
Abstract
Human induced pluripotent stem cells (iPSCs) represent a personalized stem cell source and enable research using the human as a model genetic system. Although iPSCs have been available for nearly a decade, simple application of efficient genome modification—a mainstay of genetics in the long-used mouse model—has only recently come to fruition. Recombinant and programmable nucleases induce targeted DNA damage and exploit native DNA-repair machinery to generate random mutations or designer modifications through a template-mediated process. In this review, we provide an overview of state-of-the-art nuclease technologies such as ZFN, TALEN, and CRISPR/Cas9 and their utility for genome engineering of human iPSCs. We explore how nucleases may be used to edit the genome with base-pair precision, and methods for the detection and avoidance of off-target cleavage. Finally, we highlight sources of genetic and technical variation in iPSCs, and propose resolutions to the question of appropriate isogenic controls.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Human induced pluripotent stem cell
- iPSC
- Gene targeting
- ZFN
- TALEN
- CRISPR/Cas9
- Nuclease
- Disease model
- Gene correction
- Isogenic controls
Induced Pluripotency and the Human Genetic Model Organism In Vitro
From the inner cell mass (ICM ) of fertilized embryos, James Thomson first derived human embryonic stem cells (ESCs) [1]. These novel cells have two key properties: first, they are capable of indefinite cell division in culture (self-renewal), and second, as do their biological counterparts, they maintain the capacity to differentiate into all cells and tissues of the embryo and adult (pluripotency). The unchallenged advantage of human ESCs over other experimental cell systems has been this capacity for differentiation, either in vivo via teratoma [2] or in vitro via adherent or three-dimensional (3D) cell culture [3, 4]. An application in disease research and regenerative medicine for ESCs was immediately apparent; however, the embryonic source of material has remained an ethical controversy [5].
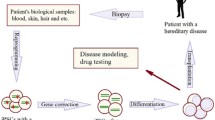
One decade has now passed since Kazutoshi Takahashi and Shinya Yamanaka first demonstrated that mouse somatic cells could be reverted through the expression of four transcription factors, Oct3/4, Sox2, Klf4, and c-Myc, to a primitive embryonic-like stem cell state [6]. Derivation of induced pluripotent stem cells (iPSCs) from human somatic cells (Fig. 1) followed shortly thereafter [7], marking a partial ethical resolution and profound technical contribution [8]. Compared to ESCs, iPSCs present an additional benefit for disease modeling and putative therapies: derived from a consenting individual, they represent personalized stem cells. Moreover, in contrast to ESCs from terminated embryos, iPSCs may be linked to the health and well-being of a living person, complemented by a recorded lifetime medical history. Thus, combined with in vitro differentiation to cells and tissues (Fig. 1), human iPSCs present a proxy by which individualized genetic variation may be accessed to understand the relevance to personal health [9].

The source, characteristics, and applications of human pluripotent stem cells. Genome engineering is applied to generate or validate human models of development and disease
Pluripotent stem cells (PSCs )—whether ESCs derived from the human embryo or iPSCs derived through reprogramming—display key properties of a tractable genetic system: a short generation time (~15 h), a high proportion of cells in S-phase [10], indefinite proliferation, ease of culture, a propensity for DNA transduction, and selection by antibiotics or genetic complementation followed by clonal isolation and expansion. With more recent advances such as Rho-kinase inhibition for improved single-cell survival [11], and feeder-free cell culture methods using defined matrices and media [12, 13], human PSC handling is more akin to murine PSC counterparts by means of single cell passage, and high-throughput 96-well clonal maintenance and expansion [14].
As is explored in the following sections, the marriage of iPSC technology with a new generation of genetic engineering tools has enabled the precise transfer of reporter or therapeutic transgenes, gene disruption, or even gene correction (Fig. 1). It is remarkable to reflect on the speed and relative ease at which both iPSC and genome engineering technologies have been adapted and merged for in vitro disease modeling and drug screening.
Rise of the Genome Editing Machines
Genetic manipulation by gene targeting is a mainstay of functional genomics. Those fundamental principles of gene targeting first outlined by Mario Cappechi using positive-negative selection in mouse ESCs [15, 16] are duly applicable to human PSCs. However, even following these guidelines, the first gene-targeting experiments in human ESCs [17] indicated that gene-targeting rates would be typically lower than observed in the mouse, leaving an obvious need for improvement.
The formation of double-strand breaks (DSBs ) in genomic DNA occurs naturally during DNA replication or in response to stresses such as ionizing radiation, and are vital to resolve recombination during meiosis and the production of immune system diversity [18]. DSB repair (DSBR) by end-resection and nonhomologous end-joining (NHEJ) can be inherently mutagenic, whereas homology-directed repair (HDR ) can faithfully restore DNA sequence in the presence of a template donor DNA (such as the sister chromatid). It was therefore hypothesized that intentional formation of DSBs at target loci could enhance gene-targeting frequencies via a custom donor DNA [19, 20]. The demonstration that the FokI nuclease domain is separable from DNA-binding domains [21] suggested a method by which nucleases could be engineered with novel specificity.
Zinc-finger nucleases (ZFNs ; Fig. 2a, top) built a foundation for recombinant endonuclease applications to enhance gene targeting through targeted DSBR, and remain a powerful tool in genome engineering even today [22]. Composed of a DNA-binding domain-encoding specificity and a FokI nuclease domain, ZFNs function as paired proteins that position and dimerize FokI monomers to cleave at a target locus [23]. The binding of zinc fingers to DNA triplets is modular [24]; however, finger–triplet interaction properties have been shown to be highly context and neighbor dependent, such that engineering custom ZFNs remains notoriously difficult. This problem has been partially addressed using validated libraries of submodules composed of two or three fingers [25]; however, screening for functional ZFNs is a resource-heavy endeavor.

Common nuclease systems used to stimulate double-strand break repair (DSBR) at target genomic sites. (a) Zinc-finger nucleases (ZFN) and TAL effector nucleases (TALEN) are composed of dimeric nuclease domains addressed by engineered DNA-binding domains. The Cas9 nuclease is addressed by a synthetic guide RNA molecule. Endonuclease components are green; targeting components are blue. (b) DSBs recruit endogenous repair machinery allowing genetic modification by nonhomologous end-joining (NHEJ) or template-mediated homology-directed repair (HDR) pathways
TAL effector nucleases (TALENs ; Fig. 2a, middle) broke the barrier between technical novelty and practical application [26], priming research laboratories through in-house nuclease design and production [27]. Plant pathogenic Xanthomonas spp. secrete TAL Effector (TALE) proteins, which activate host gene expression, resulting in a metabolic advantage to the invader [28]. The TALEs represent a unique class of proteins that bind DNA in a 1:1 modality [29, 30], making engineered design of TALENs more straightforward than that of ZFNs. The nature of this protein–DNA interaction is mediated through polymorphic protein repeats that display little degeneracy and no obvious neighbor effects. In a large-scale in vivo screen in zebrafish, TALENs were found to be more mutagenic than ZFNs [31]. Presumably, the increased tolerance of the spacer region provides a larger substrate for exonuclease activity, resulting in broad deletions compared to the conservative resection observed using tightly juxtaposed ZFNs.
CRISPR/Cas9 (Fig. 2a, bottom) has stolen the proverbial ‘show,’ capturing the attention of academia and public alike as it rapidly transcended from discovery as the hunter–killer of a potent anti-phage adaptive bacterial immune system [32], to experimental modulation and design [33] for genome engineering purposes [34]. The Cas9 protein , a general endonuclease that produces DSBs through HNH and RuvC nuclease domains, forms a ribonucleoprotein (RNP) complex with bacterially processed short CRISPR RNAs (crRNA) and trans-acting crRNA (tracrRNA) that pair with foreign genomic DNA targets to address and activate nuclease activity [35]. Biochemical characterization of the key CRISPR components, by Jennifer Doudna and Emmanuelle Charpentier [33], indicated that programmable cleavage could be achieved through the custom design of a hybrid crRNA–tracrRNA single guide RNA molecule (sgRNA ), which could be simply co-expressed or transfected as RNA along with the Streptococcus pyogenes Cas9 (SpCas9) protein. Thus, the CRISPR/Cas9 system could theoretically be programmed to cleave any 20-nt sequence upstream of a 5′-NGG-3′ protospacer adjacent motif (PAM ). This report was immediately followed by back-to-back proof-of principle experiments describing genome engineering in human cells [36, 37]. During the past 3 years, CRISPR/Cas9 technology has enabled gene knockouts across previously inaccessible genetic model organisms [38] and high-throughput genomic screens [39, 40], highlighting the simplicity of design and ease of application.
SpCas9 is by far the most commonly used CRISPR system. However, active variants from other bacterial species such as Staphylococcus aureus (SaCas9, 5′-NNGRRN-3′ PAM) [41] and Neisseria meningitidis (NmCas9, 5′-NNNNGATT-3′ PAM) [42] have been applied for genome editing with variable success. As proteins of bacterial origin, rational design of Cas9 in prokaryotes is conventional, leading to new variants of SaCas9 with modified PAM specificity, and therefore broader targeting ranges [43]. Mining prokaryotic genome databases through homology, cloning, and functional validation has yielded family members with new properties. Differing from SpCas9, Francisella novicida Cpf1 (FnCpf1) requires only a single guide RNA, and recognizes a 5′-TTN-3′-PAM, therefore accessing completely different sequence space [44]. The rich diversity of CRISPR systems in bacteria suggests that additional nucleases with distinct properties remain to be discovered.
Biochemical subtleties of DNA recognition and cleavage aside, engineered nucleases enhance random mutagenesis and gene targeting by eliciting endogenous DSBR pathways (Fig. 2b). As the NHEJ mutation spectrum is essentially random, it provides allelic depth for clonal cell panels, yet can complicate high throughput screening [45]. Under special circumstances of genomic sequence context and DSB position, DSBR can be driven by subtle regions of microhomology to produce indels in a predictable manner [46]. Bi-allelic DSBs can allow for homozygous targeting by HDR, an event achieved rarely with classic gene targeting [47]. Combinatorial events, such as HDR-mediated targeting of one allele, and NHEJ knockout of the other, can be a boon or a bane. Because of the promiscuity of the Cas9 protein, combinatorial approaches using multiplexed sgRNAs have led to multiple mutations in a mouse stem cell or embryo [48, 49], accelerating the analysis of multiple genetic interactions and emphasizing the power of the CRISPR/Cas9 nuclease system for functional genomics studies.
Adding Function to iPSCs Through Transgenesis at Safe-Harbor Loci
Gene targeting may be used to eliminate or alter endogenous genes, or introduce new functions. Transgenesis with viral or transposon systems has the advantage of being robust and rapid [14], yet as a trade-off does not directly control for integration site and therefore requires the use of populations or screening multiple clones to discern suitable or comparable gene expression levels. Nucleases permit transgenes to be introduced into defined loci and therefore minimize clonal variation by moderating position effects [50]. HDR-targeted transgenesis includes applications such as cDNA rescue of mutant genes and fluorescent knock-in alleles to report endogenous gene expression or simply label cells constitutively [51].
Perhaps the most well known “safe-harbor” locus is AAVS1, a hotspot for adeno-associated virus insertion located within intron 1 of the PPP1R12C gene [52]. AAVS1 is akin to the mouse ROSA26 locus [53], providing a reliable transgene expression with no known phenotype resulting from homozygous transgene insertions [47]. Targeting and expression of cDNAs from the AAVS1 locus has rescued monogenic diseases such as X-linked chronic granulomatous [54] and α-thalassemia [55]. Conversely, overexpression of dominant negative ion channel genes KCNQ1 and KCNH2 from the AAVS1 locus can recapitulate Long-QT syndrome for the development of an isogenic in vitro drug-screening platform [56].
Other safe harbors, such as the X-linked hypoxanthine phosphoribosyltransferase 1 (HPRT1) locus, is permissive for constitutive expression [57], yet disruption causes HPRT1-deficiency spectrum diseases ranging from gout to Lesch–Nyhan syndrome . The human l-gulono-γ-lactone oxidase (GULOP) locus is a nonfunctional pseudogene in humans [58, 59] presumed to avoid phenotypic effects. Yet, transgene expression in pluripotent and differentiated lineages is less well described. Beyond gene disruption, the local effects by potent transgenic promoters on endogenous gene expression must also be considered [60]. One such example is the citrate lyase beta-like (CLYBL ) locus that lies in a gene-deficient region of human chromosome 13 and claims to confer less severe effects on local gene expression [61]. Finally, in a mouse model of hemophilia A and B, expression of human factors VIII and IX from the endogenous albumin locus achieved long-term expression of transgenes at therapeutic levels [62]. Therefore, context-dependent safe harbors may be found in loci that are active in the target-differentiated cell type yet repressed in others, and not associated with a known haploinsufficiency phenotype.
Achieving Seamless Genome Engineering for Accurate Disease Models
In the interest of generating faithful models of human genetic disease, engineered changes that recapitulate single-nucleotide variations (SNVs ) would be preferred over crude knockouts. The de facto test for evaluating the role of candidate mutations in disease is to repair the mutation in patient iPSCs, or to recapitulate it in otherwise normal iPSCs [63]; true correction or recreation of patient-specific mutations would require approaches that are free of residual foreign genetic elements.
Classic gene targeting [16] deposits antibiotic-positive selection cassettes to enrich for HDR-mediated events (Fig. 3a). Retention of such elements is invaluable for producing knockouts, and reconcilable with the integration of reporters [47] or even therapeutic transgenes [64]. However, in the interest of modifying small regions of DNA—or in the extreme case, single nucleotides—selection cassettes and other elements can disrupt the native locus and may even cause unpredictable pleiotropic effects [65]. Removal of antibiotic selection cassettes is typically performed through site-specific recombinase-mediated excision [66]. In this approach, the recognition sites for Cre (loxP), Flp (FRT) recombinases [67] flank the selection cassette, which is introduced juxtaposed to the mutation (Fig. 3b, left). Following the selection of targeted clones, the cassette is excised by transient recombinase expression, yet a nontrivial single recombinase site (34 bp in the case of loxP) remains. Although residual elements may be positioned in “neutral” genetic regions such as introns, unexpected effects on gene expression and the predicted phenotype remain probable [64].

Derivation of human induced pluripotent stem cells (iPSCs) engineered by HDR. (a) Classic gene targeting events are enriched by positive selection. (b) Excision of antibiotic selection markers by Cre recombinase (left) leaves behind loxP sites, while PB transposase (right) removes cassettes seamlessly from endogenous or engineered TTAA tetranucleotides to deposit point mutations. (c) Mutation deposition by short single-strand oligonucleotides (ssODNs) obviates the need for excision, yet requires intensive screening
As an alternative to recombinases, the piggyBac (PB) transposon undergoes high-fidelity seamless excision from mouse and human iPSCs [68], and has been developed as an excisable positive/negative selection cassette for genome modification [69]. One caveat is that PB elements excise only from TTAA tetranucleotides, such that a TTAA must be present, or silently engineered near the mutation (Fig. 3b, right). PB provides more flexibility and a subtler footprint than recombinases, yet excision frequencies are locus dependent, and reintegration of the transposon may occur stochastically, whereas excision-prone transposase variants [70] may display higher rates of mutagenesis.
Diverging from classic targeting vector-based genome modification relying on antibiotic enrichment, short single-strand oligonucleotide (ssODN ) templates have been employed in combination with ZFNs [71, 72], TALENs [73], and CRISPR/Cas9 [48]. In this approach, ssODNs typically more than 100 nt in length carry sufficient homology to deposit point mutations into nuclease-cleaved loci in a single step without codeposition of foreign sequences (Fig. 3c), providing a clear advantage over recombinase-based methods [74]. It should be noted that ssODN -modified loci that retain the nuclease target site are potentially subject to recleavage and mutagenic NHEJ repair. Silent mutations that prevent nuclease recognition and recleavage detract from the subtlety of the method, but may be necessary to avoid additional screening. Moreover, aberrant ssODN insertions at on- or off-target sites [75] or random mutations on-target [76] may occur under normal conditions or as a reflection of oligo quality and are extremely difficult to predict and detect. Although ssODN-mediated targeting events are frequent in cell lines, the low frequency of correct targeting in iPSCs (>1 %) [48, 72, 73], compounded with possible mutagenic events, demands robust and sophisticated selection.
One advanced approach to detect correct gene editing employs serial population screening, a type of sib-selection for human iPSCs where mutation-containing populations are monitored by droplet digital polymerase chain reaction (ddPCR) and enriched using serial sub-fractionation [77]. In developing this technique, the authors successfully deposited mutations into five disease-associated genes (PHOX2B, PKP2, RBM20, PRKAG2, and BAG3). Population screening by ddPCR is robust but not trivial, requiring custom TaqMan assays, sophisticated instrumentation, and additional iPSC passages. A streamlined approach to derive gene-corrected iPSCs that combined CRISPR/Cas9 gene targeting with the somatic cell reprogramming process [76], reported gene knock-in efficiencies as high as 5 %, and ssODN-mediated gene correction rates as high as 8 %. Although useful during de novo iPSC derivation, this approach is obviously not applicable to previously established iPSC lines. Finally, frequencies of desirable targeting using ssODNs may still see improvements through lessons learned from the biochemistry of DNA opening and Cas9 cleavage. As the sgRNA nontarget (unbound) strand is released first, ssODNs with positioning and complementarity to the nontarget DNA strand are more effective at inducing HDR, up to 60 % in HEK293T cells [78]. Applications of these findings in iPSCs hold promise.
Avoiding Unwanted Outcomes: Off-Target Cleavage and Mosaicism
Nuclease cleavage of the genome is by no means infallible, and undesirable DSBR events may occur through surreptitious cleavage at sites other than the chosen target region. In these cases, DSBs repaired preferentially through NHEJ may result in subtle indels (Fig. 2b) with no capacity for counterselection. Such “off-target” effects may or may not have phenotypic consequences.
Unbiased off-target detection using whole-genome sequencing (WGS ) can evaluate genome-engineered iPSC clones [79, 80], yet the depth of data and the threshold for detecting rare mutations argue against the practicality of the approach. Exome sequencing simplifies analysis, yet provides data for only a small portion of the genome. Targeted screening methods based on degenerate sequence similarity between the sgRNA and nontarget regions of the host genome provide an off-target candidate list that may be verified using conventional NHEJ detection methods such as the T7E1 hybrid-cleavage assay [81], Sanger sequencing with decomposition [82], or deep sequencing of amplified products [46]. However, these biased approaches are time consuming and limited by the quality of prediction algorithms for candidate off-target sites.
Off-target screens relying on the functional properties of nucleases have the potential to focus screening efforts without user bias. Chromatin immunoprecipitation using Cas9 antibodies [83, 84] can detect sites of Cas9 interaction with the genome but are not related directly to DNA-cleavage events. Linear amplification-mediated high-throughput, genome-wide, translocation sequencing (LAM-PCR HTGTS) is a cumulative method that detects off-target cleavage by virtue of genomic translocations formed between nuclease-generated or even endogenous DSBs [85], indicating a two-break-minimum detection limit. On the other hand, single NHEJ events have been shown to capture foreign DNA elements such as integration defective lentiviral vectors (IDLV ) [86, 87], which can then act as tags for targeted sequencing efforts. GUIDE-seq applies this same principle, yet uses oligonucleotide tags compatible with next-generation sequencing to streamline sample processing and data integration [88]. BLESS (direct in situ breaks labeling, enrichment on streptavidin, and next-generation sequencing) attempts to capture a snapshot of the fragmented genome within cells, but requires complex fixation and manipulation steps [89]. DiGenome sequencing is an in vitro approach to genomic DSB detection using WGS to detect indels as DNA fragment ends [90]. Differences in detection profiles for these methods may reflect the methodology and must ultimately be verified experimentally.
Refining the detection of off-target cleavage is a crucial endeavor, yet does not directly prevent the causative insult to the genome. Therefore, it would be prudent to develop engineering methods that minimize off-target cleavage events or increase on-target cleavage specificity. One straightforward approach could be to temporally limit the expression of Cas9 and sgRNAs. However, simply reducing the amount of expression vector DNA transfected does not reduce the relative rates of off-target cleavage [91]. In contrast to plasmids, which can express over periods of 3 to 4 days or even integrate randomly into the genome, delivery as in vitro transcribed (IVT ) mRNA limits the nuclease expression window to 1 to 2 days and yet is still effective for on-target cleavage. An additional step toward restricted nuclease activity is to produce RNP particles through the in vitro combination of commercially available recombinant SpCas9 protein and IVT or synthetic sgRNAs, followed by delivery directly into iPSCs by electroporation or chemical transfection [92, 93]. An in-depth analysis of the off-target outcomes from such procedural changes is pending.
It is clear, however, that limiting nuclease activity temporally has the potential to reduce mosaicism under conditions normally presumed to produce clonal iPSCs. Mosaicism can arise from unique DNA cleavage and DSBR events in the daughter cells of nuclease-transfected iPSCs, resulting in two or more divergent populations in a drug-selected colony [94]. Mosaicism confounds the detection of off-target effects, which may be present below the threshold of detection in the total iPSC population. Interestingly, sib-selection procedures involving rounds of serial subcloning from the starting population [77] impose a temporal separation of nuclease treatment and physical cloning events to derive truly clonal iPSC populations.
Engineering native nuclease behavior to reduce or prevent off-target cleavage was initially proposed for recombinant FokI nuclease domains [95]. By inactivating the catalytic domain of one monomer in a ZFN dimer, ZFNickases were shown to have lower levels of off-target mutagenesis, albeit with an overall reduction in on-target HDR activity [96]. Similarly, a derivative of SpCas9 in which the RuvC nuclease domain has been inactivated by mutagenesis (SpCas9n, D10A) acts as a DNA nickase [37]. This hobbled enzyme has been used in juxtaposed pairs to produce staggered nicks, and touted as having lower off-target cleavage activity than their full active counterparts because rogue binding of a SpCas9n monomer would produce single-strand nicks rather than DSB [97]. Yet, it is important to remember that nicked DNA intermediates can still be processed by NHEJ mechanisms, resulting in an off-target indel [98], suggesting that alternative approaches still require consideration.
With solution of the DNA/RNA hybrid-bound SpCas9 protein structure [99, 100] came the possibility of rational SpCas9 engineering. Modeling revealed a positively charged groove between the nuclease and PAM interacting domains, proposed to be involved in stabilizing the nontarget DNA strand. Mutagenesis of K848A, K1003A, and R1060A residues within the groove retained approximately 60 % on-target activity, while increasing sensitivity to sgRNA mismatches, most notably outside the 7–12 nt “seed” region [44]. In another approach, diminished bonding energy through quadruple mutagenesis of DNA-contacting N497A, R661A, Q695A, and Q926A residues produced a high-fidelity variant of SpCas9 (SpCas9-HF1) with undetectable off-target activity [101]. On-target activity was reported to be 70 % of the native SpCas9, a modest compromise for higher specificity.
Modulation of the RNA component of the Cas9 RNP complex has also been shown to positively affect on- and off-target cleavage ratios. It was suggested that truncated sgRNAs (truRNAs) may gain cleavage specificity as a trade-off for activity [102], yet 16-nt versus the standard 20-nt sgRNA molecules has not become a norm for CRISPR experiments. Optimal sgRNA design has been shown to affect on-target cleavage activity [103]. More recently, revision of the rule set governing sgRNA design by Doench and colleagues suggests a predictive scoring system for increasing on-target activity while avoiding off-target cleavage, as demonstrated using a genome-wide knockout screen [104]. It remains to be seen how the community at large will adopt these bioinformatic rule sets, and if they hold true in various experimental situations.
Selection of Isogenic Clones and Technical Controls
Reprogramming technology captures the genome of the patient as a pluripotent cell resource, enabling in vitro modeling of disease that, by necessity, separates the cell from the patient. Differing from animal models, phenotyping results are therefore limited by the sophistication of in vitro cellular differentiation [105] and assay evaluation criteria. To directly link genotypes to phenotypes, appropriate control cell lines are of utmost importance.
Control iPSC lines represent a selected or engineered group of iPSCs that are genetically matched for the purpose of excluding erroneous variation and increasing the accuracy of disease studies [106]. iPSCs from unrelated normal individuals have been used to produce target cell disease controls [107], taking into account that their genetic backgrounds may vary by degrees (Fig. 4a). As such, the number of unrelated iPSC clones that must be analyzed in parallel to define a genotype–phenotype correlation increases in relation to the statistical power required (Fig. 4b). Within practical limits, the required number of control iPSC lines depends mainly on the strength and correlation of the in vitro phenotype with clinical presentation and the complex influence of genetic background variation on phenotypes observed within the patient population [108]. One standard for reducing genetic variation has been to compare disease iPSCs to normal iPSCs from unaffected siblings who share much of their genetic background with the affected donor by blood relationship (Fig. 4a). Intriguingly, as ESCs are often derived from pools of discarded in vitro fertilization (IVF) material [1], there may in fact be a higher rate of sibling relationship among publically available ESCs than iPSC lines. However, potential racial bias and an association with a higher incidence of infertility-related alleles, along with a reported marked difference in differentiation capacity between ESC lines [109], may further offset this proposed benefit. On the other hand, the documented medical background of the donor combined with a deep genetic analysis may help predict the severity of phenotypic deviation between experimental and control iPSC lines.

Appropriate sources of isogenic control iPSC clones . (a) iPSCs from unaffected siblings or normal donors are typically used as controls. (b) Multiple iPSCs may be used to reduce noise from clonal variation. (c) True isogenic controls may be produced through genome engineering. (d) To preclude phenotypic effects from off-target cleavage, different sgRNAs may be used to produce the same genomic modification
Subtle differences in the genomes and epigenomes between iPSC lines may influence in vitro phenotypes [110]. Concerns over the accumulation of mutations throughout the reprogramming process as a result of proliferative stress have been raised [111]. Conversely, more recent studies have shown that iPSC derivation is inherently stable at the genetic level [112], suggesting that the risk of genetic drift arises during extended in vitro culture and is therefore similar for both ESCs and iPSCs. However, the process of iPSC derivation itself is selective, such that preexisting somatic mutations in the patient’s donor tissue can be clonally amplified [113, 114]. It has also been proposed that reprogrammed cells might retain an epigenetic memory of their somatic source, which could influence differentiation capacity [115]. Interestingly, such epigenetic memory has been disputed by the observation that more significant variation in differentiation capacity occurs as a result of genetic background than somatic tissue source [116, 117]. Still, these uncertainties regarding inherent and acquired phenotypic variation strongly argue the case for isogenicity.
Fortunately, iPSCs themselves are inherently isogenic with their donor, and through the application of subtle nuclease-mediated genome-editing approaches described above, gene-corrected iPSCs can be derived directly from donor iPSCs (Fig. 4c) [63]. Similarly, well-characterized normal iPSCs will retain isogenicity if converted to diseased iPSCs using nuclease techniques. When patient-specific iPSCs cannot be procured, recreating mutations by genome editing provides a novel material for the study of genetic effects on disease progression and severity in a defined genetic background. Quality-controlled normal iPSCs could be accessed from one of many proposed stem cell “libraries,” which aim to generate clinical-grade and HLA haplotype-matched control iPSC lines for therapeutic applications [118]. However, it should be cautioned that in the conversion of normal iPSCs into diseased iPSCs, disease phenotypes might be masked by protective alleles. Candidate gene disruption in multiple ethnic backgrounds may therefore be necessary to exclude complex genetic effects [119].
Phenotypic variations between experimental iPSC lines and isogenic controls may have a technical origin. Off-target nuclease effects require labor-intensive screening to detect and might contribute to the observed phenotype. As an alternative to deep sequencing or comparing multiple gene-corrected clones from a single experiment (Fig. 4b, c), it is advised to instead make use of a second sgRNA with its own distinct off-target profile (Fig. 4d). In this way, it is possible to rule out common off-target events between separately derived clones as a direct influence on phenotype. Similarly, employing PB-mediated gene targeting and excision for precise editing [120], reintegration of the transposon may occur stochastically. Yet, these clones may still prove useful for validating phenotypes, because each clone should represent a novel reintegration event. Splinkerette PCR-based methods for mapping reintegrations [121] could help predict the influence on genomic integrity and possible phenotypic changes. Considering these sources of technical variation and their logical solutions, genome engineering (Fig. 4c) stands as the strictest method to maintain isogenicity within control iPSCs.
Conclusions
The combination of iPSC and nuclease technologies, particularly CRISPR/Cas9, has generated a true paradigm shift in modeling human genetics and disease. Although more accessible than ESCs, patient-specific iPSCs still require informed consent, and can prove to be morally and monetarily extravagant research materials. Applying genome-editing technologies, there is no longer a need to initiate disease modeling with the procurement of patient-specific iPSCs. Instead, candidate mutations or allelic series may be first engineered singly or in combinatorial fashion into genetically and phenotypically defined “reference” ESCs or iPSCs. Once available, the panel of iPSC “standards” may be used to refine in vitro physiological assays. Finally, as required, patient-specific iPSCs may be screened using the optimized assay system to interrogate candidate mutations and the effect of native genetic background. Future avenues of research will most certainly entail combined gene editing and reprogramming strategies, bringing to fruition both preclinical and clinical applications of stem cell technology to personalized medicine.
Abbreviations
- AAVS1:
-
Adeno-associated virus integration site 1
- CRISPR:
-
Clustered regularly interspaced short palindromic repeat
- crRNA:
-
CRISPR RNA
- ddPCR:
-
Droplet digital PCR
- DSB:
-
Double-strand break
- DSBR:
-
Double-strand break repair
- ESC:
-
Embryonic stem cell
- GFP:
-
Green fluorescent protein
- HDR:
-
Homology-directed repair
- HLA:
-
Histocompatibility leukocyte antigen
- indel:
-
Insertion or deletion
- iPSC:
-
Induced pluripotent stem cell
- ITR:
-
Inverted terminal repeat
- IVF:
-
In vitro fertilization
- MMEJ:
-
Microhomology-mediated end-joining
- NHEJ:
-
Nonhomologous end-joining
- PAM:
-
Protospacer adjacent motif
- PB:
-
piggyBac transposon
- PBase:
-
piggyBac transposase
- PCR:
-
Polymerase chain reaction
- PSC:
-
Pluripotent stem cell
- RNA:
-
Ribonucleic acid
- RNP:
-
Ribonucleoprotein
- ROCKi:
-
Rho-kinase inhibitor
- sgRNA:
-
Single guide RNA
- SNV:
-
Single nucleotide variation
- ssODN:
-
Single-strand oligonucleotide
- T7E1:
-
Bacteriophage T7 endonuclease I
- TALEN:
-
Transcription activator-like effector nuclease
- tracrRNA:
-
Trans-acting crRNA
- WGS:
-
Whole-genome sequencing
- ZFN:
-
Zinc-finger nuclease
References
Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145–7. Epub 1998/11/06.
Gertow K, Przyborski S, Loring JF, Auerbach JM, Epifano O, Otonkoski T, et al. Isolation of human embryonic stem cell-derived teratomas for the assessment of pluripotency. Curr Protoc Stem Cell Biol. 2007;Chap. 1:Unit1B 4. Epub 2008/09/12.
Keller G. Embryonic stem cell differentiation: emergence of a new era in biology and medicine. Genes Dev. 2005;19(10):1129–55. Epub 2005/05/21.
Outten JT, Gadue P, French DL, Diamond SL. High-throughput screening assay for embryoid body differentiation of human embryonic stem cells. Curr Protoc Stem Cell Biol. 2012;Chap. 1:Unit 1D 6. Epub 2012/03/15.
Zacharias DG, Nelson TJ, Mueller PS, Hook CC. The science and ethics of induced pluripotency: what will become of embryonic stem cells? Mayo Clin Proc. 2011;86(7):634–40. Epub 2011/07/02.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76. Epub 2006/08/15.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–72. Epub 2007/11/24.
Takahashi K, Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol. 2016;17(3):183–93. Epub 2016/02/18.
Inoue H, Nagata N, Kurokawa H, Yamanaka S. iPS cells: a game changer for future medicine. EMBO J. 2014;33(5):409–17. Epub 2014/02/07.
Hindley C, Philpott A. The cell cycle and pluripotency. Biochem J. 2013;451(2):135–43. Epub 2013/03/29.
Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol. 2007;25(6):681–6. Epub 2007/05/29.
Nakagawa M, Taniguchi Y, Senda S, Takizawa N, Ichisaka T, Asano K, et al. A novel efficient feeder-free culture system for the derivation of human induced pluripotent stem cells. Sci Rep. 2014;4:3594. Epub 2014/01/09.
Chen G, Gulbranson DR, Hou Z, Bolin JM, Ruotti V, Probasco MD, et al. Chemically defined conditions for human iPSC derivation and culture. Nat Methods. 2011;8(5):424–9. Epub 2011/04/12.
Kim SI, Oceguera-Yanez F, Sakurai C, Nakagawa M, Yamanaka S, Woltjen K. Inducible transgene expression in human iPS cells using versatile all-in-one piggyBac transposons. Methods Mol Biol. 2016;1357:111–31. Epub 2015/05/31.
Thomas KR, Capecchi MR. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell. 1987;51(3):503–12. Epub 1987/11/06.
Capecchi MR. Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat Rev Genet. 2005;6(6):507–12. Epub 2005/06/03.
Zwaka TP, Thomson JA. Homologous recombination in human embryonic stem cells. Nat Biotechnol. 2003;21(3):319–21. Epub 2003/02/11.
Lieber MR, Lu H, Gu J, Schwarz K. Flexibility in the order of action and in the enzymology of the nuclease, polymerases, and ligase of vertebrate non-homologous DNA end joining: relevance to cancer, aging, and the immune system. Cell Res. 2008;18(1):125–33. Epub 2007/12/19.
Jasin M. Genetic manipulation of genomes with rare-cutting endonucleases. Trends Genet. 1996;12(6):224–8. Epub 1996/06/01.
Carroll D. Using nucleases to stimulate homologous recombination. Methods Mol Biol. 2004;262:195–207. Epub 2004/02/11.
Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A. 1996;93(3):1156–60. Epub 1996/02/06.
Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11(9):636–46. Epub 2010/08/19.
Smith J, Bibikova M, Whitby FG, Reddy AR, Chandrasegaran S, Carroll D. Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Res. 2000;28(17):3361–9. Epub 2000/08/23.
Pavletich NP, Pabo CO. Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science. 1991;252(5007):809–17. Epub 1991/05/10.
Maeder ML, Thibodeau-Beganny S, Sander JD, Voytas DF, Joung JK. Oligomerized pool engineering (OPEN): an ‘open-source’ protocol for making customized zinc-finger arrays. Nat Protoc. 2009;4(10):1471–501. Epub 2009/10/03.
Mussolino C, Cathomen T. TALE nucleases: tailored genome engineering made easy. Curr Opin Biotechnol. 2012;23(5):644–50. Epub 2012/02/22.
Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39(12), e82. Epub 2011/04/16.
Bogdanove AJ, Schornack S, Lahaye T. TAL effectors: finding plant genes for disease and defense. Curr Opin Plant Biol. 2010;13(4):394–401. Epub 2010/06/24.
Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326(5959):1509–12. Epub 2009/11/26.
Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326(5959):1501. Epub 2009/11/26.
Chen S, Oikonomou G, Chiu CN, Niles BJ, Liu J, Lee DA, et al. A large-scale in vivo analysis reveals that TALENs are significantly more mutagenic than ZFNs generated using context-dependent assembly. Nucleic Acids Res. 2013;41(4):2769–78. Epub 2013/01/11.
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–12. Epub 2007/03/24.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–21. Epub 2012/06/30.
Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32(4):347–55. Epub 2014/03/04.
Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471(7340):602–7. Epub 2011/04/02.
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–6. Epub 2013/01/05.
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–23. Epub 2013/01/05.
Niu J, Zhang B, Chen H. Applications of TALENs and CRISPR/Cas9 in human cells and their potentials for gene therapy. Mol Biotechnol. 2014;56(8):681–8. Epub 2014/05/30.
Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343(6166):80–4. Epub 2013/12/18.
Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera Mdel C, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol. 2014;32(3):267–73. Epub 2014/02/19.
Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520(7546):186–91. Epub 2015/04/02.
Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF, Sontheimer EJ, et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci U S A. 2013;110(39):15644–9. Epub 2013/08/14.
Kleinstiver BP, Prew MS, Tsai SQ, Nguyen NT, Topkar VV, Zheng Z, et al. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol. 2015;33(12):1293–8. Epub 2015/11/03.
Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163(3):759–71. Epub 2015/10/01.
Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 2015;33(6):661–7. Epub 2015/05/12.
Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24(1):132–41. Epub 2013/11/21.
Oceguera-Yanez F, Kim SI, Matsumoto T, Tan GW, Xiang L, Hatani T, et al. Engineering the AAVS1 locus for consistent and scalable transgene expression in human iPSCs and their differentiated derivatives. Methods. 2015. Epub 2015/12/29.
Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154(6):1370–9. Epub 2013/09/03.
Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153(4):910–8. Epub 2013/05/07.
Papapetrou EP, Lee G, Malani N, Setty M, Riviere I, Tirunagari LM, et al. Genomic safe harbors permit high beta-globin transgene expression in thalassemia induced pluripotent stem cells. Nat Biotechnol. 2011;29(1):73–8. Epub 2010/12/15.
Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27(9):851–7. Epub 2009/08/15.
Smith JR, Maguire S, Davis LA, Alexander M, Yang F, Chandran S, et al. Robust, persistent transgene expression in human embryonic stem cells is achieved with AAVS1-targeted integration. Stem Cells. 2008;26(2):496–504. Epub 2007/11/21.
Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21(1):70–1. Epub 1999/01/23.
Zou J, Sweeney CL, Chou BK, Choi U, Pan J, Wang H, et al. Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: functional correction by zinc finger nuclease-mediated safe harbor targeting. Blood. 2011;117(21):5561–72. Epub 2011/03/18.
Chang CJ, Bouhassira EE. Zinc-finger nuclease-mediated correction of alpha-thalassemia in iPS cells. Blood. 2012;120(19):3906–14. Epub 2012/09/25.
Wang Y, Liang P, Lan F, Wu H, Lisowski L, Gu M, et al. Genome editing of isogenic human induced pluripotent stem cells recapitulates long QT phenotype for drug testing. J Am Coll Cardiol. 2014;64(5):451–9. Epub 2014/08/02.
Sakurai K, Shimoji M, Tahimic CG, Aiba K, Kawase E, Hasegawa K, et al. Efficient integration of transgenes into a defined locus in human embryonic stem cells. Nucleic Acids Res. 2010;38(7), e96. Epub 2010/01/15.
Inai Y, Ohta Y, Nishikimi M. The whole structure of the human nonfunctional l-gulono-gamma-lactone oxidase gene—the gene responsible for scurvy—and the evolution of repetitive sequences thereon. J Nutr Sci Vitaminol. 2003;49(5):315–9. Epub 2004/01/02.
Li X, Burnight ER, Cooney AL, Malani N, Brady T, Sander JD, et al. piggyBac transposase tools for genome engineering. Proc Natl Acad Sci U S A. 2013;110(25):E2279–87. Epub 2013/06/01.
Lombardo A, Cesana D, Genovese P, Di Stefano B, Provasi E, Colombo DF, et al. Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat Methods. 2011;8(10):861–9. Epub 2011/08/23.
Cerbini T, Funahashi R, Luo Y, Liu C, Park K, Rao M, et al. Transcription activator-like effector nuclease (TALEN)-mediated CLYBL targeting enables enhanced transgene expression and one-step generation of dual reporter human induced pluripotent stem cell (iPSC) and neural stem cell (NSC) lines. PLoS One. 2015;10(1), e0116032. Epub 2015/01/15.
Sharma R, Anguela XM, Doyon Y, Wechsler T, DeKelver RC, Sproul S, et al. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood. 2015;126(15):1777–84. Epub 2015/08/25.
Kim HS, Bernitz JM, Lee DF, Lemischka IR. Genomic editing tools to model human diseases with isogenic pluripotent stem cells. Stem Cells Dev. 2014;23(22):2673–86. Epub 2014/07/31.
Zou J, Mali P, Huang X, Dowey SN, Cheng L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood. 2011;118(17):4599–608. Epub 2011/09/02.
Meier ID, Bernreuther C, Tilling T, Neidhardt J, Wong YW, Schulze C, et al. Short DNA sequences inserted for gene targeting can accidentally interfere with off-target gene expression. FASEB J. 2010;24(6):1714–24. Epub 2010/01/30.
Davis RP, Costa M, Grandela C, Holland AM, Hatzistavrou T, Micallef SJ, et al. A protocol for removal of antibiotic resistance cassettes from human embryonic stem cells genetically modified by homologous recombination or transgenesis. Nat Protoc. 2008;3(10):1550–8. Epub 2008/09/20.
Grindley ND, Whiteson KL, Rice PA. Mechanisms of site-specific recombination. Annu Rev Biochem. 2006;75:567–605. Epub 2006/06/08.
Woltjen K, Michael IP, Mohseni P, Desai R, Mileikovsky M, Hamalainen R, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458(7239):766–70. Epub 2009/03/03.
Yusa K, Rashid ST, Strick-Marchand H, Varela I, Liu PQ, Paschon DE, et al. Targeted gene correction of alpha1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478(7369):391–4. Epub 2011/10/14.
Kim H, Kim K, Kim J, Kim SH, Yim J. Mutagenesis by imprecise excision of the piggyBac transposon in Drosophila melanogaster. Biochem Biophys Res Commun. 2012;417(1):335–9. Epub 2011/12/14.
Orlando SJ, Santiago Y, DeKelver RC, Freyvert Y, Boydston EA, Moehle EA, et al. Zinc-finger nuclease-driven targeted integration into mammalian genomes using donors with limited chromosomal homology. Nucleic Acids Res. 2010;38(15), e152. Epub 2010/06/10.
Chen F, Pruett-Miller SM, Huang Y, Gjoka M, Duda K, Taunton J, et al. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat Methods. 2011;8(9):753–5. Epub 2011/07/19.
Ding Q, Lee YK, Schaefer EA, Peters DT, Veres A, Kim K, et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013;12(2):238–51. Epub 2012/12/19.
Soldner F, Laganiere J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146(2):318–31. Epub 2011/07/16.
Radecke S, Radecke F, Cathomen T, Schwarz K. Zinc-finger nuclease-induced gene repair with oligodeoxynucleotides: wanted and unwanted target locus modifications. Mol Ther. 2010;18(4):743–53. Epub 2010/01/14.
Howden SE, Maufort JP, Duffin BM, Elefanty AG, Stanley EG, Thomson JA. Simultaneous reprogramming and gene correction of patient fibroblasts. Stem Cell Rep. 2015;5(6):1109–18. Epub 2015/11/21.
Miyaoka Y, Chan AH, Judge LM, Yoo J, Huang M, Nguyen TD, et al. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat Methods. 2014;11(3):291–3. Epub 2014/02/11.
Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol. 2016;34:339–44. Epub 2016/01/21.
Smith C, Gore A, Yan W, Abalde-Atristain L, Li Z, He C, et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 2014;15(1):12–3. Epub 2014/07/06.
Veres A, Gosis BS, Ding Q, Collins R, Ragavendran A, Brand H, et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 2014;15(1):27–30. Epub 2014/07/06.
Kim HJ, Lee HJ, Kim H, Cho SW, Kim J-S. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res. 2009;19:1279–88.
Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42(22), e168. Epub 2014/10/11.
Fujita T, Fujii H. Efficient isolation of specific genomic regions and identification of associated proteins by engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) using CRISPR. Biochem Biophys Res Commun. 2013;439(1):132–6. Epub 2013/08/15.
Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD, Dadon DB, et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol. 2014;32(7):670–6. Epub 2014/04/23.
Frock RL, Hu J, Meyers RM, Ho YJ, Kii E, Alt FW. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol. 2015;33(2):179–86. Epub 2014/12/17.
Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, Kaeppel C, et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol. 2011;29(9):816–23. Epub 2011/08/09.
Wang X, Wang Y, Wu X, Wang J, Wang Y, Qiu Z, et al. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat Biotechnol. 2015;33(2):175–8. Epub 2015/01/20.
Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol. 2015;33(2):187–97. Epub 2014/12/17.
Crosetto N, Mitra A, Silva MJ, Bienko M, Dojer N, Wang Q, et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat Methods. 2013;10(4):361–5. Epub 2013/03/19.
Kim D, Bae S, Park J, Kim E, Kim S, Yu HR, et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods 2015;12(3):237–243, 244 Epub 2015/02/11.
Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31(9):822–6. Epub 2013/06/25.
Kim S, Kim D, Cho SW, Kim J, Kim JS. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24(6):1012–9. Epub 2014/04/04.
Liang X, Potter J, Kumar S, Zou Y, Quintanilla R, Sridharan M, et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol. 2015;208:44–53. Epub 2015/05/25.
Merkle FT, Neuhausser WM, Santos D, Valen E, Gagnon JA, Maas K, et al. Efficient CRISPR-Cas9-mediated generation of knockin human pluripotent stem cells lacking undesired mutations at the targeted locus. Cell Rep. 2015;11(6):875–83. Epub 2015/05/06.
Sanders KL, Catto LE, Bellamy SR, Halford SE. Targeting individual subunits of the FokI restriction endonuclease to specific DNA strands. Nucleic Acids Res. 2009;37(7):2105–15. Epub 2009/02/19.
Ramirez CL, Certo MT, Mussolino C, Goodwin MJ, Cradick TJ, McCaffrey AP, et al. Engineered zinc finger nickases induce homology-directed repair with reduced mutagenic effects. Nucleic Acids Res. 2012;40(12):5560–8. Epub 2012/03/01.
Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154(6):1380–9. Epub 2013/09/03.
Certo MT, Ryu BY, Annis JE, Garibov M, Jarjour J, Rawlings DJ, et al. Tracking genome engineering outcome at individual DNA breakpoints. Nat Methods. 2011;8(8):671–6. Epub 2011/07/12.
Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156(5):935–49. Epub 2014/02/18.
Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343(6176):1247997. Epub 2014/02/08.
Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529(7587):490–5. Epub 2016/01/07.
Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32(3):279–84. Epub 2014/01/28.
Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol. 2014;32(12):1262–7. Epub 2014/09/04.
Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34(2):184–91. Epub 2016/01/19.
Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132(4):661–80. Epub 2008/02/26.
Merkle FT, Eggan K. Modeling human disease with pluripotent stem cells: from genome association to function. Cell Stem Cell. 2013;12(6):656–68. Epub 2013/06/12.
Kondo T, Asai M, Tsukita K, Kutoku Y, Ohsawa Y, Sunada Y, et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness. Cell Stem Cell. 2013;12(4):487–96. Epub 2013/02/26.
Zeltner N, Studer L. Pluripotent stem cell-based disease modeling: current hurdles and future promise. Curr Opin Cell Biol. 2015;37:102–10. Epub 2015/12/03.
Osafune K, Caron L, Borowiak M, Martinez RJ, Fitz-Gerald CS, Sato Y, et al. Marked differences in differentiation propensity among human embryonic stem cell lines. Nat Biotechnol. 2008;26(3):313–5. Epub 2008/02/19.
Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, et al. Reference maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 2011;144(3):439–52. Epub 2011/02/08.
Ronen D, Benvenisty N. Genomic stability in reprogramming. Curr Opin Genet Dev. 2012;22(5):444–9. Epub 2012/10/09.
Bhutani K, Nazor KL, Williams R, Tran H, Dai H, Dzakula Z, et al. Whole-genome mutational burden analysis of three pluripotency induction methods. Nat Commun. 2016;7:10536. Epub 2016/02/20.
Abyzov A, Mariani J, Palejev D, Zhang Y, Haney MS, Tomasini L, et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature. 2012;492(7429):438–42. Epub 2012/11/20.
Young MA, Larson DE, Sun CW, George DR, Ding L, Miller CA, et al. Background mutations in parental cells account for most of the genetic heterogeneity of induced pluripotent stem cells. Cell Stem Cell. 2012;10(5):570–82. Epub 2012/05/01.
Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467(7313):285–90. Epub 2010/07/21.
Nasu A, Ikeya M, Yamamoto T, Watanabe A, Jin Y, Matsumoto Y, et al. Genetically matched human iPS cells reveal that propensity for cartilage and bone differentiation differs with clones, not cell type of origin. PLoS One. 2013;8(1), e53771. Epub 2013/02/06.
Kyttala A, Moraghebi R, Valensisi C, Kettunen J, Andrus C, Pasumarthy KK, et al. Genetic variability overrides the impact of parental cell type and determines iPSC differentiation potential. Stem Cell Rep. 2016;6(2):200–12. Epub 2016/01/19.
Turner M, Leslie S, Martin NG, Peschanski M, Rao M, Taylor CJ, et al. Toward the development of a global induced pluripotent stem cell library. Cell Stem Cell. 2013;13(4):382–4. Epub 2013/10/08.
Rosenberg NA, Huang L, Jewett EM, Szpiech ZA, Jankovic I, Boehnke M. Genome-wide association studies in diverse populations. Nat Rev Genet. 2010;11(5):356–66. Epub 2010/04/17.
Yusa K. Seamless genome editing in human pluripotent stem cells using custom endonuclease-based gene targeting and the piggyBac transposon. Nat Protoc. 2013;8(10):2061–78. Epub 2013/09/28.
Woltjen K, Hamalainen R, Kibschull M, Mileikovsky M, Nagy A. Transgene-free production of pluripotent stem cells using piggyBac transposons. Methods Mol Biol. 2011;767:87–103. Epub 2011/08/09.
Acknowledgments
We appreciate personal communications with Dana Carroll, Bruce R. Conklin, Gregory D. Davis, J. Keith Joung, Jin-Soo Kim, and William C. Skarnes, as well as the important contributions to the field by the many scientists whose research is not cited here for the sake of brevity. This work was funded in part by grants to K.W. from the Japan Society for the Promotion of Science (Wakate B), the Kyoto University Hakubi Project, and the Research Center Network for Realization of Regenerative Medicine, Program for Intractable Diseases Research Utilizing Disease-Specific iPS Cells, of the Japan Science and Technology Agency (JST).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Woltjen, K., Oceguera-Yanez, F., Kagawa, H., Kim, SI. (2016). At the Conflux of Human Genome Engineering and Induced Pluripotency. In: Turksen, K. (eds) Genome Editing. Springer, Cham. https://doi.org/10.1007/978-3-319-34148-4_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-34148-4_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-34146-0
Online ISBN: 978-3-319-34148-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)