
Abstract
The incidence of primary malignant and nonmalignant central nervous system (CNS) tumors in children and adolescents aged 0–19 years in the US is 5.42 per 100,000, and approximately 4620 new cases are expected to be diagnosed in the US in 2015 (Ostrom et al. 2014). There is a rich variety of brain tumors found in children which is primarily a function of the patient’s age and location of origin, with the overall most common being pilocytic astrocytoma (Ostrom et al. 2014). It has been traditionally taught that approximately 60% of pediatric brain tumors are infratentorial, but the actual ratio of supratentorial to infratentorial pediatric tumors is dependent on the specific age group (Ostrom et al. 2015). Tumors can be broadly categorized as glial (e.g., astrocytomas, ependymomas), embryonal (e.g., medulloblastomas, pineoblastoma), germ cell (e.g., germinoma, teratoma), and other (e.g., choroid plexus tumors, craniopharyngiomas).
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

2.1 Introduction
The incidence of primary malignant and nonmalignant central nervous system (CNS) tumors in children and adolescents aged 0–19 years in the US is 5.42 per 100,000, and approximately 4620 new cases are expected to be diagnosed in the US in 2015 (Ostrom et al. 2014). There is a rich variety of brain tumors found in children which is primarily a function of the patient’s age and location of origin, with the overall most common being pilocytic astrocytoma (Ostrom et al. 2014). It has been traditionally taught that approximately 60% of pediatric brain tumors are infratentorial, but the actual ratio of supratentorial to infratentorial pediatric tumors is dependent on the specific age group (Ostrom et al. 2015). Tumors can be broadly categorized as glial (e.g., astrocytomas, ependymomas), embryonal (e.g., medulloblastomas, pineoblastoma), germ cell (e.g., germinoma, teratoma), and other (e.g., choroid plexus tumors, craniopharyngiomas).
Neurosurgery represents one of the main pillars of pediatric neurooncologic care, along with medical and radiation oncology, pathology, and neuroradiology. Neurosurgical interventions include management of hydrocephalus, obtaining tissue for histopathological and molecular diagnosis, and tumor resection for oncologic (i.e., survival) and/or neurologic (e.g. seizure control) benefit. In this chapter, we will take the reader through the surgical management of pediatric neurooncologic patients from the preoperative, intraoperative, and postoperative phases of care.
2.2 Initial Evaluation
2.2.1 History and Examination
Clinical presentation is variable and dependent on the location of the tumor and the age of the patient. Most children will present with hydrocephalus, symptoms of raised intracranial pressure, focal neurologic deficit, or a seizure. Some tumors will be incidentally found as part of a workup for nonspecific symptoms, such as headaches or after a minor traumatic event. A detailed neurological exam should be performed on all patients; a thorough knowledge of neuroanatomy can help qualitatively detail preoperative deficits, both minor and major. This is easier in older children, but there are specific signs and symptoms that can be revealing in younger children.
Headache is a common symptom among patients with brain tumors and occurs with, or without, elevated intracranial pressure (ICP). These headaches are classically described as being worse in the morning and exacerbated by straining, coughing, or placing the head in a dependent location. Brain tumor headaches are frequently associated with nausea and may be temporarily relieved by the hyperventilation that occurs with vomiting. In a large study examining the epidemiology of headaches associated with pediatric brain tumors, approximately two-thirds of patients had chronic or frequent headaches prior to their first admission (The epidemiology of headache among children with brain tumor. Headache in children with brain tumors. The Childhood Brain Tumor Consortium 1991). In this study, headaches tended to be triggered by straining, coughing, or sneezing, to gradually worsen over time, to cause vomiting followed by relief, and to be severe enough to wake the child from sleep. Personality changes, school problems, and focal neurologic deficits were also associated with headaches. In a similar study, the most common symptom at presentation in children with brain tumors was headache; all of the patients with headaches also had other symptoms, including mental status changes, papilledema, eye movement derangements, hemimotor or sensory abnormalities, tandem gait difficulty, or abnormal deep tendon reflexes, present at the time of diagnosis (Wilne et al. 2006).
The two cranial nerves that can be affected by hydrocephalus or elevated ICP are the trochlear (4th) and abducens (6th). The trochlear nerve innervates the superior oblique muscle, which intorts, depresses, and adducts the eye. Patients with acquired weakness of the 4th nerve report vertical and oblique diplopia that is worse in down-gaze and gaze away from the affected eye, resulting in difficulty reading. Patients will adopt a characteristic head tilt away from their affected eye to reduce their diplopia, which is called the Bielschowsky’s sign. The abducens nerve innervates the lateral rectus, which abducts the eye. Weakness of the 6th cranial nerve results in a lateral gaze palsy and horizontal diplopia that is worse with gaze toward the affected eye.
Posterior fossa tumors often present with symptoms of obstructive hydrocephalus, which in turn leads to elevated intracranial pressure. Headache and vomiting are hallmark features, particularly if present in the morning. In infants, hydrocephalus presents with a full or bulging fontanelle, separation of sutures, rapid head growth, macrocephaly, irritability, lethargy, or poor feeding/failure to thrive. Sundowning—or setting sun sign—describes downward deviation of both eyes, revealing an area of sclera above the irises. This usually occurs with advanced hydrocephalus with stretching of the third ventricle and upper brainstem. The pupils are sluggish and respond to light unequally.
Pineal region tumors can result in hydrocephalus and Parinaud’s syndrome. Parinaud’s syndrome, or dorsal midbrain syndrome, is a constellation of eye findings that includes upgaze palsy, convergence-retraction nystagmus, light-near pupillary dissociation (Argyll Robertson pupil), and lid retraction called Collier’s sign (Baloh et al. 1985). When upgaze palsy is combined with lid retraction, it produces the setting sun sign. This syndrome is often seen with pineal region tumors that place pressure on the rostral interstitial nucleus of the medial longitudinal fasciculus and the posterior commissure, which mediate upgaze and the consensual pupillary light reflex, respectively.
Diencephalic syndrome, also known as Russell’s syndrome, is characterized by progressive and severe failure to thrive (Zafeiriou et al. 2001). It is seen exclusively with suprasellar pilocytic astrocytoma tumors affecting the anterior hypothalamus. The child often appears emaciated despite being alert and active and has a “pseudohydrocephalic” face from severe loss of adipose tissue and a normal head circumference. Neurocutaneous syndromes—such as the neurofibromatoses, tuberous sclerosis, and Von Hippel-Lindau disease—are characterized by specific nervous system tumors associated with clinical exam findings. The details of these syndromes are beyond the scope of this chapter.
2.2.2 Seizures
Supratentorial tumor location, age < 2 years, and hyponatremia are independent risk factors for a first-time seizure in pediatric patients with a brain tumor (Hardesty et al. 2011). Seizures cause cerebral hyperemia and can thus precipitate a herniation event in the setting of preexisting increased intracranial pressure. They can also be the clinical manifestation of an intratumoral hemorrhage. If the patient is in status epilepticus, secondary brain damage may also occur through tissue hypoxia or acidosis. Guidelines are available that detail when imaging should be conducted in a child with a first-time nonfebrile seizure (Hirtz et al. 2000).
Antiepileptic drugs (AED)—such as phenytoin, phenobarbital, and carbamazeipine—induce the cytochrome P450 system and can reduce the efficacy of many common chemotherapeutics (Guerrini et al. 2013). Conversely, valproic acid inhibits the cytochrome P450 system and can increase levels of chemotherapeutics. Levetiracetam is a newer AED that has proven efficacious in preventing tumoral seizures with a low side-effect profile and no significant induction of the cytochrome P450 system (Zachenhofer et al. 2011). It is the first-line AED at our institution for children who suffer from seizures caused by a brain tumor.
2.2.3 Cerebral Edema
Brain tumors can cause vasogenic (i.e., interstitial) edema, which results from breakdown of the tight junctions between brain capillary endothelial cells and leakage of plasma filtrate into the interstitial space. Vasogenic edema is more marked in the white matter than the gray matter. Children are often started on steroids (e.g., dexamethasone) shortly after being diagnosed with a brain tumor. Steroids help with vasogenic edema, hydrocephalus (headaches, nausea/vomiting), and poor appetite, all of which cause the child to feel and look significantly better.
2.2.4 Preparation for Tumor Resection: Management of Hydrocephalus
For the vast majority of children, treatment of hydrocephalus is done by resecting the tumor. Prophylactic endoscopic third ventriculostomy (ETV) at the time of surgery has been shown to reduce the risk of post-resection hydrocephalus from approximately 27 to 6% in patients with posterior fossa tumors and hydrocephalus (Sainte-Rose et al. 2001). However, since resection alone effectively treats the majority of patients with posterior fossa tumor-induced hydrocephalus, pre-resection ETV is an unnecessary surgery, if tumor resection is to be carried out in a timely manner. However, if the patient’s hydrocephalus will not resolve with resection (e.g., CSF dissemination), or there is no immediate role for resection (e.g., pineal mass), or no resection at all (e.g., diffuse pontine glioma), then long-term hydrocephalus management can be achieved either by placing a ventricular shunt or by performing an ETV. The ETV Success Score was developed to help surgeons determine the likelihood of ETV succeeding in a particular child, taking into consideration age, hydrocephalus etiology, and whether the child currently has a shunt or not.
Patients who present in extremis from severe hydrocephalus may require emergent placement of an external ventricular drain (EVD) (Lin and Riva-Cambrin 2015; El-Gaidi et al. 2015). Care must be taken not to drain too much cerebrospinal fluid in patients with posterior fossa tumors as this can precipitate upward transtentorial herniation (Osborn et al. 1978). Ascending transtentorial herniation results in a clinical syndrome of nausea and vomiting, followed by progression to stupor and coma with small nonreactive pupils and loss of vertical gaze. Radiographically, there is displacement of the midbrain and cerebellum through the tentorial notch, causing flattening of the quadrigeminal cistern and a “spinning top” appearance to the midbrain from compression of the posterior aspect of the midbrain.
2.2.5 Preparation for Tumor Resection: Neuroimaging
Computed tomography (CT) scans are very useful in the initial evaluation because they are quick and sensitive for detecting hydrocephalus, hemorrhage, edema, and ectopic calcifications. Once the child is deemed stable, he or she should have a magnetic resonance image (MRI) of the brain both with and without contrast. Unless the index of suspicion is low, an MRI of the full spine (with and without contrast) should also be obtained to look for leptomeningeal—or “drop”—metastases. Standard MRI brain sequences include T1 (with and without contrast), T2, FLAIR, diffusion weighted imaging (DWI) with the apparent diffusion coefficient map (ADC), and susceptibility weighted imaging (SWI). ADC maps have been shown to correlate with tumor cellularity in pediatric brain tumors (Choudhri et al. 2015a). Sometimes brain tumors can resemble other pathologies, such as infection or demyelinating disease. Magnetic resonance (MR) perfusion and spectroscopy can help distinguish tumors from other such conditions by highlighting increased blood flow and products of cell turnover, like elevated choline and depressed N-acetylaspartate, respectively.
Vascular imaging studies, such as MR or CT angiogram/venogram, are useful if tumors involve major intracranial arteries, veins, or sinovenous structures. Traditional angiography is also a valuable preoperative tool when tumors are felt to be hypervascular and may benefit from preoperative embolization (Fig. 2.1). If such embolization is performed, resection should follow within 24–48 h as the embolization may cause new or worsening cerebral edema.

T1 weighted (T1W) MRI with contrast of an interhemispheric hemangioma (a). Angiogram demonstrates vascular supply through the pericallosal artery (b). Microcatheterization of the tumor for embolization (c). Post embolization angiogram (d)
Eloquent location of a tumor is particularly challenging for the surgeon. Functional MRI (fMRI), magnetoencephalography (MEG), transcranial magnetic stimulation (TMS), and diffusion tensor imaging (DTI) are modalities that provide further knowledge of the patient’s functional neuroanatomy (Ottenhausen et al. 2015). These imaging studies may localize eloquent regions, such as the primary motor cortex, Broca’s and Wernicke’s area, or subcortical tracts like the corticospinal, geniculocalcarine, or arcuate fasciculus. Functional MRI relies on the theory of neurovascular coupling and assumes that when functional networks within the brain are activated, perfusion-induced changes occur regionally in the blood oxygen-level that can be detected by MRI. In young children, motor mapping can be performed with passive movement (Fig. 2.2) (Choudhri et al. 2015c). MEG detects the magnetic fields created by bioelectrical currents as a result of neuronal activation and is, therefore, a direct marker of neuronal activity. Navigated TMS uses a magnetic field to induce a cortical electrical field and thus elicits or inhibits neuronal activity. A single pulse is used to elicit a motor response, or repetitive pulses are used to inhibit language function thereby mapping functional motor and language areas that are sufficient—and possibly necessary—to evoke a physiological response. DTI is the only preoperative method for visualizing subcortical white mater tracts (Fig. 2.3) (Choudhri et al. 2014b). All of these functional imaging techniques are more accurate for mapping motor areas than language areas.

Axial and coronal T2 weighted (T2W) MRI shows a low-grade glioma within the left precentral gyrus (a, b). Axial T2W image with functional MRI (fMRI) overlay from passive movement of the right lower extremity shows cortical activation along the medial margin of the tumor within the precentral gyrus near the vertex (c). Axial T2W image with fMRI overlay from passive movement of the right upper extremity shows cortical activation in the precentral gyrus inferolateral to the tumor (d). Resected tumor specimen (e). Operative setup utilizing frameless neuronavigation and a surgical microscope with the patient’s head positioned 180° away from anesthesia to facilitate intraoperative MRI (iMRI) scanning (f, g). iMRI suite and scanner (h). Coronal T2W image from initial iMRI demonstrates residual tumor (i). Coronal T2W image from second iMRI after further resection demonstrates a gross total resection (j)

Axial T2W image in a 5-year-old male shows a multicystic lesion centered in the right cerebral peduncle, consistent with a thalamopeduncular glioma (a). Axial T1W image with overlay of DTI data shows anterolateral displacement of the posterior limb of the internal capsule (red arrowheads) (b). Coronal T1W image with “tractography” overlay shows the course of the fibers of the corticospinal tract along the lateral aspect of the lesion (red arrowheads) (c)
2.2.6 Preparation for Tumor Resection: Neoadjuvant Chemotherapy
In some tumors found in newborns, infants, and young children, the risk of excessive blood loss with resection is great; the best example of this is choroid plexus carcinoma. Infants have small blood volumes; transfusing multiple blood volumes can lead to coagulopathy and electrolyte imbalance. Therefore, these patients may be best served by first treating the tumor with chemotherapy (i.e., neoadjuvant chemotherapy) before pursuing resection. Tumors will often shrink and become cystic and the reduction in vascularity is notable, resulting in safer and more complete tumor removal (Iwama et al. 2015; Van Poppel et al. 2011).
2.2.7 Preparation for Tumor Resection: Family Counseling
One of the most important steps in preparing a pediatric patient for a brain tumor resection is talking with the parents and family about the patient’s prognosis, the risks, and the goals of surgery without overwhelming and confusing them with statistics and medical terminology. While there are general risks associated with any craniotomy, such as bleeding and wound infection, it is more important to stress the potential—or even anticipated—neurologic deficits specific to the location and size of the tumor. Neurologic injury may occur as a result of the surgical approach or during extirpation of the mass. Examples include Parinaud’s syndrome with a pineoblastoma, posterior fossa syndrome in a young boy with a medulloblastoma, or cranial neuropathies with a cerebellopontine angle ependymoma. It is usually easier for the family to psychologically deal with new postoperative neurologic deficits if they’ve learned about them before surgery. It is equally important to define the expectations of surgery, such as total resection, subtotal resection, or biopsy, as well as the potential need for further surgical procedures (e.g., ventriculoperitoneal shunt, feeding tube), therapies (e.g., physical, speech), and expected length of hospital stay.
2.2.8 Preparation for Tumor Resection: Teamwork
Orchestrating a successful surgery requires the integration of multiple individuals and services, including anesthesiology, operating room nurses and technologists, and neuroradiology for intraoperative MRI cases. It is important to have a preoperative “huddle” with all team members to discuss positioning, need for vascular access, estimated length of surgery, anticipated blood loss, specific blood pressure management, need for any intraoperative neuromonitoring, and airway management (i.e., whether the patient will be extubated or remain intubated after surgery). One way to set a preoperative threshold for blood transfusion is to define the maximal allowable blood loss. Maximal allowable blood loss is the estimated blood volume of the patient multiplied by the difference between the patient’s starting and minimal allowable hematocrits, divided by the starting hematocrit. For example, a 5 kg infant with an estimated blood volume of 75 cc/kg, a starting hematocrit of 30, and a minimal acceptable hematocrit of 22 would have a maximal allowable blood loss of approximately 100 cc. If further bleeding is anticipated, transfusion of blood should be initiated.
2.3 Tumor Resection
In this section we will discuss surgical management and approaches to the more common locations and types of pediatric brain tumors, such as the pineal region/posterior third ventricle, posterior fossa, and suprasellar area. Each child’s brain tumor is unique; in many respects, its surgical management should be as well. Much of what can be done by the neurosurgeon depends on the age of the child, the type of tumor, its location and therefore the risks associated with resection, and whether or not there are local or distant metastases. For many nonmetastatic childhood intracranial neoplasms, the goal of initial surgery is complete resection (i.e., gross total resection (GTR)), defined as no conclusive evidence of residual tumor on the intra- or immediate postoperative MRI, when deemed feasible. Such philosophy applies to tumors like medulloblastoma, ependymoma (infra- and supratentorial), primitive neuroectodermal tumor (PNET), and virtually all low-grade tumors.
Intraoperative magnetic resonance imaging (iMRI) has revolutionized surgical management of pediatric brain tumors by allowing the surgeon to confirm a gross total resection, while the patient is still under general anesthesia and their wound is open (Choudhri et al. 2014a, 2015b). This high-dollar technology greatly reduces the risk of having to take the child back to the operating room for continued resection, but with the drawbacks of added operating room (OR) time, challenges in interpreting the intraoperative images, and significant new safety issues (Shah et al. 2012). It also requires close cooperation and communication with the anesthesiology team, MR technologist, OR safety officer, and neuroradiologist.
2.3.1 Posterior Fossa (Excluding Brainstem Tumors)
The posterior fossa, as mentioned previously, is a common site for pediatric tumors. The “big 3” tumors are medulloblastoma, ependymoma, and pilocytic astrocytoma. Each has their own unique imaging features. Medulloblastomas and ependymomas are typically found within the 4th ventricle, whereas pilocytic astrocytomas are most often located within the cerebellum (i.e., the vermis or hemispheres). Medulloblastomas are hypercellular and therefore appear hyperdense on the initial CT. Pilocytic astrocytomas often have a cystic component with enhancing nodule(s). Ependymomas classically project through the foramen Luschka into the cerebellopontine angle, or through the foramen magnum into the cervical spinal canal (i.e., “plastic ependymoma”). Midline or fourth ventricular tumors are approached via a standard midline suboccipital craniotomy, whereas hemispheric tumors require a lateral suboccipital approach. Although we have seen the dawn of a new era in which tumors are being classified at the molecular level, resulting in subclassification and novel “targeted” chemotherapeutic options, the surgical goal of these tumors remains maximal safe resection (Gajjar et al. 2014).
2.3.2 Brainstem Tumors
Brainstem tumors can be broadly categorized as being radiographically focal or diffuse/infiltrative (Green and Kieran 2015). The classic example of an infiltrative pediatric brainstem tumor is a diffuse intrinsic pontine glioma (DIPG). Children with these tumors are typically young and present with a combination of long-tract and cranial nerve findings. DIPG is a radiographic diagnosis, surgery is relegated to the management of hydrocephalus, and the only known treatment that has some effect, albeit temporary, is radiation (Bredlau and Korones 2014). For pontine tumors that are “atypical” in appearance, a biopsy is warranted. Focal tumors are more often low-grade, and most commonly are piloctyic astrocytomas. All focal tumors (with the exception of tectal gliomas), whether benign or malignant, should be considered for resection (Klimo et al. 2013, 2015a). Tectal gliomas have a well-known indolent biologic behavior, and like DIPG, surgery is limited to the treatment of hydrocephalus. Resection of focal brainstem tumors requires careful planning, high-quality preoperative imaging (including tractography), and detailed discussions with the parents on what neurologic deficits to expect.
2.3.3 Pineal Region/Posterior Third Ventricle
There is a wide variety of tumors that may arise in this region of the brain; examples include pineoblastoma and germ cell tumors (Fig. 2.4). Because these patients often present with obstructive hydrocephalus secondary to occlusion of the aqueduct of Sylvius, surgical management is most often directed at treating the hydrocephalus by way of an endoscopic third ventriculostomy, obtaining cerebrospinal fluid (CSF) for germ cell markers (i.e., beta human chorionic gonadotropin, alfa fetoprotein), and angling the endoscope posteriorly to obtain tissue for biopsy. If the germ cell markers are elevated, then by definition the child has a non-germinomatous germ cell tumor (e.g., choriocarcinoma, endodermal sinus tumor) and initial treatment is chemotherapy. If the germ cell markers are negative and the biopsy is consistent with a germinoma, then the child is treated with radiation with or without chemotherapy with a very high chance of cure, even with metastatic disease. A nondiagnostic biopsy with negative CSF markers usually requires an open biopsy. The three surgical approaches that we use to resect or biopsy tumors in the pineal region/posterior third ventricle are the supracerebellar-infratentorial, the occipital-transtentorial, and the posterior transcallosal (Kennedy and Bruce 2011).

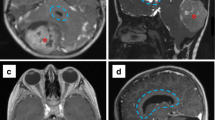
Sagittal T1W + C image in a 2.5-year-old girl with a history of bilateral retinoblastoma shows an enhancing pineal mass (red arrowhead), consistent with a “tri-lateral” retinoblastoma (a). Sagittal T1W + C image from an iMRI scan shows successful resection of the tumor (red arrowhead). Note the open craniotomy (red arrow), which would have facilitated further resection, if needed (b)
2.3.4 Sellar/Suprasellar
The two most common suprasellar tumors in children are craniopharyngiomas and optic pathway-hypothalamic astrocytomas. Children who present with diabetes insipidus (DI) and an enhancing mass along the pituitary stalk or hypothalamic region typically have one of two pathologies: germinoma or eosinophilic granuloma (histiocytosis X). It is exceedingly rare for optic pathway-hypothalamic astrocytomas or craniopharyngiomas to present with DI. Pure sellar lesions are rare, but may include craniopharyngioma, micro- or macroadenomas (functioning or non-functioning) in older children, and the nonneoplastic Rathke’s cleft cyst.
Controversy continues among neurosurgeons as to the role of surgery with craniopharyngiomas. There are those who feel that craniopharyngiomas should be maximally resected without adjuvant therapy (Elliott et al. 2010); others believe in a less aggressive surgical approach in order to avoid significant morbidity (i.e., neurologic, endocrine, or cognitive dysfunction) followed by radiotherapy (Klimo et al. 2015b). We generally ascribe to the latter philosophy. Purely cystic craniopharyngiomas can be treated with placement of an Ommaya catheter to aspirate the tumor cyst, followed by radiotherapy or the injection of intracystic chemotherapy (e.g., bleomycin), immunotherapy (e.g., interferon), or radioactive agents (e.g., P-32) (Cavalheiro et al. 2010; Mottolese et al. 2001; Zhao et al. 2010). Surgical approaches for craniopharyngiomas are dictated by the location of the tumor (Fig. 2.5) and include subfrontal, transsylvian, and anterior transcallosal approaches. Intrasellar craniopharyngiomas can be resected through a transnasal-transsphenoidal route, using a microscope or endoscope (Jane et al. 2010).

The variety of imaging appearances of craniopharyngioma. This variety underscores the need for patient-specific surgical and treatment plans. Sagittal T1W image shows a cystic suprasellar lesion (a). Sagittal T1W image shows a suprasellar cystic lesion with intrinsic T1 hyperintense signal, representing proteinaceous secretions (b). Sagittal T1W image post contrast shows a multicystic suprasellar lesion with enhancing rims, with the components having different central T1 characteristics related to different proteinaceous contents (c). There is also caudal retroclival extension. Sagittal T1W image post contrast shows a central solid enhancing component with multiple smaller cystic components (d). Sagittal T1W image post contrast shows a large central solid enhancing component, with several internal cystic areas and a single posteriorly directed cyst within the third ventricle (e)
Optic pathway-hypothalamic tumors are generally not thought to be curable by surgery alone, except in the rare case of a prechiasmatic optic nerve glioma with no functional vision. These tumors originate from non-resectable areas of the brain and can often be diagnosed by imaging alone. They are associated with neurofibromatosis type I (i.e., von Recklinghausen disease). Surgery is reserved for biopsy or subtotal resection in those cases where there is significant symptomatic mass effect or where the tumor has caused obstructive hydrocephalus by growing cephalad into the third ventricle (Goodden et al. 2014). The primary treatment modalities for these tumors are chemotherapy and/or radiotherapy. The same approaches used for craniopharyngioma can be used for this tumor, with the exception of the transnasal approach.
2.3.5 Supratentorial
The goal of surgery for most supratentorial tumors should be maximal resection. Extraaxial tumors, such as meningiomas, are rare in children. As previously discussed, functional imaging modalities should be used in cases where the tumor is in close proximity to eloquent areas (Fig. 2.2). Awake craniotomy is difficult to perform in a child, so we rely heavily on these preoperative mapping tests. For a child whose tumor cannot be completely resected but who has debilitating seizures as a result of it, surgery to resect the epileptogenic part of the tumor (e.g., temporal lobectomy) can have a substantial positive impact on the quality of that child’s life.
2.4 Postoperative Care
After tumor resection, patients are brought to the intensive care unit (ICU) for close neurologic and cardiorespiratory monitoring. Almost all patients are extubated while still deeply sedated in the OR so as to avoid any coughing or bucking as they awaken with the endotracheal tube in place and during transport to the ICU. Such reflexes can rapidly increase the patient’s systemic blood pressure and intracranial venous pressure, which could lead to hemorrhage within the fresh resection cavity, especially if there is a raw, residual tumor surface. For excessively long cases or those with high volume fluid resuscitation, extubation may be delayed until neurologic and cardiopulmonary systems are assessed and stabilized.
The most common immediate postoperative issues that require close monitoring are intracranial hemorrhage, seizure, hydrocephalus, and endocrinologic derangements. Strict blood pressure control is paramount since postoperative hypertension can result in hemorrhage within the resection cavity (Basali et al. 2000). Prompt and adequate treatment for pain and agitation often improves the patient’s blood pressure. A maximum allowable systolic blood pressure is typically set for the first 24–48 h after surgery, followed by gradual relaxation of the parameter. The blood pressure limit is age dependent, but an oft-recommended limit is less than 140 mmHg. We consider a nicardipine drip an easy and effective method of titrating the patient’s blood pressure. Hypotension is to be avoided, particularly in cases in which there was significant brainstem or spinal cord compression by the tumor, or if there was manipulation/dissection of major arteries so as to maintain adequate tissue perfusion. Patients should be kept euvolemic to mildly hypervolemic.
As discussed previously, obstructive hydrocephalus is a common presenting condition in children with brain tumors. Our general approach to such children is to resect the tumor in order to relieve the hydrocephalus, which we are successful in achieving in many cases. Mechanisms of post-resection hydrocephalus include obstruction from residual tumor and subarachnoid block caused by leptomeningeal metastasis, operative blood products, or proteinaceous CSF. All patients with preoperative hydrocephalus, or who are at risk of developing hydrocephalus postoperatively (e.g., intraventricular tumor), need to be carefully monitored for persistent or new hydrocephalus, respectively. Such evidence would include increase in ventricular size, inability to wean an external ventricular drain (EVD), development of a new or growing subdural hygroma or pseudomeningocele, and clinical changes, such as irritability, headaches, emesis, full fontanelle, or depressed level of arousal. Postoperative hydrocephalus is treated with either an EVD, a shunt, or ETV.
The Canadian Preoperative Prediction Rule for Hydrocephalus (CPPRH) was devised in an attempt to identify patients before resection who are at risk for post-resection hydrocephalus (Riva-Cambrin et al. 2009). Variables predictive of post-resection hydrocephalus include age less than 2 (score of 3), papilledema (score of 1), moderate to severe hydrocephalus (score of 2), cerebral metastasis (score of 3), and specific estimated tumor pathologies (score of 1). A total score of ≥5 places the patient at high risk. Estimated preoperative tumor pathologies based on imaging and clinical information that qualify for a score of 1 include medulloblastoma, ependymoma, and dorsally exophytic brainstem glioma. The modified CPPRH also adds the presence of transependymal edema as a risk factor (Foreman et al. 2013). For children with favorable age (>2 years), pathology (e.g., tectal glioma, pineal tumors), anatomy, and site of CSF blockage (obstruction between the third ventricle and the interpeduncular cistern), ETV is preferred over shunting as shunts are generally viewed as life-long implants that come with high risk of one or more shunt malfunction(s) (Gupta et al. 2007; Vogel et al. 2013). In cases where ETV is not appropriate or if the ETV fails, then ventricular shunting is the sole option.
If the patient has a postoperative seizure and is not already on an AED, then electrolytes and blood glucose should be checked expeditiously and any abnormalities should be promptly corrected, especially low sodium and magnesium; a non-contrast CT scan of the head should be obtained to rule out any new hemorrhage, edema, or hydrocephalus and the patient should be given a bolus of an AED, such as phosphenytoin or levetiracetam (both ~ 20 mg/kg), followed by maintenance therapy. If the patient’s seizure lasts more than 5 min or if multiple seizures occur without full neurologic recovery in the interictal period, then the patient is considered to be in status epilepticus, which is a medical emergency (Claassen et al. 2015).
Removal of sellar and suprasellar tumors, such as optic pathway gliomas or craniopharyngiomas, may lead to transient or permanent disruption of the hypothalamic-pituitary axis, and subsequent anterior and posterior pituitary lobe dysfunction. The endocrinopathies that are most problematic for neurosurgeons are the ones that can cause dramatic changes in the serum sodium level: central diabetes insipidus (DI), cerebral salt wasting (CSW), or the syndrome of inappropriate antidiuretic hormone release (SIADH). Central DI is caused by inadequate antidiuretic hormone release and results in excessive production of dilute urine and resultant hypernatremia. Urine output continuously exceeding 3 cc/kg/h with a specific gravity of 1.005 or less with a concurrent elevation in serum sodium above 145 is diagnostic. Without close monitoring of urine output and sodium levels in patients with or at risk for DI, sodium levels can easily exceed 160 mEq/L, resulting in severe dehydration, mental status changes, and seizures. The treatment is desmopressin and free water replacement titrated to the patient’s urine output.
SIADH and CSW both cause hyponatremia. With severe hyponatremia (<125 mEq/L) or rapid drops in sodium, headache, confusion, seizures, and cerebral edema can occur. SIADH results from an abnormal release of antidiuretic hormone (ADH) in the absence of a physiologic osmotic stimulus, resulting in excess water retention. Patients are either hypervolemic from the retained water or sometimes euvolemic. Serum osmolality is low (<275 mOsm/kg of water) while the urine is concentrated (>100 mOsm/kg of water). Cerebral salt wasting also produces hyponatremia and low serum osmolality in the presence of concentrated urine; but unlike SIADH, patients are hypovolemic. Intracranial disease results in failure of the kidneys to conserve sodium by an unknown mechanism. The key difference is the treatment. Fluid restriction effectively corrects the hyponatremia caused by SIADH while volume replacement with gentle sodium support treats CSW. In the setting of a malignancy, SIADH is more common. Cerebral salt wasting will also respond to a fluid challenge. Regardless of the etiology, if the hyponatremia is severe (Na < 125 mEq/L) or symptomatic (i.e., confusion, seizures or coma), then correction with hypertonic (e.g., 3%) saline is indicated. However, care must be taken not to correct the sodium too quickly. In general, if the sodium level changed rapidly then the patient can tolerate rapid correction. The serum sodium must be checked every 2–6 h. The goal is to correct the serum sodium 1–2 mEq/L/h and limit the correction to 8–10 mEq/L in 24 h. If the sodium is corrected too quickly, central pontine myelinolysis can rarely occur. Conversely, rapid correction of hypernatremia can cause or exacerbate cerebral edema.
Given the high frequency of posterior fossa tumors, posterior fossa syndrome (PFS) deserves special mention. It is a syndrome consisting of mutism, oromotor and oculomotor apraxia, emotional lability, axial hypotonia, and cerebellar/brainstem dysfunction following resection of infratentorial tumors (Robertson et al. 2006). Risk factors include young age, male sex, large midline tumors, brainstem invasion, and medulloblastoma. It is thought to result from bilateral surgical damage to the proximal efferent cerebellar pathways (Patay 2015). Most patients wake-up from surgery with intact speech but develop mutism within 1–4 days after surgery. Most recover fluent speech within 4 months with average duration of 6 weeks. Recovery begins with clumsy and broken speech slowly progressing to full sentences. However, up to one-third of children will have lasting dysarthria after surgery. Irritability, inconsolable crying, impulsiveness, and disinhibition are the most frequent changes in affect. IQ and school performance are also affected, more commonly when the deep cerebellar nuclei are damaged. Treatment generally requires prolonged rehabilitation, including physical, occupational, and speech therapy. Overall, improvement is universal but the degree of recovery is variable. Mutism and emotional lability are generally transient but long-term cognitive and motor deficits are frequently recognized in these children.
References
Anon (1991) The epidemiology of headache among children with brain tumor. Headache in children with brain tumors. The Childhood Brain Tumor Consortium. J Neuro-Oncol 10(1):31–46
Baloh RW, Furman JM, Yee RD (1985) Dorsal midbrain syndrome: clinical and oculographic findings. Neurology 35(1):54–60
Basali A, Mascha EJ, Kalfas I, Schubert A (2000) Relation between perioperative hypertension and intracranial hemorrhage after craniotomy. Anesthesiology 93(1):48–54
Bredlau AL, Korones DN (2014) Diffuse intrinsic pontine gliomas: treatments and controversies. Adv Cancer Res 121:235–259. https://doi.org/10.1016/B978-0-12-800249-0.00006-8
Cavalheiro S, Di Rocco C, Valenzuela S, Dastoli PA, Tamburrini G, Massimi L, Nicacio JM, Faquini IV, Ierardi DF, Silva NS, Pettorini BL, Toledo SR (2010) Craniopharyngiomas: intratumoral chemotherapy with interferon-alpha: a multicenter preliminary study with 60 cases. Neurosurg Focus 28(4):E12. https://doi.org/10.3171/2010.1.FOCUS09310
Choudhri AF, Klimo P Jr, Auschwitz TS, Whitehead MT, Boop FA (2014a) 3T intraoperative MRI for management of pediatric CNS neoplasms. AJNR Am J Neuroradiol 35(12):2382–2387. https://doi.org/10.3174/ajnr.A4040
Choudhri AF, Chin EM, Blitz AM, Gandhi D (2014b) Diffusion tensor imaging of cerebral white matter: technique, anatomy, and pathologic patterns. Radiol Clin North Am 52(2):413–425. https://doi.org/10.1016/j.rcl.2013.11.005
Choudhri AF, Whitehead MT, Siddiqui A, Klimo P Jr, Boop FA (2015a) Diffusion characteristics of pediatric pineal tumors. Neuroradiol J 28(2):209–216. https://doi.org/10.1177/1971400915581741
Choudhri AF, Siddiqui A, Klimo P Jr, Boop FA (2015b) Intraoperative MRI in pediatric brain tumors. Pediatr Radiol 45(Suppl 3):397–405. https://doi.org/10.1007/s00247-015-3322-z
Choudhri AF, Patel RM, Siddiqui A, Whitehead MT, Wheless JW (2015c) Cortical activation through passive-motion functional MRI. AJNR Am J Neuroradiol 36(9):1675–1681. https://doi.org/10.3174/ajnr.A4345
Claassen J, Riviello JJ Jr, Silbergleit R (2015) Emergency neurological life support: status epilepticus. Neurocrit Care 23(Suppl 2):136–142. https://doi.org/10.1007/s12028-015-0172-3
El-Gaidi MA, El-Nasr AH, Eissa EM (2015) Infratentorial complications following preresection CSF diversion in children with posterior fossa tumors. J Neurosurg Pediatr 15(1):4–11. https://doi.org/10.3171/2014.8.PEDS14146
Elliott RE, Hsieh K, Hochm T, Belitskaya-Levy I, Wisoff J, Wisoff JH (2010) Efficacy and safety of radical resection of primary and recurrent craniopharyngiomas in 86 children. J Neurosurg Pediatr 5(1):30–48. https://doi.org/10.3171/2009.7.PEDS09215
Foreman P, McClugage S 3rd, Naftel R, Griessenauer CJ, Ditty BJ, Agee BS, Riva-Cambrin J, Wellons J 3rd (2013) Validation and modification of a predictive model of postresection hydrocephalus in pediatric patients with posterior fossa tumors. J Neurosurg Pediatr 12(3):220–226. https://doi.org/10.3171/2013.5.PEDS1371
Gajjar A, Pfister SM, Taylor MD, Gilbertson RJ (2014) Molecular insights into pediatric brain tumors have the potential to transform therapy. Clin Cancer Res 20(22):5630–5640. https://doi.org/10.1158/1078-0432.CCR-14-0833
Goodden J, Pizer B, Pettorini B, Williams D, Blair J, Didi M, Thorp N, Mallucci C (2014) The role of surgery in optic pathway/hypothalamic gliomas in children. J Neurosurg Pediatr 13(1):1–12. https://doi.org/10.3171/2013.8.PEDS12546
Green AL, Kieran MW (2015) Pediatric brainstem gliomas: new understanding leads to potential new treatments for two very different tumors. Curr Oncol Rep 17(3):436. https://doi.org/10.1007/s11912-014-0436-7
Guerrini R, Rosati A, Giordano F, Genitori L, Barba C (2013) The medical and surgical treatment of tumoral seizures: current and future perspectives. Epilepsia 54(Suppl 9):84–90. https://doi.org/10.1111/epi.12450
Gupta N, Park J, Solomon C, Kranz DA, Wrensch M, Wu YW (2007) Long-term outcomes in patients with treated childhood hydrocephalus. J Neurosurg 106(5 Suppl):334–339. https://doi.org/10.3171/ped.2007.106.5.334
Hardesty DA, Sanborn MR, Parker WE, Storm PB (2011) Perioperative seizure incidence and risk factors in 223 pediatric brain tumor patients without prior seizures. J Neurosurg Pediatr 7(6):609–615. https://doi.org/10.3171/2011.3.PEDS1120
Hirtz D, Ashwal S, Berg A, Bettis D, Camfield C, Camfield P, Crumrine P, Elterman R, Schneider S, Shinnar S (2000) Practice parameter: evaluating a first nonfebrile seizure in children: report of the quality standards subcommittee of the American Academy of Neurology, The Child Neurology Society, and The American Epilepsy Society. Neurology 55(5):616–623
Iwama J, Ogiwara H, Kiyotani C, Terashima K, Matsuoka K, Iwafuchi H, Morota N (2015) Neoadjuvant chemotherapy for brain tumors in infants and young children. J Neurosurg Pediatr 15(5):488–492. https://doi.org/10.3171/2014.11.PEDS14334
Jane JA Jr, Prevedello DM, Alden TD, Laws ER Jr (2010) The transsphenoidal resection of pediatric craniopharyngiomas: a case series. J Neurosurg Pediatr 5(1):49–60. https://doi.org/10.3171/2009.7.PEDS09252
Kennedy BC, Bruce JN (2011) Surgical approaches to the pineal region. Neurosurg Clin N Am 22(3):367–380., viii. https://doi.org/10.1016/j.nec.2011.05.007
Klimo P Jr, Pai Panandiker AS, Thompson CJ, Boop FA, Qaddoumi I, Gajjar A, Armstrong GT, Ellison DW, Kun LE, Ogg RJ, Sanford RA (2013) Management and outcome of focal low-grade brainstem tumors in pediatric patients: the St. Jude experience. J Neurosurg Pediatr 11(3):274–281. https://doi.org/10.3171/2012.11.PEDS12317
Klimo P Jr, Nesvick CL, Broniscer A, Orr BA, Choudhri AF (2015a) Malignant brainstem tumors in children, excluding diffuse intrinsic pontine gliomas. J Neurosurg Pediatr 17:1–9. https://doi.org/10.3171/2015.6.PEDS15166
Klimo P Jr, Venable GT, Boop FA, Merchant TE (2015b) Recurrent craniopharyngioma after conformal radiation in children and the burden of treatment. J Neurosurg Pediatr 15(5):499–505. https://doi.org/10.3171/2014.10.PEDS14384
Lin CT, Riva-Cambrin JK (2015) Management of posterior fossa tumors and hydrocephalus in children: a review. Childs Nerv Syst 31(10):1781–1789. https://doi.org/10.1007/s00381-015-2781-8
Mottolese C, Stan H, Hermier M, Berlier P, Convert J, Frappaz D, Lapras C (2001) Intracystic chemotherapy with bleomycin in the treatment of craniopharyngiomas. Childs Nerv Syst 17(12):724–730. https://doi.org/10.1007/s00381-001-0524-5
Osborn AG, Heaston DK, Wing SD (1978) Diagnosis of ascending transtentorial herniation by cranial computed tomography. AJR Am J Roentgenol 130(4):755–760. https://doi.org/10.2214/ajr.130.4.755
Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, Wolinsky Y, Kruchko C, Barnholtz-Sloan J (2014) CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro-Oncology 16(Suppl 4):iv1–i63. https://doi.org/10.1093/neuonc/nou223
Ostrom QT, de Blank PM, Kruchko C, Petersen CM, Liao P, Finlay JL, Stearns DS, Wolff JE, Wolinsky Y, Letterio JJ, Barnholtz-Sloan JS (2015) Alex’s lemonade stand foundation infant and childhood primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro-Oncology 16(Suppl 10):x1–x36. https://doi.org/10.1093/neuonc/nou327
Ottenhausen M, Krieg SM, Meyer B, Ringel F (2015) Functional preoperative and intraoperative mapping and monitoring: increasing safety and efficacy in glioma surgery. Neurosurg Focus 38(1):E3. https://doi.org/10.3171/2014.10.FOCUS14611
Patay Z (2015) Postoperative posterior fossa syndrome: unraveling the etiology and underlying pathophysiology by using magnetic resonance imaging. Childs Nerv Syst 31(10):1853–1858. https://doi.org/10.1007/s00381-015-2796-1
Pittman T, Williams D, Weber TR, Steinhardt G, Tracy T Jr (1992) The risk of abdominal operations in children with ventriculoperitoneal shunts. J Pediatr Surg 27(8):1051–1053
Riva-Cambrin J, Detsky AS, Lamberti-Pasculli M, Sargent MA, Armstrong D, Moineddin R, Cochrane DD, Drake JM (2009) Predicting postresection hydrocephalus in pediatric patients with posterior fossa tumors. J Neurosurg Pediatr 3(5):378–385. https://doi.org/10.3171/2009.1.PEDS08298
Robertson PL, Muraszko KM, Holmes EJ, Sposto R, Packer RJ, Gajjar A, Dias MS, Allen JC, Children’s Oncology G (2006) Incidence and severity of postoperative cerebellar mutism syndrome in children with medulloblastoma: a prospective study by the Children’s Oncology Group. J Neurosurg 105(6 Suppl):444–451. https://doi.org/10.3171/ped.2006.105.6.444
Sainte-Rose C, Cinalli G, Roux FE, Maixner R, Chumas PD, Mansour M, Carpentier A, Bourgeois M, Zerah M, Pierre-Kahn A, Renier D (2001) Management of hydrocephalus in pediatric patients with posterior fossa tumors: the role of endoscopic third ventriculostomy. J Neurosurg 95(5):791–797. https://doi.org/10.3171/jns.2001.95.5.0791
Shah MN, Leonard JR, Inder G, Gao F, Geske M, Haydon DH, Omodon ME, Evans J, Morales D, Dacey RG, Smyth MD, Chicoine MR, Limbrick DD (2012) Intraoperative magnetic resonance imaging to reduce the rate of early reoperation for lesion resection in pediatric neurosurgery. J Neurosurg Pediatr 9(3):259–264. https://doi.org/10.3171/2011.12.PEDS11227
Van Poppel M, Klimo P Jr, Dewire M, Sanford RA, Boop F, Broniscer A, Wright K, Gajjar AJ (2011) Resection of infantile brain tumors after neoadjuvant chemotherapy: the St. Jude experience. J Neurosurg Pediatr 8(3):251–256. https://doi.org/10.3171/2011.6.PEDS11158
Vogel TW, Bahuleyan B, Robinson S, Cohen AR (2013) The role of endoscopic third ventriculostomy in the treatment of hydrocephalus. J Neurosurg Pediatr 12(1):54–61. https://doi.org/10.3171/2013.4.PEDS12481
Wilne SH, Ferris RC, Nathwani A, Kennedy CR (2006) The presenting features of brain tumours: a review of 200 cases. Arch Dis Child 91(6):502–506. https://doi.org/10.1136/adc.2005.090266
Zachenhofer I, Donat M, Oberndorfer S, Roessler K (2011) Perioperative levetiracetam for prevention of seizures in supratentorial brain tumor surgery. J Neuro-Oncol 101(1):101–106. https://doi.org/10.1007/s11060-010-0235-4
Zafeiriou DI, Koliouskas D, Vargiami E, Gombakis N (2001) Russell’s diencephalic syndrome. Neurology 57(5):932
Zhao R, Deng J, Liang X, Zeng J, Chen X, Wang J (2010) Treatment of cystic craniopharyngioma with phosphorus-32 intracavitary irradiation. Childs Nerv Syst 26(5):669–674. https://doi.org/10.1007/s00381-009-1025-1
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Ryan Lingo, P., Choudhri, A.F., Klimo, P. (2018). Principles of Pediatric Neurosurgery. In: Gajjar, A., Reaman, G., Racadio, J., Smith, F. (eds) Brain Tumors in Children. Springer, Cham. https://doi.org/10.1007/978-3-319-43205-2_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-43205-2_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-43203-8
Online ISBN: 978-3-319-43205-2
eBook Packages: MedicineMedicine (R0)