
Abstract
Alternative splicing of pre-mRNA is an essential event that leads to protein diversity and regulation of the cellular processes in mammals. With the advent of the next generation sequencing technologies, the role of alternative splicing is gaining a momentum. Regulation of alternative splicing is a complex process involving the core spliceosome machinery and multiple regulatory factors that enable the tightly controlled splicing of introns/exons. Any aberrant alteration in this process can result in diseases such as cancer. Indeed, accumulating evidence suggests that alternative splicing plays an important role in all hallmarks of cancer including proliferative signaling, resisting cell death, inducing angiogenesis, and activating invasion and metastasis. These changes may occur due to mutations or altered expression levels of key regulatory genes of spliceosome machinery or splicing factors. In this review, we summarize recent findings that have implicated the critical role of alternative splicing in breast cancer and discuss current understandings and its potential utility in breast cancer.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
23.1 Introduction
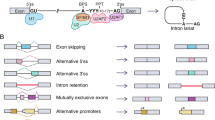
The splicing mechanism is the process in which introns are separated from the exons; the latter go on to form mature mRNAs. Alternative splicing (AS) is a mechanism by which selective inclusion/ exclusion of exons and introns during splicing of the pre-mRNAs leads to the production of more than one isoform. It plays an important role in regulating cellular processes in a tissue-specific manner (Black 2003; Pan et al. 2008). In particular, recent high-throughput sequencing technologies revealed that about 92–94 % of human genes are alternatively spliced (Blencowe 2006; Pan et al. 2008; Wang et al. 2011; Irimia and Blencowe 2012). In this process, inclusion or exclusion of exons or portions of exons or introns within a pre-mRNA transcript can result in multiple protein isoforms being encoded by a single gene. This process is tightly regulated in normal cells. Most exons are constitutive, being always spliced or included in the mature mRNA (Fig. 23.1a). However, aberrant regulation of AS may result in several diseases including cancer. The major alternative splicing patterns or events (Fig. 23.1b–f) are grouped into five types. If an exon is sometimes excluded or included, this indicates that the exon expression is regulated and also termed as cassette exon (Fig. 23.1b). In some cases, cassette exons are mutually exclusive (Fig. 23.1c); this might hold true for more than one exons. Exons can be longer or shorter affecting their splice sites. Alterations in 5′-terminal exons result in alternative promoter sites (Fig. 23.1d). On the other hand, alternative splicing of the 3′-terminal exons can lead to alternative polyadenylation sites (Fig. 23.1e). In addition, some regulatory events result in inclusion of an intron, a splicing pattern called intron retention (Fig. 23.1f).

Main alternative splicing events a Constitutive splicing; b cassette alternative exon; c mutually exclusive exons; d alternative 5′ splice site; e alternative 3′ splice site and f intron retention
Aberrant alternative splicing events in cancer may impact the alteration of genes and proteins both at the expression and functional level. These events are regulated by a complex process involving the core spliceosome machinery and multiple regulatory factors (Irimia and Blencowe 2012). A schematic was depicted in Fig. 23.2 to summarize the key regulatory players at the exon level.

Schematic representation of core spliceosomal components and its binding proteins. Splicing factors can either promote or repress splice site selection depending on the location of their binding sites with respect to splicing signals. ISE Intronic splicing\enhancer; ISS Intronic splicing silencer; ESE Exonic splicing enhancer; ESS Exonic splicing silencer; SR, Ser/Arg-repeat containing protein; hnRNP Heterogeneous ribonucleoprotein (hnRNP); and U2AF, U2 snRNP auxiliary factor. Adapted from Irimia and Blencowe Current Opinion in Cell Biology
The core spliceosome machinery is a large dynamic macromolecular RNA-protein complex composed of five small nuclear RNAs (snRNAs) and over 100 associated proteins. The association of these small RNAs with the protein factors comprise the RNA-protein-complex called small nuclear ribonucleic proteins (snRNPs). Splice sites of a gene are the binding sites for the spliceosome machinery. Splicing factors (SFs) , a subset of RNA binding proteins (RBPs), control the choice of splice sites and impact the recruitment of the spliceosome to splice sites (Chen and Weiss 2015; Liu and Cheng 2013; Zhang and Manley 2013; Cartegni et al. 2002; Irimia and Blencowe 2012). SFs exert their effect by binding specific RNA sequences, or motifs, known as exonic splicing enhancers (ESEs), exonic splicing silencers (ESS), intronic splicing enhancers (ISEs) and intronic splicing silencers (ISSs) (Cartegni et al. 2002; Irimia and Blencowe 2012). Bound SFs can either activate or inhibit the interaction between spliceosome and pre-mRNAs (McManus and Graveley 2011). Some of them can have dual function based on the location of the motifs they bind. Several splicing factors have been well established in humans (Venables et al. 2008; Twyffels et al. 2011), and categorized into two major families: serine-arginine protein (SR) and heterogeneous ribonucleoprotein (hnRNP). SRs usually promotes splicing, while hnRNPs usually inhibit the splicing process by binding to silencer sequences (Cartegni et al. 2002; David and Manley 2010; Irimia and Blencowe 2012). The decision of alternative splicing also requires cis-acting RNA splicing regulatory elements (SREs) which influence the splicing of exons/introns in the mRNA (Cho et al. 2014). Cis-acting regulatory elements are located on 200–300 nucleotides adjacent to observed splice sites. They also can alter splicing by binding to different trans-acting proteins which are remotely located and act as splicing enhancers or silencers. The ultimate decision for splicing regulation is combinatorial and context-dependent based on the cooperation and competition of splicing factors. All these factors increase the diversity and functional capacity of a gene during post-transcriptional processing and exert tight gene regulation.
Mutations of SF genes or alterations in expression levels of the proteins may contribute to aberrant AS. These proteins are guided by additional factors that can also interact with mRNAs at specific motifs to regulate the inclusion or exclusion of exons in the final transcript. Alterations in the levels and activity of these SFs thus provide another means of AS deregulation. Changes in splice sites or motifs of SFs in a given gene may also affect the alternative splicing . Besides binding to SFs, other characteristics of the protein may be altered including ligand binding, enzymatic activity, subcellular localization, and/or protein-protein interactions. This further may alter many processes that can switch cells from normal to malignant phenotype.
Deregulation of alternative splicing due to these factors may result in cancers including breast cancer . Several studies have revealed splice variants specific to tumors in several cancers including breast cancer which impact hallmarks of cancer such as proliferation, apoptosis, cell-cycle-control, metabolism, angiogenesis, and invasion (Chen and Weiss 2015; Dutertre et al. 2010; Germann et al. 2012; Swami et al. 2009; Liu and Cheng 2013; Oltean and Bates 2014; Venables et al. 2008; Zhang and Manley 2013). In this chapter, we will review the regulatory factors and alternative splicing events in breast cancer , its promises and limitations in the clinical practice.
23.2 Alternative Splicing in Breast Cancer
23.2.1 Mutations in RNA Splicing Factors
Recent next-generation sequencing technologies have revealed the presence of somatic mutations in the components of spliceosome machinery and splicing factors (Malcovati et al. 2011; Papaemmanuil et al. 2011; Yoshida and Ogawa 2014; Yoshida et al. 2011). These mutations mostly involve components that are involved in the initial steps of pre-mRNA splicing, such as 3’ splice-site recognition and occur in a mutually exclusive manner. Among the mutated splicing factors, U2AF1, SRSF2, SF3B1, and ZRSR2 genes were common mutational hotspots in myeloid neoplasms such as myelodysplastic syndrome (MDS). Although these mutations were frequent (45–85 %) in myeloid neoplasms, they exist in other hematologic malignancies and solid tumors, albeit at different frequencies (Quesada et al. 2012; Ramsay et al. 2013; Scott and Rebel 2013; Wang et al. 2011; Yoshida and Ogawa 2014). Mutations in splicing factor 3b, subunit 1 (SF3B1) occurred in 15 % of chronic lymphocytic leukemias (CLLs) (Quesada et al. 2012), and in solid cancers such as uveal melanomas (9.7 %) (Furney et al. 2013; Harbour et al. 2013), pancreatic cancers (4 %) (Biankin et al. 2012), and breast cancers (2 %) (Cancer Genome Atlas 2012; Stephens et al. 2012). Mutations in other splicing genes, such as the U2 small nuclear RNA auxiliary factor 1 gene (U2AF1), the serine/arginine-rich splicing factor 2 gene (SRSF2), and the U2 small nuclear ribonucleoprotein auxiliary factor 35 kDa subunit-related protein 2 gene (ZRSR2), have also been identified in a lower frequency than SF3B1 mutations (Yoshida and Ogawa 2014; Yoshida et al. 2011).
SF3B1 is the only splicing factor that has been reported to be among the top 35 mutated genes using next-generation sequencing on 510 breast tumors (Cancer Genome Atlas 2012). However, the frequency was low (2 % of all tumors). Of the 15 non-silent mutations, the majority were missense mutations. Patients with estrogen receptor ER+ and HER-2+ subtypes harbored the majority of these mutations. The SF3B1 was also among the 18 significantly mutated genes in untreated ER+ breast tumors from 77 patients accrued from two neo-adjuvant aromatase inhibitor clinical trials (Ellis et al. 2012). A recent study re-analyzed the mutations in spliceosomal components using public exome and whole genome sequencing data (Maguire et al. 2015). Their data also confirmed that SF3B1 was the most commonly mutated gene in the spliceosomal complex in breast cancer , in particular in ER+ breast tumors. Furthermore, SF3B1 mutations were associated with differential splicing of genes in ER+ breast tumors including TMEM14C, RPL31, DYNL11, UQCC, ABCC5 and CRNDE. Some of these splice variants have also been observed in other cancers with SF3B1 mutations (Furney et al. 2013).
23.2.2 Altered Gene Expression Levels in RNA Splicing Factors
Accumulating evidence implicates that aberrant expression of genes regulating alternative splicing is another factor that impacts the alternative splicing events in breast cancer. In our study, the splicing factor SF3B1 was upregulated in acquired endocrine resistant models as well as in cases with Oncotype DX high-recurrence scores (Gokmen-Polar et al. 2015). However, we did not observe any prognostic correlation of SF3B1 expression in our analyses using breast tumors from TCGA and Affymetrix microarray datasets. Interestingly, splicing factor 3b, subunit 3 (SF3B3), a SF3B subunit interacting with SF3B1, was also upregulated in these models. As in the case of SF3B1, high expression of SF3B3 correlated with the Oncotype DX high-recurrence cases. In contrast to SF3B1, high expression of SF3B3 correlated with poor prognosis in patients with ER+ breast cancer .
Other alterations in expression of splicing factors or components of spliceosome machinery , have also been reported in breast cancer (Grosso et al. 2008). These alterations are assumed to affect the splicing pattern of other genes that are involved in tumor development and progression. Alternatively, they might act as oncogenes. For example, splicing factor SF2/ASF is upregulated in various human tumors, and impacts alternative splicing of the tumor suppressor BIN1 and the kinases MNK2 and S6K1. While BIN1 isoforms lost their tumor-suppressor activity, the MNK2 isoform promotes MAPK-independent eIF4E phosphorylation and the S6K1 isoform has demonstrated oncogenic properties (Karni et al. 2007).
Heterogeneous ribonucleoproteins (hnRNPs) are another major group of splicing factors that are involved in different steps of pre-mRNA processing and cellular functions (Carpenter et al. 2006; Grosso et al. 2008). The hnRNP proteins are also involved in various biological processes required for tumor progression. Splicing factor SRSF1 is upregulated in human breast tumors, and its overexpression promotes transformation of mammary cells (Anczukow et al. 2015). A recent study reported the expression profile of ten splicing factors (both SRs and hnRNPs) and eight RNA-binding proteins in breast cancer cells (Silipo 2015). Taken together, these studies emphasize that alterations (mutations or altered expression) in core spliceosomal complex genes and its associated genes may contribute to aberrant alternative splicing in breast cancer progression.
23.3 Alternative Splicing Events in Breast Cancer
Aberrant alternative splicing events have been associated with the initiation and progression in breast cancer (Dutertre et al. 2010). We will enumerate some examples for each type of alternative splicing events and emphasize their contribution in breast cancer development and progression (Table 23.1).
23.3.1 Cassette Exons
23.3.1.1 Exon Skipping
The breast cancer susceptibility genes, BRCA1 and BRCA2, are good illustrative examples for exon skipping. BRCA1 RNAs from most tumors show splicing alterations (Bonnet et al. 2008; Easton et al. 2007; Lovelock et al. 2006; Tommasi et al. 2008; Caux-Moncoutier et al. 2009; Anczukow et al. 2008). For example, the full-length BRCA1 gene encodes 24 exons. Exon 18 skipping in BRCA1 can enhance (SF2/ASF) or inhibit (hnRNPA1 and hnRNPH/F) binding of splicing factors to the mRNA (Liu et al. 2001; Millevoi et al. 2010). In addition, skipping of exon 11 has been associated with cell death and proliferation. Besides exon 11 and 18 skipping, other splice variants of BRCA1 have been identified including BRCA1 full length (inclusion of all exons), partial skipping of exon 11, skipping of exons 9, 10, and partial skipping of exon 11 and IRIS isoforms (skipping of exons 12–24, but retaining a short segment from intron 11) (Tammaro et al. 2012). Additional studies are emerging regarding novel BRCA1 variants inducing splicing defects (Ahlborn et al. 2015; Romero et al. 2015; Tammaro et al. 2012). However, the clinical significance of these variants and the relevance of these mutations are unknown. With the exception of IRIS, the importance of other BRCA1 splicing events in cancer development needs to be further determined.
23.3.1.2 Exon Inclusion and Complex Splicing Patterns
CD44, a cell surface receptor, has been gained attention as a breast cancer stem cell marker and chemo-resistance and is under extensive study as a therapeutic target. CD44 has been used as biomarkers to identify and characterize the breast cancer stem cell (CSC) phenotype (Al-Hajj et al. 2003; Shipitsin et al. 2007). Breast cancer cells with CD44+/CD24- subpopulation express higher levels of pro-invasive genes and have highly invasive properties specific to ER− cell lines (Sheridan et al. 2006). However, overexpression of CD44 has been implicated in both tumor suppression and progression (Horak et al. 2008). Relevance of CD44 in breast carcinomas is still unclear in part due to the complex splice pattern observed in breast cancer.
CD44 pre-mRNA contains 19 exons, 9 of which are alternatively spliced (Loh et al. 2015). Based on the inclusion of variable exons, a number of isoforms are generated. The standard isoform of CD44 (CD44s) contains 10 constant exons (exons 1–5 and 15–20), whereas the variant CD44v isoforms includes exons 5a and 14 (exon v1–v10). Exon 5a (v1) is not expressed in humans (Screaton et al. 1993; Inoue and Fry 2015). Several groups have assessed the role of CD44 in breast cancer progression in vivo using mouse models (Brown et al. 2011; Warzecha et al. 2009). Different splice variants of CD44 have also been associated with different subtypes of breast cancer (Olsson et al. 2011). High expression of standard (CD44s) isoform was present in tumors with strong HER-2 staining and in a subgroup of basal-like tumors. Expression was associated with ALDH1 expression. In contrast, other CD44 variants are associated with luminal A subtype and with tumors with high CD44+/CD24- subpopulation. In breast cancer cell lines, the untransformed (MCF10A) and non-metastatic (MCF-7) cell lines harbor different isoform pattern (CD44v6 isoform, which includes all of the v6-containing mRNA isoforms- c5v6v7v8v9v10c6) compared to metastatic MDA-MB-231 cell lines. The splicing factor epithelial splicing regulatory protein 1 (ESRP1) and hnRNPA1 are important in controlling the CD44 isoform switch and critical for regulating the EMT phenotype in cell line models (Warzecha et al. 2009). The switch of CD44v to CD44s variants has been reported to induce EMT phenotype (Brown et al. 2011). In contrast, other studies reported that CD44v isoforms can mediate metastasis (Zhang et al. 2014, 2015; Tjhay et al. 2015). Orthotropic transplantation of a CD44v(+) subpopulation of 4T1 breast cancer cells, but not that of a CD44v(−) subpopulation, in mice results in efficient lung metastasis accompanied by expansion of stem-like cancer cells proving the role of the variant isoform in cancer metastasis (Yae et al. 2012). In summary, CD44 splicing is very complex and further analysis is necessary to understand the role of CD44 splice variants in breast cancer.
23.3.2 Mutually Exclusive Exons
Fibroblast Growth Factor Receptor 2 (FGFR2), a member of the fibroblast growth factor receptors, has been shown to be altered in breast cancer (Fletcher et al. 2013). FGFR2 is one of the examples in breast cancer where the alternative splicing of two mutually exclusive exons (FGFR2 IIIb or IIIc) alters its ligand binding ability and its biological function. Switching of FGFR2 IIIb to IIIc plays a role in EMT process and results in mammary tumor development (Cha et al. 2008; Moffa et al. 2004; Wei et al. 2012).
23.3.3 Intron Retention
Intron retention is common in most of the tumors except in breast tumors (Dvinge and Bradley 2015). Breast tumors were associated with decreased intron retention relative to normal controls. For example, Herstatin is a naturally occurring truncated HER-2 protein generated from alternative HER-2 mRNA transcripts that retain intron 8 (Jackson et al. 2013; Doherty et al. 1999). Herstatin can act as an inhibitor of full-length HER-2 by interfering with dimerization, and tyrosine phosphorylation (Guidi et al. 1997). In particular, Herstatin levels are significantly higher in noncancerous breast cells compared to carcinoma cells (Koletsa et al. 2008), p100, another truncated HER2 mRNA splice variant, exhibits the retention of intron 15 and inhibits the tumor cell proliferation and oncogenic signaling (Aigner et al. 2001). Further studies are necessary to understand its prognostic and predictive value in breast cancer .
23.3.4 Alternative 5′ Splice Sites
The apoptosis regulator gene Bcl-2-like 1 or Bcl-x, which belongs to the Bcl-2 family of proteins, can act as an anti-apoptotic (Bcl-xL) or pro-apoptotic (Bcl-xS) protein by regulating caspase activation. These two isoforms are generated based on the alternative splicing pattern of Bcl-x in the 5′ splice sites in exon 2. Overexpression of the longer isoform Bcl-xL has been reported in several cancers including breast cancer, whereas the shorter isoform Bcl-xS is downregulated in cancer (Boise et al. 1993; Adams and Cory 2007; Akgul et al. 2004; Cloutier et al. 2008; Ma et al. 2010). The alternative splicing of Bcl-x has been well documented in affecting survival or evading apoptosis, one of the key hallmarks of cancer (Hanahan and Weinberg 2000, 2011).
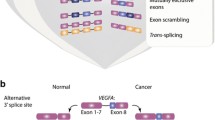
23.3.5 Alternative 3′ Splice Sites
Vascular endothelial growth factor (VEGF) is a well-known stimulator of tumor angiogenesis, tumor growth and metastasis in cancer, all of which are hallmarks of cancer. Overexpression of VEGF is an early event in breast cancer progression and a prerequisite step to tumor invasion (Guidi et al. 1997). Elevated expression of VEGF can be associated with shorter relapse-free survival and overall survival times in breast cancer patients with both positive and negative lymph nodes (Gasparini et al. 1997; Konecny et al. 2004; Relf et al. 1997). VEGF pre-mRNA is regulated by alternative splicing (Biselli-Chicote et al. 2012; Harper and Bates 2008). The VEGF gene contains eight exons having two competing 3′ splice sites (proximal and distal) in exon 8 (Houck et al. 1991). The proximal 3′ splice site of exon 8 generates the VEGF isoforms that are pro-angiogenic, whereas the distal 3′ splice site produces the VEGFβ isoforms that are anti-angiogenic. Splicing factors SRSF1 and SRSF5 (SRp40) have been shown to control the splicing of VEGF exon 8 proximal 3′ splice site and promote the production of VEGF (Nowak et al. 2008). VEGF splicing is complex and alternative splicing of other exons (exon 6 and 7) increases its functional diversity.
23.4 Future Directions; Promises and Limitations
High throughput technologies such as massively parallel RNA-sequencing have emphasized the importance of alternative splicing in biological models and human disease by providing an extensive information of small RNAs and associated proteins that are involved in RNA splicing process. Alterations of these proteins by mutations or gene expression level affect the alternative splicing events leading to altered function and protein-protein interactions of several proteins. In particular, mutations in spliceosome components have opened new therapeutic opportunities in cancer. Much work needs to be done to understand the clinical utility of key splice variants in tumor development, progression and metastasis. In particular, major challenges need to be overcome to remove significant bottlenecks for the clinical utility of cancer-specific splice variants. First, computational biology methods need to be refined and standardized among the different databases and platforms. Second, identification of gene expression alterations at the exon level need to be coupled with biological endpoints such as proliferation, apoptosis or recurrence/metastasis. For example, in breast cancer , a decrease in the proliferation rate following neoadjuvant endocrine therapies can be associated with alterations at the exon level. Exon markers can unravel the dual roles of some of the prognostic and predictive markers in breast cancer initiation, progression and metastasis. Third, experimental models need to be developed that can determine and validate the biological significance of these exon markers. However, the complexity arises when multiple exons are skipped or included. This might suggest that it is important to not only identify clinical significance at the exon level as well as at the transcript level. Fourth, databases at the transcript level need to be developed from tumors of retrospective and prospective clinical trials with the outcome follow-up. These databases are critical to understand their ultimate clinical utility both at the discovery and validation stage.
In conclusion, overcoming of all of these challenges requires the extensive collaboration of computational scientists, mathematicians, cancer biologists, pathologists and clinicians.
References
Adams JM, Cory S (2007) The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26(9):1324–1337. doi:10.1038/sj.onc.1210220
Ahlborn LB, Dandanell M, Steffensen AY, Jonson L, Nielsen FC, Hansen TV (2015) Splicing analysis of 14 BRCA1 missense variants classifies nine variants as pathogenic. Breast Cancer Res Treat 150(2):289–298. doi:10.1007/s10549-015-3313-7
Aigner A, Juhl H, Malerczyk C, Tkybusch A, Benz CC, Czubayko F (2001) Expression of a truncated 100 kDa HER2 splice variant acts as an endogenous inhibitor of tumour cell proliferation. Oncogene 20(17):2101–2111. doi:10.1038/sj.onc.1204305
Akgul C, Moulding DA, Edwards SW (2004) Alternative splicing of Bcl-2-related genes: functional consequences and potential therapeutic applications. Cell Mol Life Sci 61(17):2189–2199. doi:10.1007/s00018-004-4001-7
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF (2003) Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 100(7):3983–3988. doi:10.1073/pnas.0530291100
Anczukow O, Akerman M, Clery A, Wu J, Shen C, Shirole NH, Raimer A, Sun S, Jensen MA, Hua Y, Allain FH, Krainer AR (2015) SRSF1-regulated alternative splicing in breast cancer. Mol Cell 60(1):105–117. doi:10.1016/j.molcel.2015.09.005
Anczukow O, Buisson M, Salles MJ, Triboulet S, Longy M, Lidereau R, Sinilnikova OM, Mazoyer S (2008) Unclassified variants identified in BRCA1 exon 11: consequences on splicing. Genes Chromosomes Cancer 47(5):418–426. doi:10.1002/gcc.20546
Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, Chang DK, Cowley MJ, Gardiner BB, Song S, Harliwong I, Idrisoglu S, Nourse C, Nourbakhsh E, Manning S, Wani S, Gongora M, Pajic M, Scarlett CJ, Gill AJ, Pinho AV, Rooman I, Anderson M, Holmes O, Leonard C, Taylor D, Wood S, Xu Q, Nones K, Fink JL, Christ A, Bruxner T, Cloonan N, Kolle G, Newell F, Pinese M, Mead RS, Humphris JL, Kaplan W, Jones MD, Colvin EK, Nagrial AM, Humphrey ES, Chou A, Chin VT, Chantrill LA, Mawson A, Samra JS, Kench JG, Lovell JA, Daly RJ, Merrett ND, Toon C, Epari K, Nguyen NQ, Barbour A, Zeps N, Australian Pancreatic Cancer Genome I, Kakkar N, Zhao F, Wu YQ, Wang M, Muzny DM, Fisher WE, Brunicardi FC, Hodges SE, Reid JG, Drummond J, Chang K, Han Y, Lewis LR, Dinh H, Buhay CJ, Beck T, Timms L, Sam M, Begley K, Brown A, Pai D, Panchal A, Buchner N, De Borja R, Denroche RE, Yung CK, Serra S, Onetto N, Mukhopadhyay D, Tsao MS, Shaw PA, Petersen GM, Gallinger S, Hruban RH, Maitra A, Iacobuzio-Donahue CA, Schulick RD, Wolfgang CL, Morgan RA, Lawlor RT, Capelli P, Corbo V, Scardoni M, Tortora G, Tempero MA, Mann KM, Jenkins NA, Perez-Mancera PA, Adams DJ, Largaespada DA, Wessels LF, Rust AG, Stein LD, Tuveson DA, Copeland NG, Musgrove EA, Scarpa A, Eshleman JR, Hudson TJ, Sutherland RL, Wheeler DA, Pearson JV, McPherson JD, Gibbs RA, Grimmond SM (2012) Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491(7424):399–405. doi:10.1038/nature11547
Biselli-Chicote PM, Oliveira AR, Pavarino EC, Goloni-Bertollo EM (2012) VEGF gene alternative splicing: pro- and anti-angiogenic isoforms in cancer. J Cancer Res Clin Oncol 138(3):363–370. doi:10.1007/s00432-011-1073-2
Black DL (2003) Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72:291–336. doi:10.1146/annurev.biochem.72.121801.161720
Blencowe BJ (2006) Alternative splicing: new insights from global analyses. Cell 126(1):37–47. doi:10.1016/j.cell.2006.06.023
Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CB (1993) bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74(4):597–608
Bonnet C, Krieger S, Vezain M, Rousselin A, Tournier I, Martins A, Berthet P, Chevrier A, Dugast C, Layet V, Rossi A, Lidereau R, Frebourg T, Hardouin A, Tosi M (2008) Screening BRCA1 and BRCA2 unclassified variants for splicing mutations using reverse transcription PCR on patient RNA and an ex vivo assay based on a splicing reporter minigene. J Med Genet 45(7):438–446. doi:10.1136/jmg.2007.056895
Brown RL, Reinke LM, Damerow MS, Perez D, Chodosh LA, Yang J, Cheng C (2011) CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J Clin Invest 121(3):1064–1074. doi:10.1172/JCI44540
Cancer Genome Atlas N (2012) Comprehensive molecular portraits of human breast tumours. Nature 490(7418):61–70. doi:10.1038/nature11412
Carpenter B, MacKay C, Alnabulsi A, MacKay M, Telfer C, Melvin WT, Murray GI (2006) The roles of heterogeneous nuclear ribonucleoproteins in tumour development and progression. Biochim Biophys Acta 1765(2):85–100. doi:10.1016/j.bbcan.2005.10.002
Cartegni L, Chew SL, Krainer AR (2002) Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 3(4):285–298. doi:10.1038/nrg775
Caux-Moncoutier V, Pages-Berhouet S, Michaux D, Asselain B, Castera L, De Pauw A, Buecher B, Gauthier-Villars M, Stoppa-Lyonnet D, Houdayer C (2009) Impact of BRCA1 and BRCA2 variants on splicing: clues from an allelic imbalance study. Eur J Hum Genet 17(11):1471–1480. doi:10.1038/ejhg.2009.89
Cha JY, Lambert QT, Reuther GW, Der CJ (2008) Involvement of fibroblast growth factor receptor 2 isoform switching in mammary oncogenesis. Mol Cancer Res 6(3):435–445. doi:10.1158/1541-7786.MCR-07-0187
Chen J, Weiss WA (2015) Alternative splicing in cancer: implications for biology and therapy. Oncogene 34(1):1–14. doi:10.1038/onc.2013.570
Cho H, Davis J, Li X, Smith KS, Battle A, Montgomery SB (2014) High-resolution transcriptome analysis with long-read RNA sequencing. PLoS ONE 9(9):e108095. doi:10.1371/journal.pone.0108095
Cloutier P, Toutant J, Shkreta L, Goekjian S, Revil T, Chabot B (2008) Antagonistic effects of the SRp30c protein and cryptic 5′ splice sites on the alternative splicing of the apoptotic regulator Bcl-x. J Biol Chem 283(31):21315–21324. doi:10.1074/jbc.M800353200
David CJ, Manley JL (2010) Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev 24(21):2343–2364. doi:10.1101/gad.1973010
Doherty JK, Bond C, Jardim A, Adelman JP, Clinton GM (1999) The HER-2/neu receptor tyrosine kinase gene encodes a secreted autoinhibitor. Proc Natl Acad Sci U S A 96(19):10869–10874
Dutertre M, Vagner S, Auboeuf D (2010) Alternative splicing and breast cancer. RNA Biol 7(4):403–411
Dvinge H, Bradley RK (2015) Widespread intron retention diversifies most cancer transcriptomes. Genome Med 7(1):45. doi:10.1186/s13073-015-0168-9
Easton DF, Deffenbaugh AM, Pruss D, Frye C, Wenstrup RJ, Allen-Brady K, Tavtigian SV, Monteiro AN, Iversen ES, Couch FJ, Goldgar DE (2007) A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet 81(5):873–883. doi:10.1086/521032
Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, Ng S, Lin L, Crowder R, Snider J, Ballman K, Weber J, Chen K, Koboldt DC, Kandoth C, Schierding WS, McMichael JF, Miller CA, Lu C, Harris CC, McLellan MD, Wendl MC, DeSchryver K, Allred DC, Esserman L, Unzeitig G, Margenthaler J, Babiera GV, Marcom PK, Guenther JM, Leitch M, Hunt K, Olson J, Tao Y, Maher CA, Fulton LL, Fulton RS, Harrison M, Oberkfell B, Du F, Demeter R, Vickery TL, Elhammali A, Piwnica-Worms H, McDonald S, Watson M, Dooling DJ, Ota D, Chang LW, Bose R, Ley TJ, Piwnica-Worms D, Stuart JM, Wilson RK, Mardis ER (2012) Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 486(7403):353–360. doi:10.1038/nature11143
Fletcher MN, Castro MA, Wang X, de Santiago I, O’Reilly M, Chin SF, Rueda OM, Caldas C, Ponder BA, Markowetz F, Meyer KB (2013) Master regulators of FGFR2 signalling and breast cancer risk. Nat Commun 4:2464. doi:10.1038/ncomms3464
Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, Turajlic S, Piperno-Neumann S, de la Grange P, Roman-Roman S, Stern MH, Marais R (2013) SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov 3(10):1122–1129. doi:10.1158/2159-8290.CD-13-0330
Gasparini G, Toi M, Gion M, Verderio P, Dittadi R, Hanatani M, Matsubara I, Vinante O, Bonoldi E, Boracchi P, Gatti C, Suzuki H, Tominaga T (1997) Prognostic significance of vascular endothelial growth factor protein in node-negative breast carcinoma. J Natl Cancer Inst 89(2):139–147
Germann S, Gratadou L, Dutertre M, Auboeuf D (2012) Splicing programs and cancer. J Nucleic Acids 2012:269570. doi:10.1155/2012/269570
Gokmen-Polar Y, Neelamraju Y, Goswami CP, Gu X, Nallamothu G, Janga SC, Badve S (2015) Expression levels of SF3B3 correlate with prognosis and endocrine resistance in estrogen receptor-positive breast cancer. Mod Pathol 28(5):677–685. doi:10.1038/modpathol.2014.146
Grosso AR, Martins S, Carmo-Fonseca M (2008) The emerging role of splicing factors in cancer. EMBO Rep 9(11):1087–1093. doi:10.1038/embor.2008.189
Guidi AJ, Schnitt SJ, Fischer L, Tognazzi K, Harris JR, Dvorak HF, Brown LF (1997) Vascular permeability factor (vascular endothelial growth factor) expression and angiogenesis in patients with ductal carcinoma in situ of the breast. Cancer 80(10):1945–1953
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100(1):57–70
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. doi:10.1016/j.cell.2011.02.013
Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM (2013) Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet 45(2):133–135. doi:10.1038/ng.2523
Harper SJ, Bates DO (2008) VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat Rev Cancer 8(11):880–887. doi:10.1038/nrc2505
Horak CE, Lee JH, Marshall JC, Shreeve SM, Steeg PS (2008) The role of metastasis suppressor genes in metastatic dormancy. APMIS 116(7–8):586–601. doi:10.1111/j.1600-0463.2008.01213.x
Houck KA, Ferrara N, Winer J, Cachianes G, Li B, Leung DW (1991) The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol Endocrinol 5(12):1806–1814. doi:10.1210/mend-5-12-1806
Inoue K (2015) Fry EA (2015) Aberrant splicing of estrogen receptor, HER2, and CD44 genes in breast cancer. Genet Epigenet. 2(7):19–32. doi:10.4137/GEG.S35500
Irimia M, Blencowe BJ (2012) Alternative splicing: decoding an expansive regulatory layer. Curr Opin Cell Biol 24(3):323–332. doi:10.1016/j.ceb.2012.03.005
Jackson C, Browell D, Gautrey H, Tyson-Capper A (2013) Clinical significance of HER-2 splice variants in breast cancer progression and drug resistance. Int J Cell Biol 2013:973584. doi:10.1155/2013/973584
Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR (2007) The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol 14(3):185–193. doi:10.1038/nsmb1209
Koletsa T, Kostopoulos I, Charalambous E, Christoforidou B, Nenopoulou E, Kotoula V (2008) A splice variant of HER2 corresponding to Herstatin is expressed in the noncancerous breast and in breast carcinomas. Neoplasia 10(7):687–696
Konecny GE, Meng YG, Untch M, Wang HJ, Bauerfeind I, Epstein M, Stieber P, Vernes JM, Gutierrez J, Hong K, Beryt M, Hepp H, Slamon DJ, Pegram MD (2004) Association between HER-2/neu and vascular endothelial growth factor expression predicts clinical outcome in primary breast cancer patients. Clin Cancer Res 10(5):1706–1716
Liu HX, Cartegni L, Zhang MQ, Krainer AR (2001) A mechanism for exon skipping caused by nonsense or missense mutations in BRCA1 and other genes. Nat Genet 27(1):55–58. doi:10.1038/83762
Liu S, Cheng C (2013) Alternative RNA splicing and cancer. Wiley Interdiscip Rev RNA 4(5):547–566. doi:10.1002/wrna.1178
Loh TJ, Moon H, Cho S, Jang H, Liu YC, Tai H, Jung DW, Williams DR, Kim HR, Shin MG, Liao DJ, Zhou J, Shi W, Zheng X, Shen H (2015) CD44 alternative splicing and hnRNP A1 expression are associated with the metastasis of breast cancer. Oncol Rep 34(3):1231–1238. doi:10.3892/or.2015.4110
Lovelock PK, Healey S, Au W, Sum EY, Tesoriero A, Wong EM, Hinson S, Brinkworth R, Bekessy A, Diez O, Izatt L, Solomon E, Jenkins M, Renard H, Hopper J, Waring P, Tavtigian SV, Goldgar D, Lindeman GJ, Visvader JE, Couch FJ, Henderson BR, Southey M, Chenevix-Trench G, Spurdle AB, Brown MA, kConFab I (2006) Genetic, functional, and histopathological evaluation of two C-terminal BRCA1 missense variants. J Med Genet 43(1):74–83. doi:10.1136/jmg.2005.033258
Ma X, Zhao Y, Li Y, Lu H, He Y (2010) Relevance of Bcl-x expression in different types of endometrial tissues. J Exp Clin Cancer Res 29:14. doi:10.1186/1756-9966-29-14
Maguire SL, Leonidou A, Wai P, Marchio C, Ng CK, Sapino A, Salomon AV, Reis-Filho JS, Weigelt B, Natrajan RC (2015) SF3B1 mutations constitute a novel therapeutic target in breast cancer. J Pathol 235(4):571–580. doi:10.1002/path.4483
Malcovati L, Papaemmanuil E, Bowen DT, Boultwood J, Della Porta MG, Pascutto C, Travaglino E, Groves MJ, Godfrey AL, Ambaglio I, Galli A, Da Via MC, Conte S, Tauro S, Keenan N, Hyslop A, Hinton J, Mudie LJ, Wainscoat JS, Futreal PA, Stratton MR, Campbell PJ, Hellstrom-Lindberg E, Cazzola M, Chronic Myeloid Disorders Working Group of the International Cancer Genome C, of the Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie M (2011) Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood 118(24):6239–6246. doi:10.1182/blood-2011-09-377275
McManus CJ, Graveley BR (2011) RNA structure and the mechanisms of alternative splicing. Curr Opin Genet Dev 21(4):373–379. doi:10.1016/j.gde.2011.04.001
Millevoi S, Bernat S, Telly D, Fouque F, Gladieff L, Favre G, Vagner S, Toulas C (2010) The c.5242C> A BRCA1 missense variant induces exon skipping by increasing splicing repressors binding. Breast Cancer Res Treat 120(2):391–399. doi:10.1007/s10549-009-0392-3
Moffa AB, Tannheimer SL, Ethier SP (2004) Transforming potential of alternatively spliced variants of fibroblast growth factor receptor 2 in human mammary epithelial cells. Mol Cancer Res 2(11):643–652
Nowak DG, Woolard J, Amin EM, Konopatskaya O, Saleem MA, Churchill AJ, Ladomery MR, Harper SJ, Bates DO (2008) Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J Cell Sci 121(Pt 20):3487–3495. doi:10.1242/jcs.016410
Olsson E, Honeth G, Bendahl PO, Saal LH, Gruvberger-Saal S, Ringner M, Vallon-Christersson J, Jonsson G, Holm K, Lovgren K, Ferno M, Grabau D, Borg A, Hegardt C (2011) CD44 isoforms are heterogeneously expressed in breast cancer and correlate with tumor subtypes and cancer stem cell markers. BMC Cancer 11:418. doi:10.1186/1471-2407-11-418
Oltean S, Bates DO (2014) Hallmarks of alternative splicing in cancer. Oncogene 33(46):5311–5318. doi:10.1038/onc.2013.533
Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40(12):1413–1415. doi:10.1038/ng.259
Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, Godfrey AL, Rapado I, Cvejic A, Rance R, McGee C, Ellis P, Mudie LJ, Stephens PJ, McLaren S, Massie CE, Tarpey PS, Varela I, Nik-Zainal S, Davies HR, Shlien A, Jones D, Raine K, Hinton J, Butler AP, Teague JW, Baxter EJ, Score J, Galli A, Della Porta MG, Travaglino E, Groves M, Tauro S, Munshi NC, Anderson KC, El-Naggar A, Fischer A, Mustonen V, Warren AJ, Cross NC, Green AR, Futreal PA, Stratton MR, Campbell PJ, Chronic Myeloid Disorders Working Group of the International Cancer Genome C (2011) Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 365(15):1384–1395. doi:10.1056/NEJMoa1103283
Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, Ramsay AJ, Bea S, Pinyol M, Martinez-Trillos A, Lopez-Guerra M, Colomer D, Navarro A, Baumann T, Aymerich M, Rozman M, Delgado J, Gine E, Hernandez JM, Gonzalez-Diaz M, Puente DA, Velasco G, Freije JM, Tubio JM, Royo R, Gelpi JL, Orozco M, Pisano DG, Zamora J, Vazquez M, Valencia A, Himmelbauer H, Bayes M, Heath S, Gut M, Gut I, Estivill X, Lopez-Guillermo A, Puente XS, Campo E, Lopez-Otin C (2012) Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 44(1):47–52. doi:10.1038/ng.1032
Ramsay AJ, Rodriguez D, Villamor N, Kwarciak A, Tejedor JR, Valcarcel J, Lopez-Guillermo A, Martinez-Trillos A, Puente XS, Campo E, Lopez-Otin C, Quesada V (2013) Frequent somatic mutations in components of the RNA processing machinery in chronic lymphocytic leukemia. Leukemia 27(7):1600–1603. doi:10.1038/leu.2012.344
Relf M, LeJeune S, Scott PA, Fox S, Smith K, Leek R, Moghaddam A, Whitehouse R, Bicknell R, Harris AL (1997) Expression of the angiogenic factors vascular endothelial cell growth factor, acidic and basic fibroblast growth factor, tumor growth factor beta-1, platelet-derived endothelial cell growth factor, placenta growth factor, and pleiotrophin in human primary breast cancer and its relation to angiogenesis. Cancer Res 57(5):963–969
Romero A, Garcia-Garcia F, Lopez-Perolio I, Ruiz de Garibay G, Garcia-Saenz JA, Garre P, Ayllon P, Benito E, Dopazo J, Diaz-Rubio E, Caldes T, de la Hoya M (2015) BRCA1 Alternative splicing landscape in breast tissue samples. BMC Cancer 15:219. doi:10.1186/s12885-015-1145-9
Scott LM, Rebel VI (2013) Acquired mutations that affect pre-mRNA splicing in hematologic malignancies and solid tumors. J Natl Cancer Inst 105(20):1540–1549. doi:10.1093/jnci/djt257
Screaton GR, Bell MV, Bell JI, Jackson DG (1993) The identification of a new alternative exon with highly restricted tissue expression in transcripts encoding the mouse Pgp-1 (CD44) homing receptor. Comparison of all 10 variable exons between mouse, human, and rat. J Biol Chem 268(17):12235–12238
Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, Goulet R Jr, Badve S, Nakshatri H (2006) CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res 8(5):R59. doi:10.1186/bcr1610
Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebryiskaya T, Beroukhim R, Hu M, Halushka MK, Sukumar S, Parker LM, Anderson KS, Harris LN, Garber JE, Richardson AL, Schnitt SJ, Nikolsky Y, Gelman RS, Polyak K (2007) Molecular definition of breast tumor heterogeneity. Cancer Cell 11(3):259–273. doi:10.1016/j.ccr.2007.01.013
Silipo M, Gautrey H, Tyson-Capper A (2015) Deregulation of splicing factors and breast cancer development. J Mol Cell Biol 7(5): 388–401
Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, Butler A, Cheverton A, Gamble J, Hinton J, Jia M, Jayakumar A, Jones D, Latimer C, Lau KW, McLaren S, McBride DJ, Menzies A, Mudie L, Raine K, Rad R, Chapman MS, Teague J, Easton D, Langerod A, Oslo Breast Cancer C, Lee MT, Shen CY, Tee BT, Huimin BW, Broeks A, Vargas AC, Turashvili G, Martens J, Fatima A, Miron P, Chin SF, Thomas G, Boyault S, Mariani O, Lakhani SR, van de Vijver M, van ‘t Veer L, Foekens J, Desmedt C, Sotiriou C, Tutt A, Caldas C, Reis-Filho JS, Aparicio SA, Salomon AV, Borresen-Dale AL, Richardson AL, Campbell PJ, Futreal PA, Stratton MR (2012) The landscape of cancer genes and mutational processes in breast cancer. Nature 486(7403):400–404. doi:10.1038/nature11017
Swami V, Arteche A, Chamorro-Premuzic T, Maakip I, Stanistreet D, Furnham A (2009) Lay perceptions of current and future health, the causes of illness, and the nature of recovery: explaining health and illness in Malaysia. Br J Health Psychol 14(Pt 3):519–540. doi:10.1348/135910708X370781 308587 [pii]
Tammaro C, Raponi M, Wilson DI, Baralle D (2012) BRCA1 exon 11 alternative splicing, multiple functions and the association with cancer. Biochem Soc Trans 40(4):768–772. doi:10.1042/BST20120140
Tjhay F, Motohara T, Tayama S, Narantuya D, Fujimoto K, Guo J, Sakaguchi I, Honda R, Tashiro H, Katabuchi H (2015) CD44 variant 6 is correlated with peritoneal dissemination and poor prognosis in patients with advanced epithelial ovarian cancer. Cancer Sci 106(10):1421–1428. doi:10.1111/cas.12765
Tommasi S, Pilato B, Pinto R, Monaco A, Bruno M, Campana M, Digennaro M, Schittulli F, Lacalamita R, Paradiso A (2008) Molecular and in silico analysis of BRCA1 and BRCA2 variants. Mutat Res 644(1–2):64–70. doi:10.1016/j.mrfmmm.2008.07.005
Twyffels L, Gueydan C, Kruys V (2011) Shuttling SR proteins: more than splicing factors. FEBS J 278(18):3246–3255. doi:10.1111/j.1742-4658.2011.08274.x
Venables JP, Klinck R, Bramard A, Inkel L, Dufresne-Martin G, Koh C, Gervais-Bird J, Lapointe E, Froehlich U, Durand M, Gendron D, Brosseau JP, Thibault P, Lucier JF, Tremblay K, Prinos P, Wellinger RJ, Chabot B, Rancourt C, Elela SA (2008) Identification of alternative splicing markers for breast cancer. Cancer Res 68(22):9525–9531. doi:10.1158/0008-5472.CAN-08-1769
Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, Werner L, Sivachenko A, DeLuca DS, Zhang L, Zhang W, Vartanov AR, Fernandes SM, Goldstein NR, Folco EG, Cibulskis K, Tesar B, Sievers QL, Shefler E, Gabriel S, Hacohen N, Reed R, Meyerson M, Golub TR, Lander ES, Neuberg D, Brown JR, Getz G, Wu CJ (2011) SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med 365(26):2497–2506. doi:10.1056/NEJMoa1109016
Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP (2009) ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell 33(5):591–601. doi:10.1016/j.molcel.2009.01.025
Wei W, Liu W, Cassol CA, Zheng W, Asa SL, Ezzat S (2012) The breast cancer susceptibility gene product fibroblast growth factor receptor 2 serves as a scaffold for regulation of NF-kappaB signaling. Mol Cell Biol 32(22):4662–4673. doi:10.1128/MCB.00935-12
Yae T, Tsuchihashi K, Ishimoto T, Motohara T, Yoshikawa M, Yoshida GJ, Wada T, Masuko T, Mogushi K, Tanaka H, Osawa T, Kanki Y, Minami T, Aburatani H, Ohmura M, Kubo A, Suematsu M, Takahashi K, Saya H, Nagano O (2012) Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat Commun 3:883. doi:10.1038/ncomms1892
Yoshida K, Ogawa S (2014) Splicing factor mutations and cancer. Wiley Interdiscip Rev RNA 5(4):445–459. doi:10.1002/wrna.1222
Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, Chalkidis G, Suzuki Y, Shiosaka M, Kawahata R, Yamaguchi T, Otsu M, Obara N, Sakata-Yanagimoto M, Ishiyama K, Mori H, Nolte F, Hofmann WK, Miyawaki S, Sugano S, Haferlach C, Koeffler HP, Shih LY, Haferlach T, Chiba S, Nakauchi H, Miyano S, Ogawa S (2011) Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478(7367):64–69. doi:10.1038/nature10496
Zhang J, Manley JL (2013) Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov 3(11):1228–1237. doi:10.1158/2159-8290.CD-13-0253
Zhang P, Fu C, Bai H, Song E, Dong C, Song Y (2014) CD44 variant, but not standard CD44 isoforms, mediate disassembly of endothelial VE-cadherin junction on metastatic melanoma cells. FEBS Lett 588(24):4573–4582. doi:10.1016/j.febslet.2014.10.027
Zhang P, Fu C, Bai H, Song E, Dong C, Song Y (2015) Corrigendum to “CD44 variant, but not standard CD44 isoforms, mediate disassembly of endothelial VE-cadherin junction on metastatic melanoma cells” [FEBS Lett 588(24) (2014) 4573–4582]. FEBS Lett 589(4):553. doi:10.1016/j.febslet.2015.01.016
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Gökmen-Polar, Y. (2016). Alternative Splicing in Breast Cancer. In: Badve, S., Gökmen-Polar, Y. (eds) Molecular Pathology of Breast Cancer. Springer, Cham. https://doi.org/10.1007/978-3-319-41761-5_23
Download citation
DOI: https://doi.org/10.1007/978-3-319-41761-5_23
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-41759-2
Online ISBN: 978-3-319-41761-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)