 nuclear genes have been shown to give rise to mitochondrial disease (Lynch and Conery 2003).
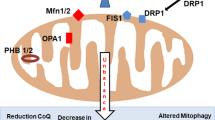
This high potential rate of abnormalities is controlled by one of the most complex mechanism: the autophagy. Autophagy is the most efficient mitochondrial turnover mechanism, providing for the complete removal of irreversibly damaged mitochondria (mitophagy). It is believed that mitochondria are normally replaced every 2–4 weeks in rat brain, heart, liver, and kidneys, although recent studies have shown that the turnover rate might be considerably higher. Mitophagy is particularly important for long-lived postmitotic cells, whose mitochondria have pronounced oxidative damage (Terman et al. 2010), and also prevents an excessive accumulation of mitochondria. It should be mentioned that the changes in mitochondriogenesis are tissue specific (Johnson et al. 2007), being more dramatic in the central nervous system, which is consistent with the generally higher susceptibility of neurons and other postmitotic cells to the aging process (Lopez-Lluch et al. 2008). Mitochondria are dynamic structures that show different morphologies: small spheres, short rods, or long tubules that depend on cell type and also cell status. In most eukaryotic cells, mitochondria move along cytoskeletal tracks, and their overall morphology depends on the balance between fusion and fission events. For example, an extent of fusion activities leads to interconnected mitochondrial networks, and on the contrary, an extent of fission events generates numerous different small spherical organelles (Westermann 2008). Mitochondrial membrane fusion/fission processes are clearly involved in mitochondria dynamics and that these events are undoubtedly critical for several cell functions and the balance cell life/cell death. Perturbations of mitochondrial dynamics can have tremendous consequences on cell metabolism and therefore on cell life/cell death (Benard and Rossignol 2008). The frequent fusion/fission events undergone by the mitochondrial network appear clearly linked to the bioenergetic state of mitochondria (Twig et al. 2008) and involve a protein machinery and a group of lipids (Furt and Moreau 2009). Disturbed mitochondrial dynamics is involved in the most important chronic disease such as neurodegenerative disorders (Burté et al. 2015), aging process (Gonzalez-Freire et al. 2015), and cardiovascular disease (Biala et al. 2015).
3 AMPK
The electron transport chain placed in the inner mitochondrial membrane has a main function of producing ATP, which is the battery of our cells. Therefore, the exact regulation of the synthesis and degradation of ATP (ATP ↔ ADP + phosphate) is essential to all forms of life. This reaction is maintained by catabolism many orders of magnitude away from equilibrium, yielding a high ratio of ATP to ADP that is used to drive energy-requiring processes. ATP generation needs to remain in balance with ATP consumption, and regulatory proteins that sense ATP and ADP levels would be a logical way to achieve this. This task is performed by the 5′ adenosine monophosphate-activated protein kinase (AMPK). It is a highly conserved sensor of cellular energy status, expressed in essentially all eukaryotic cells. AMPK is switched on by metabolic stresses and xenobiotic compounds that cause a cellular energy imbalance, which is detected as increases in the ratios of ADP–ATP and AMP–ATP (Gowans et al. 2013). Because the energy status of the cell is a crucial factor in all aspects of cell function, it is not surprising that AMPK has many downstream targets whose phosphorylation mediates dramatic changes in cell metabolism, cell growth, and other functions. In general, AMPK switches on catabolic processes that provide alternative pathways to generate ATP, while switching off anabolic pathways and other processes consuming ATP, thus acting to restore cellular energy homeostasis. The kinase evolved in single-celled eukaryotes and is still involved in multicellular organisms in regulating energy balance in a cell-autonomous manner (Hardie et al. 2012).
AMPK and its orthologues seem to exist universally as heterotrimeric complexes comprising a catalytic α-subunit and regulatory β- and γ-subunits. Each of these three subunits takes on a specific role in both the stability and activity of AMPK. Specifically, the γ-subunit includes four particular cystathionine beta synthase (CBS) domains giving AMPK its ability to sensitively detect shifts in the AMP–ATP ratio (Stapleton et al. 1996). In mammalian cells, AMPK is activated by various types of metabolic stresses (starvation for glucose or oxygen or addition of a metabolic poison, muscle contraction), drugs (metformin, phenformin, resveratrol, epigallocatechin, capsaicin, curcumin), and xenobiotics through the mechanisms described above, which involve increases in cellular AMP, ADP, or Ca2+. These can now be regarded as the classical or “canonical” AMPK activation mechanisms. However, recent work suggests that other stimuli activate AMPK via mechanisms that do not involve changes in the levels of AMP, ADP, and Ca2+, which can therefore be termed “noncanonical” mechanisms such as those triggered by ROS and DNA-damaging agents. AMPK acts as a metabolic master switch regulating several catabolic intracellular systems, including the cellular uptake of glucose and fatty acids, the β-oxidation of fatty acids, the biogenesis of glucose transporter 4 (GLUT4), mitochondrial biogenesis, and mitophagy. Also conserves ATP by switching off almost all anabolic pathways, including the biosynthesis of lipids, carbohydrates, proteins, and ribosomal RNA. It achieves this in part by phosphorylating and/or regulating enzymes or regulatory proteins that are directly involved in these pathways (Shirwany and Zou 2014). AMPK appears to have evolved early in the evolution of unicellular eukaryotes as a signaling pathway that orchestrated responses to glucose starvation in a cell-autonomous manner. But also it is intriguing that hormones that modulate energy balance at the whole-body level, which clearly arose later during the evolution of multicellular organisms, appear to have adapted to interact with the AMPK system, especially in the hypothalamus. Adiponectin, leptin, ghrelin, insulin, and triiodothyronine can influence the AMPK production in the hypothalamus (Hardie and Ashford 2014). In mammals, it also regulates metabolism and helps to maintain energy balance at the whole-body level. It does this by mediating effects of hormones and other agents acting on neurons in different hypothalamic regions, which regulate intake of food (and hence energy) and energy expenditure. AMPK also regulates diurnal rhythms of feeding and metabolism. By switching off biosynthetic pathways required for cell growth, AMPK activation exerts a cytostatic effect, helping to explain why its upstream activator, LKB1, is a tumor suppressor. Commensurate with its role in preserving cellular energy homeostasis, AMPK also downregulates ATP-requiring processes outside metabolism, including progress through the cell cycle (another potential tumor suppressor effect) and firing of action potentials in neurons (Hardie 2014). Some works demonstrate that the AMPK signaling pathway also plays a role in bone physiology. Activation of AMPK promotes bone formation in vitro and the deletion of α- or β-subunit of AMPK decreases bone mass in mice (Jeyabalan et al. 2012).
AMPK appears to be the key cellular energy sensor involved in multiple cellular mechanisms and is essential to have a good metabolic homeostasis to face all the aggression and start the inflammatory reaction. Therefore, its disturbances have been related with multiple diseases. Age-related decrease of mitochondrial biogenesis is related, at least in part, to diminished AMPK activity (Reznick et al. 2007). AMPK appears to be the key cellular energy sensor, linking decreased mitochondriogenesis to several aging-associated changes, including insulin resistance and deficient lipid metabolism (Qiang et al. 2007). AMPK activity and autophagy of monocytes were significantly decreased in Acute Coronary Syndrome patients due to a decrease in plaque vulnerability and subsequent plaque rupture (Cheng et al. 2015). Recent findings support the role of AMPK in inflammation and immunity, providing the enticing prospect of new therapeutic approaches for inflammatory diseases. All of the AMPK activators have been reported to inhibit inflammatory responses in various model systems, and AMPK-activating drugs do have anti-inflammatory actions in animal models (O’Neill and Hardie 2013). Tumor cells appear to be under selection pressure to downregulate AMPK, thus limiting its restraining influence on cell growth and proliferation. Paradoxically, however, a complete loss of AMPK function, which appears to be rare in human cancers, may be deleterious to survival of tumor cells. AMPK can therefore be either a friend or a foe in cancer, depending on the context (Hardie 2015).
4 Metabolic Syndrome and Obesity
The metabolic syndrome (MetS) is a major and escalating public health and clinical challenge worldwide in the wake of urbanization, surplus energy intake, increasing obesity, and sedentary life habits. MetS confers a fivefold increase in the risk of type 2 diabetes mellitus (T2DM) and twofold the risk of developing cardiovascular disease (CVD) over the next 5–10 years (Alberti et al. 2009). Further, patients with the MetS are at two to fourfold increased risk of stroke, a three to fourfold increased risk of myocardial infarction (MI), and twofold the risk of dying from such an event compared with those without the syndrome (Alberti and Zimmet 2005) regardless of a previous history of cardiovascular events (Olijhoek et al. 2004).
MetS is a state of chronic low-grade inflammation as a consequence of complex interplay between genetic and environmental factors. Insulin resistance, visceral adiposity, atherogenic dyslipidemia, endothelial dysfunction, genetic susceptibility, elevated blood pressure, hypercoagulable state, and chronic stress are the several factors that constitute the syndrome (Table 6.1).
There have been several definitions of MetS, but the most commonly used criteria for definition at present are from the World Health Organization (WHO) (Table 6.2).
Hypercaloric diets initially result in obesity due to the storage of extra energy in the adipose tissue. However, the continuous caloric overload eventually results in the aberrant accumulation of lipids in non-adipose tissues (Virtue et al. 2012). The direct pathological consequence of chronic hypercaloric diets is actually a multisystemic deterioration known as metabolic syndrome (Kaur 2014). Of note, the comorbidities associated with metabolic syndrome also overlap with some of the most important aging-associated diseases, namely, diabetes, cardiovascular and cerebrovascular diseases, and cancer (Gurevich-Panigrahi et al. 2009).
4.1 Inflammation in Obesity and MetS
Considering the “obesity epidemic” as mainly responsible for the rising prevalence of MetS due to the strong connection between this syndrome and, especially, abdominal obesity, it is vital to bear in mind that obesity is not only a risk factor but also a disease in itself. Obesity contributes to hypertension, high serum cholesterol, low HDL cholesterol, and hyperglycemia, and it otherwise associates with higher CDV risk. Excess adipose tissue releases several products that apparently exacerbate these risk factors. They include free fatty acids (FFA), cytokines: interleukin-6 and Tumor Necrosis Factor-α (IL-6 and TNF-α), plasminogen activator inhibitor 1 (PAI-1), and adiponectin. A high plasma FFA level overloads muscle and liver with lipids, which is an event commonly found in abdominal obesity (Miles and Jensen 2005), enhancing insulin resistance (Boden et al. 2001). Evidence suggests that TNF-α induces adipocytes apoptosis (Xydakis et al. 2004) and promotes insulin resistance by the inhibition of the insulin receptor substrate 1 signaling pathway (Lau et al. 2005). Plasma TNF-α is positively associated with the body weight and triglycerides (TGs), while a negative association exists between plasma TNF-α levels and high-density lipoprotein-cholesterol (HDL-C) (Xydakis et al. 2004). In addition, IL-6 has been shown to be positively associated with body mass index (BMI), fasting insulin, and the development of T2DM (Pradhan et al. 2001) and negatively associated with HDL-C (Zuliani et al. 2007).
Plasminogen activator inhibitor-1 (PAI-1) levels are increased in abdominally obese subjects (Cigolini et al. 1996), and an elevated PAI-1 contributes to a prothrombotic state, whereas low adiponectin levels that accompany obesity correlate with worsening of metabolic risk factors, since it regulates the lipid and glucose metabolism, increases insulin sensitivity, regulates food intake and body weight, and protects against a chronic inflammation (Liu and Liu 2010). Adiponectin is seen to be “protective,” not only in its inverse relationship with the features of MetS (Engeli et al. 2003) but also through its antagonism of TNF-α action (Ouchi et al. 2000). Considering all these aspects, new therapies for metabolic diseases have been investigated.
4.2 Mitochondrial DNA Alterations in Obesity and MetS
Obesity results from an energy surplus, greater energy intake than expenditure. Excess energy is stored as fat in white adipose tissue (WAT), dysfunction of which lies at the core of obesity and associated metabolic disorders. By contrast, brown adipose tissue (BAT) burns fat and dissipates chemical energy as heat. The development and activation of “brown-like” adipocytes, also known as beige cells, result in WAT browning and thermogenesis (heat production). Both “white” and “brown” adipose cells (color is due to the quantity of mitochondria) play a crucial role in thermogenesis. “Brown-like” adipocytes, which have a rich sympathetic innervation, are present in rodents and infants, whereas in adults are usually interspersed with WAT. Due to the numerous mitochondria existing in BAT, these cells are an important heat source, generating more heat and less ATP than “white-like” adipocytes. This event occurs because of the presence of mitochondrial uncoupling proteins (UCP), as it has already been demonstrated in murine models. Three isoforms have been identified, UCP1, 2, and 3, which are differently distributed in BAT.
These proteins “uncouple” oxidative phosphorylation, and those way mitochondria continues oxidizing substrates while producing minor quantities of ATP levels, which favors the net loss of energy in the form of heat. As one would expect, cold exposure or leptin administration increases UCP activity and quantity in BAT.
Noradrenalin is a hormone which acts on β3-adrenergic receptors in BAT and boosts peroxisome proliferator-activated receptor gamma (PPAR-γ) activity, which activates, at the same time, the gene that encodes UCP-1. Curiously, β3-adrenergic receptors expression is lower in mice that are genetically obese.
Until recently, studies on mtDNA biology in adipose tissue have been limited to the analysis of mitochondrial biogenesis in BAT. However, recent studies have highlighted the importance of mitochondrial biogenesis in WAT and the potential for mitochondrial alterations to disturb white adipocyte development and function. Studies by Corvera and collaborators (Wilson-Fritch et al. 2003) have shown that mitochondrial biogenesis is directly associated with white adipocyte differentiation; genetically obese mice (ob/ob) displayed impaired mitochondrial mass and function in white fat and thiazolidinediones, PPAR-γ activators that favor adipocyte differentiation, ameliorated these alterations (Wilson-Fritch et al. 2004). It has been also shown that white adipocyte differentiation is associated with increases in the relative abundance of mtDNA, and upregulation of components of the mtDNA replication and transcription machinery, such as TFAM (Mitochondrial Transcription Factor A) (Shi et al. 2008), and components of deoxynucleotide metabolism are required for mtDNA replication (Rylova et al. 2005). Agents that promote white adipocyte differentiation in vitro, such as glitazones, also increase mtDNA levels in human adipocytes in vitro (Bogacka et al. 2005a).
Studies of potential alterations in WAT mtDNA as they relate to obesity have focused on two aspects: changes in mtDNA levels that underlie obese phenotypes and the occurrence of mutated, polymorphic, forms of mtDNA that are specifically associated with obesity. In experimental models of obesity, such as ob/ob or db/db mice, abnormally low levels of mtDNA have been reported (Choo et al. 2006; Rong et al. 2007). As noted above for in vitro studies, treatment of obese mice with glitazones increases mtDNA levels in white fat (Rong et al. 2007). A study in which mice were treated with a diet enriched in polyunsaturated fatty acids identified mtDNA encoded transcripts and proteins among the most upregulated genes in WAT; this upregulation was associated with an enhancement of fatty acid oxidation in WAT (Flachs et al. 2006).
In humans, the scenario appears to be more complex. It has been reported that mtDNA levels in adipose tissue are lowered in type 2 diabetic patients (Bogacka et al. 2005b), and studies by Arner and collaborators (Dahlman et al. 2006) have confirmed that mtDNA levels are not associated with obesity per se, but rather with type 2 diabetes phenotypes. Moreover, mtDNA levels were found to be strongly related to lipogenesis in WAT, rather than to BMI.
The mechanism by which mtDNA copy number in white adipose tissue could affect lipogenesis rate remains to be established, but it stands in contrast to the expected relationship between mtDNA level variations and energy expenditure and fat oxidation. Moreover, in humans, as in rodents, pioglitazone treatment causes an increase in mtDNA levels in WAT of type 2 diabetes patients (Bogacka et al. 2005b), but not in nondiabetic obese individuals (Bogacka et al. 2007). These findings highlight the potential role of mtDNA levels in WAT mass.
4.3 Homeostatic Control of Energy Balance in Obesity
The arcuate nucleus (ARC) is a key hypothalamic nucleus that regulates appetite, eating behavior, and energy state (Schwartz et al. 2000). It receives afferent pathways from digestive tract and contains leptin, an adipokine involved in the regulation of satiety and energy intake (Lau et al. 2005), and other significant hormone receptors. A reduction in leptin levels activates orexigenic neurons, leading to an increase in food intake, fat synthesis, and storage (anabolism) and to a decrease in energy expenditure. In reverse, when increases in leptin levels take place, another group of neurons are activated, triggering an anorexigenic effect and catabolism.
Energy balance depends directly on food intake, energy storage in adipose tissue, and energy expenditure (Spiegelman and Flier 2001). For the majority of people, this process is closely connected by an homeostatic system which integrates different hormones, such as leptin (Ahima and Flier 2000), considered as an indicator of lipid reserves, since an augmentation in lipid deposits enhances leptin releasing in plasma by adipocytes. Ghrelin, which is released to the intestine during food intake, provides us the feeling of hunger, and cholecystokinin (CCK), secreted into the duodenum as a response to the process of food intake and digestion, providing satiety, as well.
Since homeostatic control in energy balance is extremely complex, it is not easy to precisely determine what does not work in obesity. In fact, when leptin was discovered (Zhang et al. 1994), it was thought that an alteration in its kinetics would provide a simple explanation to this disease, but there is a notable variability in leptin sensibility among individuals, and some people seem to synthesize insufficient amounts of this hormone. However, plasmatic leptin usually reaches higher levels in obese than in normal weight people. What prevail in obese is a greater resistance to leptin and not an insufficient production of it (Hutley and Prins 2005). This resistance may obey to alterations in blood circulation leptin transport, in its transport to Central Nervous System (CNS), or in hypothalamic leptin receptors (like db/db mice).
Obesity, could be involved in other alteration in mediators apart from leptin, for example, TNF-α, cytokine that transmits information from adipose tissue to brain, is increased in insulin-resistant obese adipose tissue. Another common pathophysiological alteration in obesity is a decreasing insulin sensibility in skeletal muscle and adipose tissue.
Thus, events causing obesity depends on diet, exercise, social, economic and cultural factors, and genetic predisposition (Barsh et al. 2000). Although other causes have been related to alterations in leptin action or synthesis, such as a thermogenesis decreasing in adipocytes or a reduction the energy metabolic consumption. A key player at regulating energy balance is AMP-activated protein kinase (AMPK), at both cellular and whole-body levels, placing it at the center stage in studies of obesity, diabetes, and the metabolic syndrome (Hardie 2008).
4.4 AMPK: A Master Metabolic Regulator
4.4.1 Lipid Metabolism Controlled by AMPK
AMPK plays a key role in lipid metabolism, being involved in Acetyl-CoA carboxylase (ACC) phosphorylation and inactivation. ACC catalyzes the transformation from Acetyl-CoA into malonyl-CoA, which is the very first reaction in fatty acid biosynthesis in liver and adipose tissue. By inactivating ACC, AMPK is then responsible for the inhibition of fatty acid synthesis in lipogenic tissues. AMPK also has long-term effects on transcriptional genes involved in lipogenesis, ACC and fatty acid synthesis, interfering with the expression, and the activity of transcriptional factors, such as sterol regulatory element-binding protein 1c (SREBP1c) (Hardie 2008; Foretz et al. 2005) and carbohydrate response element-binding protein (ChREBP) (Zhou et al. 2001).
In addition, in both liver and striated muscle (skeletal and myocardial), malonyl-CoA produced by ACC plays a regulatory role. It blocks, in fact, fatty acids transport from cytosol to mitochondria by inhibiting carnitine palmityl transferase (CPT-1). AMPK activation in those tissues triggers a decrease in cytosolic concentration of malonyl-CoA, enabling that way the fatty acids penetration into the mitochondria and its consequent oxidation. By this mechanism, recent data reveal that the adipokines, leptin, and adiponectin stimulate fatty acid oxidation in both liver and skeletal muscle secondarily to the AMPK activation in these tissues (Minokoshi et al. 2002; Tomas et al. 2002). So, the effect of lipid depletion on these tissues improves metabolic parameters in different insulin-resistant rodent models. In fact, the accumulation of triglycerides in liver and skeletal muscle is linked to the pathophysiology of insulin resistance in human and animals, as well as lipid depletion in these tissues ameliorates insulin sensitivity (lipotoxicity concept). So here lies the metabolic interest of the AMPK activation on lipotoxicity reduction.
AMPK may be also involved in the hypothalamus satiety central control; this brain region plays an essential role in energy homeostasis by controlling food intake and energy expenditure. Hypothalamic AMPK activity varies according to the nutritional status; AMPK is activated in fasting and inhibited in satiety period (Minokoshi et al. 2004). At the same time, it is also interesting that the existing relationship between the activity of hypothalamic AMPK and food intake in response to different hormones or metabolites is known to be modulated by nutritional state. For example, ghrelin and endocannabinoids activate AMPK and induce food intake while insulin, glucose, or leptin acts in the opposite manner (Andersson et al. 2004; Kola et al. 2005). Several studies have demonstrated that the variations in hypothalamic energetic state are able to directly modulate AMPK activity, which suggests that hypothalamic AMPK may be a therapeutic target in the numerous factors that affect eating behavior. But, it is important to bear in mind that AMPK is differently regulated in the hypothalamus and in peripheral tissues. For example, leptin activates AMPK in skeletal muscle and inhibits it in the hypothalamus, which suggests that regulation mechanisms are not the same. Therefore, drawbacks in peripheral tissues must be taken into account when considering AMPK as a pharmacological inhibitor for the treatment of metabolic disorders (Fig. 6.1).

Hypothalamic AMPK regulation by hormonal and nutritional signals
The obesity epidemic and its complications continue rising as a global health challenge, despite the increasing public awareness and the use of lifestyle and medical interventions; the main treatment for obesity consists of lifestyle modifications based on a suitable diet and physical exercise. Since the current antiobesity drugs, such as Orlistat (lipase inhibitor), suffer from numerous disadvantages, gastrointestinal symptoms, elevated blood pressure, abdominal pain, dyspepsia, diarrhea, flatulence, etc (Bray and Tartaglia 2000), the biomedical community is urged to develop new treatments for the metabolic diseases. AMP-activated protein kinase (AMPK), as a master regulator of energy homeostasis, is a potential target for therapeutic agents that may meet this challenge. For these reasons, novel drugs activating AMPK may have potential for the treatment of obesity, T2DM, and MetS.
Some compounds have already shown to have an effect on AMPK. 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) is a known activator of AMPK that induces allosteric changes in AMPK conformation and thereby leading to kinase activation. In 3T3L1 cells, AICAR inhibits adipocyte differentiation by downregulating key transcriptional factors such as sterol regulatory element-binding protein 1 (SREBP1), CCAAT-enhancer binding proteins (C/EBPα), and peroxisome proliferator-activated receptor gamma (PPARγ), which strictly regulates adipocyte differentiation. AICAR proved remarkably effective in maintaining body weight and epididymal fat content, improving insulin sensitivity and glucose tolerance in diet-induced obese mice models. However, in rats, the chronic administration of AICAR resulted in significant changes, in skeletal muscle that included an increase in GLUT4 and glycogen stores, and increased activity of hexokinase and mitochondrial oxidative enzymes (Giri et al. 2004, 2006).
The potential to reduce hypertriglyceridemia and elevated storage of triglycerides by inhibiting triglyceride and fatty acid synthesis and stimulating fatty acid oxidation and also the ability to lower blood glucose by activation of AMPK suggest that modulators of AMPK kinase activity might prove effective remedy for treating obesity and related metabolic disorders. Reports have also shown that certain strains of mouse that are resistant to diet-induced obesity (mice overexpressing uncoupling protein-1 in white adipocytes, stearoyl-CoA desaturase-1 knockouts, and mice overexpressing uncoupling protein-3 in skeletal muscle) exhibit increased basal level of AMPK activity (Matejkova et al. 2004). These findings have led to an intense interest in designing AMPK activators as potential therapies for type II diabetes and obesity.
Thus, AMPK plays a key role in regulating a wide range of activities in lipid and glucose metabolism; it appears as a promising strategy for the treatment of obesity and related metabolic disorders.
Keeping in view the epidemic of obesity, the role of AMPK pharmacotherapy seems to be the most obvious tool to combat the disease and attempts to develop novel therapies via AMPK-mediated mechanisms are worthy of pursuit.
5 Diabetes
The incidence of diabetes is increasing worldwide approaching epidemic proportions. According to National Diabetes Statistics, the number of people diagnosed and undiagnosed with diabetes in the USA reached 29.1 million people, which is 9.3 % of the general population in 2012.
Diabetes is considered a metabolic disease that is characterized by high blood sugar levels over a prolonged period (hyperglycemia). We can differentiate three main types: type 1 which results from the pancreas failure to produce enough insulin, type 2 when cells fail to respond to insulin properly (insulin resistance), and gestational diabetes occurs when pregnant women without a previous history of diabetes develop a high blood sugar level.
Type 2 diabetes (T2DM) is the most common form of this pathology and is directly related to obesity and metabolic syndrome. When obesity is established and body weight increases with age, a parallel state of chronic inflammation, characterized by an elevation of proinflammatory cytokines, can induce changes and switch the metabolic homeostatic set points, leading to T2DM (Medzhitov 2010). In T2DM, the major insulin-resistant organs include liver, muscle, and adipose tissue. In a state of insulin resistance, glucose uptake and utilization are dramatically decreased, and skeletal muscle becomes metabolically inflexible, unable to switch between glucose and fatty acid use. The main complications of diabetes are the microvascular and macrovascular complications such as retinopathy, nephropathy, and cardiovascular diseases, which are mediated by inflammatory processes.
5.1 Inflammation in Diabetes
Nowadays, it is well known that inflammation plays a key role in the natural history of diabetes (Agrawal and Kant 2014). Oxidative stress and inflammation are key players in insulin resistance progression and the establishment of T2DM. We know that inflammation is able to increase insulin resistance. The major cell involved in inflammation and insulin resistance is the adipocyte. Insulin regulates glucose uptake and triglyceride storage by adipocytes. The various adipocytokines, especially leptin, adiponectin, omentin, resistin, and visfatin, contribute to beta cell dysfunction in the pancreas. Adipose tissue also secretes dipeptidyl peptidase-4 (DDP-4) which enhances the degradation of glucagon like peptide-1 (GLP-1) and has an insulinotropic effect on beta cells (insulin secreting cells) (Lamers et al. 2011). Cytokines including tumor necrosis factor-alpha (TNF-α), interleukin beta (IL-1β), and interferon-gamma (IFN-ɣ) disrupt the regulation of intracellular calcium in the beta cells and hence insulin release. TNF-α acts on beta cells leading to their accelerated death (Cai et al. 2011).
Oxidative stress is another pathway that leads to inflammation through activation of cytokines (Lamb and Goldstein 2008). Pancreatic islets have low antioxidant defense and are hence vulnerable to oxidative stress. Marselli found differential regulation of oxidative stress genes in cells of T2DM subject compared with healthy controls (Marselli et al. 2010).
An emerging body of evidence also suggests that insulin suppresses the inflammatory process not only through preventing hyperglycemia but also by modulating key inflammatory molecules (Hyun et al. 2011). With all this in mind, the search for anti-inflammatory therapies for diabetes was started.
5.2 Mitochondrion and Insulin Resistance
The concept of insulin resistance was introduced like the previous step of T2DM. Although, it became evident that insulin resistance was not confined to T2DM, has been regarded as the centerpiece of the pathophysiologic mechanism of T2DM.
The relation between mitochondrial dysfunction and insulin resistance is known since the 1960s. To demonstrate the importance of this relationship the assays of Jucker and his colleagues (Jucker et al. 2001), who using a sophisticated nuclear magnetic resonance spectroscopy, measure the mitochondrial function in the human liver and muscle. They showed that the insulin resistance in elderly people can be explained about 40 % reduction in the mitochondrial oxidative phosphorylation capability compared to young people and demonstrated that T2DM patients showed approximately 30 % reduction in mitochondrial phosphorylation activity compared to the insulin-sensitive control subjects (Petersen et al. 2003, 2004). A recent review established a direct relation between defective mitochondrial dysfunction, reduction on fatty acid oxidation, and inhibition of glucose transport. This is the hallmark of insulin resistance and T2DM. The chronic production of excess ROS and inflammation result in mitochondrial dysfunction potentially inducing lipid accumulation and the endless vicious cycle of insulin resistance (Hernández-Aguilera et al. 2013). Altered mitochondrial function is the major factor that leads to increased muscular lipid accumulation and decreased insulin sensitivity. Mitochondrial dysfunction could provide important implications for the diagnosis and treatment of T2DM and other related disease like obesity and metabolic syndrome (Lee et al. 2010).
5.3 Role of AMPK in Diabetes
A number of physiological processes have been shown to stimulate AMPK, including conditions that lead to alterations of the intracellular AMP/ATP ratio (e.g., hypoxia, glucose deprivation) and calcium concentration, as well as the action of various hormones, cytokines, and adipokines. The activated form of the enzyme is responsible for metabolic changes via phosphorylation of various downstream substrates. The net effect is a change in local and whole-body energy utilization from an energy-consuming state to an energy-producing state in order to restore energy balance.
We know AMPK plays a key role in the interconversion of glucose (the primary cellular energy substrate) and its storage forms by affecting the transcription and translocation of the GLUT4 glucose transporter, glycogen synthesis, glycolysis, and gluconeogenesis.
Beginning with mice clinical trials, Hardie was the first to relate a possible role of disturbed AMPK signaling in diabetes (Winder and Hardie 1999). After that, a growing body of evidence has begun to validate this idea. For example, studies in AMPKα2 knockout mice observed hyperglycemia, glucose intolerance, and increased hepatic glucose production (Andreelli et al. 2006). A recent review suggests that AMPK mediates glucose uptake and is complementary to insulin as well as possibly independent of this hormone, thereby implicating the kinase in diabetes pathophysiology (Shirwany and Zou 2014).
AMPK is a key regulator of energy balance and plays many roles in human health and disease. Activation of AMPK by pharmacological agents holds a considerable potential to reverse the metabolic abnormalities associated with T2DM. So, AMPK could be a potential target for novel agents that may meet this global epidemic (Zhang et al. 2009).
5.4 AMPK and Inflammation: Targets of Treatment
A growing body of evidence is emerging to show that metabolic diseases are intimately related to chronic inflammation. The new pharmacological strategies are focused to reduce silent inflammation (Scheen et al. 2015). The considered insulin sensitizers or glucose-lowering agents appear to have greater anti-inflammatory activity than insulin-secreting agents (Pfützner et al. 2005).
Several glucose-lowering agents currently used as antidiabetic medications exert anti-inflammatory actions that may contribute to improved T2DM patients’ outcomes. This effect may result from correction of hyperglycemia, but may also be due to direct effects of the drug, independent of improvement of glucose control (Scheen et al. 2015). This is demonstrated for metformin and thiazolidinediones (TZDs).
5.4.1 Metformin
This is the first-choice drug for the management of T2DM. This biguanide acts as an AMP-activated protein kinase (AMPK) activator (Foretz et al. 2014). Activation of AMPK has a high number of potentially antiatherosclerotic effects, including reducing inflammatory cell adhesion to blood vessel endothelium, reducing lipid accumulation, proliferation of inflammatory cells caused by oxidized lipids, stimulation of gene expression responsible for cellular antioxidant defenses, and stimulation of enzymes responsible for nitric oxide formation (Ewart and Kennedy 2011).
Metformin can inhibit proinflammatory responses and cytokine-induced nuclear factor kappab (NF-κB) activation via AMPK activation in vascular endothelial cells (Hattori et al. 2006; Isoda et al. 2006) and also inhibit inflammatory responses via the AMPK-phosphatase and tensin homologue (PTEN) pathway in vascular smooth muscle cells (Kim and Choi 2012). AMPK activity can also inhibit monocyte-to-macrophage differentiation (Vasamsetti et al. 2015), while other anti-inflammatory mechanisms have been proposed with lysosomes as a target of metformin (Lockwood 2010).
Although, surveys with metformin in human showed less pronounced efficacy than TZDs in reducing various inflammatory markers (Erem et al. 2014; Hanefeld et al. 2011), it seems clear that metformin may confer benefits in chronic inflammatory diseases independent of its ability to normalize blood glucose. There is now growing interest in identifying and exploiting AMPK anti-inflammatory effects with the development of new compounds that are currently under investigation (Scheen et al. 2015).
5.4.2 Thiazolidinediones
TZDs or glitazones are a group of agonists of peroxisome proliferator-activated receptor gamma (PPARɣ). PPARs have been implicated as a molecular pathway in insulin resistance, T2DM, and atherosclerosis (Szanto and Nagy 2008). When the rat muscle cells were isolated and incubated in culture medium containing TDZ for 15 min, they significantly increased phosphorylation of AMPK (LeBrasseur et al. 2006), as well as the AMP/ATP ratio. It is suggested that TDZs can activate AMPK by a mechanism that is likely independent of PPARɣ-regulated gene transcription. However, the major effect of TDZs is likely to be on the release of adiponectin by adipocytes, leading to activation of AMPK in liver to reduce glucose production (Kubota et al. 2007). Several studies have compared the anti-inflammatory effects of glitazones with other glucose-lowering agents and found glitazones to be superior (Marfella et al. 2006).
Many other specific anti-inflammatory approaches have been investigated in recent years to treat T2DM and its associated vascular complications, but none has yet emerged for use in clinical practice (Esser et al. 2015).
6 Conclusions
Energy production and its control are main tasks in every form of lives and are essential to have a proper homeostasis to adapt to the environment and to face every aggression. The Metabolic Disorders are considered pandemics and they need an adequate control and prevention. One important way to achieve it is to deepen the pathogenic mechanisms. Mitochondria and AMPK are the key elements through which it happens; their knowledge and research allow us to a better management. The discovery and use of drugs that can modulate them are imperative to improve our way of managing the metabolic disorders.
References
Agrawal NK, Kant S (2014) Targeting inflammation in diabetes: newer therapeutic options. World J Diabetes 5:697–710
Ahima RS, Flier JS (2000) Leptin. Annu Rev Physiol 62:413–437
Alberti KGMM, Zimmet P (2005) The metabolic syndrome—a new worldwide definition. Lancet 366:1059–1062
Alberti KGMM, Eckel RH, Grundy SM et al (2009) Harmonizing the metabolic syndrome: a joint interim statement of the international diabetes federation task force on epidemiology and prevention; National heart, lung, and blood institute; American heart association; World heart federation; International atherosclerosis society; And international association for the study of obesity. Circulation 120:1640–1645
Andersson U, Filipsson K, Abbott CR et al (2004) AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem 279:12005–12008
Andreelli F, Foretz M, Knauf C, Cani PD, Perrin C, Iglesias MA, Pillot B, Bado A, Tronche F, Mithieux G, Vaulont S, Burcelin R, Viollet B (2006) Liver adenosine monophosphate-activated kinasealpha2 catalytic subunit is a key target for the control of hepatic glucose production by adiponectin and leptin but not insulin. Endocrinology 147:2432–2441
Archibald JM (2015) Evolution: gene transfer in complex cells. Nature 524:423–424
Barsh GS, Farooqi IS, O’Rahilly S (2000) Genetics of body-weight regulation. Nature 404:644–651
Benard G, Rossignol R (2008) Ultrastructure of the mitochondrion and its bearing on function and bioenergetics. Antioxid Redox Signal 10:1313–13142
Biala AK, Dhingra R, Kirshenbaum LA (2015) Mitochondrial dynamics: orchestrating the journey to advanced age. J Mol Cell Cardiol 83:37–43
Boden G, Lebed B, Schatz M, Homko C, Lemieux S (2001) Effects of acute changes of plasma free fatty acids on intramyocellular fat content and insulin resistance in healthy subjects. Diabetes 50:1612–1617
Bogacka I, Ukropcova B, McNeil M, Gimble JM, Smith SR (2005a) Structural and functional consequences of mitochondrial biogenesis in human adipocytes in vitro. J Clin Endocrinol Metab 90:6650–6656
Bogacka I, Xie H, Bray GA, Smith SR (2005b) Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes 54:1392–1399
Bogacka I, Gettys TW, de Jonge L et al (2007) The effect of β-adrenergic and peroxisome proliferator-activated receptor-γ stimulation on target genes related to lipid metabolism in human subcutaneous adipose tissue. Diabetes Care 30:1179–1186
Bray GA, Tartaglia LA (2000) Medicinal strategies in the treatment of obesity. Nature 404:672–677
Brown CL, Halvorson EE, Cohen GM, Lazorick S, Skelton JA (2015) Addressing childhood obesity: opportunities for prevention. Pediatr Clin North Am 62:1241–1261
Burté F, Carelli V, Chinnery PF, Yu-Wai-Man P (2015) Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol 11:11–24
Cai K, Qi D, Wang O, Chen J, Liu X, Deng B, Qian L, Liu X, Le Y (2011) TNF-α acutely upregulates amylin expression in murine pancreatic beta cells. Diabetologia 54:617–626
Cheng J, Qiao L, Xu X, Zhai C, Zhao K, Ji X, Chen W (2015) Lower AMP-activated protein kinase level is associated with the vulnerability of coronary atherosclerotic plaques by attenuating the expression of monocyte autophagy. Coron Artery Dis 26:322–327
Choo HJ, Kim JH, Kwon OB et al (2006) Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia 49:784–791
Cigolini M, Targher G, Andreis IAB, Tonoli M, Agostino G, De Sandre G (1996) Visceral fat accumulation and its relation to plasma hemostatic factors in healthy men. Arterioscler Thromb Vasc Biol 16:368–374
Dahlman I, Forsgren M, Sjögren A et al (2006) Downregulation of electron transport chain genes in visceral adipose tissue in type 2 diabetes independent of obesity and possibly involving tumor necrosis factor-α. Diabetes 55:1792–1799
Engeli S, Feldpausch M, Gorzelniak K et al (2003) Association between adiponectin and mediators of inflammation in obese women. Diabetes 52:942–947
Erem C, Ozbas HM, Nuhoglu I, Deger O, Civan N, Ersoz HO (2014) Comparison of effects of gliclazide, metformin and pioglitazone monotherapies on glycemic control and cardiovascular risk factors in patients with newly diagnosed uncontrolled type 2 diabetes mellitus. Exp Clin Endocrinol Diabetes 122:295–302
Esser N, Paquot N, Scheen AJ (2015) Anti-inflammatory agents to treat or prevent type 2 diabetes, metabolic syndrome and cardiovascular disease. Expert Opin Investig Drugs 24:283–307
Ewart MA, Kennedy S (2011) AMPK and vasculoprotection. Pharmacol Ther 131:242–253
Ezzati M, Lopez AD, Rodgers A, Vander Hoorn S, Murray CJ, Comparative Risk Assessment Collaborating Group (2002) Selected major risk factors and global and regional burden of disease. Lancet 360:1347–1360
Flachs P, Mohamed-Ali V, Horakova O et al (2006) Polyunsaturated fatty acids of marine origin induce adiponectin in mice fed a high-fat diet. Diabetologia 49:394–397
Foretz M, Ancellin N, Andreelli F et al (2005) Short-term over expression of a constitutively active form of AMP-activated protein kinase in the liver leads to mild hypoglycemia and fatty liver. Diabetes 54:1331–1339
Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B (2014) Metformin: from mechanisms of action to therapies. Cell Metab 20:953–966
Furt F, Moreau P (2009) Importance of lipid metabolism for intracellular and mitochondrial membrane fusion/fission processes. Int J Biochem Cell Biol 41:1828–1836
Giri S, Nath N, Smith B, Viollet B, Singh AK, Singh I (2004) 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: a possible role of AMP-activated protein kinase. J Neurosci 24:479–487
Giri S, Rattan R, Haq E et al (2006) AICAR inhibits adipocyte differentiation in 3T3L1 and restores metabolic alterations in diet-induced obesity mice model. Nutr Metab 3:1743–7075
Gonzalez-Freire M, de Cabo R, Bernier M, Sollott SJ, Fabbri E, Navas P, Ferrucci L (2015) Reconsidering the role of mitochondria in aging. J Gerontol A Biol Sci Med Sci 70:1334–1342
Gowans GJ, Hawley SA, Ross FA, Hardie DG (2013) AMP is a true physiological regulator of AMP-activated protein kinase, both by allosteric activation and by enhancing net phosphorylation. Cell Metab 18:556–566
Gurevich-Panigrahi T, Panigrahi S, Wiechec E, Los M (2009) Obesity: pathophysiology and clinical management. Curr Med Chem 16:506–521
Hanefeld M, Pfutzner A, Forst T, Kleine I, Fuchs W (2011) Double-blind, randomized, multicentre, and active comparator controlled investigation of the effect of pioglitazone, metformin, and the combination of both on cardiovascular risk in patients with type 2 diabetes receiving stable basal insulin therapy: the Piocomb study. Cardiovasc Diabetol 10:65
Hardie DG (2008) AMPK: a key regulator of energy balance in the single cell and the whole organism. Int J Obes 32:S7–S12
Hardie DG (2014) AMP-activated protein kinase: maintaining energy homeostasis at the cellular and whole-body levels. Annu Rev Nutr 34:31–55
Hardie DG (2015) Molecular pathways: is AMPK a friend or a foe in cancer? Clin Cancer Res 21:3836–3840
Hardie DG, Ashford ML (2014) AMPK: regulating energy balance at the cellular and whole body levels. Physiology (Bethesda) 29:99–107
Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Bio 13:251–262
Hattori Y, Suzuki K, Hattori S, Kasai K (2006) Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension 47:1183–1188
Hernández-Aguilera A, Rull A, Rodríguez-Gallego E, Riera-Borrull M, Luciano-Mateo F, Camps J, Menéndez JA, Joven J (2013) Mitochondrial dysfunction: a basic mechanism in inflammation-related non-communicable diseases and therapeutic opportunities. Mediators Inflamm 2013:135698
Hutley L, Prins JB (2005) Fat as an endocrine organ: relationship to the metabolic syndrome. Am J Med Sci 330:280–289
Hyun E, Ramachandran R, Hollenberg MD, Vergnolle N (2011) Mechanisms behind the anti-inflammatory actions of insulin. Crit Rev Immunol 31:307–340
Isoda K, Young JL, Zirlik A, MacFarlane LA, Tsuboi N, Gerdes N, Schönbeck U, Libby P (2006) Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler Thromb Vasc Biol 26:611–617
Jeyabalan J, Shah M, Viollet B, Chenu C (2012) AMP-activated protein kinase pathway and bone metabolism. J Endocrinol 212:277–290
Johnson DT, Harris RA, French S, Blair PV, You J, Bemis KG, Wang M, Balaban R (2007) Tissue heterogeneity of the mammalian mitochondrial proteome. Am J Physiol Cell Physiol 292:C689–C697
Jucker BM, Dufour S, Ren J, Cao X, Previs SF, Underhill B, Cadman KS, Shulman GI (2001) Assessment of mitochondrial energy coupling in vivo by 13C/31P NMR. Proc Natl Acad Sci USA 97:6880–6884
Kaur J (2014) A comprehensive review on metabolic syndrome. Cardiol Res Pract 2014:943162
Kim SA, Choi HC (2012) Metformin inhibits inflammatory response via AMPK-PTEN pathway in vascular smooth muscle cells. Biochem Biophys ResCommun 425:866–872
Kola B, Hubina E, Tucci SA et al (2005) Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated protein kinase. J Biol Chem 280:25196–25201
Kubota N, Yano W, Kubota T, Yamauchi T, Itoh S, Kumagai H, Kozono H, Takamoto I, Okamoto S, Shiuchi T, Suzuki R, Satoh H, Tsuchida A, Moroi M, Sugi K, Noda T, Ebinuma H, Ueta Y, Kondo T, Araki E, Ezaki O, Nagai R, Tobe K, Terauchi Y, Ueki K, Minokoshi Y, Kadowaki T (2007) Adiponectin stimulates AMP activated protein kinase in the hypothalamus and increases food intake. Cell Metab 6:55–68
Lamb RE, Goldstein BJ (2008) Modulating an oxidative-inflammatory cascade: potential new treatment strategy for improving glucose metabolism, insulin resistance, and vascular function. Int J Clin Pract 62:1087–1095
Lamers D, Famulla S, Wronkowitz N, Hartwig S, Lehr S, Ouwens DM, Eckardt K, Kaufman JM, Ryden M, Müller S, Hanisch FG, Ruige J, Arner P, Sell H, Eckel J (2011) Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes 60:1917–1925
Lane N (2006) Powerhouse of disease. Nature 440:600–602
Lane N, Martin W (2010) The energetics of genome complexity. Nature 21:929–934
Lau DCW, Dhillon B, Yan H, Szmitko PE, Verma S (2005) Adipokines: molecular links between obesity and atheroslcerosis. Am J Physiol Heart Circ Physiol 288:H2031–H2041
LeBrasseur N, Kelly M, Tsao TS, Farmer SR, Saha AK, Ruderman NB, Tomas E (2006) Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. Am J Physiol Endocrinol Metab 291:E175–E181
Lee HK, Cho YM, Kwak SH, Lim S, Park KS, Shim EB (2010) Mitochondrial dysfunction and metabolic syndrome—looking for environmental factors. Biochim Biophys Acta 1800:282–289
Liu M, Liu F (2010) Transcriptional and post-translational regulation of adiponectin. Biochem J 425:41–52
Lockwood TD (2010) The lysosome among targets of metformin: new anti-inflammatory uses for an old drug? Expert Opin Ther Targets 14:467–478
Lopez-Lluch G, Irusta PM, Navas P, de Cabo R (2008) Mitochondrial biogenesis and healthy aging. Exp Gerontol 43:813–819
Lynch M, Conery JS (2003) The origins of genome complexity. Science 302:1401–1404
Marfella R, D’Amico M, Esposito K, Baldi A, Di Filippo C, Siniscalchi M, Sasso FC, Portoghese M, Cirillo F, Cacciapuoti F, Carbonara O, Crescenzi B, Baldi F, Ceriello A, Nicoletti GF, D’Andrea F, Verza M, Coppola L, Rossi F, Giugliano D (2006) The ubiquitin-proteasome system and inflammatory activity in dia-betic atherosclerotic plaques: effects of rosiglitazone treatment. Diabetes 55:622–632
Marselli L, Thorne J, Dahiya S, Sgroi DC, Sharma A, Bonner-Weir S, Marchetti P, Weir GC (2010) Gene expression profiles of Beta-cell enriched tissue obtained by laser capture microdissection from subjects with type 2 diabetes. PLoS One 5:e11499
Matejkova O, Mustard KJ, Sponarova J et al (2004) Possible involvement of AMP-activated protein kinase in obesity resistance induced by respiratory uncoupling in white fat. FEBS Lett 569:245–248
Medzhitov R (2010) Inflammation 2010: new adventures of an old flame. Cell 140:771–776
Miles JM, Jensen MD (2005) Counterpoint: visceral adiposity is not causally related to insulin resistance. Diabetes Care 28:2326–2328
Minokoshi Y, Kim YB, Peroni OD et al (2002) Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 415:339–343
Minokoshi Y, Alquier T, Furukawa N et al (2004) AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428:569–574
Mitchell P (1961) Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191:144–148
O’Neill LA, Hardie DG (2013) Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493:346–355
Olijhoek JK, Van Der Graaf Y, Banga J-D, Algra A, Rabelink TJ, Visseren FLJ (2004) The metabolic syndrome is associated with advanced vascular damage in patients with coronary heart disease, stroke, peripheral arterial disease or abdominal aortic aneurysm. Eur Heart J 25:342–348
Ouchi N, Kihara S, Arita Y et al (2000) Adiponectin, an adipocytederived plasma protein, inhibits endothelial NF-kB signaling through a cAMP-dependent pathway. Circulation 102:1296–1301
Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI (2003) Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300:1140–1142
Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI (2004) Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 350:664–671
Pfützner A, Marx N, Lübben G, Langenfeld M, Walcher D, Konrad T, Forst T (2005) Improvement of cardiovascular risk markers by pioglitazone is independent from glycemic control: results from the pioneer study. J Am Coll Cardiol 45:1925–1931
Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM (2001) C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 286:327–334
Qiang W, Weiqiang K, Qing Z, Pengju Z, Yi L (2007) Aging impairs insulin-stimulated glucose uptake in rat skeletal muscle via suppressing AMPKalpha. Exp Mol Med 39:535–543
Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, Liu ZX, Dong J, Mustard KJ, Hawley SA, Befroy D, Pypaert M, Hardie DG, Young LH, Shulman GI (2007) Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab 5:151–156
Rich PR (2003) The cost of living. Nature 421:583
Rocher C, Taanman JW, Pierron D, Faustin B, Benard G, Rossignol R, Malgat M, Pedespan L, Letellier T (2008) Influence of mitochondrial DNA level on cellular energy metabolism: implications for mitochondrial diseases. J Bioenerg Biomembr 40:59–67
Rong JX, Qiu Y, Hansen MK et al (2007) Adipose mitochondrial biogenesis is suppressed in db/db and high-fat diet-fed mice and improved by rosiglitazone. Diabetes 56:1751–1760
Rylova SN, Albertioni F, Flygh G, Eriksson S (2005) Activity profiles of deoxynucleoside kinases and 5′-nucleotidases in cultured adipocytes and myoblastic cells: insights into mitochondrial toxicity of nucleoside analogs. Biochem Pharmacol 69:951–960
Schapira AH (2006) Mitochondrial disease. Lancet 368:70–82
Scheen AJ, Esser N, Paquot N (2015) Antidiabetic agents: potential anti-inflammatory activity beyond glucose control. Diabetes Metab 41:183–194
Schwartz MW, Woods SC, Porte DJ et al (2000) Central nervous control of food intake. Nature 404:661–671
Shi X, Burkart A, Nicoloro SM et al (2008) Paradoxical effect of mitochondrial respiratory chain impairment on insulin signaling and glucose transport in adipose cells. J Biol Chem 283:30658–30667
Shirwany NA, Zou MH (2014) AMPK: a cellular metabolic and redox sensor. A minireview. Front Biosci (Landmark Ed) 19:447–474
Spiegelman BM, Flier JS (2001) Obesity regulation and energy balance. Cell 104:531–543
Stapleton D, Mitchelhill KI, Gao G, Widmer J, Michell BJ, Teh T, House CM, Fernandez CS, Cox T, Witters LA, Kemp BE (1996) Mammalian AMP-activated protein kinase subfamily. J Biol Chem 271:611–614
Strong K, Mathers C, Leeder S, Beaglehole R (2005) Preventing chronic diseases: how many lives can we save? Lancet 366:1578–1582
Szanto A, Nagy L (2008) The many faces of PPARgamma: anti-inflammatory by any means? Immunobiology 213:789–803
Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT (2010) Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal 12:503–535
Tomas E, Tsao TS, Saha AK et al (2002) Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation. Proc Natl Acad Sci 99:16309–16313
Twig G, Hyde B, Shirihai OS (2008) Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta 1777:1092–1097
Vasamsetti SB, Karnewar S, Kanugula AK, Thatipalli AR, Kumar JM, Kotamraju S (2015) Metformin inhibits monocyte-to-macrophage differentiation via AMPK mediated inhibition of STAT3 activation: potential role in atherosclerosis. Diabetes 64:2028–2041
Virtue S, Even P, Vidal-Puig A (2012) Below thermoneutrality, changes in activity do not drive changes in total daily energy expenditure between groups of mice. Cell Metab 16:665–671
Westermann B (2008) Molecular machinery of mitochondrial fusion and fission. J Biol Chem 283:13501–13505
Williams RS (1986) Mitochondrial gene expression in mammalian striated muscle: evidence that variation in gene dosage is the major regulatory event. J Biol Chem 261:12390–12394
Wilson-Fritch L, Burkart A, Bell G et al (2003) Mitochondrial biogenesis and remodeling during adipogenesis and in response to the insulin sensitizer rosiglitazone. Mol Cell Biol 23:1085–1094
Wilson-Fritch L, Nicoloro S, Chouinard M et al (2004) Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J Clin Invest 114:1281–1289
Winder WW, Hardie DG (1999) AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes. Am J Physiol 277:E1–E10
Xydakis AM, Case CC, Jones PH et al (2004) Adiponectin, inflammation, and the expression of the metabolic syndrome in obese individuals: the impact of rapid weight lose through caloric restriction. J Clin Endocrinol Metab 89:2697–2703
Zhang Y, Proenca R, Maffei M et al (1994) Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432
Zhang BB, Zhou G, Li C (2009) AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab 5:407–416
Zhou G, Myers R, Li Y et al (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108:1167–1174
Zuliani G, Volpato S, Blé A, Bandinelli S, Corsi AM, Lauretani F, Paolisso G, Fellin R, Ferrucci L (2007) High interleukin-6 plasma levels are associated with low HDL-C levels in communitydwelling older adults: the InChianti study. Atherosclerosis 192:384–390
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Bullon, P., Marin-Aguilar, F., Roman-Malo, L. (2016). AMPK/Mitochondria in Metabolic Diseases. In: Cordero, M., Viollet, B. (eds) AMP-activated Protein Kinase. Experientia Supplementum, vol 107. Springer, Cham. https://doi.org/10.1007/978-3-319-43589-3_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-43589-3_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-43587-9
Online ISBN: 978-3-319-43589-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)