
Abstract
Proteogenomics is a multi-omics research field that has the aim to efficiently integrate genomics, transcriptomics and proteomics. With this approach it is possible to identify new patient-specific proteoforms that may have implications in disease development, specifically in cancer. Understanding the impact of a large number of mutations detected at the genomics level is needed to assess the effects at the proteome level. Proteogenomics data integration would help in identifying molecular changes that are persistent across multiple molecular layers and enable better interpretation of molecular mechanisms of disease, such as the causal relationship between single nucleotide polymorphisms (SNPs) and the expression of transcripts and translation of proteins compared to mainstream proteomics approaches. Identifying patient-specific protein forms and getting a better picture of molecular mechanisms of disease opens the avenue for precision and personalized medicine. Proteogenomics is, however, a challenging interdisciplinary science that requires the understanding of sample preparation, data acquisition and processing for genomics, transcriptomics and proteomics. This chapter aims to guide the reader through the technology and bioinformatics aspects of these multi-omics approaches, illustrated with proteogenomics applications having clinical or biological relevance.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
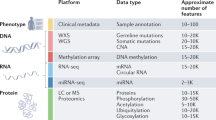


Keywords
3.1 Introduction
Genome sequencing technology aims to reveal the nucleotide sequence of the genome and stage-specific transcriptome states across different cells and tissues. The proteome is defined as “the protein complement of the genome”. Proteins are the product of the translated part of the genome and transcriptome. Proteins are biologically active molecules, while genomes and transcriptomes, besides exerting a regulatory role, hold information on possible protein primary sequences that the cells of an organism can express and use to fulfill their molecular activities and biological functions.
Sequencing DNA or mRNA requires an analytical system that distinguishes precisely between the nucleobases cytosine, guanine adenine (DNA, RNA), thymine (DNA) and uracil (RNA). Combinations of these five bases represent a much simpler chemical system compared to the chemical space spanned by the twenty amino acids and their possible chemical modifications, e.g. through post-translational modifications that form the proteins (Chuh and Pratt 2015; Walsh et al. 2005; Markiv et al. 2012; Bischoff and Schlüter 2012). This larger and more diverse chemical space and the currently available peptides and protein sequencing technologies are not sensitive and powerful with respect to sequencing length compared with current state-of-the-art DNA and RNA sequencing technologies. Additionally, the information content at the genomics and transcriptomics level can be easily amplified, but no such technology exists for proteomics . The main difference between mainstream next generation sequencing technology and shotgun bottom-up LC-MS/MS proteomics is that the former provides hypothesis-free de novo sequencing data, from which the sequence of base pairs can be determined without prior information. However, proteomics analysis determines the primary amino acid sequence from an often incomplete list of fragment ions resulting from the fragmentation of peptides constituting the initial protein. Not all the obtained fragment ion or MS/MS spectra are suitable for a hypothesis-free de novo sequence determination of the fragmented peptide. Therefore the most popular approach to analyze shotgun LC-MS/MS spectra are based on targeted database search (DBS) algorithms, which uses a list of protein sequences that are expected to be present in the analyzed sample. This approach is therefore hypothesis-driven and the success of the identification relies on the accurate prediction of the protein sequence that is expected to be present in the sample. In order to provide accurate sequence information, the proteomics community uses sequences assembled by consortia or large groups that have been quality-controlled either manually (SwissProt) or computationally (TrEMBL and Ensembl). The definition of canonical sequences according to the most widely used UniProtKB/SwissProt database (Consortium 2015) is:
-
1.
The protein sequence of all the protein products encoded by one gene in a given species is represented in a single entry to reduce protein sequence redundancy
-
2.
The canonical sequence includes the protein sequence that has the highest occurrence
-
3.
The canonical protein sequence shows the highest similarity to orthologous sequences found in other species
-
4.
The length of the sequence or amino acid composition allows the clearest description of protein domains, isoforms, polymorphisms and post-translational modifications (PTMs)
-
5.
In the absence of any other information the longest sequence is chosen
For organisms – amongst them humans, for which the genome sequence is completed – the protein sequence derived from genome translation is used, unless there is clear evidence that a different polymorphism is more frequent at a given position.
It is clear from the definition of the canonical sequence that it represents an average sequence of the proteome, but it cannot be used to detect peptides specific for low frequency variants or new variants. The proteogenomics approach performs next generation sequencing of a genome and/or transcriptome in the same sample and composes the protein sequence used during DBS of peptide and protein inference. This composition is not a trivial task and gene models that predict the translation of genomics sequences into proteins are used. In the early days of the genomics era, proteogenomics was defined as a description of “studies in which proteomic data are used for improved genome annotation and characterization of the protein-coding potential” (Nesvizhskii 2014; Menschaert and Fenyo 2015; Bischoff et al. 2015). Therefore in the early days the proteomics dataset helped to provide accurate genome annotation . Nowadays it is more frequent to use the genomic sequence information to obtain sample, or in clinical research patient-specific protein sequence information and predict which protein forms are present in a given sample. Therefore, proteogenomics data analysis allows better and more accurate protein identification and better reflects the biological processes that are active in the cell and/or tissue of the analyzed sample. Since a high quality patient specific database is used for peptide and protein identification, proteogenomics enables a personalized approach to identify patient specific molecular heterogeneity and novel patient phenotypes within a disease. Furthermore, it allows discovery of biomarkers for its specific diagnosis, as well as the discovery of new drug targets that allow more precision and personalized treatment. Importantly, proteogenomics analysis has become more affordable by the reduction of sequencing costs, which has enabled the generation of more precise information of clinical samples, and thus patient specific proteomes, when compared to mainstream proteomics analysis using public databases.
This chapter has the primary aim to provide an overview of the main characteristics of data obtained with next generation sequencing technology combined with the shotgun LC-MS/MS proteomics approach, to describe the key data processing steps and the integrated data interpretation of these two molecular layers. The chapter is intended for readers interested in the data analysis and interpretation of one or both -omics fields with the ultimate goal to perform a proteogenomics analysis. Best practice in data acquisition, data processing approaches and challenges with respect to data and analysis tools will be thoroughly discussed.
3.2 RNA and DNA Sequencing
3.2.1 Genomic Sequencing Technologies
The translated protein sequence can be deduced from full genome, exome and transcriptome sequencing data but the most widely used approach is polyadenylated transcriptome sequencing (RNA-Seq ). Figure 3.1 summarizes the starting molecular level (DNA, mRNA), and the applied protocols and factors that should be taken into account during sequencing. Sequencing the full genome costs an order of magnitude more than sequencing exomes or transcriptomes. For DNA sequencing, the two main options are Whole Genome Sequencing (WGS) and Whole Exome Sequencing (WES). While the first gives a complete overview of variations in the genome, the second covers the coding part of the genome (exome), which accounts only for several percent of the complete genome. For these reasons WES could be a good choice as source of genetic information for a proteogenomics approach. Sequencing polyadenylated mRNA has the advantage that the majority of the transcripts were already processed by the splicing machinery, resulting in a high fraction (>90 %) of mature transcripts with spliced introns, which provide the highest quality of sequence information to predict the sequence of the translated proteome. The other alternative is the removal of highly abundant ribosome mRNA with special kits (Ribo-zero kits), which enables the user to sequence the complete transcriptome that contains the translated and non-translated transcripts.

Chart showing the different molecular classes that can be sequenced using next generation sequencing (RNA red, DNA green), showing the starting material, the sequencing protocols and experimental factors (purple rectangles) that can be set by the user. WGS represents whole genome sequencing and WES represents whole exome sequencing
There are different technologies for transcriptome and genome sequencing. In the early days of sequencing technology, DNA sequencing was used to obtain the first complete genomes. The most important milestones were deciphering of the genome of the bacteriophage ΦX 174 (Sanger et al. 1977) (first complete genome) by Frederick Sanger and the human genome by the Human Genome Project (Lander et al. 2001). The technique developed by Frederick Sanger was the first to be automated and is considered as “first generation” of sequencing technology. The “second generation” (also called Next Generation Sequencing ) started with MPSS (Massively Parallel Signature Sequencing) from Lynx Therapeutics and was characterized by cheaper, faster and more efficient sequencing, which led to the acquisition of an enormous amount of genomic information. Nowadays, the most widely used sequencing technology is short-read based sequencing, with the Illumina HiSeq sequencing machines. Figure 3.1 shows the standard protocol for DNA sequencing, which technology has not only drastically reduced the time necessary for sequencing but also the cost of each analysis run, leading to complete transcriptome sequencing in a matter of hours. Typical fragment sizes range from 100 bps up to 600 bps. Fragments are then read from one (single-end) or both sides (paired-end) up to 250 bps.
There are various options when preparing samples for the sequencing run(s), based on different protocols that focus on different types of transcripts or different ways to analyze them. While at first only the coding messages of the transcriptome were sequenced, through a selection of polyadenylated transcripts (the mRNAs that are most likely to be translated into proteins), the growing interest in the non-coding transcriptome has led to a different approach, where only the major non-coding RNA type, the ribosomal RNA (rRNA), is depleted. This protocol is defined as rRNA depletion or the Ribo-zero approach and is achieved with special ribosomal mRNA removal kits. For a proteogenomics approach, it is often considered a good choice to use the polyadenylated mRNA protocol and thus focus only on protein-generating transcripts, to minimize the error rate and provide the most accurate protein sequence information that is supposed to be expressed in the cells of the target organism. Conversely, the rRNA-depletion protocol retains long non-coding RNAs (or lncRNAs) and other non-polyadenylated transcripts, which are thought to have a regulatory function. However, there is growing evidence that some of the lncRNAs might be translated. lncRNAs are lineage-specific and it is hypothesized that they show similar characteristics as evolutionary young protein coding genes (Ruiz-Orera et al. 2014). Proteomics identification of lncRNA from large public database such as the PRIDE repository (http://www.ebi.ac.uk/pride/archive/) showed high FDR rates of translated lncRNA sequences and therefore the results should be taken with care (Volders et al. 2013, 2015).
In addition to the decision of what should be sequenced, there are various factors that should be taken into account when designing an RNA or DNA-sequencing experiment. For example, the sequencing depth or the number of reads for each sample determine the quality of data and influence important properties such as the quality of the alignment to a reference genome, the number of identified sequence variants that differ from the reference genome and affect the reliability of quantifications. The optimal sequencing level should be determined based on the aim of the experiment. However, it is obvious that a complex sample (for example from biopsies that typically contain different types of cell from different tissues) requires higher sequencing depth when compared to a simpler sample consisting of one cell or tissue type.
In a similar fashion, the length of the reads may have a consistent effect on the quality of the post-sequencing alignment to a reference genome and thus the ability to correctly determine the transcripts structure and amount. Longer reads tend to minimize the effect of sequencing errors and capture splicing events or multi-nucleotide deletions and insertions more efficiently. On the other hand, if the intention is only to quantify the amount of transcript(s) present, short reads (such as 50 bp) may be sufficient, leading to reduced cost and analysis time.
Sequencing can be performed with two approaches concerning the reading directions of 500 base pair transcript fragments, these are known as single and paired-ends. The effect of longer reads is magnified when paired-end reads are used. There are sample preparation kits that cannot discriminate whether a sequence is read in forward or reverse direction (e.g., TruSeq from Illumina) and there are kits that can deliver this information (e.g., BioO Scientific’s NextFlex). When strandedness information is lacking, it is still possible to predict from which strand the reads originate by exploiting the unique sequence of introns and exons of each transcript. Information on the exact sequenced strand is important when identifying variants, as each strand may carry a different allele (a different base in the corresponding position on each strand), could be coding (contain the translated amino acid sequence) or template (contains the complementary nucleotide base sequence) and strands originate from maternal and paternal chromosomes. In paired-end sequencing the sequence is first read in one direction and then from the opposite direction having around 500 bps of distance between the two ends (Fig. 3.2). It is important to note that one read covers a relatively small part of a fragment, but taking the fragment length into account for alignment to a reference genome provides more accurate alignment than with single-end reading. The two reads in paired-end sequencing are also called a “mate-pair”. Paired-end reads provide more accurate data when trying to detect large modifications of the genome and transcriptome, like large insertions, deletions and translocations (also called Copy Number Variations, or CNVs). Single-end reads are less potent in this respect, but their generation is less expensive and requires shorter analysis time. Single-end sequencing is a suitable approach when transcript quantification is the sole aim of the experiment.

Paired-end sequencing of fragments using the Illumina sequencer and alignment of the sequence reads to a reference genome matching the reads at both ends and taking the length of fragments into account. Reference genome sequence part with two exons and one intron is shown
3.2.2 Sequencing Technology
A typical protocol for short read sequencers is shown in Fig. 3.3 and is composed of the following steps:

Main step of the library preparation (a) and of the DNA/mRNA fragment sequencing (b). Further details are described in details in the main text (Figure adapted with permission from (Metzker 2010)) Copyright (2010) Nature Publishing Group
-
1.
DNA/RNA extraction and isolation from sample to retrieve the DNA/polyadenylated mRNA fraction. Extraction can be performed with different protocols using chemical approaches such as phenol-based extractions, direct lysis of DNA and RNA strands or using a mechanical approach such as centrifugation trough molecular filters of defined size and recovery of the nucleic acids with magnetic beads coated with DNA/RNA binding molecules.
-
2.
This is followed by fragmentation of the extracted DNA and mRNA to obtain shorter pieces that can be efficiently sequenced. Fragmentation can be achieved with different methods. The most common is using enzymes that cut the nucleic acids randomly (though the sites where each enzyme cuts is known, it is therefore possible to infer the fragment distribution), by sonication (use of high amplitude sound waves to break DNA and RNA strands) or by intense heating. These steps are followed by selecting fragments of desired length, which is usually performed using a size exclusion gel electrophoresis. There are alternative approaches for size selection such as using magnetic beads by adjusting the concentration of the nucleic acid-binding agents present on the surface of the beads and thus selecting shorter or longer fragments. Extraction, fragmentation and fragment selection with a desired size are often performed by standardized protocols using commercial kits such as the widely used TruSeq Sample Preparation Kit from Illumina.
In the case of mRNA analysis, transcript fragments are reverse-transcribed into cDNA, which turns the mRNA sequence into a DNA sequence (Fig. 3.4). Adapters of 6–8 nucleotides in length are ligated to each end of the fragments, which permits them to be immobilized on the surface of a flow-cell, which is a container where the sequence amplification and sequencing reaction take place. The adapters are complementary to primers already present and fixed on the surface of the flow-cell where they act as anchors when a transcript is fixed on the surface. Adapters may contain a short signature which is unique for each sample, and is called “barcode” (4–12 nucleotides long, with unique sequence for each sample). This allows multiple samples to be sequenced at the same time.

Scheme of DNA and mRNA sample preparation for sequencing (left column) and pre-processing of raw data (right column) (Figure is reproduced with permission from (Martin and Wang 2011). Copyright (2011) Nature Publishing Group)
-
3.
Polymerase-based amplification takes place and creates clusters of clones of the same fragment in a limited area called “spot”. Fragments are flexible and can bend in a way that the “free-end” of the adapter binds to another immobilized primer on the cell-flow surface. The polymerase can still bind to the immobilized primer and produce the second strand for each fragment. Due to this behavior this step is also called “bridge amplification”. This is necessary to create a cluster that can provide a signal strong enough to be measured by the light-sensitive sensor of the sequencer.
-
4.
At this point, it is possible to start sequencing. To do so specially modified nucleotides called “labeled reversible terminators” are used. Four terminators are needed for each base (Adenine, Cytosine, Thymine and Guanine). Each is labeled with a different fluorescent fluorophore group, a light-sensitive molecule that will emit light at specific wavelength (red, green, blue and yellow) when excited by lasers of different wavelengths. The flow-cell is made of glass allowing the emitted fluorescence to be detected by a photo-sensitive detector. Terminators stop the polymerase chain reaction as well without requiring an extra chemical reaction with this purpose that led to the name of “terminators”. Reading the intensity of the emitted fluorescence at the three wavelengths emitted by the 4 terminators allow to “read” which base was added by the polymerase. Following fluorescence measurement a chemical reaction is performed to cleave the dye and the terminator group from the incorporated nucleotide allowing to continue the polymerase chain reaction. This cycle is then repeated for a fixed number of times (determined by the read length), which is typically 100 or 125 in an Illumina short read sequencer.
The sequencer uses internal software to transform the measured raw fluorescence information to base pairs and includes parameters that reflect the quality of the reads. The measured base sequence is collected and saved in FastQ format (Fig. 3.5). FastQ is a simple, text based format, composed of 4 parts per entry (read): the first line starts with an “@” symbol and is an identifier of the read, which may include various kinds of information such as the length of the read, a batch ID and a read individual ID; the second part is the read itself, which may occupy more than one line depending on its length; the third part is a single line of comment starting with a ‘ + ’ symbol and which may repeat the first line, report additional information or left blank; the last part is a string of symbols, one for each letter in the read sequence. These symbols encode for numbers that represent an estimate of the quality of the resulting sequence. The numbers are calculated through a –log10 (estimated error rate) where the estimated error rate is the probability that the letter in a single position is correct.

Example of a sequence read of transcript fragment in FastQ format (Adapted with permission from Cock et al. (2010). Copyright (2010) Oxford University Press)
The described Illumina short read sequencing technology has the advantage to deliver large amounts of sequencing data but the relatively short sequencing length of 100/125 base pairs, which information is insufficient to reconstruct large repeated regions in the genome and to reconstruct the exact transcript profile for genes with a high number of splicing events. The obtained data require significant efforts to reconstruct the transcriptome through a complex bioinformatics pipeline. Third generation sequencers are emerging, such as the PacBio sequencing technology, which provide much longer sequence reads, up to tens of thousands of consecutive bases, an approach that should resolve these issues.
3.2.3 Bioinformatics Processing of Raw Data for Proteogenomics Application
The obtained raw short sequence read data in FastQ format is processed with complex bioinformatics workflows. A workflow typically consists of the following steps:
-
1.
Quality assessment of the reads and trimming (removing) of low quality reads
-
2.
Assembly of short reads and alignment to the reference genome
-
3.
Variant calling and transcript quantification
-
4.
Prediction of translated protein sequence by finding open reading frames (ORFs) and stop codons and saving the results in protein sequence Fasta file format
The obtained protein sequence information is then used for protein and peptide identification using DBS of LC-MS/MS proteomics data, while the quantitative transcript profile is used to determine the differentially expressed transcripts in a group of samples, such as controls and samples from different stages of disease. Different tools are available for each processing step. The alignment of the raw data to the reference genome can be replaced with de novo hypothesis-free transcript assembly. Bioinformatics processing and the subsequent statistical analysis is an error-prone process and the quality of the obtained results should be thoroughly assessed. Each tool in the bioinformatics workflow makes different assumptions which are based on different mathematical models and algorithmic approaches, which in turn tend to capture only a part of the biological significance contained in the data. With respect to proteogenomics, the best performance assessment is to check the number of identified peptides and proteins. This assessment can be performed for different workflows built from different tools and parameters. The sample preparation protocol and the bioinformatics workflow to process RNA-Seq data is presented in Fig. 3.4:
-
1.
The quality assessment and trimming is performed with the FastQC (Patel and Jain 2012), FastX Toolkit (Pearson et al. 1997) and Trimmomatic tools (Bolger et al. 2014) which provide a quality control report in html format for each raw FastQ files. Obviously trimming is performed only if the quality-control reports indicate that this is necessary due to low quality of the sequence. A very common case is a drop in quality in the final part of the read due to the degradation of the efficiency of the chemical reaction of the sequencing process. These lower quality bases are generally removed at this step. The FastQC, FastX Toolkit and Trimmomatic tools are easy to use and require low computation power.
-
2.
The trimmed FastQ files are either aligned to the reference genome using aligner tools such as STAR (Dobin et al. 2013) or Tophat2 (Kim et al. 2013). The output is then the alignment of the reads to the reference genome and the result is stored in a Binary Alignment/Map (BAM) file format. A BAM file is a compressed or binary version of a SAM or Sequence Alignment/Map format file. The SAM format follows precise specifications (see details in Lee et al. (2009)), which give the format a fixed scheme and defines where a read maps on a reference genome/transcriptome. It is composed of several lines of TAB separated fields in a fixed order, preceded by a header that gives general information on the alignment. The other option is to perform de novo assembly of the short reads without the use of a reference genome. This task is typically performed with tools such as ABySS (Simpson et al. 2009) and Trinity (Grabherr et al. 2011). De novo assembly is a computationally intensive task, as the tool needs to calculate several possible combinations of reads (grouped together in “contigs”). However reference genomes or transcriptomes are not perfect, they do contain errors, and the use of a reference genome also restricts the possibility to discover novel transcripts. Using a reference genome is a conservative choice, and can be sufficient when the analysis does not have the goal to attempt to capture all the possible transcripts in a sample or aims for maximum reliability of the assembled sequence of the transcripts.
-
3.
The BAM file is processed by an assembler tool , which has the aim to identify the full transcript constitution in the measured sample and estimate the amount of each transcript. Commonly used transcriptome assemblers include genome reference-guided tools, such as Cufflinks (Trapnell et al. 2010), and reference-free or de novo transcriptome assemblers, for example Trinity (Grabherr et al. 2011). In addition a BAM file can be used as input for a genomic viewer tool, such as IGV (Integrative Genome Viewer) (Robinson et al. 2011) or Savant (Fiume et al. 2010). These genome browsers can show exactly how the reads are aligned and distributed through an easy-to-use graphical user interface, which may also include peptide abundance which data is available in a proteogenomics study.
-
4.
At this point it is also possible to discover sequence variations in the analyzed samples. This operation is performed through the use of dedicated tools, the “variant callers”, such as the HaplotypeCaller algorithm of GATK (Genome Analysis Toolkit) (McKenna et al. 2010) or the SNP Caller which is part of SAMTools (Li et al. 2009). These algorithms are able to efficiently evaluate if a SNP or insertion and deletion (indel) is present at a certain position and calculate the probability of the correctness of the findings.
-
5.
The final steps consist of prediction of transcripts that are most likely translated into proteins and obtain the corresponding protein sequence. For this operation specialized tools, such as Transdecoder are used. This tool was conceived as an additional step to the Trinity pipeline but it can also be used as a standalone program. Transdecoder accepts files in General Transfer Format (GTF), which is a text-based TAB separated scheme used to describe genomic entities, such as transcripts or genes. GTF files are normally used for annotation of the transcripts. An alternative tool is the recently developed GeneMarkS-T (Tang et al. 2015), which is an adaptation of GeneMarkS (Besemer et al. 2001), where prokaryotic-only ORF predictor implemented in GeneMarkS was modified to translate eukaryotic transcriptomes. Transdecoder output results in a Fasta formatted protein sequence list derived directly from the transcript list used as input. Fasta is a very simple text format for biological sequences, similar to FastQ but with only two parts, an identifier line preceded by a ‘ > ’ symbol and the sequence itself in amino acid or nucleotide sequence of the transcripts/ proteins. Example of fasta file format showing the nucleotide base sequence of Apex nuclease 1 gene and corresponding amino acid sequence of the translated protein highlighting single amino acid variant (SAAV) is shown in Fig. 3.6. The sample-specific predicted amino acid sequence of translated proteins is subsequently used in DBS to identify peptides and proteins in raw LC-MS/MS data and to determine the proteome constitution of the samples. After pre-processing the transcript sequence, transcript identity and quantity is obtained. The bioinformatics workflow used to process transcripts including concrete tools with the input data is shown in Fig. 3.7.
Fig. 3.6 
Example of fasta format showing nucleotide base sequence of APEX nuclease 1 gene (upper part) and the corresponding protein sequence (lower part) of transcript ENST00000398030_D148E. The header line contains the gene, transcript or protein ID and description of the transcript is followed by a line containing the base sequence of the transcript. This gene contains a SNP of G → T at position 712 leading SAAV by replacing aspartic acid to glutamic acid at position 148 in the translated protein sequence. MS/MS spectra of peptide holding the SAAV and highlighted in bold in both sequences is shown in Fig. 3.11b. Non protein coding part is highlighted in green, the replaced D → E amino acid and GAG → GAT codon is highlighted in red, while the stop codon is shown in blue (highlights are only used to visualize different aspect of the sequence and is no part of the fasta format definition). In transcript T (thymine) is replaced by U (uracil)
Fig. 3.7 
Flow chart of bioinformatics workflow to pre-process sequencing data to make them ready for statistical analysis and provide the amino acid sequence of predicted translated proteins


NCBI Gene Expression Omnibus (GEO) (Barrett et al. 2013) provides repositories for raw sequencing data that can be mined and reanalyzed, for example to obtain additional information on genome or transcript expression profiles of the same or similar cell and tissue that is the aim of the study.
3.3 Proteomics Analysis
As mentioned in the introduction, the most popular shotgun bottom-up LC-MS/MS based proteomics technology is not a sequencing technology, but is based on the fragmentation of protein-derived peptides. Intact large proteins cannot be fragmented efficiently and large proteins show problems for separation by liquid chromatography (LC), a step that is required to reduce sample complexity prior to analysis with mass spectrometry . Fragmenting and separating by LC is much more efficient for smaller peptides even though the enzymatic cleavage of proteins leads to much higher sample complexity.
The first problem related to shotgun LC-MS/MS proteomics is that the original sample protein composition with respect of protein species and quantities should be reconstructed from the primary amino acid sequences and quantities of the identified peptides. This operation is called protein inference (Farrah et al. 2011; Nesvizhskii 2007; Nesvizhskii and Aebersold 2005) and cannot be performed accurately because information on the intact protein species composition of the sample is lost during the enzymatic cleavage step. During protein identification, proteins that cannot be distinguished from each other based on the set of identified peptides are grouped in protein groups. Therefore, the quantity of a given protein in one group is always the same. Protein inference raises the question of how to determine the amount of single proteins included in the same group. Some methods only use the quantity of peptides that uniquely map to a protein group. Others split the quantities of shared peptides between protein groups according to the ratio of unique peptides, this fractional quantity of shared peptides is then used together with the complete quantities of unique peptides to calculate protein quantity. MaxQuant (Tyanova et al. 2015; Cox and Mann 2008) assigns the shared peptides (so called razor peptides) to a protein group with the largest number of identified peptides and uses the quantity of razor peptides in the assigned protein group to calculate the quantity of proteins present in that particular protein group. The many existing protein isoforms detected by RNA-Seq , which are included in the protein sequence Fasta file used for DBS, result in many identified proteins in protein groups as the outcome of a proteogenomics experiments. This outcome is better summarized as aggregate quantitative information of all protein products per gene, especially when only spectral counts are available, which only give semi-quantitative information. Further quantitative details should be explored at the peptide level, preferably using single-stage quantification, especially when single amino acid variants (SAAV) or short indels affect only one or two peptides of a target protein.
Peptide and protein quantification in comprehensive bottom-up LC-MS/MS experiments can be performed using stable isotope labelling and label-free approaches. Stable isotope labeling uses either metabolically incorporated stable isotopes such as the stable isotope labeling by amino acids in cell culture (SILAC) approach to incorporate 13C and 15N -labelled amino acids that cannot be synthesized de novo by cells in culture such as lysine and arginine, or the incorporation of 15N into newly synthetized amino acids and thus into the complete newly synthetized proteome. Chemical labels may introduce moieties with different stable isotope constitutions that result in different MS signals either in single stage (e.g., ICAT) or after fragmentation (e.g., iTRAQ and TMT) for peptides originating from different samples. Stable isotope labeling techniques have the advantage of multiplexing, i.e., reducing the number of analyses and instrument time by analyzing mixed samples, where sample specific information is obtained from ions with the same chemical but different isotopic constitution. This goes at the expense of the dynamic measurable concentration range according to the multiplexing factor. In label-free quantification the user has the choice between spectral count-based analyses based on counting the number of peptide-spectrum matches (PSMs) for each protein, which provide semi-quantitative peptide and protein quantification. The other option is to use the more accurate single-stage-MS-based quantification approach, which calculates the peak height, peak area or peak volume of isotopologue peaks in the single-stage MS map. For more information, the reader is advised to read specialized reviews on label-free (Christin et al. 2011; Horvatovich and Bischoff 2010; Horvatovich et al. 2006) and stable isotope-based quantification approaches (Bantscheff et al. 2007, 2012).
3.3.1 Raw Data
Mass spectrometry raw data is collected in scans, which is in nature one dimensional data with two parameters: m/z and ion intensity. However, the information content of scans depends on the applied mass spectrometry method. Nowadays, the untargeted comprehensive bottom-up data dependent acquisition (DDA) LC-MS/MS approach is the most commonly used approach. In DDA, a non-fragmented scan is first acquired that holds quantitative information on all compounds detected by the instrument at the time of the mass spectra acquisition. A single-stage scan is followed by 3 to 20 fragment ions scans obtained with a small precursor ion isolation window which is typically 1–2 Da wide and is centered to the most intense single-stage ions. The cycle containing single-stage scan and the 3–20 fragment ion scans with different precursor isolation windows is then repeated for the whole experiment, adopting dynamically to the actual peptide composition eluting from the LC column during the analysis, and results in fragmentation of the most abundant ions entering the mass spectrometer. The selected ions are then excluded with twice the peak width at half maximum to enable other lower abundant not yet fragmented peaks to be selected. Despite the m/z exclusion, DDA is biased towards high abundant peptides. The obtained fragment spectra (or MS/MS spectra) are then used for peptide identification , which means that MS/MS spectra are assigned to peptide primary amino acid sequences. Recently, data independent acquisition (DIA) (Sajic et al. 2015) is gaining popularity in which the precursor isolation window is larger, typically of 20–25 Da. The non-fragmented scan is followed by successive fragmented scans that have a precursor isolation window targeting different consecutive precursor m/z ranges. One full instrument duty cycle covers a large range of m/z ratios (typically between 300 and 2000 Da in proteomics applications) leading to 2–3 s of duty cycle. In theory, DIA data contains all the information that is possible to collect with an instrument that includes one stage fragmentation. DIA data is more complex and is more challenging to analyze and interpret than DDA fragment spectra obtained with small isolation windows that have low probability to have interferences i.e. fragment ions from multiple co-fragmented peptides. The bioinformatics community is currently developing new solutions to analyze such data, such as OpenSWATH (Rost et al. 2014) or DIANA (Teleman et al. 2015). This chapter does not discuss the differences and properties of the different types of mass spectrometers and the reader is invited to visit reviews on this topics (Bensimon et al. 2012; Gstaiger and Aebersold 2009; Domon and Aebersold 2006).
The raw mass spectrometry data is generally saved by vendor data acquisition software in vendor specific binary formats, which are different from each other. To harmonize data storage, the HUPO protein standardization initiative (PSI) has established an xml based format for raw mass spectrometry data , such as mzXML (Pedrioli et al. 2004), mzData (Orchard et al. 2004) and mzML (Turewicz and Deutsch 2011), but older ASCII format such as Mascot Generic Format or mgf (Kirchner et al. 2010) are still used e.g. as input format for various data processing tools. Standardization of processed data for different purposes, such as to store peptide identification and protein inference results in mzIdentML format, to store quantification data in mzTab (Griss et al. 2014) and mzQuant (Walzer et al. 2013) formats and to exchange quality control metrics in qcML (Walzer et al. 2014) format have been developed by the proteomics community. The proteoWizard (Chambers et al. 2012; Kessner et al. 2008) toolset contains libraries and tools to convert raw vendor specific mass spectrometry data to HUPO PSI standard formats and enable the user to perform basic mass spectrometry signal processing operations. Raw mass spectrometry data can be visualized by multiple tools such as BatMass (Nesvizhskii and Avtonomov), TOPPView (Sturm and Kohlbacher 2009) from OpenMS (Bertsch et al. 2011) or PView (Khan et al. 2009).
3.3.2 Peptide Identification and Protein Inference
Primary peptide sequences are determined from fragment (MS/MS) spectra. The most widely used fragmentation approach is collision induced dissociation (CID), when ions of intact peptides are accelerated in a vacuum and collided with neutral gase. The collision is transferring energy to the peptides leading to cleavage of bonds in the peptide backbones Another type of fragmentation is electron transfer dissociation (ETD), which uses a negatively charged poly-aromatic compound such as fluoranthene, anthracene or azobenzene to transfer an electron to the positively charged peptide. The transferred electron conveys energy to the peptide backbone, which leads to fragmentation. There are three bonds that can lead to fragmentation on the peptide backbone leading to six types of fragments: a, b, c containing N-terminal and x, y and z containing the C-terminal of the peptide (Fig. 3.8a). However, not all fragments have the same probability to be observed in an MS/MS spectrum, for example, CID mainly leads to the formation of y ions, also resulting in lower abundance b ions and a ions can sometimes be observed. ETD fragmentation mainly leads to the formation of c and z ions. The ionization and fragmentation efficiency of intact peptides can be influenced by chemical modifications, for example by using chemical labels that contain basic residues or residues that can provide a mobile proton (Bischoff et al. 2015). In the fragmentation process, the lower energy bonds will be cleaved, which often results in an incomplete fragment ion series (Fig. 3.8b). This prevents the de novo interpretation of the mass spectra, which prevent identification of the MS/MS spectra if the user does not have any presumptions on the peptides sequence. Additional fragment mass spectra may contain considerable noise, which further complicates the identification process. For this reason, the best approach to interpret such data is to use a list of protein sequences that are supposed to be present in the analyzed samples. Such protein sequence can be predicted from the genome of the host organism, which contains the most prevalent protein sequences or the so-called canonical sequences. UniProt (Consortium 2015) and Ensembl (Herrero et al. 2016) provide high quality canonical sequences that are used for peptide and protein identification during normal proteomics data analysis. One must note that the canonical sequence contains the sequence of most prevalent protein form, which is the most similar orthologue sequence to other species and the length of the protein form that allow the clearest description of the protein sequence variability (see Introduction Sect. 3.1). This protein sequence does not allow identification of all protein sequence variants, especially those that are specific to individuals and may bear importance in disease mechanisms.

(a) schematic representation of fragment ion series (a, b, c for N-terminal and x, y, z for C terminal) generated during peptide backbone fragmentation. y, b and a ions are generated mainly during CID (purple) fragmentation, while c, z and with lower abundance y ions are generated during ETD (blue) fragmentation. (b) CID MS/MS spectra of KIQVLQQQADDAEER peptide showing complete y and almost complete b ion series, which spectra is suitable for de novo interpretation. (c) MS/MS of EANFDINQLYDCNWVVVNCSTPGNFFHVLR peptides, which shows incomplete y and b ion series and gaps in y ion series are highlighted with red arrow indicating the missing sequence part. These gaps prevent de novo interpretation since the exact amino acid sequence information is missing. In MS/MS spectra non-identified signals are highlighted in grey. These signals may correspond to noise, non-interpreted fragment ions or fragment ions from co-eluting peptides that fell into the precursor ion selection window. Visualisation made with PeptideShaker (Vaudel et al. 2015) and the figure is adapted with permission from Bischoff et al. (2015) (Copyright (2015) Elsevier)
In an LC-MS/MS dataset not all MS/MS spectra are identified during DBS, due to the following reasons:
-
1.
The fragment spectra is too noisy
-
2.
The fragmentation efficiency is too low to perform accurate identification
-
3.
The absence of the peptide sequence in the protein sequence database
-
4.
The presence of PTMs not searched during DBS
Sharing raw LC-MS/MS data and reusing it by several bioinformatics portals e.g., to catalogue identified peptides and provide high quality spectral libraries such as PeptideAtlas (Deutsch et al. 2008; Farrah et al. 2011) is promoted by ProteomeXchange (Ternent et al. 2014; Cote et al. 2012), which is an initiative of the European Bioinformatics Institute to store raw proteomics mass spectrometry data.
Due to gaps in fragment ion series and noise in fragment spectra, the most successful strategy is DBS. In this process the sequence of proteins supposed to be present in the sample are digested in silico with the protease used for the protein cleavage in the experiment and peptides that have the same theoretical mass (with certain mass tolerance) than the precursor ion are selected. The mass of high abundant ion series of the selected peptides are in silico calculated and the obtained mass list is compared with the mass list of the MS/MS spectra using score specific to the DBS algorithm. The peptide with the highest score if it pass the threshold with given false discovery rate (FDR) is then considered to be the identity of the MS/MS spectra (Fig. 3.9). Scores are generally dependent from multiple parameters, such as the size of the search space, i.e., how well does the protein sequence database match the measured proteome (Shanmugam and Nesvizhskii 2015), the considered PTMs of peptides, the mass resolution of the precursor and fragment ions, and the fragmentation efficiency and quality (noise content) of the MS/MS spectra. Additionally, not all MS/MS spectra will have a corresponding match in the search space; such spectra will be matched and scored erroneously. For this reason, the goal is to find scores that can separate correct identifications from the incorrect ones with well described statistics such as false discovery rate (FDR).

Schematic representation of bioinformatics algorithms performing peptide spectrum matches (PSM). Acquired raw MS/MS spectra are either submitted to (1). DBS that match list of fragments ions predicted from sequence that supposed to be present in the sample, (2). submitted to spectral library search, which match the raw MS/MS to a library of annotated MS/MS spectra, (3). submitted to sequence tag search (or mass-tag) algorithm or (4). de novo sequencing. The output of the search is a ranked list of peptides, where the peptide sequence with best score is considered as best match and peptide sequence of the analyzed MS/MS spectra. The scores of the best matches are submitted to FDR calculation either using empirical expectation-maximization algorithm or target-decoy approach (Figure adapted with permission from (Nesvizhskii 2010). Copyright (2010) Elsevier)
The score distribution of correct and incorrect identifications should be determined to calculate FDR, and there are two main widely used approaches:
-
1.
Expectation-Maximization (EM) (Keller et al. 2002) approach based on empirical Bayesian statistics
-
2.
Target-decoy approach (TD) (Elias and Gygi 2010)
EM tries to identify the score distribution of the correct and incorrect hits by calculating two distinct distributions based on the mixture model. While the TD approach tries to determine the distribution of the incorrect identifications based on decoy peptide sequences generally obtained by in silico digestion of reversed protein sequences used for the DBS. These approaches allow the user to obtain a list of identified PSMs, which can be used to derive a list of identified unique peptides, which can then be used to perform protein inference.
Since peptides are measured and identified in shotgun LC-MS/MS experiments, the original protein constitution of the samples should be reconstructed based on the identified set of peptides (Fig. 3.10). This is not a trivial task, since identified peptides sequence may map uniquely to a protein sequence or be shared between multiple ones. The other difference between sequencing and proteomics data is the scale of the number of entries. The number of identified peptide sequences is much lower (typically 10,000–30,000 unique sequences) than the number of uniquely mapping reads (typically 20 millions reads). The overlap between the peptide sequences is low, which generally occurs between peptides having missed cleavages (locations in the protein where the enzyme should cut in theory, but did not cut to produce a peptide).

Schematic representation of shotgun LC-MS/MS analysis of a proteomics samples and followed bioinformatics data interpretation. Original protein constitution of a samples is disrupted by enzymatic cleavage resulting in a highly complex peptide mixture analyzed by LC-MS/MS. The obtained MS/MS of peptides are then identified with DBS or by other tools and the set if highly confidently identified peptides are used to construct back the original protein constitution of the sample by performing protein inference. Black squares in peptide identifications represent wrong PSM, which lead to include incorrectly identified peptides and proteins (Figure adapted with permission from Nesvizhskii et al. (2003) Copyright (2003) American Chemistry Society)
Identified proteins are grouped together when they cannot be distinguished from each other based on the set of observed peptide sequences in the dataset. The sequence coverage of the identified protein is an important parameter. The sequence coverage depends on the abundance of the protein and the peptide composition. The most abundant proteins have higher sequence coverage than lower abundant proteins. The average protein sequence coverage is low, with a medium of 10–20 % in a typical proteomics dataset. This means that peptides that could distinguish various protein isoforms, due to for instance splice junction differences, SAAV or small indels, are incomplete even when deep sequencing is performed (Ruggles et al. 2015; Tay et al. 2015; Sheynkman et al. 2013). The low sequence coverage is caused by multiple factors:
-
1.
Proteins and peptide signals cannot be amplified (as is the case with DNA and RNA signals)
-
2.
Not all MS/MS fragment spectra are identified
-
3.
The applied protease (e.g., trypsin) does not provide unique or enough protein sequence specific peptides for the complete sequence of the analyzed proteins
Sequence coverage can be improved by deep sequencing that use multilevel fractionation, e.g., by applying multidimensional chromatographic separation (Horvatovich et al. 2010), by using different peptide fragmentation approaches in the mass spectrometer and chemical labels that enhance fragmentation efficiency (Bischoff et al. 2015) and by using multiple proteases for enzymatic cleavage (Low et al. 2013; Trevisiol et al. 2015).
The false identification of peptides may lead to the incorrect identification of a protein and the fact that multiple correctly identified peptides map to a single protein, while incorrectly identified peptides map randomly to single proteins in the database leads to an enrichment of false protein identifications compared to PSM or peptide identification errors. For this reason, the FDR rate should be calculated not only for the PSM and peptide but also at the protein level (Vaudel et al. 2015).
Beside DBS, other approaches can be used to perform PSM. The short sequence tag approach tries to identify consecutive amino acid sequence in the MS/MS spectra and uses the precursor ion mass and the masses of the fragments from the N, and the C terminus of peptides for the identification. MS/MS spectra that include low noise content and shows complete fragment ion series could be used for hypothesis-free de novo sequencing, without the use of any assumptions on protein sequence that should be present in the analyzed sample. A more and more popular approach is the use of spectral similarities between the MS/MS spectra of interest and high quality identified MS/MS spectra (so called consensus spectra) often averaged from multiple MS/MS spectra of different experiments. This approach is called spectral library search (Lam 2011) and has the advantage that it does not only use the mass list of the fragment ions, but that it also includes their intensity, which is a parameter that is difficult to predict in silico and which is not or only partially included in DBS. More and more high quality peptide spectral libraries are available that can be used to perform spectral library searches. High quality annotated spectral libraries are available such as NIST, the PeptideAtlas (Deutsch et al. 2008) and the Global Proteome Machine Database (GPMDB) (Craig et al. 2006). Figure 3.9 provides a summary of the most important PSM identification strategies.
In proteogenomics, the FDR rate of MS/MS identifications of novel peptide differs from peptides derived from canonical sequences of public databases, such as UniProt (Consortium 2015). For this reason the best PSM scoring strategy is cascade identification, which includes consecutive steps of identification as follow:
-
1.
Filter out all low quality MS/MS spectra
-
2.
DBS identification using UniProt database (SwissProt and TrEMBL) or Ensembl
-
3.
Identification of the remaining non-identified MS/MS spectra with novel peptide or protein sequences (Nesvizhskii 2014)
Similar cascade identification strategies have been implemented for different types of rare peptides, such as non- and semi-tryptic peptides, terminal peptides and PTM searches as described in Kertesz-Farkas et al. (Kertesz-Farkas et al. 2015).
Many software tools exist to perform PSM, protein inference using a given set of FDR at PSM, peptide and protein levels, these include the Trans Proteomic Pipeline (Deutsch et al. 2010; Deutsch et al. 2015) (TPP, open source), the TOPPAS workflow, which is based on OpenMS for label-free quantification and identification (Weisser et al. 2013), MaxQuant (Cox and Mann 2008) (open source), SearchGUI (Vaudel et al. 2011) / PeptideShaker (Vaudel et al. 2015) (open source) and PEAKS (commercial) (Zhang et al. 2012). Many individual tools exist for DBS (Eng et al. 2013; Kim and Pevzner 2014; Bjornson et al. 2008; Geer et al. 2004), de novo sequencing (Muth et al. 2014; Jeong et al. 2013; Frank and Pevzner 2005) and FDR calculations at PSM, at peptide and protein levels (Kall et al. 2007). For further details on these tools, the reader is invited to read specialized reviews on the topic (Hoopmann and Moritz 2013; Eng et al. 2011; Hughes et al. 2010; Kapp and Schutz 2007).
3.4 Applications, Conclusion and Future Perspectives
Acquiring genomics (mainly polyadenylated mRNA) and shotgun proteomics data from the same sample and evaluate it in a proteogenomics data integration pipeline allows to gain information at both molecular levels but also to identify novel protein forms that would not be identified using public databases with DBS. As an example, we present the data of the proteogenomics analysis of the human lung fibroblast cell line MRC5. Using the standard identification of UniProt we identified 11,936 peptides and when we used the RNA sequence information of the same cells we could identify an additional 282 peptides, which represent the sample specific peptide sequence. Figure 3.11a shows a number of peptide sequences that has been identified with canonical sequences of UniProt, peptides that match to SAAVs due to non-synonymous SNPs, peptides matching to new isoforms and peptides that match to non-annotated new gene models. Figure 3.11b shows an example of a high quality MS/MS spectrum presented with complete y and b ion annotation of peptides (VSYGIG(D → E)EEHDQEGR) holding SAAV that replaces an aspartic acid (D) to glutamic acid (E) at positon 148. This peptide is mapping uniquely to APEX nuclease, which is a multifunctional DNA repair enzyme. This peptide cannot be identified in the human UniProt protein sequence, but can be found in the APEX nuclease sequence of many other species, which indicates that this mutation may alter the activity of this protein in the MRC5 human cell line.

Identification of peptides that match uniquely to sample specific protein forms in human MRC5 fibroblast cell line. (a) Venn diagram representing the number of peptides that has been identified using protein sequence database containing sequence from UniProt, non-synonymous isoforms, new isoforms and new gene models. (b) example of CID MS/MS spectra of VSYGIG(D → E)EEHDQEGR with annotation of y and b ion series of peptides that hold SAAV replacing aspartic acid (D) to Glutamic acid (E) at position 148. Base sequence of the corresponding gene and the amino acid sequence of the corresponding protein is shown in Fig. 3.6, highlighting the gene structure, the presented peptide sequence and the position of SAAV
The moderate spearman correlation of 0.4 between the amount of transcript coding proteins and proteins shows that there is an additional level of regulation which includes post-transcriptional and post-translational effects (Schwanhausser et al. 2013, 2011). Therefore, the information at the two molecular levels differs and should be considered to be complementary. Both levels may deliver large amount of information, which is difficult to interpret, such as number of differentially expressed proteins. In this case, focusing on the intersection of genes and transcripts / proteins that show the same trend at both molecular levels may provide a useful focus to interpret the outcome of a proteogenomics study.
An example of considering joint changes at the transcriptomics and proteomics levels is shown in Fig. 3.12. This figure shows a pseudo Volcano plot indicating a fold change and t-test significance at transcript and protein levels. The result was obtained in a proteogenomics study performed to identify molecular changes in liver of hypertensive SHR rats when compared to a control BN-Lx rat strain. The study by Low et al. (2013) shows genome and polyadenylated transcriptome sequencing for eight rats (about 100 million of reads/sample) and deep proteomics analysis for two rats using a two dimensional LC-MS/MS experiment. To obtain the highest possible sequence coverage and the largest measured dynamic concentration range, five proteases (trypsin, chymotrypsin, LysC, GluC and AspN) and strong cation exchange (SCX) as first liquid chromatography and reversed phase C18 (RPC18) with low pH as second dimension were used. This setup led to 36 fractions / samples and 180 RPC18 analysis using high resolution Orbitrap instrument and nearly 2 weeks of analysis time. From the acquired 12 million MS/MS spectra, two million were identified using Mascot / PEAKS DBS searches and resulted in 175,000 non-redundant peptide sequences matching to 26,463 rat proteins. In this experiment, 1195 predicted new genes, 83 splicing events, 126 proteins with non-synonymous variants and 20 isoforms with non-synonymous RNA editing were identified.

Outcome of proteogenomics study in hypertensive SHR and control BN-Lx rats. (a) pseudo Volcano plot showing fold changes in transcript on the horizontal axis and fold changes of proteins in the vertical axis. Blue dots (n = 59) represent significant changes at transcriptome level only and red dots (n = 54) represent significant changes at transcript and protein levels. The most significantly down-regulated Cyp17a1 gene is highlighted with red circle. (b) transcript and protein expression level at location of gene Cyp17a1 showing the position of incorrectly annotated start site (TSS black arrow) and the real start site (TSS grey arrow). (c) expression quantitative trait loci (eQTL) showing the transcript expression regulation by the SNP at the real start site (Adapted with permission from Low et al. (2013) Copyright (2013) Elsevier)
Differential gene expression analysis at both molecular layers revealed that genes related to cytochrome P450 (CYP450) are mainly differentially expressed in the same direction. Particularly, the gene Cyp17a1 was the strongest down-regulated in hypertensive SHR rats. Having both genomics and transcriptomics data in hand, it was demonstrated that the transcription start site was incorrectly annotated in the reference rat genome and that the correct start site was 2 kb further upstream from the current annotation on the 5’ exon. The correct start site in SHR rats included a SNP, which prevented transcription and translation of the protein coded by the Cyp17a1 gene (Fig. 3.11b). In this case the proteogenomics analysis helped to identify a gene related to the hypertensive rat phenotype, but also to correct the genome annotation , revealing the cause of the down-regulation of the transcript and the protein product by a SNP at the starting site of the Cyp17a1 gene.
Proteogenomics still requires important efforts to collect data at genomic and/or transcriptomic and proteomics levels and the correct analysis of the obtained data, which requires expertise from both omics fields as well as from bioinformatics . Despite the significant improvement of high-throughput proteomics peptide identification technology in recent years, proteomics still does not provide clean data for de novo sequencing and is unable to deliver the same coverage of peptide sequence information when compared to genomics sequencing technology. Additional improvement will be possible by combining ribosomal sequencing data, the so-called translatome, with transcriptomics, since it helps to filter out transcripts that have a low potential for translation and may include potentially translated lncRNA – despite the fact that this technology delivers only 30 nucleotide base length sequences (Gawron et al. 2014; Chang et al. 2014). Proteogenomics analysis can be completed with the PUromycin-associated Nascent CHain Proteomics (PUNCH-P) technology that aims to identify newly synthetized proteins by capturing ribosome-nascent chain complexes from cells followed by incorporation of biotinylated puromycin (Aviner et al. 2013).
Further impetus for proteogenomics is evident in the Chromosome-Centric Human Proteome Project (C-HPP) (Horvatovich et al. 2015), which aims to catalogue all human protein products and make them searchable on the basis of genomics location. Proteogenomics data acquisition and data integration plays a central role in C-HPP, which promotes the development of new technologies and bioinformatics workflows with strong quality control, and aims to provide a powerful technology platform for clinical application and personalized medicin e.
References
Aviner, R., Geiger, T., & Elroy-Stein, O. (2013). PUNCH-P for global translatome profiling: Methodology, insights and comparison to other techniques. Translation (Austin), 1(2), e27516. doi:10.4161/trla.27516
Bantscheff, M., Schirle, M., Sweetman, G., Rick, J., & Kuster, B. (2007). Quantitative mass spectrometry in proteomics: A critical review. Analytical and Bioanalytical Chemistry, 389(4), 1017–1031. doi:10.1007/s00216-007-1486-6.
Bantscheff, M., Lemeer, S., Savitski, M. M., & Kuster, B. (2012). Quantitative mass spectrometry in proteomics: Critical review update from 2007 to the present. Analytical and Bioanalytical Chemistry, 404(4), 939–965. doi:10.1007/s00216-012-6203-4.
Barrett, T., Wilhite, S. E., Ledoux, P., Evangelista, C., Kim, I. F., Tomashevsky, M., Marshall, K. A., Phillippy, K. H., Sherman, P. M., Holko, M., Yefanov, A., Lee, H., Zhang, N., Robertson, C. L., Serova, N., Davis, S., & Soboleva, A. (2013). NCBI GEO: Archive for functional genomics data sets–update. Nucleic Acids Research, 41(Database issue), D991–D995. doi:10.1093/nar/gks1193.
Bensimon, A., Heck, A. J., & Aebersold, R. (2012). Mass spectrometry-based proteomics and network biology. Annual Review of Biochemistry, 81, 379–405. doi:10.1146/annurev-biochem-072909-100424.
Bertsch, A., Gropl, C., Reinert, K., & Kohlbacher, O. (2011). OpenMS and TOPP: Open source software for LC-MS data analysis. Methods in Molecular Biology, 696, 353–367. doi:10.1007/978-1-60761-987-1_23.
Besemer, J., Lomsadze, A., & Borodovsky, M. (2001). GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Research, 29(12), 2607–2618.
Bischoff, R., & Schlüter, H. (2012). Amino acids: Chemistry, functionality and selected non-enzymatic post-translational modifications. Journal of Proteomics, 75(8), 2275–2296. doi:10.1016/j.jprot.2012.01.041.
Bischoff, R., Permentier, H., Guryev, V., & Horvatovich, P. (2015). Genomic variability and protein species – Improving sequence coverage for proteogenomics. Journal of Proteomics. doi:10.1016/j.jprot.2015.09.021.
Bjornson, R. D., Carriero, N. J., Colangelo, C., Shifman, M., Cheung, K. H., Miller, P. L., & Williams, K. (2008). X!!Tandem, an improved method for running X! Tandem in parallel on collections of commodity computers. Journal of Proteome Research, 7(1), 293–299. doi:10.1021/pr0701198.
Bolger, A. M., Lohse, M., & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120. doi:10.1093/bioinformatics/btu170.
Chambers, M. C., Maclean, B., Burke, R., Amodei, D., Ruderman, D. L., Neumann, S., Gatto, L., Fischer, B., Pratt, B., Egertson, J., Hoff, K., Kessner, D., Tasman, N., Shulman, N., Frewen, B., Baker, T. A., Brusniak, M. Y., Paulse, C., Creasy, D., Flashner, L., Kani, K., Moulding, C., Seymour, S. L., Nuwaysir, L. M., Lefebvre, B., Kuhlmann, F., Roark, J., Rainer, P., Detlev, S., Hemenway, T., Huhmer, A., Langridge, J., Connolly, B., Chadick, T., Holly, K., Eckels, J., Deutsch, E. W., Moritz, R. L., Katz, J. E., Agus, D. B., MacCoss, M., Tabb, D. L., & Mallick, P. (2012). A cross-platform toolkit for mass spectrometry and proteomics. Nature Biotechnology, 30(10), 918–920. doi:10.1038/nbt.2377.
Chang, C., Li, L., Zhang, C., Wu, S., Guo, K., Zi, J., Chen, Z., Jiang, J., Ma, J., Yu, Q., Fan, F., Qin, P., Han, M., Su, N., Chen, T., Wang, K., Zhai, L., Zhang, T., Ying, W., Xu, Z., Zhang, Y., Liu, Y., Liu, X., Zhong, F., Shen, H., Wang, Q., Hou, G., Zhao, H., Li, G., Liu, S., Gu, W., Wang, G., Wang, T., Zhang, G., Qian, X., Li, N., He, Q. Y., Lin, L., Yang, P., Zhu, Y., He, F., & Xu, P. (2014). Systematic analyses of the transcriptome, translatome, and proteome provide a global view and potential strategy for the C-HPP. Journal of Proteome Research, 13(1), 38–49. doi:10.1021/pr4009018.
Christin, C., Bischoff, R., & Horvatovich, P. (2011). Data processing pipelines for comprehensive profiling of proteomics samples by label-free LC-MS for biomarker discovery. Talanta, 83(4), 1209–1224. doi:10.1016/j.talanta.2010.10.029.
Chuh, K. N., & Pratt, M. R. (2015). Chemical methods for the proteome-wide identification of posttranslationally modified proteins. Current Opinion in Chemical Biology, 24, 27–37. doi:10.1016/j.cbpa.2014.10.020.
Cock, P. J., Fields, C. J., Goto, N., Heuer, M. L., & Rice, P. M. (2010). The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Research, 38(6), 1767–1771. doi:10.1093/nar/gkp1137/ConsortiumN.
Consortium U. (2015). UniProt: A hub for protein information. Nucleic Acids Research, 43(Database issue), D204–D212. doi:10.1093/nar/gku989.
Cote, R. G., Griss, J., Dianes, J. A., Wang, R., Wright, J. C., van den Toorn, H. W., van Breukelen, B., Heck, A. J., Hulstaert, N., Martens, L., Reisinger, F., Csordas, A., Ovelleiro, D., Perez-Rivevol, Y., Barsnes, H., Hermjakob, H., & Vizcaino, J. A. (2012). The PRoteomics IDEntification (PRIDE) Converter 2 framework: An improved suite of tools to facilitate data submission to the PRIDE database and the ProteomeXchange consortium. Molecular & Cellular Proteomics, 11(12), 1682–1689. doi:10.1074/mcp.O112.021543.
Cox, J., & Mann, M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology, 26(12), 1367–1372. doi:10.1038/nbt.1511.
Craig, R., Cortens, J. C., Fenyo, D., & Beavis, R. C. (2006). Using annotated peptide mass spectrum libraries for protein identification. Journal of Proteome Research, 5(8), 1843–1849. doi:10.1021/pr0602085.
Deutsch, E. W., Lam, H., & Aebersold, R. (2008). PeptideAtlas: A resource for target selection for emerging targeted proteomics workflows. EMBO Reports, 9(5), 429–434. doi:10.1038/embor.2008.56.
Deutsch, E. W., Mendoza, L., Shteynberg, D., Farrah, T., Lam, H., Tasman, N., Sun, Z., Nilsson, E., Pratt, B., Prazen, B., Eng, J. K., Martin, D. B., Nesvizhskii, A. I., & Aebersold, R. (2010). A guided tour of the trans-proteomic pipeline. Proteomics, 10(6), 1150–1159. doi:10.1002/pmic.200900375.
Deutsch, E. W., Mendoza, L., Shteynberg, D., Slagel, J., Sun, Z., & Moritz, R. L. (2015). Trans-proteomic pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics Clinical Applications, 9(7–8), 745–754. doi:10.1002/prca.201400164.
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M., & Gingeras, T. R. (2013). STAR: Ultrafast universal RNA-seq aligner. Bioinformatics, 29(1), 15–21. doi:10.1093/bioinformatics/bts635.
Domon, B., & Aebersold, R. (2006). Mass spectrometry and protein analysis. Science, 312(5771), 212–217. doi:10.1126/science.1124619.
Elias, J. E., & Gygi, S. P. (2010). Target-decoy search strategy for mass spectrometry-based proteomics. Methods in Molecular Biology, 604, 55–71. doi:10.1007/978-1-60761-444-9_5.
Eng, J. K., Searle, B. C., Clauser, K. R., & Tabb, D. L. (2011). A face in the crowd: Recognizing peptides through database search. Molecular & Cellular Proteomics, 10(11), R111.009522. doi:10.1074/mcp.R111.009522.
Eng, J. K., Jahan, T. A., & Hoopmann, M. R. (2013). Comet: An open-source MS/MS sequence database search tool. Proteomics, 13(1), 22–24. doi:10.1002/pmic.201200439.
Farrah, T., Deutsch, E. W., Omenn, G. S., Campbell, D. S., Sun, Z., Bletz, J. A., Mallick, P., Katz, J. E., Malmstrom, J., Ossola, R., Watts, J. D., Lin, B., Zhang, H., Moritz, R. L., & Aebersold, R. (2011). A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Molecular & Cellular Proteomics, 10(9), M110 006353. doi:10.1074/mcp.M110.006353.
Fiume, M., Williams, V., Brook, A., & Brudno, M. (2010). Savant: Genome browser for high-throughput sequencing data. Bioinformatics, 26(16), 1938–1944. doi:10.1093/bioinformatics/btq332.
Frank, A., & Pevzner, P. (2005). PepNovo: De novo peptide sequencing via probabilistic network modeling. Analytical Chemistry, 77(4), 964–973.
Gawron, D., Gevaert, K., & Van Damme, P. (2014). The proteome under translational control. Proteomics, 14(23–24), 2647–2662. doi:10.1002/pmic.201400165.
Geer, L. Y., Markey, S. P., Kowalak, J. A., Wagner, L., Xu, M., Maynard, D. M., Yang, X., Shi, W., & Bryant, S. H. (2004). Open mass spectrometry search algorithm. Journal of Proteome Research, 3(5), 958–964. doi:10.1021/pr0499491.
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., Adiconis, X., Fan, L., Raychowdhury, R., Zeng, Q., Chen, Z., Mauceli, E., Hacohen, N., Gnirke, A., Rhind, N., di Palma, F., Birren, B. W., Nusbaum, C., Lindblad-Toh, K., Friedman, N., & Regev, A. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology, 29(7), 644–652. doi:10.1038/nbt.1883.
Griss, J., Jones, A. R., Sachsenberg, T., Walzer, M., Gatto, L., Hartler, J., Thallinger, G. G., Salek, R. M., Steinbeck, C., Neuhauser, N., Cox, J., Neumann, S., Fan, J., Reisinger, F., Xu, Q. W., Del Toro, N., Perez-Riverol, Y., Ghali, F., Bandeira, N., Xenarios, I., Kohlbacher, O., Vizcaino, J. A., & Hermjakob, H. (2014). The mzTab data exchange format: Communicating mass-spectrometry-based proteomics and metabolomics experimental results to a wider audience. Molecular & Cellular Proteomics, 13(10), 2765–2775. doi:10.1074/mcp.O113.036681.
Gstaiger, M., & Aebersold, R. (2009). Applying mass spectrometry-based proteomics to genetics, genomics and network biology. Nature Reviews Genetics, 10(9), 617–627. doi:10.1038/nrg2633.
Herrero, J., Muffato, M., Beal, K., Fitzgerald, S., Gordon, L., Pignatelli, M., Vilella, A. J., Searle, S. M., Amode, R., Brent, S., Spooner, W., Kulesha, E., Yates, A., & Flicek, P. (2016). Ensembl comparative genomics resources. Database: The Journal of Biological Databases and Curation. doi:10.1093/database/bav096.
Hoopmann, M. R., & Moritz, R. L. (2013). Current algorithmic solutions for peptide-based proteomics data generation and identification. Current Opinion in Biotechnology, 24(1), 31–38. doi:10.1016/j.copbio.2012.10.013.
Horvatovich, P. L., & Bischoff, R. (2010). Current technological challenges in biomarker discovery and validation. European Journal of Mass Spectrometry, 16(1), 101–121. doi:10.1255/ejms.1050.
Horvatovich, P., Govorukhina, N., & Bischoff, R. (2006). Biomarker discovery by proteomics: Challenges not only for the analytical chemist. The Analyst, 131(11), 1193–1196. doi:10.1039/b607833h.
Horvatovich, P., Hoekman, B., Govorukhina, N., & Bischoff, R. (2010). Multidimensional chromatography coupled to mass spectrometry in analysing complex proteomics samples. Journal of Separation Science, 33(10), 1421–1437. doi:10.1002/jssc.201000050.
Horvatovich, P., Lundberg, E. K., Chen, Y. J., Sung, T. Y., He, F., Nice, E. C., Goode, R. J., Yu, S., Ranganathan, S., Baker, M. S., Domont, G. B., Velasquez, E., Li, D., Liu, S., Wang, Q., He, Q. Y., Menon, R., Guan, Y., Corrales, F. J., Segura, V., Casal, J. I., Pascual-Montano, A., Albar, J. P., Fuentes, M., Gonzalez-Gonzalez, M., Diez, P., Ibarrola, N., Degano, R. M., Mohammed, Y., Borchers, C. H., Urbani, A., Soggiu, A., Yamamoto, T., Salekdeh, G. H., Archakov, A., Ponomarenko, E., Lisitsa, A., Lichti, C. F., Mostovenko, E., Kroes, R. A., Rezeli, M., Vegvari, A., Fehniger, T. E., Bischoff, R., Vizcaino, J. A., Deutsch, E. W., Lane, L., Nilsson, C. L., Marko-Varga, G., Omenn, G. S., Jeong, S. K., Lim, J. S., Paik, Y. K., & Hancock, W. S. (2015). Quest for missing proteins: Update 2015 on chromosome-centric human proteome project. Journal of Proteome Research, 14(9), 3415–3431. doi:10.1021/pr5013009.
Hughes, C., Ma, B., & Lajoie, G. A. (2010). De novo sequencing methods in proteomics. Methods in Molecular Biology, 604, 105–121. doi:10.1007/978-1-60761-444-9_8.
Jeong, K., Kim, S., & Pevzner, P. A. (2013). UniNovo: A universal tool for de novo peptide sequencing. Bioinformatics, 29(16), 1953–1962. doi:10.1093/bioinformatics/btt338.
Kall, L., Canterbury, J. D., Weston, J., Noble, W. S., & MacCoss, M. J. (2007). Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nature Methods, 4(11), 923–925. doi:10.1038/nmeth1113.
Kapp, E., & Schutz, F. (2007). Overview of tandem mass spectrometry (MS/MS) database search algorithms. Current protocols in protein science / editorial board, John E Coligan [et al] Chapter 25:Unit25 22. doi:10.1002/0471140864.ps2502s49.
Keller, A., Nesvizhskii, A. I., Kolker, E., & Aebersold, R. (2002). Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Analytical Chemistry, 74(20), 5383–5392.
Kertesz-Farkas, A., Keich, U., & Noble, W. S. (2015). Tandem mass spectrum identification via cascaded search. Journal of Proteome Research, 14(8), 3027–3038. doi:10.1021/pr501173s.
Kessner, D., Chambers, M., Burke, R., Agus, D., & Mallick, P. (2008). ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics, 24(21), 2534–2536. doi:10.1093/bioinformatics/btn323.
Khan, Z., Bloom, J. S., Garcia, B. A., Singh, M., & Kruglyak, L. (2009). Protein quantification across hundreds of experimental conditions. Proceedings of the National Academy of Sciences of the United States of America, 106(37), 15544–15548. doi:10.1073/pnas.0904100106.
Kim, S., & Pevzner, P. A. (2014). MS-GF+ makes progress towards a universal database search tool for proteomics. Nature Communications, 5, 5277. doi:10.1038/ncomms6277.
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., & Salzberg, S. L. (2013). TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 14(4), R36. doi:10.1186/gb-2013-14-4-r36.
Kirchner, M., Steen, J. A., Hamprecht, F. A., & Steen, H. (2010). MGFp: An open Mascot Generic Format parser library implementation. Journal of Proteome Research, 9(5), 2762–2763. doi:10.1021/pr100118f.
Lam, H. (2011). Building and searching tandem mass spectral libraries for peptide identification. Molecular & Cellular Proteomics, 10(12), R111.008565. doi:10.1074/mcp.R111.008565.
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., Funke, R., Gage, D., Harris, K., Heaford, A., Howland, J., Kann, L., Lehoczky, J., LeVine, R., McEwan, P., McKernan, K., Meldrim, J., Mesirov, J. P., Miranda, C., Morris, W., Naylor, J., Raymond, C., Rosetti, M., Santos, R., Sheridan, A., Sougnez, C., Stange-Thomann, Y., Stojanovic, N., Subramanian, A., Wyman, D., Rogers, J., Sulston, J., Ainscough, R., Beck, S., Bentley, D., Burton, J., Clee, C., Carter, N., Coulson, A., Deadman, R., Deloukas, P., Dunham, A., Dunham, I., Durbin, R., French, L., Grafham, D., Gregory, S., Hubbard, T., Humphray, S., Hunt, A., Jones, M., Lloyd, C., McMurray, A., Matthews, L., Mercer, S., Milne, S., Mullikin, J. C., Mungall, A., Plumb, R., Ross, M., Shownkeen, R., Sims, S., Waterston, R. H., Wilson, R. K., Hillier, L. W., McPherson, J. D., Marra, M. A., Mardis, E. R., Fulton, L. A., Chinwalla, A. T., Pepin, K. H., Gish, W. R., Chissoe, S. L., Wendl, M. C., Delehaunty, K. D., Miner, T. L., Delehaunty, A., Kramer, J. B., Cook, L. L., Fulton, R. S., Johnson, D. L., Minx, P. J., Clifton, S. W., Hawkins, T., Branscomb, E., Predki, P., Richardson, P., Wenning, S., Slezak, T., Doggett, N., Cheng, J. F., Olsen, A., Lucas, S., Elkin, C., Uberbacher, E., Frazier, M., Gibbs, R. A., Muzny, D. M., Scherer, S. E., Bouck, J. B., Sodergren, E. J., Worley, K. C., Rives, C. M., Gorrell, J. H., Metzker, M. L., Naylor, S. L., Kucherlapati, R. S., Nelson, D. L., Weinstock, G. M., Sakaki, Y., Fujiyama, A., Hattori, M., Yada, T., Toyoda, A., Itoh, T., Kawagoe, C., Watanabe, H., Totoki, Y., Taylor, T., Weissenbach, J., Heilig, R., Saurin, W., Artiguenave, F., Brottier, P., Bruls, T., Pelletier, E., Robert, C., Wincker, P., Smith, D. R., Doucette-Stamm, L., Rubenfield, M., Weinstock, K., Lee, H. M., Dubois, J., Rosenthal, A., Platzer, M., Nyakatura, G., Taudien, S., Rump, A., Yang, H., Yu, J., Wang, J., Huang, G., Gu, J., Hood, L., Rowen, L., Madan, A., Qin, S., Davis, R. W., Federspiel, N. A., Abola, A. P., Proctor, M. J., Myers, R. M., Schmutz, J., Dickson, M., Grimwood, J., Cox, D. R., Olson, M. V., Kaul, R., Shimizu, N., Kawasaki, K., Minoshima, S., Evans, G. A., Athanasiou, M., Schultz, R., Roe, B. A., Chen, F., Pan, H., Ramser, J., Lehrach, H., Reinhardt, R., McCombie, W. R., de la Bastide, M., Dedhia, N., Blocker, H., Hornischer, K., Nordsiek, G., Agarwala, R., Aravind, L., Bailey, J. A., Bateman, A., Batzoglou, S., Birney, E., Bork, P., Brown, D. G., Burge, C. B., Cerutti, L., Chen, H. C., Church, D., Clamp, M., Copley, R. R., Doerks, T., Eddy, S. R., Eichler, E. E., Furey, T. S., Galagan, J., Gilbert, J. G., Harmon, C., Hayashizaki, Y., Haussler, D., Hermjakob, H., Hokamp, K., Jang, W., Johnson, L. S., Jones, T. A., Kasif, S., Kaspryzk, A., Kennedy, S., Kent, W. J., Kitts, P., Koonin, E. V., Korf, I., Kulp, D., Lancet, D., Lowe, T. M., McLysaght, A., Mikkelsen, T., Moran, J. V., Mulder, N., Pollara, V. J., Ponting, C. P., Schuler, G., Schultz, J., Slater, G., Smit, A. F., Stupka, E., Szustakowki, J., Thierry-Mieg, D., Thierry-Mieg, J., Wagner, L., Wallis, J., Wheeler, R., Williams, A., Wolf, Y. I., Wolfe, K. H., Yang, S. P., Yeh, R. F., Collins, F., Guyer, M. S., Peterson, J., Felsenfeld, A., Wetterstrand, K. A., Patrinos, A., Morgan, M. J., de Jong, P., Catanese, J. J., Osoegawa, K., Shizuya, H., Choi, S., & Chen, Y. J. (2001). Initial sequencing and analysis of the human genome. Nature, 409(6822), 860–921. doi:10.1038/35057062.
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., & Durbin, R. (2009). The sequence alignment/Map format and SAMtools. Bioinformatics, 25(16), 2078–2079. doi:10.1093/bioinformatics/btp352.
Low, T. Y., van Heesch, S., van den Toorn, H., Giansanti, P., Cristobal, A., Toonen, P., Schafer, S., Hubner, N., van Breukelen, B., Mohammed, S., Cuppen, E., Heck, A. J., & Guryev, V. (2013). Quantitative and qualitative proteome characteristics extracted from in-depth integrated genomics and proteomics analysis. Cell Reports, 5(5), 1469–1478. doi:10.1016/j.celrep.2013.10.041.
Markiv, A., Rambaruth, N. D., & Dwek, M. V. (2012). Beyond the genome and proteome: Targeting protein modifications in cancer. Current Opinion in Pharmacology, 12(4), 408–413. doi:10.1016/j.coph.2012.04.003.
Martin, J. A., & Wang, Z. (2011). Next-generation transcriptome assembly. Nature Reviews Genetics, 12(10), 671–682. doi:10.1038/nrg3068.
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S., Daly, M., & DePristo, M. A. (2010). The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research, 20(9), 1297–1303. doi:10.1101/gr.107524.110.
Menschaert, G., & Fenyo, D. (2015). Proteogenomics from a bioinformatics angle: A growing field. Mass Spectrometry Reviews. doi:10.1002/mas.21483.
Metzker, M. L. (2010). Sequencing technologies – The next generation. Nature Reviews Genetics, 11(1), 31–46. doi:10.1038/nrg2626.
Muth, T., Weilnbock, L., Rapp, E., Huber, C. G., Martens, L., Vaudel, M., & Barsnes, H. (2014). DeNovoGUI: An open source graphical user interface for de novo sequencing of tandem mass spectra. Journal of Proteome Research, 13(2), 1143–1146. doi:10.1021/pr4008078.
Nesvizhskii, A. I. (2007). Protein identification by tandem mass spectrometry and sequence database searching. Methods in Molecular Biology, 367, 87–119. doi:10.1385/1-59745-275-0:87.
Nesvizhskii, A. I. (2010). A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. Journal of Proteomics, 73(11), 2092–2123. doi:10.1016/j.jprot.2010.08.009.
Nesvizhskii, A. I. (2014). Proteogenomics: Concepts, applications and computational strategies. Nature Methods, 11(11), 1114–1125. doi:10.1038/nmeth.3144.
Nesvizhskii, A., & Avtonomov, D. http://www.batmass.org/
Nesvizhskii, A. I., & Aebersold, R. (2005). Interpretation of shotgun proteomic data: The protein inference problem. Molecular & Cellular Proteomics, 4(10), 1419–1440. doi:10.1074/mcp.R500012-MCP200.
Nesvizhskii, A. I., Keller, A., Kolker, E., & Aebersold, R. (2003). A statistical model for identifying proteins by tandem mass spectrometry. Analytical Chemistry, 75(17), 4646–4658.
Orchard, S., Taylor, C., Hermjakob, H., Zhu, W., Julian, R., & Apweiler, R. (2004). Current status of proteomic standards development. Expert Review of Proteomics, 1(2), 179–183. doi:10.1586/14789450.1.2.179.
Patel, R. K., & Jain, M. (2012). NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PloS One, 7(2), e30619. doi:10.1371/journal.pone.0030619.
Pearson, W. R., Wood, T., Zhang, Z., & Miller, W. (1997). Comparison of DNA sequences with protein sequences. Genomics, 46(1), 24–36. doi:10.1006/geno.1997.4995.
Pedrioli, P. G., Eng, J. K., Hubley, R., Vogelzang, M., Deutsch, E. W., Raught, B., Pratt, B., Nilsson, E., Angeletti, R. H., Apweiler, R., Cheung, K., Costello, C. E., Hermjakob, H., Huang, S., Julian, R. K., Kapp, E., McComb, M. E., Oliver, S. G., Omenn, G., Paton, N. W., Simpson, R., Smith, R., Taylor, C. F., Zhu, W., & Aebersold, R. (2004). A common open representation of mass spectrometry data and its application to proteomics research. Nature Biotechnology, 22(11), 1459–1466. doi:10.1038/nbt1031.
Robinson, J. T., Thorvaldsdottir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., & Mesirov, J. P. (2011). Integrative genomics viewer. Nature Biotechnology, 29(1), 24–26. doi:10.1038/nbt.1754.
Rost, H. L., Rosenberger, G., Navarro, P., Gillet, L., Miladinovic, S. M., Schubert, O. T., Wolski, W., Collins, B. C., Malmstrom, J., Malmstrom, L., & Aebersold, R. (2014). OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nature Biotechnology, 32(3), 219–223. doi:10.1038/nbt.2841.
Ruggles, K. V., Tang, Z., Wang, X., Grover, H., Askenazi, M., Teubl, J., Cao, S., McLellan, M. D., Clauser, K. R., Tabb, D. L., Mertins, P., Slebos, R., Erdmann-Gilmore, P., Li, S., Gunawardena, H. P., Xie, L., Liu, T., Zhou, J. Y., Sun, S., Hoadley, K. A., Perou, C. M., Chen, X., Davies, S. R., Maher, C. A., Kinsinger, C. R., Rodland, K. D., Zhang, H., Zhang, Z., Ding, L., Townsend, R. R., Rodriguez, H., Chan, D., Smith, R. D., Liebler, D. C., Carr, S. A., Payne, S., Ellis, M. J., & Fenyo, D. (2015). An analysis of the sensitivity of proteogenomic mapping of somatic mutations and novel splicing events in cancer. Molecular & Cellular Proteomics. doi:10.1074/mcp.M115.056226.
Ruiz-Orera, J., Messeguer, X., Subirana, J. A., & Alba, M. M. (2014). Long non-coding RNAs as a source of new peptides. eLife, 3, e03523. doi:10.7554/eLife.03523.
Sajic, T., Liu, Y., & Aebersold, R. (2015). Using data-independent, high-resolution mass spectrometry in protein biomarker research: Perspectives and clinical applications. Proteomics Clinical Applications, 9(3–4), 307–321. doi:10.1002/prca.201400117.
Sanger, F., Air, G. M., Barrell, B. G., Brown, N. L., Coulson, A. R., Fiddes, C. A., Hutchison, C. A., Slocombe, P. M., & Smith, M. (1977). Nucleotide sequence of bacteriophage phi X174 DNA. Nature, 265(5596), 687–695.
Schwanhausser, B., Busse, D., Li, N., Dittmar, G., Schuchhardt, J., Wolf, J., Chen, W., & Selbach, M. (2011). Global quantification of mammalian gene expression control. Nature, 473(7347), 337–342. doi:10.1038/nature10098.
Schwanhausser, B., Busse, D., Li, N., Dittmar, G., Schuchhardt, J., Wolf, J., Chen, W., & Selbach, M. (2013). Corrigendum: Global quantification of mammalian gene expression control. Nature, 495(7439), 126–127. doi:10.1038/nature11848.
Shanmugam, A. K., & Nesvizhskii, A. I. (2015). Effective leveraging of targeted search spaces for improving peptide identification in tandem mass spectrometry based proteomics. Journal of Proteome Research, 14(12), 5169–5178. doi:10.1021/acs.jproteome.5b00504.
Sheynkman, G. M., Shortreed, M. R., Frey, B. L., & Smith, L. M. (2013). Discovery and mass spectrometric analysis of novel splice-junction peptides using RNA-Seq. Molecular & Cellular Proteomics, 12(8), 2341–2353. doi:10.1074/mcp.O113.028142.
Simpson, J. T., Wong, K., Jackman, S. D., Schein, J. E., Jones, S. J., & Birol, I. (2009). ABySS: A parallel assembler for short read sequence data. Genome Research, 19(6), 1117–1123. doi:10.1101/gr.089532.108.
Sturm, M., & Kohlbacher, O. (2009). TOPPView: An open-source viewer for mass spectrometry data. Journal of Proteome Research, 8(7), 3760–3763. doi:10.1021/pr900171m.
Tang, S., Lomsadze, A., & Borodovsky, M. (2015). Identification of protein coding regions in RNA transcripts. Nucleic Acids Research, 43(12), e78. doi:10.1093/nar/gkv227.
Tay, A. P., Pang, C. N., Twine, N. A., Hart-Smith, G., Harkness, L., Kassem, M., & Wilkins, M. R. (2015). Proteomic validation of transcript isoforms, including those assembled from RNA-Seq data. Journal of Proteome Research, 14(9), 3541–3554. doi:10.1021/pr5011394.
Teleman, J., Rost, H. L., Rosenberger, G., Schmitt, U., Malmstrom, L., Malmstrom, J., & Levander, F. (2015). DIANA–algorithmic improvements for analysis of data-independent acquisition MS data. Bioinformatics, 31(4), 555–562. doi:10.1093/bioinformatics/btu686.
Ternent, T., Csordas, A., Qi, D., Gomez-Baena, G., Beynon, R. J., Jones, A. R., Hermjakob, H., & Vizcaino, J. A. (2014). How to submit MS proteomics data to ProteomeXchange via the PRIDE database. Proteomics, 14(20), 2233–2241. doi:10.1002/pmic.201400120.
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., Salzberg, S. L., Wold, B. J., & Pachter, L. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology, 28(5), 511–515. doi:10.1038/nbt.1621.
Trevisiol, S., Ayoub, D., Lesur, A., Ancheva, L., Gallien, S., & Domon, B. (2015). The use of proteases complementary to trypsin to probe isoforms and modifications. Proteomics. doi:10.1002/pmic.201500379.
Turewicz, M., & Deutsch, E. W. (2011). Spectra, chromatograms, metadata: mzML-the standard data format for mass spectrometer output. Methods in Molecular Biology, 696, 179–203. doi:10.1007/978-1-60761-987-1_11.
Tyanova, S., Temu, T., Carlson, A., Sinitcyn, P., Mann, M., & Cox, J. (2015). Visualization of LC-MS/MS proteomics data in MaxQuant. Proteomics, 15(8), 1453–1456. doi:10.1002/pmic.201400449.
Vaudel, M., Barsnes, H., Berven, F. S., Sickmann, A., & Martens, L. (2011). SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X! Tandem searches. Proteomics, 11(5), 996–999. doi:10.1002/pmic.201000595.
Vaudel, M., Burkhart, J. M., Zahedi, R. P., Oveland, E., Berven, F. S., Sickmann, A., Martens, L., & Barsnes, H. (2015). PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nature Biotechnology, 33(1), 22–24. doi:10.1038/nbt.3109.
Volders, P. J., Helsens, K., Wang, X., Menten, B., Martens, L., Gevaert, K., Vandesompele, J., & Mestdagh, P. (2013). LNCipedia: A database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Research, 41(Database issue), D246–D251. doi:10.1093/nar/gks915.
Volders, P. J., Verheggen, K., Menschaert, G., Vandepoele, K., Martens, L., Vandesompele, J., & Mestdagh, P. (2015). An update on LNCipedia: A database for annotated human lncRNA sequences. Nucleic Acids Research, 43(Database issue), D174–D180. doi:10.1093/nar/gku1060.
Walsh, C. T., Garneau-Tsodikova, S., & Gatto, G. J., Jr. (2005). Protein posttranslational modifications: The chemistry of proteome diversifications. Angewandte Chemie International Edition, 44(45), 7342–7372. doi:10.1002/anie.200501023.
Walzer, M., Qi, D., Mayer, G., Uszkoreit, J., Eisenacher, M., Sachsenberg, T., Gonzalez-Galarza, F. F., Fan, J., Bessant, C., Deutsch, E. W., Reisinger, F., Vizcaino, J. A., Medina-Aunon, J. A., Albar, J. P., Kohlbacher, O., & Jones, A. R. (2013). The mzQuantML data standard for mass spectrometry-based quantitative studies in proteomics. Molecular & Cellular Proteomics, 12(8), 2332–2340. doi:10.1074/mcp.O113.028506.
Walzer, M., Pernas, L. E., Nasso, S., Bittremieux, W., Nahnsen, S., Kelchtermans, P., Pichler, P., van den Toorn, H. W., Staes, A., Vandenbussche, J., Mazanek, M., Taus, T., Scheltema, R. A., Kelstrup, C. D., Gatto, L., van Breukelen, B., Aiche, S., Valkenborg, D., Laukens, K., Lilley, K. S., Olsen, J. V., Heck, A. J., Mechtler, K., Aebersold, R., Gevaert, K., Vizcaino, J. A., Hermjakob, H., Kohlbacher, O., & Martens, L. (2014). qcML: An exchange format for quality control metrics from mass spectrometry experiments. Molecular & Cellular Proteomics, 13(8), 1905–1913. doi:10.1074/mcp.M113.035907.
Weisser, H., Nahnsen, S., Grossmann, J., Nilse, L., Quandt, A., Brauer, H., Sturm, M., Kenar, E., Kohlbacher, O., Aebersold, R., & Malmstrom, L. (2013). An automated pipeline for high-throughput label-free quantitative proteomics. Journal of Proteome Research, 12(4), 1628–1644. doi:10.1021/pr300992u.
Zhang, J., Xin, L., Shan, B., Chen, W., Xie, M., Yuen, D., Zhang, W., Zhang, Z., Lajoie, G. A., & Ma, B. (2012). PEAKS DB: De novo sequencing assisted database search for sensitive and accurate peptide identification. Molecular & Cellular Proteomics, 11(4), M111 010587. doi:10.1074/mcp.M111.010587.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Barbieri, R., Guryev, V., Brandsma, CA., Suits, F., Bischoff, R., Horvatovich, P. (2016). Proteogenomics: Key Driver for Clinical Discovery and Personalized Medicine. In: Végvári, Á. (eds) Proteogenomics. Advances in Experimental Medicine and Biology, vol 926. Springer, Cham. https://doi.org/10.1007/978-3-319-42316-6_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-42316-6_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-42314-2
Online ISBN: 978-3-319-42316-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)