
Abstract
Despite extensive research, the etiology of childhood acute lymphoblastic leukemia (ALL) remains largely unknown. There is growing evidence that childhood ALL arises from in utero chromosomal abnormalities that can lead to clonal expansion of pre-leukemic precursor cells. The risk factors for ALL in children are multiple, most notably common germline polymorphisms and rare genetic syndromes that directly influence hematopoiesis and cell cycling, as well as infection-related aberrant DNA editing.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Acute Lymphoblastic Leukemia
- Fanconi Anemia
- Standardize Incidence Ratio
- Nijmegen Breakage Syndrome
- Childhood Acute Lymphoblastic Leukemia
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1.1 Introduction
Despite extensive research, the etiology of childhood acute lymphoblastic leukemia (ALL) remains largely unknown. There is growing evidence that this cancer may arise from in utero chromosomal abnormalities that can lead to clonal expansion of pre-leukemic precursor cells. The risk factors for ALL in children are multiple, most notably common germline polymorphisms and rare genetic syndromes that directly influence hematopoiesis and cell cycling, as well as possibly infection-related aberrant DNA editing.
1.2 General Epidemiology
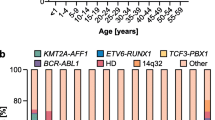
The incidence of ALL varies by age, ethnicity, geographic region, and also differs by immunologic and molecular subtypes. In both the United States and the Nordic countries, the overall incidence rate is 3.9 per 100,000/year before the age of 15 years [1, 2]. The incidence is higher in Hispanic Americans (4.1 per 100,000/year), and is lower in African American children (2.1 per 100,000/year) [3, 4]. In general, low-income countries have lower incidences of ALL than high-income countries, with a few exceptions such as Costa Rica (4.6 per 100,000/year), however these differences may be the result of incomplete registration [4–7]. The incidence of ALL shows a characteristic peak between 2 and 5 years after birth [2, 7], but age-related ALL risk differs substantially by cytogenetic subtype (Fig. 1.1). ALL in infants (<1 year) is in most cases characterized by MLL gene rearrangements (rMLL), which are rare in older children [8–10]. Between 2 and 5-year olds, ALL is dominated by high-hyperdiploid (HeH, modal chromosome number >50) and t(12;21)[ETV6-RUNX1] karyotypes, while T-cell ALL has a less pronounced peak around 4–9 years [2, 10, 11]. In low-income countries, the 2–5 year age peak is much less obvious, with a higher proportion of T-ALL [5–7, 12–15]. Interestingly, some studies noted incremental increase in ALL incidences specifically in this age range as a function of economic growth and improving living conditions [16–19]. Taken together, these observations (1) suggest that different ALL subtypes may have distinctive etiological mechanisms and (2) point toward possible effects of economic development-related environmental factors on ALL risk.

Age distribution of childhood ALL cases by immunologic and molecular subtypes. Numbers represented are all children diagnosed with ALL in Denmark, Sweden, Norway, Finland, and Iceland between 1992–2007. Upper panel: bar heights represent the number of cases in each age group relative to the total number of cases between 0 and 14 years. Lower panel: relative distribution of subtypes within each age group. The testing for t(12;21)[ETV6-RUNX1] by fluoresence in situ hybridization was gradually introduced during this period, and accordingly some amp(21) patients have been missed. Ph+, Philadelphia chromosome-positive; HeH, high-hyperdiploid
1.3 Natural History
Monozygotic twins have a 10–20% concordance rate for ALL, and concordant cases have been shown to harbor identical and clonotypic molecular signatures (e.g. ETV6-RUNX1 fusion sequence, or T-cell receptor (TCR), immunoglobuline (IGH) gene and MLL rearrangements) possibly because a leukemic or preleukemic clone arose prenatally in one twin and spread to the other through placental vascular anastomoses [20–25]. Further evidence for a prenatal initiation is provided by studies backtracking disease-specific molecular markers in both twin and singleton leukemias in dried blood spot samples (DBSS) from birth (Table 1.1).
For infant rMLL ALL, rearrangement has been identified in DBSS in the vast majority of cases, suggesting that this disease almost always arises prenatally. Older patients with rMLL are usually DBSS negative, but the translocation has been successfully backtracked in one case diagnosed at 6 years. Similar findings have been reported for ETV6-RUNX1 ALL. This translocation causes a fusion of the ETV6 and RUNX1 genes, and the resulting chimeric protein has been shown to promote cell survival in mice and human cells [44–46]. Three studies on concordant (monozygotic) twins revealed identical ETV6-RUNX1 fusion sequences in both twins and clonal expansion of fusion-positive precursors at a minimum level of 10−4 preleukemic cells at birth. Prenatally initiated ETV6-RUNX1 + cases have had a latency of up to 14 years before overt leukemia occurred [47]. Furthermore, two ALL-discordant twin pairs have been described in which the healthy twin also harbored an ETV6-RUNX1 + clone at birth [30] or at age 3 [44], suggesting that the translocation in itself is insufficient for leukemia development. Leukemic ETV6-RUNX1 + cells harbor a variable number of additional mutations; often a deletion of the wildtype ETV6 allele or other genes involved in B-lymphocyte development and differentiation [48–51]. Molecular studies of concordant monozygotic twins showed that these mutations are unique to each twin and thus occur as secondary postnatal events [51, 52]. An often cited study found that 1% of all healthy newborns harbored ETV6-RUNX1 at birth (i.e. 100-fold of the incidence of ETV6-RUNX1 ALL) [53], but subsequent validation studies have raised questions about the reliability of the initial finding [54–58]. Thus, while healthy children in general may harbor ETV6-RUNX1 + cells without developing ALL, the exact prevalence of such an event has yet to be determined.
HeH ALL cases frequently have detectable clonotypic IGH rearrangements in neonatal blood spots (17 of 29) and the hyperdiploidy in itself can also arise prenatally [38]. Importantly, a hyperdiploid clone has been found in a healthy twin sibling of a child with HeH ALL [59]. Recently, a whole-genome sequencing approach has further supported the notion that gross chromosomal gains occur early in life as the sentinel event and that additional postnatal events are also necessary for leukemia development [60].
Clonal development of T-ALL is much less understood and leukemic genomic aberration is rarely detected at birth in children with this ALL subtype, suggesting an entirely different etiology compared to B-ALL.
In summary, rMLL, ETV6-RUNX1 +, and HeH ALL show the most convincing evidence of prenatal initiation, while other subtypes such as T-ALL, BCR-ABL and TCF3-PBX1 are less frequently or never prenatally initiated.
1.4 Environmental Risk Factors
1.4.1 Infectious Disease and Immune Stimulation
It has long been hypothesized that infectious disease plays a role in the development of ALL. In 1988, Leo Kinlen postulated that mixing of previously isolated populations could cause epidemics of an unidentified pathogen to which leukemia was a rare response [61]. This hypothesis was based on observed spatial and temporal clustering of ALL cases, which occurs at an exceedingly rare frequency [62]. The same year, Mel Greaves suggested that children with little early-life immune stimulation can develop leukemia as an aberrant response to a delayed exposure to common infections [63]. This ‘delayed-infection hypothesis’, which in many ways is similar to the ‘hygiene hypothesis’ concerning allergies and atopic disease, is particularly relevant to ALL risk in the 2–5 year age peak [64–67]. In these cases, the prenatal formation of a preleukemic clone may constitute a commonly occurring ‘first hit’, and an aberrant immune response due to delayed immune maturation and subsequent uncontrolled proliferative stress on exposure to a common childhood infection occur subsequently will in rare cases cause a second hit and initiate malignant transformation [64, 65].
A substantial body of evidence has been gathered in support of an association between infections and ALL risk. Since the actual number of childhood infections is difficult to measure, proxy measures such as daycare-attendance (children in daycare are more exposed to common infections early in life) are typically examined [68]. A meta-analysis from 2010 by Urayama et al. included 14 studies and a total of 6108 cases and found a significantly reduced risk of ALL among children in daycare (OR = 0.76; 95% CI: 0.67–0.87) [69]. A recent study confirmed this finding and furthermore indicated that the protective effect of daycare is even stronger with earlier start of attendance [70]. Another measure of early immune stimulation is breastfeeding, for which two meta-analyses consistently found an association with a reduced risk of ALL; subsequently a large case-control study with 7,399 ALL cases and 11,181 controls also reported an OR of 0.86 (95% CI: 0.79–0.94) for children breastfed for 6 months or more [70–72]. Other proxies for immune stimulation include birth order and vaccinations, but epidemiological findings on these exposures are inconsistent [70, 73–79]. More direct attempts at measuring actual number of infections during early childhood have included patient registries [80–84], questionnaires [70, 85, 86], and interviews [87–89]. Generally, studies using parentally reported measures found an inverse or no association between infections and ALL risk, while the patient registry-based methods, which have the strength of eliminating recall bias, found either positive or null associations. Interpreting data from these studies is difficult for a number of reasons, most notably the heterogeneity of exposure definitions and the timing of infections in relation to ALL diagnosis. According to the delayed infection hypothesis, children prone to ALL-development should have fewer infections in early life and subsequently start developing aberrant responses to common infections, most likely resulting in symptomatic infectious disease. However, in the months leading up to ALL diagnosis the disease itself also becomes a risk factor for infections, and thus the expected direction of causality between infection and leukemia becomes difficult to identify in such epidemiologic studies [84].
Recent molecular studies have shed new lights on the role of infection in ALL development. Whole-genome sequencing of ETV6-RUNX1 ALL cells revealed that most of the somatic deletions commonly seen in this subtype are mediated by the RAG enzymes, the main function of which is V(D)J recombination in normal pre-B cells [90], potentially as a result of infection-related hyperactivation of RAG. Subsequently, Swaminathan et al. showed that premature activation of the AID enzyme (which normally mediates somatic hypermutation and class-switch recombination in mature B-cells) resulting in inappropriate, synchronous activation of AID and RAG increases genetic instability in pre-B cells, especially those with the ETV6-RUNX1 fusion [91]. The authors furthermore showed that while infectious stimuli (mimicked by lipopolysaccharide) could induce leukemic transformation of ETV6-RUNX1 + cells, this development was delayed or prevented in mice without functional AID or RAG, respectively. Another example highlighting a molecular mechanism involved in infection-mediated ALL development is PAX5, a gene commonly mutated in B-ALL. A recent study showed that PAX5 heterozygous mice were prone to develop ALL, but only if they were exposed to common infections [92]. It is important to note that these molecular studies show that infections are likely involved in ALL development, but provide no direct evidence of how early vs. late infection alters the risk of ALL during childhood.
1.4.2 Other Risk Factors
Despite a large number of epidemiological studies and meta-analyses, most findings regarding proposed environmental risk factors remain inconclusive. The only confirmed association is high birth weight, although the underlying mechanism is unknown [93]. Other factors such as ionizing radiation, electromagnetic fields and maternal smoking during pregnancy remain uncertain (Table 1.2). A common limitation is that the vast majority of these studies address ALL as a single disease entity and thus may have missed associations with specific ALL subtypes.
1.5 Heritability of ALL
Studies addressing the risk of leukemia among offspring of childhood leukemia survivors have been hampered by small sample sizes [123–126]. More reliable estimates of ALL heritability come from studies on risk in siblings of affected children. These studies have two important limitations: first, because of preleukemic cells’ ability to spread in utero, twins with leukemia need to be excluded before estimating disease heritability, and secondly it is difficult to distinguish genetic effects from shared environmental risk factors between siblings. A recent Nordic population-based study reported a standardized incidence ratio (SIR) of 3.2 for ALL risk among siblings [127]. Furthermore, one study investigating 54 sibships with two or more cases of ALL found an unexpectedly high subtype concordance, pointing to a genetic basis of ALL etiology [128]. On the basis of genome-wide SNP data, it was estimated that inherited genetic polymorphisms account for at least 24% (95% CI: 6–42%) of variation in ALL risk [129]. In conclusion, these reports provide evidence for a genetic component in disease susceptibility, although reliable quantitative estimates of genetic contribution to ALL risk are not available.
1.6 High-Penetrance Genetic Predisposition
Out of more than 125 known cancer predisposition genes (CPGs), only 27 genes (associated with 9 rare syndromes and two non-heritable congenital disorders) are convincingly linked to childhood ALL (Table 1.3 and Fig. 1.2) [130, 131].

Effect sizes and frequencies for known ALL genetic risk factors. Syndrome risks and frequencies are based on best available evidence as described in Table 1.3; SNP odds ratios are based on references in Table 1.4, and SNP risk allele frequencies are based on worldwide populations from the 1000 Genomes Project. CMMRD, constitutional mismatch repair-deficiency; AT ataxia-telangiectasia, LFS Li-Fraumeni syndrome, DS Down syndrome, FA Fanconi anemia, NF neurofibromatosis type 1
In a 2015 a registry study of 4939 childhood ALL cases, only 29 subjects were diagnosed with non-Down syndrome (DS) predisposition syndromes (0.6%) [161]. However, a recent comprehensive study of whole genome or whole exome sequencing in 588 non-DS childhood leukemia cases found germline mutations in known CPGs in 26 cases (4.4%) [161, 162]. This suggests that high-penetrance Mendelian genetics, discussed in detail below, may play a larger role in ALL etiology than previously appreciated.
1.6.1 Syndromes Where ALL Is a Dominant Cancer Phenotype
DS is one of the most common congenital abnormalities (1 in 691 live births) and also the most recognizable ALL-predisposition syndrome [140, 163]. ALL and AML risk is significantly increased, with SIR before 30 years of 24.4 and 20.3, respectively. Interestingly, individuals with DS have significantly lower incidence of solid cancers than the background population [142, 164]. DS-associated ALL is more likely to have somatic rearrangements involving the CRLF2 gene and almost always has B-cell immunophenotype. DS patients represent the only known group where ALL is the most common malignancy at any age. Taken together DS-ALL constitutes 2–3% of ALL [131, 165].
While the driver of leukemogenesis remains uncertain for DS it is likely that chromosome 21 is involved, as an acquired extra copy of chromosome 21 is also seen in hyperdiploid ALL and the intracromosomal amplification of chromosome 21 seen in the iAMP21-ALL subtype [139].
In fact, iAMP21-ALL has recently been found to be more frequent in individuals with the germline translocation rob(15;21)(q10;q10)c, a rare constitutional genetic abnormality. Amplification of the genes involved in the translocation duplicates the entire abnormal chromosome and confers an estimated 2,700-fold increased risk of iAMP21-ALL [138]. However, considering the rarity of both iAMP21-ALL and rob(15;21)c, <1 in 1,500 ALL cases are likely to be related to rob(15;21)c associated.
PAX5 is known to be somatically mutated or deleted in approximately 30% of B-ALL cases [166]. In 2013, one germline PAX5 mutation was found in three kindreds of familial ALL [159, 160]. The 3 families had 18 documented and 3 obligate mutation carriers with 11 cases of B-ALL, with another 2 ALLs in untested children. These PAX5 mutations may be exclusively related to ALL risk, but further study is warranted.
ETV6, like PAX5, is known to be recurrently mutated or translocated in leukemic cells [166, 167]. In 2015, three studies independently reported nine families with ETV6 germline mutations, all having a dominantly heritable thrombocytopenia and high incidence of ALL among mutation carriers [143, 144, 146]. Collectively, 35 documented and 4 obligate carriers have developed a total of 14 leukemias (mostly ALL), with another 2 occurring in untested children. One systematic sequencing study targeting germline ETV6 in 4,405 ALL cases, identified 31 ETV6 variants potentially related to 35 ALL cases, with carriers found to be significantly older than non-carriers (mean age 10.2 vs. 4.7) [144]. Thus, ETV6 mutations may be present in nearly 1% of all ALL cases, and perhaps higher in patients over 5 years of age.
1.6.2 Syndromes Where ALL Is Part of a Mixed Cancer Phenotype
Li-Fraumeni Syndrome (LFS) is a rare cancer predisposition syndrome, in which germline TP53 mutation confers a ~90% lifetime risk of developing cancer in a spectrum of tissues with one third being diagnosed before 18 years of age. The increased ALL risk is largely restricted to cases with low hypodiploid leukemia karyotype (underlying TP53 mutation present in 43.3% of low hypodiploid ALL) [168].
Ataxia-telangiectasia (A-T) is a rare syndrome caused by recessive mutations in the ATM gene and typically presents with progressive cerebellar ataxia before 4 years of age [134]. A-T patients have a high risk of leukemias (especially T-cell ALL) and lymphomas, as well as hypersensitivity to ionizing radiation and chemotherapy related to the role of ATM in DNA repair [132, 134].
Bloom Syndrome is characterized by pre- and postnatal growth deficiency (stature typically <1.5 m), skin lesions and high risk of ALL, AML, lymphoma, and epithelial carcinomas [135]. Twelve ALLs were found in less than the 300 cases registered world-wide and in at least two cases ALL preceded Bloom Syndrome diagnosis [169, 170].
Nijmegen Breakage Syndrome (NBS) is another very rare recessive syndrome, which mainly occurs in Slavic populations [157] (a Slavic founder deletion of five bases in the NBN gene is found in >90% of NBS cases), yet NBS has also been described in >8 other countries with private mutations [157, 158, 171, 172]. Patients display microcephaly, intrauterine growth retardation with short stature, recurrent sinopulmonary infections and increased risk of cancers, especially lymphoma and leukemia [157, 173].
Fanconi Anemia (FA) is a rare recessive syndrome with a high risk of AML, MDS and other hematological diseases set at ~10%/year [147, 174]. In a registry with 1300 FA patients only 7 ALLs were reported and FA-leukemias are predominantly myeloid (96%) [147, 148]. While skeletal deformations and classic hematological findings often lead to diagnosis early in life, malignancies including ALL can be the presenting feature [175, 176].
There is a long string of genetic syndromes for which sporadic reports described ALL as a possible cancer manifestation, although the matter has not been systematically examined. The most common are RASopathies (e.g. NF1) [177–179], where 6 ALLs were seen among 1176 mutation carriers in 1 study [180]. Others include: Bruton’s Agammaglobulinemia [181], Familial Platelet Disorder with Associated Myeloid Malignancies [182, 183], Weaver syndrome [184], Sotos syndrome [185], Rubinstein-Taybi syndrome [186], Börjeson-Forssman-Lehmann Syndrome [187] and SH2B3 deficiency [188].
It should be noted that ALL predisposition syndrome may not be symptomatic prior to leukemia diagnosis with only non-specific clinical features such as growth failure and microcephaly. Family history needs to be carefully examined to identify possible underlying genetic causes in a pediatric oncology setting.
1.7 Low-Penetrance Genetic Predisposition
Emerging from the ‘common disease—common variant’ hypothesis, the past two decades have seen the application of first candidate gene-driven and later genome-wide association studies in ALL etiology research [189, 190].
Single nucleotide polymorphism (SNP)-based candidate gene studies (CGSs) have explored ALL etiology by focusing on genes involved in carcinogen metabolism, folate metabolism, and DNA repair pathways. A 2010 systematic review identified 47 CGSs on 25 variations in 16 genes all tested for association with ALL, showing pooled significance (P < 0.05) in only 8 variants (OR range; 0.73–1.78) with an apparent false positive report probabilities of at least 20% [191]. Other studies have focused on human leukocyte antigen (HLA) genes, particularly class II loci HLA-DR and HLA-DP, with the latter showing evidence of significantly different associations between ALL subtypes as well as interactions with proxies for immune stimulation [192, 193]. However, a larger study has cast doubt on the validity of these findings [194].
2009 saw the first two genome-wide association studies (GWAS) independently demonstrating associations between ALL susceptibility and SNPs in ARID5, IKZF1 and CEBPE [195, 196]. Subsequently, SNPs in four other genes have been found to be associated with either overall ALL risk or subtype-specific risk, with a total of 13 SNPs in 6 genes having been widely validated thus far (Table 1.4) [197–201].
The heterogeneity of ALL is reflected in the GWAS findings, with some SNPs showing a stronger association with specific subtypes. ARID5B, for instance, is most strongly associated with HeH ALL. SNPs in TP63 and GATA3, on the other hand, show isolated associations with ETV6-RUNX1 ALL and Ph-like ALL, respectively [196, 199, 201, 204, 205].
While there is little doubt that the GWAS findings identified genuine inherited risk factors for ALL, there is a paucity of studies describing the molecular mechanisms underlying these associations. Somatic deletions in both CDKN2A and IKZF are frequent in ALL, and these two genes play important roles in tumor suppression and lymphocyte development, respectively [200, 206]. In one recent study, 35 tumors from CDKN2A risk variant rs3731217 carriers preferentially retained the risk allele, suggesting that the SNP is advantageous during tumor growth [204]. ARID5B is also involved in lymphocyte differentiation, but its mechanism in ALL development is poorly understood.
Within the validated risk variants, no significant gene–gene interactions have been reported [195, 196, 203]. The effects of these risk alleles are relatively stable across ethnicities, and risk allele frequencies correlate well with population differences in ALL incidence [197]. One pathway-based GWAS on ALL risk was recently described but these results have yet to be reproduced [207]. Inspired by the observations that ALL subtypes differ across both environmental and genetic risk factors, other researchers have attempted to identify interactions between the two by combining genotypes with data on various environmental exposures [208–211]. These studies, however, have so far failed to reliably identify gene-environment interactions.
Studies on childhood ALL etiology will improve knowledge of the pathogenesis, predict disease risk, and provide new targets for treatment.
The low-penetrance genetic predispositions discussed above (e.g., risk alleles identified by GWAS) constitute a minor increase in the absolute risk of developing ALL, e.g. from 1 in 2,000 to 1 in 1,500. While the effects of these variants individually are modest with limited clinical implication, their cumulative impact can be comparable to those of the highly penetrant genetic predisposition syndromes. However, it is debatable whether early diagnosis of an aggressive cancer like ALL can lead to improved outcome [212]. Hence, clinical surveillance aimed at early diagnosis of ALL may not necessarily benefit at-risk subjects and may in fact lead to uncertainty and anxiety for the families [213].
Still, many of the genetic syndromes discussed above may modify health conditions other than the risk of developing ALL. Preemptive surveillance for non-ALL cancers (e.g. TP53 carriers) and/or treatment modification (e.g. avoidance of radiation therapy in cases with A-T) can lead to lower mortality and morbidity for the children and their at-risk family members [214–218]. For this reason, recognition and diagnosis of predisposition syndromes in pediatric oncology is crucial. In fact, it has been suggested that pediatric cancer patients under the age of five should be evaluated for A-T before starting chemotherapy and/or radiotherapy because of potentially fatal adverse effects of conventional doses due to defective DNA repair in these cases [134, 219].
1.8 Future Directions
While substantial progress has been made in identifying risk factors for ALL (especially the role of inherited genetic variants), our understanding of ALL disease etiology is far from complete. An important field of research in the coming years will be to identify gene-gene and gene-environment interactions that contribute to ALL leukemogenesis, and whether approaches can be developed to target these processes and reduce disease risk and burden in genetically predisposal children.
References
SEER Cancer Statistics review 1975–2012 [Internet]. National Cancer Institute; [cited 2016 Feb 29]. Available from: http://seer.cancer.gov/csr/1975_2012/browse_csr.php.
Hjalgrim LL, Rostgaard K, Schmiegelow K, Soderhall S, Kolmannskog S, Vettenranta K, et al. Age- and sex-specific incidence of childhood leukemia by immunophenotype in the nordic countries. JNCI J Natl Cancer Inst. 2003;95(20):1539–44.
Linabery AM, Ross JA. Trends in childhood cancer incidence in the U.S. (1992–2004). Cancer. 2008;112(2):416–32.
Parkin DM, Kramárová E, Draper GJ, Masuyer E, et al. International incidence of childhood cancer. Lyon: IARC Scientific Publications; 1998.
Stiller CA, Parkin DM. Geographic and ethnic variations in the incidence of childhood cancer. Br Med Bull. 1996;52(4):682–703.
Schmiegelow K, Vestergaard T, Nielsen SM, Hjalgrim H. Etiology of common childhood acute lymphoblastic leukemia: the adrenal hypothesis. Leukemia. 2008;22(12):2137–41.
Greaves MF, Colman SM, Beard ME, Bradstock K, Cabrera ME, Chen PM, et al. Geographical distribution of acute lymphoblastic leukaemia subtypes: second report of the collaborative group study. Leukemia. 1993;7(1):27–34.
Biondi A, Cimino G, Pieters R, Pui CH. Biological and therapeutic aspects of infant leukemia. Blood. 2000;96(1):24–33.
Pieters R, Schrappe M, De Paola L, Hann I, De Giulio R, Felice M, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukemia (Interfant-99): an observational study and a multicenter randomized trial. Lancet. 2007;370(9583):240–50.
Szczepanski T, Harrison CJ, van Dongen JJM. Genetic aberrations in paediatric acute leukaemias and implications for management of patients. Lancet Oncol Elsevier Ltd. 2010;11(9):880–889.
Forestier E, Schmiegelow K. The incidence peaks of the childhood acute leukemias reflect specific cytogenetic aberrations. J Pediatr Hematol Oncol. 2006;28(8):486–95.
Rajalekshmy KR, Abitha AR, Pramila R, Gnanasagar T, Shanta V. Immunophenotypic analysis of T-cell acute lymphoblastic leukaemia in Madras, India. Leuk Res. 1997;21(2):119–24.
Gmidène A, Sennana H, Elghezal H, Ziraoui S, Youssef YB, Elloumi M, Issaoui L, Harrabi I, Raynaud SSA. Cytogenetic analysis of 298 newly diagnosed cases of acute lymphoblastic leukaemia in Tunisia. Hematol Oncol. 2008;26:91–7.
Williams CK. Childhood leukemia and lymphoma: African experience supports a role for environmental factors. Cancer Res. 2012;72(8 Suppl):5484.
Liang D-C, Shih L-Y, Yang C-P, Hung I-J, Liu H-C, Jaing T-H, et al. Frequencies of ETV6–RUNX1 fusion and hyperdiploidy in pediatric acute lymphoblastic leukemia are lower in far East than West. Pediatr Blood Cancer. 2010;55:430–3.
McNally RJQ, Birch JM, Taylor GM, Eden OB. Incidence of childhood precursor B-cell acute lymphoblastic leukaemia in north-west England. Lancet. 2000;356:484–5.
Hrusak O, Trka J, Zuna J, Polouckova A, Kalina T, Stary J. Acute lymphoblastic leukemia incidence during socioeconomic transition: selective increase in children from 1 to 4 years. Leukemia. 2002;16:720–5.
Fraumeni JF, Miller RW. Epidemiology of human leukemia. J Natl Cancer Inst. 1967;38(4):593–603.
Spix C, Eletr D, Blettner M, Kaatsch P. Temporal trends in the incidence rate of childhood cancer in Germany 1987–2004. Int J Cancer. 2008;122(8):1859–67.
Greaves MF, Maia AT, Wiemels JL, Ford AM. Leukemia in twins: lessons in natural history. Blood. 2013;102(7):2321–33.
Ford AM, Ridge SA, Cabrera ME, Mahmoud H, Steel CM, Chan LC, et al. In utero rearrangements in the trithorax-related oncogene in infant leukaemias. Nature. 1993;363(6427):358–60.
Gill Super HJ, Rothberg PG, Kobayashi H, Freeman AI, Diaz MO, Rowley JD. Clonal, nonconstitutional rearrangements of the MLL gene in infant twins with acute lymphoblastic leukemia: in utero chromosome rearrangement of 11q23. Blood. 1994;83(3):641–4.
Maia AT, van der Velden VHJ, Harrison CJ, Szczepanski T, Williams MD, Griffiths MJ, et al. Prenatal origin of hyperdiploid acute lymphoblastic leukemia in identical twins. Leukemia. 2003;17(11):2202–6.
Kadan-Lottick NS, Kawashima T, Tomlinson G, Friedman DL, Yasui Y, Mertens AC, et al. The risk of cancer in twins: a report from the childhood cancer survivor study. Pediatr Blood Cancer. 2006;46(5):476–81.
Ford AM, Pombo-de-Oliveira MS, McCarthy KP, MacLean JM, Carrico KC, Vincent RF, et al. Monoclonal origin of concordant T-cell malignancy in identical twins. Blood. 1997;89(1):281–5.
Gale KB, Ford AM, Repp R, Borkhardt A, Keller C, Eden OB, et al. Backtracking leukemia to birth: identification of clonotypic gene fusion sequences in neonatal blood spots. Proc Natl Acad Sci U S A. 1997;94(25):13950–4.
Wiemels JL, Cazzaniga G, Daniotti M, Eden OB, Addison GM, Masera G, et al. Prenatal origin of acute lymphoblastic leukemia in children. Lancet. 1999;354(9189):1499–503.
Fasching K, Panzer S, Haas OA, Marschalek R, Gadner H, Panzer-Grümayer ER. Presence of clone-specific antigen receptor gene rearrangements at birth indicates an in utero origin of diverse types of early childhood acute lymphoblastic leukemia. Blood. 2000;95(8):2722–4.
Yagi T, Hibi S, Tabata Y, Kuriyama K, Teramura T, Hashida T, et al. Detection of clonotypic IGH and TCR rearrangements in the neonatal blood spots of infants and children with B-cell precursor acute lymphoblastic leukemia. Blood. 2000;96(1):264–8.
Maia AT, Ford AM, Reza Jalali G, Harrison CJ, Malcolm Taylor G, Eden OB, et al. Molecular tracking of leukemogenesis in a triplet pregnancy. Blood. 2001;98(2):478–82.
Renate Panzer-Grümayer E, Fasching K, Panzer S, Hettinger K, Schmitt K, Stöckler-lpsiroglu S, et al. Nondisjunction of chromosomes leading to hyperdiploid childhood B-cell precursor acute lymphoblastic leukemia is an early event during leukemogenesis. Blood. 2002;100(1):347–9.
Taub JW, Konrad MA, Ge Y, Naber JM, Scott JS, Matherly LH, et al. High frequency of leukemic clones in newborn screening blood samples of children with B-precursor acute lymphoblastic leukemia. Blood. 2002;99(8):2992–6.
Hjalgrim LL, Madsen HO, Melbye M, Jørgensen P, Christiansen M, Andersen MT, et al. Presence of clone-specific markers at birth in children with acute lymphoblastic leukaemia. Br J Cancer. 2002;87(9):994–9.
Wiemels JL, Leonard BC, Wang Y, Segal MR, Hunger SP, Smith MT, et al. Site-specific translocation and evidence of postnatal origin of the t(1;19) E2A-PBX1 fusion in childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2002;99(23):15101–6.
McHale CM, Wiemels JL, Zhang L, Ma X, Buffler PA, Guo W, et al. Prenatal origin of TEL-AML1-positive acute lymphoblastic leukemia in children born in California. Genes Chromosom Cancer. 2003;37(1):36–43.
Teuffel O, Betts DR, Dettling M, Schaub R, Schäfer BW, Niggli FK. Prenatal origin of separate evolution of leukemia in identical twins. Leukemia. 2004;18(10):1624–9.
Maia AT, Koechling J, Corbett R, Metzler M, Wiemels JL, Greaves M. Protracted postnatal natural histories in childhood leukemia. Genes Chromosom Cancer. 2004;39(4):335–40.
Maia AT, Tussiwand R, Cazzaniga G, Rebulla P, Colman S, Biondi A, et al. Identification of preleukemic precursors of hyperdiploid acute lymphoblastic leukemia in cord blood. Genes Chromosom Cancer. 2004;40(1):38–43.
Fischer S, Mann G, Konrad M, Metzler M, Ebetsberger G, Jones N, et al. Screening for leukemia- and clone-specific markers at birth in children with T-cell precursor ALL suggests a predominantly postnatal origin. Blood. 2007;110(8):3036–8.
Gruhn B, Taub JW, Ge Y, Beck JF, Zell R, Häfer R, et al. Prenatal origin of childhood acute lymphoblastic leukemia, association with birth weight and hyperdiploidy. Leukemia. 2008;22(9):1692–7.
Wiemels JL, Kang M, Chang JS, Zheng L, Kouyoumji C, Zhang L, et al. Backtracking RAS mutations in high hyperdiploid childhood acute lymphoblastic leukemia. Blood Cells Mol Dis. 2010;45(3):186–91.
Eguchi-ishimae M, Eguchi M, Kempski H, Greaves M, Dc W. NOTCH 1 mutation can be an early, prenatal genetic event in T-ALL brief report NOTCH1 mutation can be an early, prenatal genetic event in T-ALL. Blood. 2011;111(1):376–8.
Mansur MB, van Delft FW, Colman SM, Furness CL, Gibson J, Emerenciano M, et al. Distinctive genotypes in infants with T-cell acute lymphoblastic leukaemia. Br J Haematol. 2015;171(4):574–84.
Hong D, Gupta R, Ancliff P, Atzberger A, Brown J, Soneji S, Green J, Colman S, Piacibello W, Buckle V, Tsuzuki S, Mel Greaves TE. Initiating and cancer-propagating cells in TEL-AML1–associated childhood leukemia. Science (80-). 2008;339:1095–9.
Tsuzuki S, Seto M, Greaves M, Enver T. Modeling first-hit functions of the t(12;21) TEL-AML1 translocation in mice. Proc Natl Acad Sci U S A. 2004;101(22):8443–8.
Fischer M, Schwieger M, Horn S, Niebuhr B, Ford A, Roscher S, et al. Defining the oncogenic function of the TEL/AML1 (ETV6/RUNX1) fusion protein in a mouse model. Oncogene. 2005;24(51):7579–91.
Wiemels JL, Ford AM, Van Wering ER, Postma A, Greaves M. Protracted and variable latency of acute lymphoblastic leukemia after TEL-AML1 gene fusion in utero. Blood. 1999;94(3):1057–62.
Lilljebjorn H, Heidenblad M, Nilsson B, Lassen C, Horvat A, Heldrup J, et al. Combined high-resolution array-based comparative genomic hybridization and expression profiling of ETV6/RUNX1-positive acute lymphoblastic leukemias reveal a high incidence of cryptic Xq duplications and identify several putative target genes within the c. Leukemia. 2007;21(10):2137–44.
Tsuzuki S, Karnan S, Horibe K, Matsumoto K, Kato K, Inukai T, et al. Genetic abnormalities involved in t(12;21) TEL-AML1 acute lymphoblastic leukemia: analysis by means of array-based comparative genomic hybridization. Cancer Sci. 2007;98(5):698–706.
Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446(7137):758–64.
Bateman CM, Colman SM, Chaplin T, Young BD, Eden TO, Bhakta M, et al. Acquisition of genome-wide copy number alterations in monozygotic twins with acute lymphoblastic leukemia. Leukemia. 2010;115(17):3553–8.
Ma Y, Dobbins SE, Sherborne AL, Chubb D, Galbiati M, Cazzaniga G, et al. Developmental timing of mutations revealed by whole-genome sequencing of twins with acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2013;110(18):7429–33.
Mori H, Colman SM, Xiao Z, Ford AM, Healy LE, Donaldson C, et al. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci U S A. 2002;99(12):8242–7.
Greaves M, Colman SM, Kearney LFA. To the editor: fusion genes in cord blood. Blood. 2011;117(1):369–70.
Schmiegelow K, Lausten-Thomsen U, Madsen HO, Nersting J, Hjalgrim H. Challenges and pitfalls in the mapping of the natural history of t(12;21)–positive childhood ALL. Blood. 2011;117(1):370–2.
Kusk MS, Lausten-Thomsen U, Andersen MK, Olsen M, Hjalgrim H, Schmiegelow K. False positivity of ETV6/RUNX1 detected by FISH in healthy newborns and adults. Pediatr Blood Cancer. 2014;61:1704–6.
Lausten-thomsen U, Madsen HO, Vestergaard TR, Hjalgrim H, Dc W, Nersting J. Prevalence of t(12;21)[ETV6-RUNX1]-positive cells in healthy neonates. Blood. 2012;117(1):186–9.
Zuna J, Madzo J, Krejci O, Zemanova Z, Kalinova M, Muzikova K, Zapotocky M, Starkova J, Hrusak O, Horak J, Trka J. ETV6/RUNX1 (TEL/AML1) is a frequent prenatal first hit in childhood leukemia. Blood. 2011;117(1):369–70.
Bateman CM, Alpar D, Ford AM, Colman SM, Wren D, Morgan M, et al. Evolutionary trajectories of hyperdiploid ALL in monozygotic twins. Leukemia. Nature Publishing Group. 2014;29(1):58–65.
Paulsson K, Lilljebjörn H, Biloglav A, Olsson L, Rissler M, Castor A, et al. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat Genet Nature Publishing Group. 2015;47(6):672–677.
Kinlen L. Evidence for an infective cause of childhood leukaemia: comparison of a Scottish new town with nuclear reprocessing sites in Britain. Lancet. 1988;332(8624):1323–7.
Kreis C, Grotzer M, Hengartner H, Spycher BD, Spycher BD. Space-time clustering of childhood cancers in Switzerland: a nationwide study. Int J Cancer. 2016;2135:2127.
Greaves MF. Speculations on the cause of childhood acute lymphoblastic leukemia. Leukemia. 1988;2(2):120–5.
Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer. 2006;6(3):193–203.
Wiemels J. Perspectives on the causes of childhood leukemia. Chem Biol Interact. Elsevier Ireland Ltd. 2012;196(3):59–67.
Strachan DP. Hay fever, hygiene, and household size. Br Med J. 1989;299(November):1259–60.
Greaves M. The “delayed infection” (aka “hygiene”) hypothesis for childhood leukaemia. In: The hygiene hypothesis and darwinian medicine. Birkhauser Verlag Basel, Switzerland. 2009. p. 239–55.
Osterholm MT. Infectious disease in child day care: an overview. Pediatrics. 1994;94:987–90.
Urayama KY, Buffler PA, Gallagher ER, Ayoob JM, Ma X. A meta-analysis of the association between day-care attendance and childhood acute lymphoblastic leukaemia. Int J Epidemiol. 2010;39(3):718–32.
Rudant J, Lightfoot T, Urayama KY, Petridou E, Dockerty JD, Magnani C, et al. Childhood acute lymphoblastic leukemia and indicators of early immune stimulation: a childhood leukemia international consortium study. Am J Epidemiol. 2015;181(8):549–62.
Kwan ML, Buffler PA, Abrams B, Kiley VA. Breastfeeding and the risk of childhood leukemia: a meta-analysis. Public Health Rep. 2004;119(6):521–35.
Martin RM, Gunnell D, Owen CG, Smith GD. Breast-feeding and childhood cancer: a systematic review with metaanalysis. Int J Cancer. 2005;117(6):1020–31.
Altieri A, Castro F, Bermejo JL, Hemminki K. Number of siblings and the risk of lymphoma, leukemia, and myeloma by histopathology. Cancer Epidemiol Biomark Prev. 2006;15(7):1281–6.
Von Behren J, Spector LG, Mueller BA, Carozza SE, Chow EJ, Fox EE, et al. Birth order and risk of childhood cancer: a pooled analysis from five US States. Int J Cancer. 2011;128(11):2709–16.
Ma X, Does MB, Metayer C, Russo C, Wong A, Buffler PA. Vaccination history and risk of childhood leukaemia. Int J Epidemiol. 2005;34(5):1100–9.
Auvinen A, Hakulinen T, Groves F. Haemophilus influenzae type B vaccination and risk of childhood leukaemia in a vaccine trial in Finland. Br J Cancer. 2000;83(7):956–8.
Groves FD, Gridley G, Wacholder S, Shu XO, Robison LL, Neglia JP, et al. Infant vaccinations and risk of childhood acute lymphoblastic leukaemia in the USA. Br J Cancer. 1999;81(1):175–8.
MacArthur AC, McBride ML, Spinelli JJ, Tamaro S, Gallagher RP, Theriault GP. Risk of childhood leukemia associated with vaccination, infection, and medication use in childhood: the Cross-Canada childhood leukemia study. Am J Epidemiol. 2008;167(5):598–606.
Mallol-Mesnard N, Menegaux F, Auvrignon A, Auclerc MF, Bertrand Y, Nelken B, et al. Vaccination and the risk of childhood acute leukaemia: the ESCALE study (SFCE). Int J Epidemiol. 2007;36(1):110–6.
Chang JS, Tsai CR, Tsai YW, Wiemels JL. Medically diagnosed infections and risk of childhood leukaemia: a population-based case-control study. Int J Epidemiol. 2012;41(4):1050–9.
Cardwell CR, McKinney PA, Patterson CC, Murray LJ. Infections in early life and childhood leukaemia risk: a UK case-control study of general practitioner records. Br J Cancer. 2008;99(9):1529–33.
Roman E, Simpson J, Ansell P, Kinsey S, Mitchell CD, McKinney PA, et al. Childhood acute lymphoblastic leukemia and infections in the first year of life: a report from the United Kingdom Childhood Cancer Study. Am J Epidemiol. 2007;165(5):496–504.
Vestergaard T, Rostgaard K, Grau K, Schmiegelow K, Hjalgrim H. Hospitalisation for infection prior to diagnosis of acute lymphoblastic leukaemia in children. Pediatr Blood Cancer. 2013;60:428–32.
Crouch S, Lightfoot T, Simpson J, Smith A, Ansell P, Roman E. Infectious illness in children subsequently diagnosed with acute lymphoblastic leukemia: modeling the trends from birth to diagnosis. Am J Epidemiol. 2012;176(5):402–8.
Jourdan-Da Silva N, Perel Y, Méchinaud F, Plouvier E, Gandemer V, Lutz P, et al. Infectious diseases in the first year of life, perinatal characteristics and childhood acute leukaemia. Br J Cancer. 2004;90(1):139–45.
Rosenbaum PF, Buck GM, Brecher ML. Allergy and infectious disease histories and the risk of childhood acute lymphoblastic leukaemia. Paediatr Perinat Epidemiol. 2005;19(2):152–64.
Neglia JP, Linet MS, Shu XO, Severson RK, Potter JD, Mertens AC, et al. Patterns of infection and day care utilization and risk of childhood acute lymphoblastic leukaemia. Br J Cancer. 2000;82(1):234–40.
Perrillat F, Clavel J, Auclerc MF, Baruchel A, Leverger G, Nelken B, Philippe N, Schaison G, Sommelet D, Vilmer EHD. Day-care, early common infections and childhood acute leukaemia: a multicentre French case-control study. Br J Cancer. 2002;86(7):1064–9.
Urayama KY, Ma X, Selvin S, Metayer C, Chokkalingam AP, Wiemels JL, et al. Early life exposure to infections and risk of childhood acute lymphoblastic leukemia. Int J Cancer. 2011;128(7):1632–43.
Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet Nature Publishing Group. 2014;46(2):116–125.
Swaminathan S, Klemm L, Park E, Papaemmanuil E, Ford A, Kweon S-M, et al. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat Immunol. 2015;16(7):766–74.
Martin-Lorenzo A, Hauer J, Vicente-Duenas C, Auer F, Gonzalez-Herrero I, Garcia-Ramirez I, et al. Infection exposure is a causal factor in B-precursor acute lymphoblastic leukemia as a result of Pax5 inherited susceptibility. Cancer Discov. 2015;5:1328–43.
Hjalgrim LL, Westergaard T, Rostgaard K, Schmiegelow K, Melbye M, Hjalgrim H, et al. Birth weight as a risk factor for childhood leukemia: a meta-analysis of 18 epidemiologic studies. Am J Epidemiol. 2003;158(8):724–35.
Doll R, Wakeford R. Risk of childhood cancer from fetal irradiation. Br J Radiol. The British Institute of Radiology. 1997;70(830):130–139.
Mathews JD, Forsythe AV, Brady Z, Butler MW, Goergen SK, Byrnes GB, et al. Cancer risk in 680,000 people exposed to computed tomography scans in childhood or adolescence: data linkage study of 11 million Australians. BMJ. 2013;346:f2360.
Wakeford R. The cancer epidemiology of radiation. Oncogene. 2004;23(38):6404–28.
Brenner DJ, Doll R, Goodhead DT, Hall EJ, Land CE, Little JB, et al. Cancer risks attributable to low doses of ionizing radiation: assessing what we really know. Proc Natl Acad Sci U S A. 2003;100(24):13761–6.
Wakeford R, Kendall GM, Little MP. The proportion of childhood leukaemia incidence in Great Britain that may be caused by natural background ionizing radiation. Leukemia Nature Publishing Group. 2009;23(4):770–776.
Raaschou-Nielsen O, Andersen CE, Andersen HP, Gravesen P. Domestic radon and childhood cancer in Denmark. Epidemiology. 2008;19(4):536–43.
Tong J, Qin L, Cao Y, Li J, Zhang J, Nie J, et al. Environmental radon exposure and childhood leukemia. J Toxic Environ Health B. 2012;15(5):332–47.
Hauri D, Spycher B, Huss A, Zimmermann F, Grotzer M, von der Weid N, Weber D, Spoerri A, Kuehni CRM. Domestic radon exposure and risk of childhood cancer: a prospective census-based cohort study. Environ Health Perspect. 2013;121(10):1239–44.
Schüz J, Ahlbom A. Exposure to electromagnetic fields and the risk of childhood leukaemia: a review. Radiat Prot Dosim. 2008;132(2):202–11.
Schüz J. Exposure to extremely low-frequency magnetic fields and the risk of childhood cancer: update of the epidemiological evidence. Prog Biophys Mol Biol. 2011;107(3):339–42.
Schüz J, Dasenbrock C, Ravazzani P, Röösli M, Schär P, Bounds P, et al. Extremely low-frequency magnetic fields and the risk of childhood leukemia: a risk assessment by the ARIMMORA consortium. Environ Health Perspect. 2015;37:1–7.
Merzenich H, Schmiedel S, Bennack S, Brüggemeyer H, Philipp J, Blettner M, et al. Childhood leukemia in relation to radio frequency electromagnetic fields in the vicinity of TV and radio broadcast transmitters. Am J Epidemiol. 2008;168(10):1169–78.
Elliott P, Toledano MB, Bennett J, Beale L, de Hoogh K, Best N, et al. Mobile phone base stations and early childhood cancers: case-control study. BMJ. 2010;340:c3077.
Sergentanis TN, Thomopoulos TP, Gialamas SP, Karalexi MA, Biniaris-Georgallis SI, Kontogeorgi E, et al. Risk for childhood leukemia associated with maternal and paternal age. Eur J Epidemiol. Springer Netherlands. 2015;30(12):1229–1261.
Yan K, Xu X, Xu X, Liu X, Wang X, Hua S, et al. The associations between maternal factors during pregnancy and the risk of childhood acute lymphoblastic leukemia: a, meta-analysis. Pediatr Blood Cancer. 2015;14(5):1526–31.
Orsi L, Rudant J, Ajrouche R, Leverger G, Baruchel A, Nelken B, et al. Parental smoking, maternal alcohol, coffee and tea consumption during pregnancy, and childhood acute leukemia: the ESTELLE study. Cancer Causes Control Springer International Publishing. 2015;26(7):1003–1017.
Ferreira JD, Couto AC, Emerenciano M, Pombo-de-Oliveira MS, Koifman S. Maternal alcohol consumption during pregnancy and early age leukemia risk in Brazil. Biomed Res Int. 2015;2015:732495.
Milne E, Greenop KR, Scott RJ, de Klerk NH, Bower C, Ashton LJ, et al. Parental alcohol consumption and risk of childhood acute lymphoblastic leukemia and brain tumors. Cancer Causes Control. 2013;24(2):391–402.
Liu R, Zhang L, McHale CM, Hammond SK. Paternal smoking and risk of childhood acute lymphoblastic leukemia: systematic review and meta-analysis. J Oncol. 2011;2011:16.
Dockerty JD, Herbison P, Skegg DCG, Elwood M. Vitamin and mineral supplements in pregnancy and the risk of childhood acute lymphoblastic leukaemia: a case-control study. BMC Public Health. 2007;7:136.
Milne E, Royle JA, Miller M, Bower C, De Klerk NH, Bailey HD, et al. Maternal folate and other vitamin supplementation during pregnancy and risk of acute lymphoblastic leukemia in the offspring. Int J Cancer. 2010;126(11):2690–9.
Goh YI, Bollano E, Einarson TR, Koren G. Prenatal multivitamin supplementation and rates of pediatric cancers: a meta-analysis. Clin Pharmacol Ther. 2007;81(5):685–91.
Fear NT, Roman E, Ansell P, Simpson J, Day N, Eden OB. Vitamin K and childhood cancer: a report from the United Kingdom Childhood Cancer Study. Br J Cancer. 2003;89:1228–31.
Parker L, Cole M, Craft AW, Hey EN. Neonatal vitamin K administration and childhood cancer in the north of England: retrospective case-control study. BMJ. 1998;316:189–93.
Infante-Rivard C, Weichenthal S. Pesticides and childhood cancer: an update of Zahm and Ward’s 1998 review. J Toxic Environ Health B. 2007;10(1–2):81–99.
Nasterlack M. Pesticides and childhood cancer: An update. Int J Hyg Environ Health. 2007;210(5):645–57.
Bailey HD, Infante-Rivard C, Metayer C, Clavel J, Lightfoot T, Kaatsch P, et al. Home pesticide exposures and risk of childhood leukemia: findings from the childhood leukemia international consortium. Int J Cancer. 2015;137(11):2644–63.
Alexander FE, Patheal SL, Biondi A, Fusion MLLG, Brandalise S, Cabrera M, et al. Transplacental chemical exposure and risk of infant leukemia with MLL gene. Fusion. 2001;61:2542–6.
Spector LG, Xie Y, Robison LL, Spector LG, Xie Y, Robison LL, et al. Maternal diet and infant leukemia: the DNA topoisomerase II inhibitor hypothesis: a report from the children ’ s oncology group. Cancer Epidemiol Biomark Prev. 2005;14:651–5.
Sankila R, Olsen JH, Anderson H, Garwicz S, Glattre E, Hertz H, Langmark F, Lanning M, Møller T, Hraf P. Risk of cancer among offspring of childhood-cancer survivors. N Engl J Med. 1998:1339–44.
Hawkins MM, Draper GJ, Winter DL. Cancer in the offspring of survivors of childhood leukaemia and non-Hodgkin lymphomas. Br J Cancer. 1995;71(6):1335–9.
Mulvihill JJ, Connelly RR, Austin DF, Cook JW, Holmes FF, Krauss MR, et al. Cancer in offspring of long-term survivors of childhood and adolescent cancer. Lancet. 1987;2283:813–7.
Hawkins M, Draper G, Smith R. Cancer among 1,348 offspring of survivors of childhood cancer. Int J Cancer. 1989;43:975–8.
Kharazmi E, da Silva Filho MI, Pukkala E, Sundquist K, Thomsen H, Hemminki K. Familial risks for childhood acute lymphocytic leukaemia in Sweden and Finland: far exceeding the effects of known germline variants. Br J Haematol. 2012;159:585–8.
Schmiegelow K, Lausten Thomsen U, Baruchel A, Pacheco CE, Pieters R, Pombo-de-Oliveira MS, et al. High concordance of subtypes of childhood acute lymphoblastic leukemia within families: a from sibships with multiple cases of leukemia. Leukemia. 2012;26(4):675–81.
Enciso-Mora V, Hosking FJ, Sheridan E, Kinsey SE, Lightfoot T, Roman E, et al. Common genetic variation contributes significantly to the risk of childhood B-cell precursor acute lymphoblastic leukemia. Leukemia. Nature Publishing Group. 2012;26(10):2212–2215.
Rahman N. Realizing the promise of cancer predisposition genes. Nature. 2014;505(7483):302–8.
Kratz CP, Stanulla M, Cavé H. Genetic predisposition to acute lymphoblastic leukemia: overview on behalf of the I-BFM ALL Host Genetic Variation Working Group. Eur J Med Genet. 2015;59:1–5.
Morrell D, Cromartie E, Swift M. Mortality and cancer incidence in 263 patients with ataxia-telangiectasia. J Natl Cancer Inst. 1986;77(1):89–92.
Liberzon E, Avigad S, Stark B, Zilberstein J, Freedman L, Gorfine M, et al. Germ-line ATM gene alterations are associated with susceptibility to sporadic T-cell acute lymphoblastic leukemia in children. Genes Chromosom Cancer. 2004;39(2):161–6.
Gatti R. Ataxia-Telangiectasia. 1999 Mar 19 [Updated 2010 Mar 11]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 1993–2016.
Sanz MM, German J. Bloom’s syndrome. 2006 Mar 22 [Updated 2013 Mar 28]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 1993–2016.
Wimmer K, Kratz CP, Vasen HFA, Caron O, Colas C, Entz-Werle N, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium “care for CMMRD” (C4CMMRD). J Med Genet. 2014;51(6):355–65.
Malkin D, Nichols KE, Zelley K, Schiffman JD. Predisposition to pediatric and hematologic cancers: a moving target. Am Soc Clin Oncol Educ Book. 2014;34:e44–55.
Li Y, Schwab C, Ryan SL, Papaemmanuil E, Robinson HM, Jacobs P, et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature. 2014;508(7494):98–102.
Harrison CJ, Schwab C. Constitutional abnormalities of chromosome 21 predispose to iAMP21-acute lymphoblastic leukaemia. Eur J Med Genet. Elsevier Masson SAS. 2016;59(3):162–165.
Hickey F, Hickey E, Summar KL. Medical update for children with down syndrome for the pediatrician and family practitioner. Adv Pediatr Elsevier Inc. 2012;59(1):137–157.
Santoro SL, Martin LJ, Hopkin RJ. Screening for hematological disorders in mosaic down syndrome: parent report of experiences. Clin Pediatr (Phila). 2016;55(5):421–7.
Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with down’s syndrome. Lancet. 2000;355(9199):165–9.
Topka S, Vijai J, Walsh MF, Jacobs L, Maria A, Villano D, et al. Germline ETV6 mutations confer susceptibility to acute lymphoblastic leukemia and thrombocytopenia. PLoS Genet. 2015;11(6):e1005262.
Moriyama T, Metzger ML, Wu G, Nishii R, Qian M, Devidas M, et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: a systematic genetic study. Lancet Oncol Elsevier Ltd. 2015;16(16):1659–1666.
Zhang MY, Churpek JE, Keel SB, Walsh T, Lee MK, Loeb KR, et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet. 2015;47(2):180–5.
Noetzli L, Lo RW, Lee-Sherick AB, Callaghan M, Noris P, Savoia A, et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet Nature Publishing Group. 2015;47(5):535–538.
Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010;24:101–22.
Alter BP. Cancer in Fanconi anemia, 1927–2001. Cancer. 2003;97(2):425–40.
Alter BP, Kupfer G. Fanconi Anemia. 2002 Feb 14 [Updated 2013 Feb 7]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 1993–2016.
Swift M. Fanconi’s anaemia in the genetics of neoplasia. Nature. 1971;230(5293):370–3.
Bougeard G, Renaux-Petel M, Flaman JM, Charbonnier C, Fermey P, Belotti M, et al. Revisiting Li-Fraumeni syndrome from TP53 mutation carriers. J Clin Oncologia. 2015;33(21):2345–52.
Lalloo F, Varley J, Ellis D, Moran A, O’Dair L, Pharoah P, et al. Prediction of pathogenic mutations in patients with early-onset breast cancer by family history. Lancet. 2003;361(9363):1101–2.
Gonzalez KD, Noltner KA, Buzin CH, Gu D, Wen-Fong CY, Nguyen VQ, et al. Beyond li fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol. 2009;27(8):1250–6.
Stiller CA, Chessells JM, Fitchett M. Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer. 1994;70(5):969–72.
Evans DG, Howard E, Giblin C, Clancy T, Spencer H, Huson SM, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet Part A. 2010;152(2):327–32.
Lammert M, Friedman JM, Kluwe L, Mautner VF. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch Dermatol. 2005;141(1):71–4.
Pastorczak A, Górniak P, Sherborne A, Hosking F, Trelińska J, Lejman M, et al. Role of 657del5 NBN mutation and 7p12.2 (IKZF1), 9p21 (CDKN2A), 10q21.2 (ARID5B) and 14q11.2 (CEBPE) variation and risk of childhood ALL in the Polish population. Leuk Res. 2011;35(11):1534–6.
Ochs H, Smith E, Puck J. Primary immunodeficiency diseases: a molecular and cellular approach. New York: Oxford University Press; 2013. 911 p.
Shah S, Schrader KA, Waanders E, Timms AE, Vijai J, Miething C, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet. 2013;45(10):1226–31.
Auer F, Rüschendorf F, Gombert M, Husemann P, Ginzel S, Izraeli S, et al. Inherited susceptibility to pre B-ALL caused by germline transmission of PAX5 c.547G>A. Leukemia. 2014;28(5):1136–8.
Schütte P, Möricke A, Zimmermann M, Bleckmann K, Reismüller B, Attarbaschi A, et al. Preexisting conditions in pediatric ALL patients: spectrum, frequency and clinical impact. Eur J Med Genet. 2016;59(3):143–51.
Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373(24):2336–46.
Palomaki GE, Kloza EM, Lambert-Messerlian GM, Haddow JE, Neveux LM, Ehrich M, et al. DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation study. Genet Med. 2011;13(11):913–20.
Tabarés-Seisdedos R, Dumont N, Baudot A, Valderas JM, Climent J, Valencia A, et al. No paradox, no progress: Inverse cancer comorbidity in people with other complex diseases. Lancet Oncol Elsevier Ltd. 2011;12(6):604–608.
Lundin C, Forestier E, Klarskov Andersen M, Autio K, Barbany G, Cavelier L, et al. Clinical and genetic features of pediatric acute lymphoblastic leukemia in down syndrome in the Nordic countries. J Hematol Oncol. 2014;7(1):32.
Kuiper RP, Schoenmakers EFPM, van Reijmersdal SV, Hehir-Kwa JY, van Kessel AG, van Leeuwen FN, et al. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. 2007;21(6):1258–66.
Van Vlierberghe P, Ambesi-Impiombato A, Perez-Garcia A, Haydu JE, Rigo I, Hadler M, et al. ETV6 mutations in early immature human T cell leukemias. J Exp Med. 2011;208(13):2571–9.
Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45(3):242–52.
Adams M, Jenney M, Lazarou L, White R, Birdsall S. Europe PMC funders Group Acute myeloid leukaemia after treatment for acute lymphoblastic leukaemia in girl with Bloom syndrome. J Genet Syndr Gene Ther. 2014;4(8).
Werner-Favre C, Wyss M, Cabrol C, Felix F, Guenin R, Laufer D, et al. Cytogenetic study in a mentally retarded child with bloom syndrome and acute lymphoblastic leukemia. Am J Med Genet. 1984;18(2):215–21.
Pasic S, Vujic D, Fiorini M, Notarangelo LD. T-cell lymphoblastic leukemia/lymphoma in Nijmegen breakage syndrome. Haematologica. 2004;89(8):91–2.
Varon R, Vissinga C, Platzer M, Cerosaletti KM, Chrzanowska KH, Saar K, et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell. 1998;93(3):467–76.
The International NBS Study Group. Nijmegen breakage syndrome. Arch Dis Child. 2000;82(5):400–6.
Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anemia. Blood. 2003;101(3):822–6.
Spinella J-F, Healy J, Saillour V, Richer C, Cassart P, Ouimet M, et al. Whole-exome sequencing of a rare case of familial childhood acute lymphoblastic leukemia reveals putative predisposing mutations in Fanconi anemia genes. BMC Cancer. 2015;15:539.
Mushtaq N, Wali R, Fadoo Z, Saleem AF. Acute lymphoblastic leukemia in a child with fanconi’s anaemia. J Coll Physicians Surg Pakistan. 2012;22(7):458–60.
Kratz CP, Rapisuwon S, Reed H, Hasle H, Rosenberg PS. Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet Part C Semin Med Genet. 2011;157(2):83–9.
Kratz CP, Franke L, Peters H, Kohlschmidt N, Kazmierczak B, Finckh U, Bier A, Eichhorn B, Blank C, Kraus C, Kohlhase J, Pauli S, Wildhardt G, Kutsche K, Auber B, Christmann A, Bachmann N, Mitter D, Cremer FW, Mayer K, Duamer-Haas A, Zenker M. Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br J Cancer. 2015;Apr 14;112:1392–7.
Smpokou P, Zand DJ, Rosenbaum KN, Summar ML. Malignancy in Noonan syndrome and related disorders. Clin Genet. 2015;88(6):516–22.
Cavé H, Caye A, Strullu M, Aladjidi N, Vignal C, Ferster A, et al. Acute lymphoblastic leukemia in the context of RASopathies. Eur J Med Genet. 2016;59(3):173–8.
Hoshino A, Okuno Y, Migita M, Ban H, Yang X, Kiyokawa N, et al. X-Linked agammaglobulinemia associated with B-precursor acute lymphoblastic leukemia. J Clin Immunol. 2015;35(2):108–11.
Prebet T, Carbuccia N, Raslova H, Favier R, Rey J, Arnoulet C, et al. Concomitant germ-line RUNX1 and acquired ASXL1 mutations in a T-cell acute lymphoblastic leukemia. Eur J Haematol. 2013;91(3):277–9.
Linden T, Schnittger S, Groll AH, Juergens H, Rossig C. Childhood B-cell precursor acute lymphoblastic leukaemia in a patient with familial thrombocytopenia and RUNX1 mutation. Br J Haematol. 2010;151(5):528–30.
Basel-Vanagaite L. Acute lymphoblastic leukemia in weaver syndrome. Am J Med Genet A. 2010;152(2):383–6.
Tatton-Brown K, Rahman N. The NSD1 and EZH2 overgrowth genes, similarities and differences. Am J Med Genet Part C Semin Med Genet. 2013;163(2):86–91.
Report C. Rubinstein- Taybi syndrome and acute leukemia Pseudohernaturia in neonates. J Pediatr. 1978;92(5):8–9.
Chao MM, Todd MA, Kontny U, Neas K, Sullivan MJ, Hunter AG, et al. T-cell acute lymphoblastic leukemia in association with Borjeson-Forssman-Lehmann syndrome due to a mutation in PHF6. Pediatr Blood Cancer. 2010;55(4):722–4.
Perez-Garcia A, Ambesi-Impiombato A, Hadler M, Rigo I, LeDuc CA, Kelly K, et al. Genetic loss of SH2B3 in acute lymphoblastic leukemia. Blood. 2013;122(14):2425–32.
Lander ES. The new genomics: global views of biology. Science. 1996;274(5287):536–9.
Gibson G. Rare and common variants: twenty arguments. Nat Rev Genet Nature Publishing Group. 2012;13(2):135–145.
Vijayakrishnan J, Houlston RS. Candidate gene association studies and risk of childhood acute lymphoblastic leukemia: a systematic review and meta-analysis. Haematologica. 2010;95(8):1405–14.
Urayama KY, Thompson PD, Taylor M, Trachtenberg EA, Chokkalingam AP. Genetic variation in the extended major histocompatibility complex and susceptibility to childhood acute lymphoblastic leukemia: a review of the evidence. Front Oncol. 2013;3:300.
Urayama KY, Chokkalingam AP, Metayer C, Ma X, Selvin S, Barcellos LF, et al. HLA-DP genetic variation, proxies for early life immune modulation and childhood acute lymphoblastic leukemia risk. Blood. 2012;120(15):3039–47.
Hosking FJ, Leslie S, Dilthey A, Moutsianas L, Wang Y, Sara E, et al. MHC variation and risk of childhood B-cell precursor acute lymphoblastic leukemia. Blood. 2012;117(5):1633–40.
Papaemmanuil E, Hosking FJ, Vijayakrishnan J, Price A, Olver B, Sheridan E, et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1006–10.
Treviño LR, Yang W, French D, Hunger SP, Carroll WL, Devidas M, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1001–5.
Xu H, Yang W, Perez-Andreu V, Devidas M, Fan Y, Cheng C, et al. Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst. 2013;105(10):733–42.
Migliorini G, Fiege B, Hosking FJ, Ma Y, Kumar R, Sherborne AL, et al. Variation at 10p12. 2 and 10p14 in fl uences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood. 2013;122(19):3298–307.
Perez-Andreu V, Roberts KG, Harvey RC, Yang W, Cheng C, Pei D, et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet. 2013;45(12):1494–8.
Xu H, Zhang H, Yang W, Yadav R, Morrison AC, Qian M, et al. Inherited coding variants at the CDKN2A locus influence susceptibility to acute lymphoblastic leukaemia in children. Nat Commun. Nat Publ Group. 2015;6:7553.
Ellinghaus E, Stanulla M, Richter G, Ellinghaus D, te Kronnie G, Cario G, et al. Identification of germline susceptibility loci in ETV6-RUNX1-rearranged childhood acute lymphoblastic. Leukemia. 2012;26(5):902–9.
Prasad RB, Hosking FJ, Vijayakrishnan J, Papaemmanuil E, Koehler R, Greaves M, et al. Verification of the susceptibility loci on 7p12.2, 10q21.2, and 14q11.2 in precursor B-cell acute lymphoblastic leukemia of childhood. Blood. 2010;115(9):1765–7.
Sherborne AL, Hosking FJ, Prasad RB, Kumar R, Koehler R, Vijayakrishnan J, et al. Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet. 2010;42(6):492–4.
Walsh KM, de Smith AJ, Hansen HM, Smirnov IV, Gonseth S, Endicott AA, et al. A heritable missense polymorphism in CDKN2A confers strong risk of childhood acute lymphoblastic leukemia and is preferentially selected during clonal evolution. Cancer Res. 2015;75(22):4884–94.
Moriyama T, Relling MV, Yang JJ. Inherited genetic variation in childhood acute lymphoblastic. Leukemia. 2015;125(26):3988–96.
Mullighan CG, Su X, Zhang J, Radtke I, Phillips LAA, Miller CB, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. New England Journal of Medicine. 2009;360:470–80.
Hsu L-I, Briggs F, Shao X, Metayer C, Wiemels JL, Chokkalingam AP, et al. Pathway analysis of genome-wide association study in childhood leukemia among Hispanics. Cancer Epidemiol Biomark Prev [Internet]. 2016; Available from: http://cebp.aacrjournals.org/cgi/doi/10.1158/1055-9965.EPI-15-0528.
Evans TJ, Milne E, Anderson D, De Klerk NH, Jamieson SE, Talseth-Palmer BA, et al. Confirmation of childhood acute lymphoblastic leukemia Variants, ARID5B and IKZF1, and interaction with parental environmental exposures. PLoS One. 2014;9(10):e110255.
Hsu LI, Chokkalingam AP, Briggs FBS, Walsh K, Crouse V, Fu C, et al. Association of genetic variation in IKZF1, ARID5B, and CEBPE and surrogates for early-life infections with the risk of acute lymphoblastic leukemia in Hispanic children. Cancer Causes Control. 2015;26(4):609–19.
Linabery AM, Blommer CN, Spector LG, Davies SM, Robison LL, Ross JA. ARID5B and IKZF1 variants, selected demographic factors, and childhood acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Leuk Res. Elsevier Ltd. 2013;37(8):936–942.
Rudant J, Orsi L, Bonaventure A, Goujon-Bellec S, Baruchel A, Petit A, et al. ARID5B, IKZF1 and non-genetic factors in the etiology of childhood acute lymphoblastic leukemia: the ESCALE study. PLoS One. 2015;10(3):1–16.
Baker JM, To T, Beyene J, Zagorski B, Greenberg ML, Sung L. Influence of length of time to diagnosis and treatment on the survival of children with acute lymphoblastic leukemia: a population-based study. Leuk Res. Elsevier Ltd. 2014;38(2):204–209.
Strahm B, Malkin D. Hereditary cancer predisposition in children: genetic basis and clinical implications. Int J Cancer. 2006;119(9):2001.
Villani A, Tabori U, Schiffman J, Shlien A, Beyene J, Druker H, et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study. Lancet Oncol. 2011;12(6):559–67.
Knapke BS, Zelley K, Nichols KE, Kohlmann W, Schiffman JD. Identification, management, and evaluation of children with cancer-predisposition syndromes. Am Soc Clin Oncol Educ Book 2012:576–84.
Durno CA, Aronson M, Tabori U, Malkin D, Gallinger S, Chan HSL. Oncologic surveillance for subjects with biallelic mismatch repair gene mutations: 10 year follow-up of a kindred. Pediatr Blood Cancer. 2012;59(4):652–6.
Pastorczak A, Stolarska M, Trelińska J, Zawitkowska J, Kowalczyk J, Mlynarski W. Nijmegen breakage syndrome (NBS) as a risk factor for CNS involvement in childhood acute lymphoblastic leukemia. Pediatr Blood Cancer. 2011;57(1):160–2.
Pastorczak A, Szczepanski T, Mlynarski W. Clinical course and therapeutic implications for lymphoid malignancies in Nijmegen breakage syndrome. Eur J Med Genet. Elsevier Masson SAS. 2016;59(3):126–132.
Hersby DS, Sehested A, Kristensen K, Schmiegelow K. T-cell ALL in ataxia telangiectasia cured with only 7 weeks of anti-leukemic therapy. J Pediatr Hematol Oncol. 2014;37(2):154–5.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Tulstrup, M., Stoltze, U.K., Schmiegelow, K., Yang, J.J. (2017). Epidemiology and Etiology of Childhood ALL. In: Vora, A. (eds) Childhood Acute Lymphoblastic Leukemia. Springer, Cham. https://doi.org/10.1007/978-3-319-39708-5_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-39708-5_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-39707-8
Online ISBN: 978-3-319-39708-5
eBook Packages: MedicineMedicine (R0)