
Abstract
The identification of small proteins and peptides has consistently proven to be challenging. However, technological advances as well as multi-omics endeavors facilitate the identification of novel small coding sequences, leading to new insights. Specifically, the application of next generation sequencing technologies (NGS), providing accurate and sample specific transcriptome / translatome information, into the proteomics field led to more comprehensive results and new discoveries. This book chapter focuses on the inclusion of RNA-Seq and RIBO-Seq also known as ribosome profiling, an RNA-Seq based technique sequencing the +/− 30 bp long fragments captured by translating ribosomes. We emphasize the identification of micropeptides and neo-antigens, two distinct classes of small translation products, triggering our current understanding of biology. RNA-Seq is capable of capturing sample specific genomic variations, enabling focused neo-antigen identification. RIBO-Seq can identify translation events in small open reading frames which are considered to be non-coding, leading to the discovery of micropeptides. The identification of small translation products requires the integration of multi-omics data, stressing the importance of proteogenomics in this novel research area.
Volodimir Olexiouk and Gerben Menschaert equally contributed to the book chapter as first authors
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
4.1 Introduction
Unraveling protein biosynthesis is undoubtedly a multi-omics integration endeavor. From the DNA template (genomics ) a region is transcribed (transcriptomics) and subsequently translated (translatomics) into protein products (proteomics ). The aforementioned omics fields definitely intertwine, but are likewise considered self-sufficient, demonstrated by their vast complexity. Integration of matching multi-omics datasets, although challenging, can lead to more sound results and even new insights. Advances in bioinformatics have facilitated this multi-omics integration and expert tools became available to tackle specific parts of proteogenomics analyses (e.g., PROTEOFORMER (Crappé et al. 2014a), PEPTIDESHAKER (Vaudel et al. 2015b), also see (Menschaert and Fenyö 2015) for a review of bioinformatics tools available in the proteogenomics field). An intriguing multi-omics empowered field tries to identify novel protein coding sequences. Direct assessment of proteins through mass spectrometry based proteomics analysis, combined with genomics , transcriptomics and translatomics information provides the necessary means to unravel the information flow from DNA to proteins (Wang and Zhang 2014). Particularly, the identification of micropeptides , translation products of small open reading frames , and neo-antigens, peptides resulting from proteins variants conceivably recognized by the immune system, are discussed in this book chapter. First, we will briefly describe the MS-based proteomics technology, highlighting the necessity for multi-omics integration in the research fields mentioned above.
As mentioned, the preferred methodology for protein / peptide identification is mass spectrometry (MS) , a technique with high sensitivity and specificity (Cheng et al. 2014; Ryu 2014), capable of detecting up to 10,000 proteins from a single sample (Nagaraj et al. 2011). The global workflow in MS consists of enzymatic digestion of proteins extracted from the sample into peptides that are subsequently fragmented and analyzed by a mass spectrometer, providing peptide fragmentation spectra by registering the mass-to-charge ratio of ionized peptide fragments. Peptides are identified through database search engines (e.g., X!tandem (Craig and Beavis 2004), Myrimatch (Tabb et al. 2007), MS-GF+ (Kim and Pevzner 2014; Granholm et al. 2014), Comet (Eng et al. 2015), MS Amanda (Dorfer et al. 2014)). A peptide-spectrum match (PSM) score is calculated by comparing experimental spectra against theoretical spectra, generated after in silico digestion of all proteins provided in a sequence database. Statistical validation methods in MS -based proteomics compute the false discovery rate (FDR) by means of a target-decoy approach assuming the reference database to contain the “true” pool of sequences represented in the sample (Hernandez et al. 2014). Consequently, deviation from this assumption impairs validation, implying that the main paradigm here is not to use the most exhaustive reference database, but to adversely focus on the most suitable reference database representing the true nature of the biological sample (Gupta et al. 2011; Nesvizhskii 2010; Wang et al. 2009a; Keller et al. 2002). Obviously, small proteins (micropeptides ) produce less cleaved peptides and are often not present in reference protein databases , implicating their MS identification. Also, distinguishing resembling peptides can be complicated, as is frequently the case for neo-antigen identification.
Search engines and algorithms will definitely influence the peptide identification rate, but the reference database construction is pivotal, as inclusion is a prerequisite for identification. Uniprot-KB (EMBL et al. 2013; Apweiler et al. 2014) is mostly used as the reference database in the MS -based proteomics identification process. This database is incomplete as it (partly) lacks information on novel proteoforms (isoforms), single nucleotide variation (SNV), indels (insertions and deletions), and gene fusion products. A more suitable reference database for novel protein identification is constructed containing all ORFs from the translation of the genome in its six reading frames. This strategy makes that all possible protein forms except for peptides spanning the exon junctions are included. That is why these are widely used for prokaryotes by virtue of a small genome and lack of splicing (Baudet et al. 2010). Since 98 % of the human genome is predicted to be non-coding (Lander et al. 2001), this approach would massively increase the search space resulting in an unattractive approach in terms of both computation time and error rate, while also omitting mutations , small open reading frames and non-AUG start sites.
Considering the 6 frame translation approach, only one sixth are true candidates, impairing the statistical validation model used (Hernandez et al. 2014; Blakeley et al. 2012). Furthermore, splice isoforms, single nucleotide variation and indels remain undetectable in a 6-frame translated reference database. A smaller reference database can be constructed from cDNA libraries or expressed sequence tags (EST), ensuring that the corresponding sequences are transcribed as they are derived from RNA (Hernandez et al. 2014). Furthermore, as the reference database has been constructed from RNA, alternative splice proteoforms may be included. Implementing such strategy in human has succeeded to compress the database to 3 % compared to a 6-frame reference database, with minimal sacrifices to the peptide sequence content (Edwards 2007). Another study using the Ensembl (Cunningham et al. 2014) database, including all isoforms, observed a 7 % increase in peptide identification compared to the non-redundant Swiss-Prot database (Fei et al. 2011). Tools as GENQUEST reduce the search space by filtering peptides on their mass and isoelectric point (Sevinsky et al. 2008). Although the aforementioned database choices have proven to be useful, the generated reference database contains sequences on a species wide level, where sample specific genomic (SNVs, indels) and RNA splice variations remain unregistered. Next generation sequencing (NGS) techniques enable the user to capture the transcriptome and/or translatome relatively accurate, fast and cost-efficient, thus enabling sample-specific reference database construction (Bahassi and Stambrook 2014). This review discusses how the integration of NGS techniques with MS -based proteomics enables the identification of novel, small proteins, strongly focusing on ribosome profiling and RNA-Seq . To illustrate the relevance of these techniques in current novel research fields, RNA-Seq mediated neo-antigen discovery and RIBO-Seq empowered micropeptide identification are discussed.
4.2 RNA-Seq
The majority of MS -based proteomic studies consist of comparing the obtained spectra against protein databases of known / predicted proteins, resulting in a high number of unidentified spectra. These unidentified spectra may map to novel peptides absent from the used protein database , represent splice variants, alternative open reading frames (e.g., stop codon read-through, alternative start sites) or genetic variations (Ning and Nesvizhskii 2010). RNA-Seq provides a comprehensive profile of the transcriptome and enables the construction a database reflecting the native transcript composition, including those novel sequences (Woo et al. 2014; Marguerat and Bähler 2010; Wang et al. 2009b). A study performed by Wang et al. (2012) describes a workflow to derive a protein database from RNA-Seq data and records a substantial increase in peptide identifications in comparison to searches against an Ensembl database. Furthermore, RNA-Seq data allowed the detection of peptides containing SNPs associated with cancer. A workflow designed by Sheynkman et al. (2013), establishing a database focusing on splice junctions derived from RNA-Seq, identified unannotated transcript junctions from Jurkat cells. Compared to cDNA and EST libraries, RNA-Seq provides a more advanced and comprehensive methodology to identify novel splice junctions (Sheynkman et al. 2013). Moreover, RNA-Seq enables proteomics studies on non-model organisms with limited genome annotation (Lopez-Casado et al. 2012; Song et al. 2012; Armengaud 2013). Many RNA-Seq datasets are publically available (e.g., in the Sequence Read Archive (Leinonen et al. 2011b) or European Nucleotide Archive (Leinonen et al. 2011a)) and can be utilized in proteogenomics applications. It is advised to pool multiple RNA-Seq experiments cumulatively (Woo et al. 2014) to construct a search space when non-matching proteomics and transcriptomics datasets are used.
4.2.1 Neo-antigens
The immune system recognizes an extensive range of antigens, which are distinguished as either ‘self’ or ‘non-self’ molecules. All human cells present peptide antigens on major histocompatibility complex (MHC) molecules, which interact with T-cell receptors (TCR), present on the plasma membrane of T-cells. When a peptide presented on the MHC is not recognized as ‘self’, this elicits a T-cell response, causing apoptosis or inactivation of the corresponding target cell. The presentation of ‘non-self’ peptide antigens may be induced by various reasons, ranging from viral infection to disturbed homeostasis (Singhal et al. 2013; Attaf et al. 2015). As tumor cells evolve from ordinary cells, they develop distinct characteristics recognizable by the immune system. Hence, the immune system is clearly of great importance in cancer development. The immune system can promote tumor growth by impairing tumor cell immunogenicity or act as a tumor suppressor by destroying or restraining tumor expansion (Koebel et al. 2007; Shankaran et al. 2001; Dunn et al. 2002). Immunotherapy, where T-cell activity is stimulated through the inhibition of the T-cell deactivation pathway (checkpoint blockade (Gubin et al. 2014)), has been shown to be an effective treatment in a variety of human malignancies (Wolchok and Chan 2014; Sharma and Allison 2015). For instance, Rosenberg (Hinrichs and Rosenberg 2014) demonstrated how infusion of tumor-infiltrating lymphocytes can be an effective treatment option in metastatic melanoma and antibody treatment sensitizing T-cell activation improved overall survival of metastatic melanoma patients (Hodi et al. 2010). The ability of T-cells to elicit a T-cell response based on the interaction with MHC molecules on tumor cells indicates the existence of tumor specific epitopes on antigens. These antigens can be derived from native proteins for which T-cell tolerance is incomplete (e.g., tissue / time restricted proteins being expressed) or they can be formed from proteins absent from the human genome (e.g., mutated proteins), called neo-antigens. Neo-epitopes are a product of tumor-specific DNA alterations and thus result in novel protein sequences (Schumacher and Schreiber 2015).
Studies in mouse models indicate that vaccination with neo-antigens increased tumor control in immunotherapy (Gubin et al. 2014; Yadav et al. 2014). However neo-antigen identification is tedious and limitations in MS sensitivity result in a substantial fraction of false negatives. Also, the identification of genomic variations in proteins does not guarantee MHC presentation. Combining transcriptomics sequencing techniques (RNA-Seq) to identify mutated proteins absent in native cells with proteomics identification of MHC presented antigens provides a feasible workflow useable in clinical studies. The global design of this workflow consists of the identification of tumor-specific genomic variation trough RNA-Seq, followed by an optional in silico filtering by algorithms to predict MHC antigen presentation and the construction of a database consisting of possible neo-antigen (Lu et al. 2014; Linnemann et al. 2014; Robbins et al. 2013). Next MS -based proteomics matches the experimentally identified MHC bound antigens against the RNA-Seq derived database, selecting high confidence neo-antigen. Functional essays can be performed to experimentally identify neo-antigens as demonstrated in mouse models, successfully treating cancer (Rizvi et al. 2015; Yadav et al. 2014; Bassani-Sternberg et al. 2015). Figure 4.1 provides a summary of the neo-antigen identification workflow.

A simplified neo-antigen identification workflow. Tumor cells are sequenced to identify genomic variations specific to these tumor cells, next a database is generated consisting of neo-antigen candidates. Optionally, in silico algorithms can be used to predict MHC antigen presentation, resulting in a more confident dataset. Next, MS-based proteomics identifies MHC bound antigens followed by functional analysis confirming candidate neo-antigens
4.3 RIBO-Seq
In the late 1960s, the ability of ribosomes to protect mRNA from endonuclease digestion was demonstrated (Steitz 1969). Despite this early discovery, it was not until the advent of NGS and the accompanying bioinformatics toolsets, that genome-wide translatome profiling became attainable. At the end of the twentieth century a technique named polysome profiling emerged (Johannes et al. 1999), yielding large scale analysis of translation. In summary, polysome profiling captures mRNA immobilized on translating ribosomes, separates these polyribosomes (e.g., ultracentrifugation on a sucrose gradient) and subsequently sequences the obtained RNA fragments (Faye et al. 2014). This technique, identifying mRNA with ribosomal occupancy, saw various use-cases throughout the years and is still frequently applied (Piccirillo et al. 2014). However, it was with the advent of RIBO-Seq, enabling massive parallel sequencing of the +/− 30 nt mRNA fragments protected by ribosomes (RPFs), that in-depth assessment of the translatome was empowered (Ingolia et al. 2009, 2012, 2014). The main advantage of RIBO-Seq over polysome profiling is the ability to retrieve positional information obtained from these RPFs with sub-codon resolution, enabling accurate prediction of the ribosome A-site positions. The RIBO-Seq technique diverged into two complementary implementations, capturing either elongating ribosomes or initiating ribosomes. RIBO-Seq of elongating ribosomes is feasible through the addition of antibiotics inhibiting ribosome translocation (e.g., cycloheximide (Ingolia et al. 2009) and emetine (Ingolia et al. 2012)), peptidyl transferase (e.g., chloramphenicol) or by thermal freezing (Oh et al. 2011). Initiating ribosomes, allowing the deduction of translation initiation sites (TIS), is achieved through the addition of initiation blocking antibiotics (e.g., harringtonine (Ingolia et al. 2012) or lactimidomycin (Lee et al. 2012)). Figure 4.2 sketches an overview of RIBO-Seq protocol.

A general overview of the RIBO-Seq protocol. First, cell lysates are prepared in conditions accurately reflecting in vivo translation. Secondly, addition of nucleases will digest RNA (nuclease footprinting), however the +/−30 nt mRNA fragments encapsulated by ribosomes are protected from digestion (ribosome footprints). Next, ribosome-footprints are separated from cell lysates followed by purification of ribosome protected RNA. Ligation of single-stranded adaptors enables reverse transcription. Subsequently, first strand reverse transcription products are circularized and transcript products hybridized to rRNA probes are depleted. Finally, PCR amplifies the remaining sequences that are subsequently sequenced. An in depth description of the protocol is provided by Ignolia et al. (2012b)
4.3.1 RIBO-Seq Unravels the Translatome
Although many variations are attributable to changes in gene transcripts, RIBO-Seq likewise reveals pervasive translational regulation (Michel and Baranov 2013). For example, Ignolia et al. (2009b) examined the ability of ribosome profiling to monitor changes in protein synthesis in response to starvation in yeast, observing translation changes in approximately one-third of the genes. Two other studies examining the translatome in response to heatshock (Shalgi et al. 2013) and proteotoxic stress (Liu et al. 2013) revealed interesting properties of the influence of chaperones on elongating ribosomes in response these stresses. In a study performed by Brar et al. (2012), exploring changes in expression during meiosis in yeast by performing RIBO-Seq over stage-specific time points, numerous dynamic events (including translation products of small open reading frames ) were captured, unidentified by other techniques. A study performed by Stern-Ginossar et al. (2012) analyzed gene expression changes of human foreskin fibroblasts during cytomegalovirus infection. Measurements across different time-stamps revealed prominent viral gene translational regulation, where translation varied at least fivefold in 82 % of ORFs.
Furthermore, RIBO-Seq can identify novel translated regions, until now undetectable with other techniques. For instance several 5’-UTR ORFs, associated to a regulatory function (Ingolia et al. 2009, 2011; Brar et al. 2012), have been identified by ribosome profiling . The ORFs in 5’ untranslated regions are difficult to identify due to their specific characteristics: short length, limited coverage, non-AUG initiation, sometimes overlapping with canonical ORFs. Michel et al. (2012) demonstrated that given sufficient ribosome coverage, alternative reading frames are discernible by analyzing the triplet codon periodicity characteristic to translation and observable with the ribosome profiling technique. They reported on 5’-UTR ORFs with higher RPF intensity than the main canonical downstream ORF. In many cases these upstream ORFs (uORFs) partly overlapped with the canonical ORF. Furthermore Michel et al. identified frame transitions in translation, confirming well-known cases of frame shifts in humans. In a study performed by Gerashchenko et al. (2012) in yeast, four novel frame shift events were identified that correlated to oxidative stress. Also, the start site determination with the ribosome profiling technique enables the identification of ORFs with non-AUG start sites, resulting in numerous identified near-cognate initiation sites. Wan and Qian (2014) developed a database containing alterative translation initiation sites and their associated ORF identified by RIBO-Seq. Ribosomal activity was also observed in non-coding regions, revealing putative novel protein coding regions (Ingolia et al. 2012; Lee et al. 2012).
4.3.2 RIBO-Seq, a Bridge Between RNA-Seq and Proteomics
Protein inference from transcript abundance assumes constant RNA stability as well as stable translation rates. This assumption is erroneous as RNA stability can be highly variable and translation rates are volatile across transcripts. RIBO-Seq bridges the gap between RNA-Seq and proteomics by providing translational information, enabling improved inference from the transcriptome to the proteome and vice versa. RIBO-Seq is capable of detecting coding transcripts, but no direct evidence is provided whether these translated sequences ultimately yield stable protein products. Ribosomal occupancy could yield regulatory functions, but couls also point to unstable protein products or noise (Ingolia et al. 2014; Guttman and Rinn 2012). Several in silico tools and metrics were devised to predict the coding potential of ORFs (based on ribosome protected fragment length (Ingolia et al. 2014), triplet periodicity (Bazzini et al. 2014) and conservation (Lin et al. 2011)). However, MS -based validation remains a crucial confirmation technique in most cases. In turn, MS-based proteomics requires a database consisting of sample specific protein sequences. RIBO-Seq assisted database generation has several advantages over RNA-Seq generated databases. Novel proteoforms can be identified thus optimizing the search space (Calviello et al. 2015; Menschaert et al. 2013; Van Damme et al. 2014; Koch et al. 2014). This approach has been used by Fritsch et al. (2012) to identify 546 N-terminal protein extension in human, Menschaert et al. (2013) observed a 2.5 % increase in the overall protein identification rate using this approach. In a recent study performed by Fields et al. (2015), 1990 protein isoforms, 696 truncations, 341 extension and 1379 upstream ORFs were identified by RIBO-Seq. Automated pipelines facilitating RIBO-Seq integration in MS -based experiments, such as PROTEOFORMER (Crappé et al. 2014a), are readily available and easy to implement. Moreover Xie et al. (2015) developed an online database to query, analyze, visualize and download RIBO-Seq dataset s.
4.4 Micropeptides
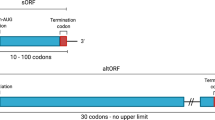
Micropeptides are defined as functional translation products originating from small open reading frames (sORFs) . No consensus was reached regarding the sORF size and some studies consider an upper threshold of 200–250 codons (Hayden and Bosco 2008; Yang et al. 2011). However, the most widespread sORF size limit is 100 codons, a rule that we endorse here. A pioneering genome-wide study in 2003 on yeast suggested the functional importance of sORFs (Kessler et al. 2003), describing functionally conserved sORFs discovered by means of cross-species BLAST analysis. Only a few years later, Savard et al. (2006) identified mille-pattes in the red flour beetle by means of EST screening, a polycistronic peptide encoding four sORFs regulating HOX-genes. Kondo et al. (2007) and Galindo et al. (2007) examined mille-pattes analogs in Drosophila melanogaster resulting in the discovery of the tarsal-less (tal) and polished rice (pri) genes, respectively. This polycistronic mRNA, previously categorized as being non-coding, apparently was miss-annotated based on the ORFs size (Tupy et al. 2005). At the moment of writing, the tal and pri translation products are among the best characterized examples of micropeptides, regulating embryonic development throughout numerous insect species (Chanut-Delalande et al. 2014). The discovery of these tal and pri genes, together with the advent of ribosome profiling , boosted the research into sORF-encoded micropeptides. Several different research groups reported on the discovery of putatively coding sORFs using various techniques, pointing to novel functional micropeptides (Saghatelian and Couso 2015; Chu et al. 2015; Bazzini et al. 2014; Magny et al. 2013; Slavoff et al. 2013; Tonkin and Rosenthal 2015; Crappé et al. 2013; Pauli et al. 2014). Toddler, for example, is an embryonic signal that promotes cell movement (Pauli et al. 2014), Myoregulin regulates Ca2+ handling in muscle cells (Magny et al. 2013) and Sarcolipin regulates muscle-based thermogenesis in mammals (Tonkin and Rosenthal 2015). This is a relatively new research field (Crappé et al. 2014b; Andrews and Rothnagel 2014; Albuquerque et al. 2015), where the results of many in silico based studies and proteogenomics endeavors need further experimental validation.
4.4.1 In Silico Micropeptide Identification
Automated gene annotation systems correctly identify the majority of verified protein coding ORFs based on recognizable genomic sequence characteristics (e.g., canonical initiation codons, splice sites, promoter sequences) (Sleator 2010). Most gene annotation algorithms set a lower threshold of 100 base triplets to exclude false positive annotations (Carninci et al. 2005; Frith et al. 2006a, b; Dinger et al. 2008). Recently, studies suggest that applying this lower threshold precludes the identification of numerous small proteins (Pauli et al. 2014; Bazzini et al. 2014; Ma et al. 2014; Frith et al. 2006a, b; Chng et al. 2013; Galindo et al. 2007; Crappé et al. 2013). Some computational approaches have been developed, such as uPEPperoni (Skarshewski et al. 2014) and sORF finder (Hanada et al. 2009), providing in silico assessment of putatively coding sORFs , based on phylogenetic conservation. While the identification of sORFs is relatively straightforward, it does require a start and stop codon separated by at most 98 codons, the discrimination of coding vs. non-coding sORFs of this excessive pool of sORFs has proved to be more difficult. Due to their small size, many sORFs lacking any coding potential occur by chance. Cross-species conservation can be used as a proxy to function, but solely relying on phylogenetic conservation could prevent the identification of biologically relevant species-specific sORFs (Clamp et al. 2007). PhyloCSF (Lin et al. 2011) models phylogenetic relations between species by analyzing conservation at the amino acid level, rather than the nucleotide level and is most regularly used for small open reading frame assessment. It outperforms other methodologies (Reading Frame Conservation metrics, the regular CSF method or a d n /d s test) and is capable of identifying micropeptide coding sORFs as short as 13 amino acids (Guttman and Rinn 2012). Using mainly conservation as a criterion, Mackowiak et al. (2015) identified numerous conserved sORFs in different species (831 in H. sapiens, 350 in M. musculus, 211 in D. rerio, 194 in D. melanogaster, and 416 in C. elegans), some of which have been described and characterized previously.
4.4.2 RIBO-Seq Enables the Identification of Translated sORFs
RNA-based transcriptomics is ignorant to ORF delineation; therefore most studies rely on conservation and pattern recognition for sORF identification. A recent study in yeast identified several micropeptides, one of which was also functionally characterized in influencing osmotic stress. The technique was based on using a 6-frame translation database derived from RNA-Seq data as a search space for subsequent MS fragmentation spectra matching (Yagoub et al. 2015). However, RNA-Seq does not indicate translation of the sORFs as opposed to RIBO-Seq. On top of pinpointing translated mRNA regions, RIBO-Seq can also reveal TIS, enabling the detection of non-AUG sORFs. In silico detection of non-AUG sORFs is laborious and difficult, since the search space becomes extensively larger, but from previous RIBO-Seq studies it has become clear that non-canonical start codons are more common than previously expected (Ingolia et al. 2011). Also, Slavoff et al. (2013) identified translation products from sORFs having non-AUG start sites using an MS -based proteogenomics approach. Recently, Fields et al. (2015) used a regression method on ribosome profiling data to identify sORFs that demonstrate an RPF length pattern and resemble that of annotated protein-coding ORFs. They discovered numerous sORFs, of which a subset shows very weak sequence conservation.
sORFs can be located in coding sequences (CDS), in 5’-untranslated regions (5’-UTR), in 3’-untranslated regions (3’-UTR), in intergenic regions (in-between genes) or in non-coding RNA regions. A first proof of 5’-UTR sORFs being translated was observed by Crowe et al. (2006). They revealed that 20 % of human 5’-UTR ORFs have TIS in an optimal Kozak sequence context, competent of ribosomal recognition. Follow-up studies revealed approximately 6750 conserved upstream TIS in mice (Lee et al. 2012) and approximately 3000 novel 5-UTR sORFs in human (Fritsch et al. 2012). A few 5’-UTR sORFs were identified encoding micropeptides (e.g., MKKS in human (Akimoto et al. 2013), CPA1 in yeast (Werner et al. 1987)) with regulatory functions. Jorgenson (Jorgensen and Dorantes-Acosta 2012) claimed that 5’-UTR sORFs can regulate the downstream translation of the canonical ORF (also called the peptoswitch mechanism) as exemplified by CPA1. The discovery of dually coding transcripts (transcripts where more than one overlapping ORF can be translated), enabled the discovery of CDS-overlapping sORFs (e.g., CASP1 (Ronsin et al. 1999) and altPrP (Vanderperre et al. 2011) in human). Most 3’-UTR sORFs are considered non-coding and are confirmed by the RIBO-Seq profiles that closely resemble those of non-coding ORFs. Still, a limited set of 3’-UTR sORFs was identified by MS -based techniques (e.g., Bazzini et al. (2014) identified ten 3’-UTR sORFs using MS in combination with RIBO-Seq in a proteogenomics approach). Both sORFs in intergenic as well as in non-coding regions have been observed with RIBO-Seq (Lee et al. 2012). In particular, ribosomal activity on long non-coding RNA (lncRNA) fuelled a debate in the scientific community (Pauli et al. 2015) on whether or not lncRNAs are truly non-coding (Ruiz-Orera et al. 2014; Smith et al. 2014). Figure 4.3 provides an overview of sORFs identified in different (annotated) genomic region s.

sORFs classification. sORFs can be classified according to their genomic location, here an overview is provided of the different sORF classifications
4.4.3 Multi-omics Integration Is Still Indispensable
Ribosome occupancy does not necessarily mean translation into functional protein products; furthermore, RIBO-Seq is susceptible to noise. Besides conservation, several tools and metrics were developed to distinguish coding from non-coding sORFs . For example Ignolia et al. (2014) observed that the ribosome protected fragment (RPF) length distribution differs significantly between truly coding and non-coding ORFs and developed the FLOSS-score to distinguish between both categories (Fig. 4.4). Bazzini et al. (2014) developed the ORFscore, which calculates the preference of RPFs to accumulate in the first frame of coding sequences (Fig. 4.5), making full use of the triplet periodicity in the RIBO-Seq signal. The Ribosome Release Score (RRS) examines the release of translating ribosomes after hitting a stop codon (Guttman and Rinn 2012) (Fig. 4.4). More complex statistical methods are based on learning algorithms such as Coding Potential calculator (Kong et al. 2007), CRITICA (Badger and Olsen 1999), CSTMiner (Castrignanò et al. 2004) and the recently described ORF-RATER (Fields et al. 2015) and RiboTaper (Calviello et al. 2015). ORF-RATER, a regression based translating ORF identifier based on RIBO-Seq data, discovered numerous novel ORFs, including sORFs with MS -evidence (Fields et al. 2015). Likewise, RiboTaper exploits a statistical approach to identify translated ORFs based on the nucleotide periodicity of RIBO-Seq data and correctly identified annotated protein coding sORFs , such as the aforementioned Toddler sORF (Calviello et al. 2015). However, in the novel field of micropeptide discovery, MS -based identification still remains indispensable. A proteogenomics approach generating a database of putatively coding sORFs derived from RIBO-Seq (or RNA-Seq ) information, followed by MS-based proteomics identification creates an ideal setting for sORF discovery. Numerous sORFs have been identified using this approach (Ma et al. 2014; Bazzini et al. 2014; Mackowiak et al. 2015). A public database for sORFs (http://www.sorfs.org) exists, gathering multi-omics (RIBO-Seq and MS ) evidence and in silico metrics. The resource currently harbors 266,342 sORFs across three model species (human, mouse, fruit fly) (Olexiouk et al. 2015), but will expand in the near future, with more data on other organism and cell types and including the latest “coding potential” metrics. Figure 4.4 provides an overview of the micropeptide identification workflow .

Overview of coding potential assessment methods based on RIBO-seq . The FLOSS score compares the RPF-length distribution of sORFs with the RPF-length distribution of canonical protein-coding transcripts; strong disagreement between the two RPF-length distributions indicates non-coding behavior. The ORFscore calculates the preference of RPFs of coding ORFs to accumulate in the first frame of the coding sequence and the RRS provides a score based on the tendency of ribosome to dissociate from RNA after hitting a stop coding in coding ORFs

A simplified micropeptide identification workflow. First, translating sORFs are identified using RIBO-seq . Next, candidate protein coding sORFs are predicting using methods described in the “Multi-omics integration is still indispensable” section and a database of translated sORFs is generated for proteomics identification. Results from both pathways can be combined in order to select micropeptides for functional analysis
4.5 Conclusion and Future Perspectives
A multi-omics identification workflow for translation products is certainly advantageous, and is indispensable for novel (small) proteoform identifications. Such a proteogenomics approach is in many cases sample specific, enabling the analysis of sample specific variations. In cancer research , where variations obtained in a single cell may result in tumorous behavior and where these variations are frequently distinct between different tumor types, capturing such sample specific variations is crucial. Identification of neo-antigens in essence holds the identification of sample specific variation, obtainable by transcriptome sequencing technologies. However MS -based proteomics identification remains essential in order to perceive whether these transcript changes yield non-synonymous peptide variations. While still in its infancy, neo-antigen research increases the overall understanding of the immune system and moreover holds important therapeutic value.
The RIBO-Seq enabled genome-wide assessment of translation (translatomics) bridges two omics fields: transcriptomics and proteomics . Genome wide analysis of this ribosome profiling information already resulted in the identification of numerous sORFs with coding potential, questioning the non-coding character of sORFs. Follow-up analyses observed sORFs that resemble canonical coding ORFs and some are in the mean fully characterized as being coding. Over the last years, various tools and metrics were devised to assess the coding potential of sORFs (both conservation and sequence based). Also, workflows aiding the integration of RIBO-Seq information and MS -based proteomics are becoming available, e.g., PROTEOFORMER (Crappé et al. 2014a). The scientific community is becoming aware of sORFs as potentially protein coding units. As a result, public sORF databases, such as http://www.sorfs.org, will be highly useful in the experimental design of future experiments (Olexiouk et al. 2015). Moreover, already conducted experiments (with an emphasis on MS -based proteomics studies) must be reprocessed to account for micropeptides . The scientific community is becoming aware of the large amount of publically available proteomics data accumulated over the past years that is currently being left untouched, while our scientific knowledge and technology evolved tremendously (Vaudel et al. 2015a, b; Verheggen et al. 2015). The sORFs .org database already holds a pilot study where 1172 publically available MS datasets from PRIDE were reprocessed, providing MS-evidence for more than 5000 micropeptides. Cumulative evidence that sORFs are able to encode functional micropeptides has been gathered, but their exact biological relevance often remains to be determined. Undoubtedly, future research on overexpression or knock-down will reveal more about the functional roles of specific sORF-encoded micropeptides.
4.6 Funding
Postdoctoral Fellows of the Research Foundation – Flanders (FWO-Vlaanderen) [G.M.,12A7813N]. Research Foundation – Flanders (FWO-Vlaanderen) [V.O, G0D3114N].
References
Akimoto, C., et al. (2013). Translational repression of the McKusick-Kaufman syndrome transcript by unique upstream open reading frames encoding mitochondrial proteins with alternative polyadenylation sites. Biochimica et Biophysica Acta - General Subjects, 1830(3), 2728–2738.
Albuquerque, J. P., Tobias-santos, V., & Rodrigues, A. C. (2015). small ORFs: A new class of essential genes for development. Genetics and Molecular Biology, 283, 278–283.
Andrews, S. J., & Rothnagel, J. a. (2014). Emerging evidence for functional peptides encoded by short open reading frames. Nature Reviews Genetics, 15(3), 193–204.
Apweiler, R., et al. (2014). Activities at the Universal Protein Resource (UniProt). Nucleic Acids Research, 42(D1), D191–D198.
Armengaud, J. (2013). Microbiology and proteomics, getting the best of both worlds! Environmental Microbiology, 15(1), 12–23.
Attaf, M., et al. (2015). The T cell antigen receptor: The Swiss Army knife of the immune system. Clinical & Experimental Immunology, 181(1), 1–18.
Badger, J. H., & Olsen, G. J. (1999). CRITICA: Coding region identification tool invoking comparative analysis. Molecular Biology and Evolution, 16(4), 512–524.
Bahassi, E. M., & Stambrook, P. J. (2014). Next-generation sequencing technologies: Breaking the sound barrier of human genetics. Mutagenesis, 29(5), 303–310.
Bassani-Sternberg, M., et al. (2015). Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Molecular & Cellular Proteomics, 14(3), 658–673.
Baudet, M., et al. (2010). Proteomics-based refinement of Deinococcus deserti genome annotation reveals an unwonted use of non-canonical translation initiation codons. Molecular & Cellular Proteomics, 9(2), 415–426.
Bazzini, A. A., et al. (2014). Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO Journal, 33(9), 981–993.
Blakeley, P., Overton, I. M., & Hubbard, S. J. (2012). Addressing statistical biases in nucleotide-derived protein databases for proteogenomic search strategies. Journal of Proteome Research, 11(11), 5221–5234.
Brar, G. a., et al. (2012). High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science, 335(6068), 552–557.
Calviello, L. et al. (2015, December). Detecting actively translated open reading frames in ribosome profiling data. Nature Methods, 13(2), 1–9.
Carninci, P., et al. (2005). The transcriptional landscape of the mammalian genome. Science, 309(5740), 1559–1563.
Castrignanò, T. et al. (2004). CSTminer: A web tool for the identification of coding and noncoding conserved sequence tags through cross-species genome comparison. Nucleic Acids Research, 32(Web Server issue), W624–W627.
Chanut-Delalande, H., et al. (2014). Pri peptides are mediators of ecdysone for the temporal control of development. Nature Cell Biology, 16(11), 1035–1044.
Cheng, K., et al. (2014). Fit-for-purpose curated database application in mass spectrometry-based targeted protein identification and validation. BMC Research Notes, 7, 444.
Chng, S. C., et al. (2013). ELABELA: A hormone essential for heart development signals via the apelin receptor. Developmental Cell, 27(6), 672–680.
Chu, Q., Ma, J., & Saghatelian, A. (2015). Identification and characterization of sORF-encoded polypeptides. Critical Reviews in Biochemistry and Molecular Biology, 50(2), 134–141.
Clamp, M., et al. (2007). Distinguishing protein-coding and noncoding genes in the human genome. Proceedings of the National Academy of Sciences of the United States of America, 104(49), 19428–19433.
Craig, R., & Beavis, R. C. (2004). TANDEM: Matching proteins with tandem mass spectra. Bioinformatics, 20(9), 1466–1467.
Crappé, J., et al. (2013). Combining in silico prediction and ribosome profiling in a genome-wide search for novel putatively coding sORFs. BMC Genomics, 14, 648.
Crappé, J., Ndah, E., et al. (2014a). PROTEOFORMER: Deep proteome coverage through ribosome profiling and MS integration. Nucleic Acids Research, 10, 1–10.
Crappé, J., Van Criekinge, W., & Menschaert, G. (2014b). Little things make big things happen: A summary of micropeptide encoding genes. EuPA Open Proteomics, 3, 128–137.
Crowe, M. L., Wang, X.-Q., & Rothnagel, J. a. (2006). Evidence for conservation and selection of upstream open reading frames suggests probable encoding of bioactive peptides. BMC Genomics, 7, 16.
Cunningham, F., et al. (2014). Ensembl 2015. Nucleic Acids Research, 43(D1), D662–D669.
Dinger, M. E., et al. (2008). Differentiating protein-coding and noncoding RNA: Challenges and ambiguities. PLoS Computational Biology, 4(11), e1000176.
Dorfer, V., et al. (2014). MS Amanda, a universal identification algorithm optimized for high accuracy tandem mass spectra. Journal of Proteome Research, 13(8), 3679–3684.
Dunn, G. P., et al. (2002). Cancer immunoediting: From immunosurveillance to tumor escape. Nature Immunology, 3(11), 991–998.
Edwards, N. J. (2007). Novel peptide identification from tandem mass spectra using ESTs and sequence database compression. Molecular Systems Biology, 3(1), 102.
EMBL, SIB Swiss Institute of Bioinformatics, & Protein Information Resource (PIR). (2013). UniProt. Nucleic Acids Research, 41, D43–D47.
Eng, J. K., et al. (2015). A deeper look into comet—Implementation and features. Journal of The American Society for Mass Spectrometry, 26(11), 1865–1874.
Faye, M. D., Graber, T. E., & Holcik, M. (2014). Assessment of selective mRNA translation in mammalian cells by polysome profiling. Journal of Visualized Experiments, 92, 1–8.
Fei, S. S., et al. (2011). Protein database and quantitative analysis considerations when integrating genetics and proteomics to compare mouse strains. Journal of Proteome Research, 10(7), 2905–2912.
Fields, A. P., et al. (2015). A regression-based analysis of ribosome-profiling data reveals a conserved complexity to mammalian translation. Molecular Cell, 60(5), 816–827.
Frith, M. C., et al. (2006a). Discrimination of non-protein-coding transcripts from protein-coding mRNA. RNA Biology, 3(1), 40–48.
Frith, M. C., et al. (2006b). The abundance of short proteins in the mammalian proteome. PLoS Genetics, 2(4), 515–528.
Fritsch, C., et al. (2012). Genome-wide search for novel human uORFs and N-terminal protein extensions using ribosomal footprinting. Genome Research, 22(11), 2208–2218.
Galindo, M. I., et al. (2007). Peptides encoded by short ORFs control development and define a new eukaryotic gene family. PLoS Biology, 5(5), 1052–1062.
Gerashchenko, M. V., Lobanov, a. V., & Gladyshev, V. N. (2012). Genome-wide ribosome profiling reveals complex translational regulation in response to oxidative stress. Proceedings of the National Academy of Sciences, 109(43), 17394–17399.
Granholm, V., et al. (2014). Fast and accurate database searches with MS-GF + Percolator. Journal of Proteome Research, 13(2), 890–897.
Gubin, M. M., et al. (2014). Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature, 515(7528), 577–581.
Gupta, N., et al. (2011). Target-decoy approach and false discovery rate: When things may go wrong. Journal of The American Society for Mass Spectrometry, 22(7), 1111–1120.
Guttman, M., & Rinn, J. L. (2012). Modular regulatory principles of large non-coding RNAs. Nature, 482(7385), 339–346.
Hanada, K., et al. (2009). sORF finder: A program package to identify small open reading frames with high coding potential. Bioinformatics, 26(3), 399–400.
Hayden, C. a., & Bosco, G. (2008). Comparative genomic analysis of novel conserved peptide upstream open reading frames in Drosophila melanogaster and other dipteran species. BMC Genomics, 9, 61.
Hernandez, C., Waridel, P., & Quadroni, M. (2014). Database construction and peptide identification strategies for proteogenomic studies on sequenced genomes. Current Topics in Medicinal Chemistry, 14(3), 425–434.
Hinrichs, C. S., & Rosenberg, S. a. (2014). Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunological Reviews, 257(1), 56–71.
Hodi, F. S., et al. (2010). Improved survival with ipilimumab in patients with metastatic melanoma. The New England Journal of Medicine, 363(8), 711–723.
Ingolia, N. T. et al. (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science (New York, N.Y.), 324(5924), 218–223.
Ingolia, N. T., Lareau, L. F., & Weissman, J. S. (2011). Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell, 147(4), 789–802.
Ingolia, N. T., et al. (2012). The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nature Protocols, 7(8), 1534–1550.
Ingolia, N. T., et al. (2014). Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Reports, 8(5), 1365–1379.
Johannes, G., et al. (1999). Identification of eukaryotic mRNAs that are translated at reduced cap binding complex eIF4F concentrations using a cDNA microarray. Proceedings of the National Academy of Sciences of the United States of America, 96(23), 13118–13123.
Jorgensen, R. A., & Dorantes-Acosta, A. E. (2012, August). Conserved peptide upstream open reading frames are associated with regulatory genes in Angiosperms. Frontiers in Plant Science, 3, 1–11.
Keller, A., et al. (2002). Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Analytical Chemistry, 74(20), 5383–5392.
Kessler, M. M., et al. (2003). Systematic discovery of new genes in the Saccharomyces cerevisiae genome. Genome Research, 13(2), 264–271.
Kim, S., & Pevzner, P. a. (2014). MS-GF+ makes progress towards a universal database search tool for proteomics. Nature Communications, 5, 5277.
Koch, A., et al. (2014). A proteogenomics approach integrating proteomics and ribosome profiling increases the efficiency of protein identification and enables the discovery of alternative translation start sites. Proteomics, 14, 2688–2698.
Koebel, C. M., et al. (2007). Adaptive immunity maintains occult cancer in an equilibrium state. Nature, 450(7171), 903–907.
Kondo, T., et al. (2007). Small peptide regulators of actin-based cell morphogenesis encoded by a polycistronic mRNA. Nature Cell Biology, 9(6), 660–665.
Kong, L. et al. (2007). CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Research, 35(Web Server issue), W345–W349.
Lander, E. S., et al. (2001). Initial sequencing and analysis of the human genome. Nature, 409(6822), 860–921.
Lee, S. S., et al. (2012). Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proceedings of the National Academy of Sciences of the United States of America, 109(37), E2424–E2432.
Leinonen, R., Akhtar, R., et al. (2011a). The European nucleotide archive. Nucleic Acids Research, 39(Database issue), D28–D31.
Leinonen, R., Sugawara, H., & Shumway, M. (2011b). The sequence read archive. Nucleic Acids Research, 39(Database issue), D19–D21.
Lin, M. F., Jungreis, I., & Kellis, M. (2011). PhyloCSF: A comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics, 27(13), 275–282.
Linnemann, C., et al. (2014). High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nature Medicine, 21(1), 81–85.
Liu, B., Han, Y., & Qian, S. B. (2013). Cotranslational response to proteotoxic stress by elongation pausing of ribosomes. Molecular Cell, 49(3), 453–463.
Lopez-Casado, G., et al. (2012). Enabling proteomic studies with RNA-Seq: The proteome of tomato pollen as a test case. Proteomics, 12, 761–774.
Lu, Y. C., et al. (2014). Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clinical Cancer Research, 20(13), 3401–3410.
Ma, J., et al. (2014). Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. Journal of Proteome Research, 13(3), 1757–1765.
Mackowiak, S. D., et al. (2015). Extensive identification and analysis of conserved small ORFs in animals. Genome Biology, 16(1), 179.
Magny, E. G. et al. (2013). Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science, 341(6150), 1116–1120.
Marguerat, S., & Bähler, J. (2010). RNA-Seq: From technology to biology. Cellular and Molecular Life Sciences, 67(4), 569–579.
Menschaert, G., & Fenyö, D. (2015). Proteogenomics from a bioinformatics angle: A growing field. Mass Spectrometry Reviews, 34(1), 16.
Menschaert, G., et al. (2013). Deep proteome coverage based on ribosome profiling aids mass spectrometry-based protein and peptide discovery and provides evidence of alternative translation products and near-cognate translation initiation events. Molecular & Cellular Proteomics, 12(7), 1780–1790.
Michel, A. M., & Baranov, P. V. (2013). Ribosome profiling: A Hi-Def monitor for protein synthesis at the genome-wide scale. Wiley Interdisciplinary Reviews: RNA, 4(5), 473–490.
Michel, A. M., et al. (2012). Observation of dually decoded regions of the human genome using ribosome profiling data. Genome Research, 22(11), 2219–2229.
Nagaraj, N., et al. (2011). Deep proteome and transcriptome mapping of a human cancer cell line. Molecular Systems Biology, 7(548), 1–8.
Nesvizhskii, A. I. (2010). A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. Journal of Proteomics, 73(11), 2092–2123.
Ning, K., & Nesvizhskii, A. I. (2010). The utility of mass spectrometry-based proteomic data for validation of novel alternative splice forms reconstructed from RNA-Seq data: A preliminary assessment. BMC Bioinformatics, 11(Suppl 11), S14.
Oh, E., et al. (2011). Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo. Cell, 147(6), 1295–1308.
Olexiouk, V. et al. (2015). sORFs.org: A repository of small ORFs identified by ribosome profiling. Nucleic Acids Research, p.gkv1175.
Pauli, A. et al. (2014). Toddler: An embryonic signal that promotes cell movement via Apelin receptors. Science (New York, N.Y.), 343(6172), 1248636.
Pauli, A., Valen, E., & Schier, A. F. (2015). Identifying (non-)coding RNAs and small peptides: Challenges and opportunities. BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology, 37(1), 103–112.
Piccirillo, C. a., et al. (2014). Translational control of immune responses: From transcripts to translatomes. Nature Immunology, 15(6), 503–511.
Rizvi, N. A., et al. (2015). Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science, 348(6230), 124–128.
Robbins, P. F., et al. (2013). Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nature Medicine, 19(6), 747–752.
Ronsin, C. et al. (1999). A non-AUG-defined alternative open reading frame of the intestinal carboxyl esterase mRNA generates an epitope recognized by renal cell carcinoma-reactive tumor-infiltrating lymphocytes in situ. Journal of Immunology (Baltimore, Md. : 1950), 163(1), 483–490.
Ruiz-Orera, J., et al. (2014). Long non-coding RNAs as a source of new peptides. eLife, 3, e03523.
Ryu, S. Y. (2014). Bioinformatics tools to identify and quantify proteins using mass spectrometry data. Advances in Protein Chemistry and Structural Biology, 94, 1–17.
Saghatelian, A., & Couso, J. P. (2015). Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology, 11(12), 909–916.
Savard, J., et al. (2006). A segmentation gene in tribolium produces a polycistronic mRNA that codes for multiple conserved peptides. Cell, 126(3), 559–569.
Schumacher, T. N., & Schreiber, R. D. (2015). Neoantigens in cancer immunotherapy. Science (New York, N.Y.), 348(6230), 69–74.
Sevinsky, J. R., et al. (2008). Whole genome searching with shotgun proteomic data: Applications for genome annotation. Journal of Proteome Research, 7(1), 80–88.
Shalgi, R., et al. (2013). Widespread regulation of translation by elongation pausing in heat shock. Molecular Cell, 49(3), 439–452.
Shankaran, V., et al. (2001). IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature, 410(6832), 1107–1111.
Sharma, P., & Allison, J. P. (2015). The future of immune checkpoint therapy. Science (New York, N.Y.), 348(6230), 56–61.
Sheynkman, G. M., et al. (2013). Discovery and mass spectrometric analysis of novel splice-junction peptides using RNA-Seq. Molecular & Cellular Proteomics, 12(8), 2341–2353.
Singhal, A., Mori, L., & De Libero, G. (2013). T cell recognition of non-peptidic antigens in infectious diseases. The Indian Journal of Medical Research, 138(5), 620–631.
Skarshewski, A., et al. (2014). uPEPperoni: An online tool for upstream open reading frame location and analysis of transcript conservation. BMC Bioinformatics, 15, 36.
Slavoff, S. a., et al. (2013). Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nature Chemical Biology, 9(1), 59–64.
Sleator, R. D. (2010). An overview of the current status of eukaryote gene prediction strategies. Gene, 461(1–2), 1–4.
Smith, J. E., et al. (2014). Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Reports, 7(6), 1858–1866.
Song, J., et al. (2012). An improvement of shotgun proteomics analysis by adding next-generation sequencing transcriptome data in orange. PloS One, 7(6), 5–10.
Steitz, J. a. (1969). Nucleotide sequences of the ribosomal binding sites of bacteriophage R17 RNA. Cold Spring Harbor Symposia on Quantitative Biology, 34, 621–630.
Stern-Ginossar, N. et al. (2012). Decoding human cytomegalovirus. Science (New York, N.Y.), 338(6110), 1088–1093.
Tabb, D. L., Fernando, C. G., & Chambers, M. C. (2007). MyriMatch: Highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. Journal of Proteome Research, 6(2), 654–661.
Tonkin, J., & Rosenthal, N. (2015). One small step for muscle: A new micropeptide regulates performance. Cell Metabolism, 21(4), 515–516.
Tupy, J. L., et al. (2005). Identification of putative noncoding polyadenylated transcripts in Drosophila melanogaster. Proceedings of the National Academy of Sciences of the United States of America, 102(15), 5495–5500.
Van Damme, P., et al. (2014). N-terminal proteomics and ribosome profiling provide a comprehensive view of the alternative translation initiation landscape in mice and men. Molecular & Cellular Proteomics, 13(5), 1245–1261.
Vanderperre, B., et al. (2011). An overlapping reading frame in the PRNP gene encodes a novel polypeptide distinct from the prion protein. The FASEB Journal, 25(7), 2373–2386.
Vaudel, M., & Verheggen, K. et al. (2015). Exploring the potential of public proteomics data. Proteomics, (January 2016), 1–30.
Vaudel, M., Burkhart, J. M., et al. (2015b). PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nature Biotechnology, 33(1), 22–24.
Verheggen, K. et al. (2015). Pladipus enables universal distributed computing in proteomics bioinformatics. Journal of Proteome Research, p.acs.jproteome.5b00850.
Wan, J., & Qian, S. B. (2014). TISdb: A database for alternative translation initiation in mammalian cells. Nucleic Acids Research, 42(November 2013), 845–850.
Wang, X., & Zhang, B. (2014). Integrating genomic, transcriptomic, and interactome data to improve peptide and protein identification in shotgun proteomics. Journal of Proteome Research, 13(6), 2715–2723.
Wang, G., et al. (2009a). Decoy methods for assessing false positives and false discovery rates in shotgun proteomics. Analytical Chemistry, 81(1), 146–159.
Wang, Z., Gerstein, M., & Snyder, M. (2009b). RNA-Seq: A revolutionary tool for transcriptomics. Nature Reviews Genetics, 10(1), 57–63.
Wang, X., et al. (2012). Protein identification using customized protein sequence databases derived from RNA-Seq data. Journal of Proteome Research, 11(2), 1009–1017.
Werner, M., et al. (1987). The leader peptide of yeast gene CPA1 is essential for the translational repression of its expression. Cell, 49(6), 805–813.
Wolchok, J., & Chan, T. (2014). Cancer: Antitumour immunity gets a boost. Nature, 515, 496–498.
Woo, S., et al. (2014). Proteogenomic database construction driven from large scale RNA-Seq data. Journal of Proteome Research, 13(1), 21–28.
Xie, S.-Q. et al. (2015). RPFdb: A database for genome wide information of translated mRNA generated from ribosome profiling. Nucleic Acids Research, p.gkv972.
Yadav, M., et al. (2014). Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature, 515(7528), 572–576.
Yagoub, D. et al. (2015). Proteogenomic discovery of a small, novel protein in yeast reveals a strategy for the detection of unannotated short open reading frames. Journal of Proteome Research, p.acs.jproteome.5b00734.
Yang, X., et al. (2011). Discovery and annotation of small proteins using genomics, proteomics, and computational approaches. Genome Research, 21(4), 634–641.
Conflict of Interest Statement
None declared.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Olexiouk, V., Menschaert, G. (2016). Identification of Small Novel Coding Sequences, a Proteogenomics Endeavor. In: Végvári, Á. (eds) Proteogenomics. Advances in Experimental Medicine and Biology, vol 926. Springer, Cham. https://doi.org/10.1007/978-3-319-42316-6_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-42316-6_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-42314-2
Online ISBN: 978-3-319-42316-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)