
Abstract
Recent advances in understanding the role of the immune system in ovarian cancer have culminated in the introduction of multiple promising immunotherapeutic treatment strategies. These include the adoptive transfer of immune effectors such as monoclonal antibodies and T cells, vaccination, and immunomodulatory therapy. In this chapter, we discuss the various therapeutic strategies, their mechanisms of action, and their key clinical trials in ovarian cancer. We also highlight promising combinatorial treatment regimens and present the challenges that are being critically addressed by clinicians and researchers to enhance the efficacy of immunotherapy.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Immunotherapy
- Monoclonal antibody
- Adoptive transfer
- Vaccination
- Checkpoint blockade
- Combination immunotherapies
Introduction
Recent advances in understanding the role of the immune system in ovarian cancer have culminated in the introduction of multiple promising immunotherapeutic treatment strategies. Mounting clinical evidence suggested that ovarian cancers are immunogenic, with the early observation that patients with tumor-infiltrating CD3+ T cells had improved responses to chemotherapy and increased overall survival [1]. Other studies subsequently confirmed tumor-infiltrating lymphocytes (TILs), specifically CD8 T cells, as predictors of favorable clinical outcome [2–4]. On the other hand, the presence of immunosuppressive CD4+CD25+Foxp3+ regulatory T cells (Tregs) in tumors correlated with poor outcome [5]. Furthermore, several tumor-associated antigens (TAAs) recognized by peripheral blood T cells or TILs have been identified, including mutated cell cycle regulatory proteins (p53), cancer-testis antigens (NY-ESO-1), cancer antigens (CA-125), growth-activating receptors (EGFR and HER2/neu), and folate receptors (folate receptor alpha, FRα) [6–9]. These TAAs serve not only as markers of disease progression but also as potential therapeutic targets for several immunotherapies. Lastly, the expression of immune inhibitory receptors such as programmed death 1 (PD-1) on TILs and its ligand (PD-L1) on tumor cells has created opportunities for combination therapies with checkpoint inhibitors [10–12].
This book chapter will review various immunotherapeutic approaches, their mechanisms of action, and their key clinical trials in ovarian cancer. Therapeutic strategies are divided into three categories: adoptive transfer of immune effectors, vaccination, and immunomodulatory therapy.
Immunotherapies for Ovarian Cancer
Adoptive Transfer of Immune Effectors
Immune effectors employed in adoptive transfer include monoclonal antibodies (mAbs) against antigenic targets expressed by tumor cells or within the tumor microenvironment, as well as autologous or allogeneic antitumor T cells (also known as adoptive T-cell therapy or ACT). Both of these approaches bypass the need for in vivo antigen presentation and immune effector proliferation [13].
Monoclonal Antibodies (mAbs)
With the US Food and Drug Administration (FDA) approval of rituximab (anti-CD20) in 1997 for chemotherapy-resistant non-Hodgkin lymphoma, a new era of cancer therapy dawned. The FDA has since approved more than 20 mAbs for clinical use in oncologic care. Based on their antigenic target, mAbs can be classified into mAbs that target tumor cells (direct tumor cell killers), mAbs that target the tumor microenvironment (TME) (TME modifiers), and mAbs that target immune checkpoints (checkpoint inhibitors), among others [14, 15]. mAbs have shown promise in ovarian cancer and are increasingly being examined in clinical trials. The latter category of mAbs (checkpoint inhibitors) will be discussed in section “Depleting Tregs”.
In addition to neutralizing the function of their antigenic targets by inhibiting their signaling pathways (such as tumor growth and angiogenesis), mAbs can modulate the immune response against tumor cells, such as increasing dendritic cell (DC) maturation, priming effector cells (T cells and natural killer “NK” cells), and activating complement-dependent cytotoxicity (CDC) and antibody-dependent cell-mediated cytotoxicity (ADCC) pathways. Conjugation to antineoplastic toxins in antibody-drug conjugates (ADCs) bestows additional cytotoxic activity to mAbs and allows more precise delivery of chemotherapy following their binding to target antigen and subsequent internalization into tumor cells [16, 17].
mAbs Targeting Tumor Cells
Anti-EGFR (Cetuximab and Panitumumab)
The epidermal growth factor receptor (EGFR) is overexpressed by 9–62 % of ovarian cancers and has been associated with high tumor grade and poor patient outcome [18]. Cetuximab (Erbitux®, BMS and Eli Lilly) is an FDA-approved chimeric IgG1 mAb that binds to the extracellular domain of EGFR, preventing EGFR signaling and promoting receptor internalization and ubiquitin-mediated degradation [19, 20]. Single-agent studies of cetuximab reported minimal activity. In a phase II trial of weekly cetuximab monotherapy in patients with persistent/recurrent ovarian or primary peritoneal carcinoma, none of the 25 patients achieved complete response (CR), 9 patients had stable disease (SD), and only one patient achieved a partial response (PR) [21]. On the other hand, cetuximab in combination with chemotherapy showed only modest activity. In a phase II trial of cetuximab and carboplatin in patients with relapsed, platinum-sensitive ovarian cancer, 9 of the 26 patients with EGFR-positive tumors developed an objective response (OR) and eight had SD. Additionally, response to this treatment regimen did not correlate with tumor EGFR expression and was associated with dermatologic toxicity in the majority of patients [22]. These observations highlight the need for developing effective combination therapies with chemotherapy and for determining more robust predictors for patient responsiveness in order to improve responses to anti-EGFR therapy and patient outcomes [18].
Panitumumab (Vectibix®, Amgen) is another FDA-approved anti-EGFR mAb of the IgG2 isotype that has shown encouraging results in a recent phase II clinical trial. In patients with platinum-resistant ovarian cancer, the combination of panitumumab and the chemotherapeutic pegylated liposomal doxorubicin (PLD) demonstrated 9 % PR and 19 % SD [23]. It should be mentioned that in the intent-to-treat population, the overall response rate (18.7 %) was similar to that reported in other phase II clinical trials of monotherapy with PLD in patients with platinum-refractory/platinum-resistant disease (19.7 %) [24].
Anti-mesothelin (Amatuximab) and Anti-CA-125 (Abagovomab and Oregovomab)
The high frequency of expression of the TAAs mesothelin and CA-125 in ovarian cancer has made them potential targets for mAb therapy. Mesothelin is a glycosylphosphatidylinositol (GPI)-anchored cell surface protein that is involved in tumor resistance to several chemotherapeutic drugs and in promoting tumor metastasis through its interaction with the mucin CA-125 [25]. CA-125 (also known as MUC16) is a TAA that is also overexpressed in ovarian cancer. CA-125 can be proteolytically cleaved from the tumor cell surface and has been employed as a serum biomarker to screen for ovarian cancer as well as to monitor responses to therapy. In addition to promoting tumor invasion and metastasis, CA-125 exerts immunosuppressive activity through protecting tumor cells from NK cell attack [26]. Attempts at targeting the mesothelin-MUC16 interaction using mAbs have met limited success. In a phase I trial in patients with mesothelin-expressing tumors (including four with ovarian cancer) receiving amatuximab (anti-mesothelin chimeric IgG1 mAb, MORAb-009, Morphotek), no CR or PR was seen [27]. Amatuximab is currently in a phase II trial for mesothelioma patients. Abagovomab (anti-idiotypic CA-125 murine IgG1 mAb) and oregovomab (anti-CA-125 murine IgG1 mAb, OvaRex®, AltaRex) failed to show a survival benefit in large clinical trials [28–30]. Oregovomab is currently in a phase II trial in combination with chemotherapy for patients with advanced epithelial ovarian cancer (NCT01616303). ADCs may prove a more promising strategy and are being explored in preclinical studies and ongoing clinical trials, as will be discussed.
Anti-FRα (Farletuzumab)
FRα is widely expressed on epithelial ovarian cancers, especially in platinum-resistant patients, but not on normal ovarian tissues [31, 32]. Farletuzumab (Morphotek) is an investigational humanized IgG1 anti-FRα mAb that mediates tumor cell cytotoxicity via CDC and ADCC rather than blocking folate transport [32]. In a phase II trial in platinum-sensitive ovarian cancer patients experiencing a first relapse, farletuzumab alone was poorly effective but when combined with chemotherapy (carboplatin and taxanes) improved objective response rates (ORR) to 75 %. Additionally, 80.9 % of patients normalized CA-125 [33]. However, a recent phase III trial was discontinued after farletuzumab in combination with paclitaxel failed to meet its end point of improving progression-free survival (PFS) in platinum-resistant ovarian cancer patients. Since a trend toward improved PFS was observed, additional analyses will be required to determine whether farletuzumab may improve outcome for patients [34]. In 2015, a phase II trial was launched to assess the combination of farletuzumab with carboplatin and paclitaxel or PLD in patients with low CA-125 platinum-sensitive ovarian cancer (NCT02289950).
mAbs Targeting Tumor Microenvironment
Anti-VEGF (Bevacizumab)
The vascular endothelial growth factor (VEGF) binds to its receptors on endothelial cells and activates signaling pathways that regulate normal development of the vasculature as well as pathologic angiogenesis in cancer [35]. In ovarian cancer, tumor VEGF gene expression correlates with a poor prognosis [36]. Bevacizumab (Avastin®, Roche) is a humanized IgG1 anti-VEGF mAb that can neutralize all isoforms of VEGF. In addition to its antiangiogenic activity, bevacizumab can also modulate the immune response by increasing DC maturation and priming of T cells, as demonstrated in multiple myeloma and melanoma [37, 38]. Bevacizumab is active in platinum-resistant ovarian cancer, both as monotherapy and in combination with chemotherapy [39–42]. In the phase III AURELIA trial in platinum-resistant ovarian cancer, combining bevacizumab with chemotherapy improved PFS (increased from 3.4 to 6.7 months) and ORR (increased from 11 to 27 %) [43].
Anti-TAMs (Anti-CSF-1R, Anti-CCL22, and Anti-B7-H4)
Similar to DCs, macrophages are phagocytic innate immune cells that can, to a lesser extent, induce T-cell activation. Macrophages are broadly classified into classical (M1-polarized) and alternative (M2-polarized) phenotypes. M1 macrophages are involved in Th1 responses through antigen presentation and secretion of immunostimulatory cytokines such as interleukins 6 and 12 (IL-6 and IL-12), while M2 macrophages are involved in Th2 responses through secretion of immunosuppressive cytokines such as IL-10 and transforming growth factor β (TGF-β). Tumor-associated macrophages (TAMs) are a major component of the TME and, in agreement with the M2 signature, have been associated with enhanced tumor progression, angiogenesis, and immunosuppression [44]. M2 TAMs are abundantly present in ovarian cancers and malignant ascites, and their numbers correlate with malignancy, while elevated M1 to M2 TAM ratios correlate with improved 5-year prognosis [45–47].
The macrophage colony-stimulating factor-1 receptor (CSF-1R) binds CSF-1 (also known as macrophage CSF or M-CSF) and is involved in regulating macrophage migration, proliferation, survival, and function [48]. Inhibiting CSF-1R activation using an antagonistic mAb has been shown in preclinical murine tumor models of high TAM infiltration to strongly reduce TAMs and enhance the CD8/CD4 T-cell ratio [49]. RG7155 (by Roche) is an investigational humanized anti-CSF-1R mAb that has recently entered clinical trials. In an ongoing phase Ia/Ib trial in patients with tenosynovial giant cell tumor (NCT01494688), RG7155 markedly reduced TAMs and was well tolerated [50]. CSF-1R blockade may thus be a promising strategy for depleting TAMs in ovarian cancer.
Another promising strategy is to modulate TAM-T-cell interactions in the TME. TAMs can recruit Tregs to the TME through the chemokine CCL22, which in turn suppresses tumor-specific T-cell immunity. In xenograft models of primary human ovarian tumors, neutralizing CCL22 using anti-CCL22 mAb inhibited Treg migration to tumors [5]. Tregs can also secrete IL-10, which can stimulate the expression of the checkpoint B7-H4 on macrophages. B7-H4 is expressed by >70 % of freshly isolated TAMs and negatively regulates T-cell responses [51, 52]. It is also expressed by ovarian cancer tumor cells, but only B7-H4+ macrophages suppress T-cell immunity and are negatively associated with patient outcome [52, 53]. Blocking B7-H4 interactions with single-chain fragments of antibody variable regions (scFvs) rescued tumor antigen-specific T-cell activation in vitro and delayed the growth of established tumors in mice [54]. The use of mAbs to reverse TAM-mediated immunosuppression represents a promising therapeutic approach to enhance T-cell tumor immunity in ovarian cancer.
Bispecific Antibodies
Anti-EpCAM × Anti-CD3 (Catumaxomab)
The epithelial cell adhesion molecule (EpCAM) is a transmembrane glycoprotein mediating calcium-independent cell-cell adhesion in the epithelium. It is overexpressed in primary, metastatic, and recurrent epithelial ovarian cancers across subtypes and has been associated with poor prognosis [55–58]. In ovarian cancer-associated malignant ascites, EpCAM is expressed by tumor cells in 70–100 % of cases [59]. Catumaxomab (Removab®, Fresenius Biotech GmbH) is a chimeric, bispecific, trifunctional antibody that binds to epithelial tumor cells via EpCAM and to T cells via CD3, facilitating the localization of T cells to the tumor tissue. Additionally, catumaxomab has a functional Fc domain (composed of mouse IgG2a and rat IgG2b) that can activate Fc receptor-expressing NK cells and mediate tumor cell cytotoxicity via ADCC [60, 61]. In a randomized phase II/III trial in patients with malignant ascites (including 129 ovarian cancer patients), catumaxomab prolonged puncture-free survival (PuFI: time to first need for paracentesis after treatment or time to death, whichever occurred first) [59] and received market approval by the European Medicines Agency (EMA) for this indication. In a recent phase II trial in chemotherapy-refractory ovarian cancer patients with malignant ascites, catumaxomab prolonged both the PuFI and the time to first therapeutic puncture (TTPu) and had a beneficial effect on the quality of life through improving ascites symptoms [62].
ADCs
Anti-mesothelin Conjugated to DM4 (Anetumab Ravtansine) and Anti-CA-125 Conjugated to MMAE (Sofituzumab Vedotin)
Anetumab ravtansine (BAY 94–9343, Bayer) is an ADC that consists of a human anti-mesothelin IgG1 mAb conjugated to the microtubule-targeting drug DM4 via a reducible disulfide linker. Following binding and internalization by tumor cells, degradation of the linker releases a cytotoxic DM4 metabolite. Anetumab ravtansine was superior to standard-of-care treatments in patient-derived xenograft models of ovarian cancer and led to complete eradication. Furthermore, its efficacy correlated with the expression level of mesothelin [63, 64]. Currently, anetumab ravtansine is being evaluated in a phase I clinical trial (NCT01439152).
Sofituzumab vedotin (RG7458 or DMUC5754A, Roche/Genentech) consists of a humanized anti-CA-125 IgG1 mAb conjugated to the microtubule-targeting drug monomethyl auristatin E (MMAE) via a protease-cleavable peptide linker. In a phase I trial in 44 patients with platinum-resistant ovarian cancer (NCT01335958), sofituzumab vedotin demonstrated a toxicity profile that was comparable to other current therapeutics and led to 1 CR and 4 PR. Similar to anetumab ravtansine, its efficacy correlated with the TAA expression level [65].
Anti-FRα Conjugated to DM4 (Mirvetuximab Soravtansine)
Mirvetuximab soravtansine (IMGN853, ImmunoGen) consists of a chimeric anti-FRα IgG1 mAb conjugated to DM4 via a reducible disulfide linker. Preclinical studies in xenograft models showed that the ADC efficiently targeted FRα+ tumors and was also cytotoxic to adjacent FRα− tumor cells (bystander effect), reflecting an ability to eradicate tumors with heterogeneous expression of FRα [66]. In an ongoing phase I trial in patients with platinum-resistant epithelial ovarian cancer (NCT01609556), mirvetuximab soravtansine as a single agent demonstrated promising preliminary clinical activity with an ORR of 53 % in the overall cohort and 80 % in the high FRα expression subset. Preliminary analysis suggested that FRα expression correlates well with ADC activity [67, 68]. Other ongoing trials are comparing the efficacy of mirvetuximab soravtansine to chemotherapy in patients with FRα+ advanced epithelial ovarian cancer (NCT02631876) or combining it with chemotherapy (NCT02606305).
Adoptive T-Cell Therapy (ACT)
Adoptive T-cell therapy (ACT) involves using ex vivo activated tumor-specific T cells that are either derived from tumors (TILs) and enriched for particular antigen specificity or are genetically engineered to express either tumor-specific T-cell receptors (TCRs) or chimeric antigen receptors (CARs). Prior to reinfusion into the cancer patient, cells are expanded with IL-2, and lymphodepleting chemotherapy and/or radiotherapy is administered to promote the in vivo survival and expansion of adoptively transferred T cells by increasing cytokines and antigen-presenting cell (APC) activity and eliminating immunosuppressive cells [69, 70]. Ongoing intensive research aims to improve this attractive (albeit labor-intensive and expensive) approach by improving T-cell constructs, automating T-cell generation, and optimizing toxicity management [13].
TILs
The use of TILs for ACT benefits from the natural selection of patient TMEs to polyclonal, tumor-specific T cells which have escaped thymic deletion and homed to tumors [71]. In the 1990s, the adoptive transfer of TILs expanded ex vivo with IL-2 was examined in ovarian cancer [72–74]. Aoki et al. reported that in 17 patients with advanced or recurrent ovarian cancer, ACT alone administered to seven patients led to 1 CR and 4 PR, while ACT administered to ten patients in conjunction with cisplatin led to 7 CR and 2 PR [73]. In a pivotal trial, Fujita et al. reported that in patients with advanced-stage epithelial ovarian cancer, ACT after optimal debulking surgery and cisplatin chemotherapy improved the 3-year overall survival rates to 100 % versus 67.5 % for patients not receiving ACT [74]. The drawbacks of ACT using TILs are numerous, including tolerance to self-antigens and the low yield of tumor-specific lymphocytes for ex vivo expansion [75]. Attempts to overcome these drawbacks have led to the use of genetically engineered T cells from peripheral blood for ACT.
Genetically Engineered T Cells
To redirect the T-cell specificity of normal peripheral blood lymphocytes (PBLs), T cells are genetically modified to recognize TAA using viral vectors encoding for either TCRs (which are MHC-restricted) or CARs. In CARs, TCR intracellular signaling domains are coupled with surface variable regions of antibodies; CARs can thus recognize TAA in an MHC-unrestricted fashion and their activation is enhanced upon TAA contact [70]. A phase I/IIa trial is currently ongoing for ACT with TCRs recognizing the TAA NY-ESO-1 in patients with recurrent or treatment-refractory ovarian cancer carrying the HLA-A201 allele (NCT01567891). In addition, several CAR trials are under way. The first trial was conducted in 2006 in 14 patients with advanced FRα+ ovarian cancer using FRα-specific CARs. However, transferred CARs were undetectable at 1 month, and no clinical benefit was observed [76]. The addition of costimulatory signaling capabilities to the intracytoplasmic domain of CARs (such as CD137) has improved in vivo CAR persistence and activity [77, 78]. Mesothelin-specific CARs are also being pursued in ovarian cancer. In an ongoing phase I trial in patients with mesothelin-expressing tumors (including two with ovarian cancer), CARs were adoptively transferred without lymphodepletion and were found to traffic to tumor sites and to persist in the blood for 3–4 weeks post infusion [79]. Other trials are also ongoing (NCT02159716 and NCT01583686).
Vaccination
Therapeutic cancer vaccines aim to “teach” the immune system to recognize tumor cells through supplying whole tumor cells or tumor-derived peptides. These are provided together with immune adjuvants, including pattern recognition receptor ligands (such as poly-ICLC and the incomplete Freund’s adjuvant Montanide) and granulocyte-macrophage colony-stimulating factor (GM-CSF), to promote DC activity. Unlike passive immunotherapy with adoptively transferred mAbs or T cells, vaccines are an active immunotherapy strategy that can generate long-lasting immunological memory. The convenience and low toxicity have made vaccines an attractive approach in ovarian cancer as in other types of cancer. Nonetheless, limited efficacy has been observed [6, 80]. Efforts to improve performance include optimizing target antigens, improving vaccine platforms by using DCs and oncolytic viruses, and developing combinatorial approaches with immunomodulatory therapy (the latter will be discussed in section “Combination Therapies”).
Vaccination Based on Tumor Peptides or Tumor Cells
Peptide Vaccines
Peptide vaccines employ short peptides from TAAs that can directly bind to exact HLA class I molecules on DCs, bypassing the need for antigen processing and generating CD8 T-cell responses (albeit short-lived). In addition to using adjuvants to increase peptide immunogenicity, recent advances in improving the efficacy of peptide vaccines include the use of synthetic or overlapping long peptides, which require antigen processing by DCs but are efficiently presented to both CD4 and CD8 T cells [81].
NY-ESO-1
NY-ESO-1 is an immunogenic TAA that is expressed by 40 % of epithelial ovarian cancers and generates antibody and cellular immune responses in multiple cancer patients [82, 83]. In a pilot study of patients with advanced ovarian cancer and minimal disease burden, administration of NY-ESO-1 peptide of HLA class I/II specificities with Montanide induced both NY-ESO-1-specific CD4 and CD8 T-cell responses in the majority of patients and improved PFS. Importantly, a patient who experienced complete regression had a recurrence later with an NY-ESO-1-negative tumor, highlighting the drawback of immune escape tumor variants with peptide (single target) vaccines [84]. In a phase I trial in high-risk ovarian cancer patients in their first remission, NY-ESO-1 peptide with Montanide led to NY-ESO-1-specific CD8 T-cell responses in both NY-ESO-1-positive and NY-ESO-1-negative tumors and CR in 33 % of patients [85]. A phase I trial in 28 patients with advanced ovarian cancer in second or third remission examined overlapping long peptides (OLPs) from NY-ESO-1 either alone or in combination with Montanide or Montanide and poly-ICLC. Antibody and CD8 T-cell responses specific to NY-ESO-1 were undetectable with OLP alone but were detected in 91 % of patients receiving OLP and both adjuvants, where each had a distinct effect for the induction of NY-ESO-1-specific Th1 cells [86, 87]. Recently, a phase I trial in 12 patients with relapsed ovarian cancer examined the effect of adding decitabine (a DNA methyltransferase inhibitor) as an epigenetic modifier to NY-ESO-1 peptide vaccine administered with the adjuvants Montanide and GM-CSF and the chemotherapeutic liposomal doxorubicin. Increased NY-ESO-1 serum antibodies and T-cell responses were observed in the majority of patients, while SD or PR was noted in six out of ten evaluable patients [88].
p53
p53 is a protein that is encoded by the tumor suppressor gene TP53 and regulates the fate of cells upon DNA damage [89]. p53 is overexpressed in 50–60 % of ovarian cancers, and the presence of p53 antibodies has been identified as a positive prognostic factor in ovarian cancer patients [90, 91]. In addition, circulating and tumor-infiltrating p53-specific memory T cells were detected in patients with ovarian cancer [92]. In a phase II trial in patients with advanced-stage ovarian cancer, a p53 peptide vaccine administered with IL-2, GM-CSF, and Montanide as adjuvants led to immune responses (as measured by interferon γ (IFN-γ) production and tetramer assays). However, IL-2 administration increased toxicity and induced Treg expansion, leading the authors to suggest the removal of IL-2 from this vaccine regimen. Importantly, the trial found that the subcutaneous p53 peptide vaccine had a similar efficacy to an intravenous vaccine of DCs pulsed with p53 peptides, suggesting that the peptide vaccine is a superior choice given its simpler approach in preparation and administration [93]. Another phase II trial examined p53 synthetic long peptides (p53-SLP) with Montanide in patients with recurrent ovarian cancer. While IFN-γ-producing p53-specific CD4 T cells were induced, Th2 cytokines dominated the p53-specific response, and no improvement in clinical outcome was observed [94, 95].
HER-2/neu
A phase I trial in 19 patients with breast or ovarian cancer showed that vaccination with HER-2/neu-derived MHC class II “helper” peptides, which contain MHC class I epitopes, administered with GM-CSF as adjuvant induced potent and long-lasting HER-2-specific IFN-γ-producing CD8 T cells. A larger, phase I trial examined HER-2/neu peptides with GM-CSF in patients with advanced HER-2/neu+ cancers (including five patients with ovarian cancer). The vaccine induced HER-2/neu-specific T-cell responses in 92 % of patients. Importantly, the responses were long-lived, and epitope spreading to additional HER-2/neu epitopes and to p53, was observed in some patients [96, 97].
Tumor Cell Vaccines
Personalized vaccines based on whole tumor cells represent an alternative to peptide vaccines that allow the generation of a diverse immune response directed at multiple TAAs. Because they incorporate both MHC class I and class II epitopes, tumor cell vaccines can limit tumor escape variants. On the other hand, using whole tumor cells carries the risk of stimulating tolerogenic or autoimmune, rather than immunogenic, responses due to the significant presence of self-antigens [98]. The FANG vaccine represents an elegant approach to enhance the immunogenicity of whole tumor cells. It is composed of autologous tumor cells genetically modified to encode GM-CSF (as an adjuvant) and a bifunctional short hairpin RNAi that inhibits TGF-β by targeting furin transferase. In a phase I trial that included five patients with ovarian cancer, the vaccine was safe and elicited an immune response correlating with prolonged survival [99]. A follow-up phase II/III trial is currently ongoing in patients with advanced ovarian cancer achieving CR following primary surgical debulking and chemotherapy (NCT02346747). Preliminary results show that 92 % of vaccinated patients developed immunity (as measured by IFN-γ production), and the median regression-free survival (RFS) was 399 days versus 94 days for control patients [99, 100].
Vaccination Based on DCs
DC-based vaccines were developed to overcome the low number and/or defective ability of DCs in cancer patients to process and present tumor antigens. Autologous DCs are generated ex vivo from peripheral blood monocytes in the presence of cytokine and growth factor cocktails that induce DC expansion and maturation. DCs are then loaded with TAAs or whole tumor lysates prior to reinfusion into patients [101].
A promising TAA for DC-based vaccines is mucin 1 (MUC-1), a heavily glycosylated surface protein that is overexpressed and aberrantly glycosylated in a large number of cancers including ovarian cancer [102]. In a phase I trial in advanced-stage ovarian cancer patients, DCs pulsed with MUC-1 peptides generated tumor-specific CD8 T cells [103]. A phase II study examined the CVac® vaccine (Prima BioMed) of MUC-1-loaded DCs in 63 patients with epithelial ovarian cancer in complete remission. In patients who had achieved a remission after second-line therapy, PFS and OS were improved with the CVac vaccine as compared to patients receiving standard-of-care therapy [104]. Another potential TAA is HER-2/neu. A phase I trial involving four ovarian cancer patients evaluated lapuleucel-T (Neuvenge®, Dendreon), a DC-based vaccine composed of autologous peripheral blood mononuclear cells (including APCs) cultured ex vivo with HER-2/neu peptides linked to GM-CSF. The vaccine generated HER-2-specific T-cell responses and led to short-term SD in two of the four patients [105].
DCs loaded with whole tumor lysates have also shown promise in ovarian cancer. In a phase I trial in six patients with recurrent advanced ovarian cancer, patients received DCs pulsed with whole tumor lysates and keyhole limpet hemocyanin (KLH) as an adjuvant. The treatment was well tolerated, and three of the six patients showed PFS of 25–45 weeks [106]. A phase II trial further examined the tumor lysate-pulsed DCs and KLH that was administered with low-dose IL-2 as an adjuvant in ten ovarian cancer patients with minimal residual disease. The vaccine resulted in 3 CR for 38–83 months and induced tumor-related immunity in responders, including NK cell activity and IFN-γ-producing T cells [107]. In another pilot study in five patients with recurrent ovarian cancer, DCs pulsed with tumor lysates oxidized with hypochlorous acid (which enhances immunogenicity), two patients had a PFS of 24 months or more [108].
Vaccination Based on Viruses
Recombinant viral vaccines are attractive TAA delivery systems due to their inherent immunogenicity and ability to exploit DC trafficking to the injection site for enhanced TAA uptake and presentation. Commonly employed viral vectors are members of the Poxviridae family and include vaccinia and fowlpox viruses. The vaccinia vector induces strong cellular and humoral immune responses to the transgene it encodes but is limited by the development of host-induced neutralizing antibodies to the vector itself and by the exclusion of use in immunocompromised patients. On the other hand, fowlpox viruses can be administered in booster doses due to absence of neutralizing antibody development but are less efficient than vaccinia vectors in inducing immune responses [109].
In a pilot study involving three ovarian cancer patients, the PANVAC vaccine regimen was evaluated. It consisted of the transgenes for the TAAs carcinoembryonic antigen (CEA) and MUC-1 along with the transgenes for the TRICOM adjuvant (the costimulatory molecule CD80, intercellular adhesion molecule-1 (ICAM-1), and leukocyte function-associated antigen-3 (LFA-3)) engineered into vaccinia (PANVAC-V) as a prime and fowlpox (PANVAC-F) as booster vaccinations. Immune responses to MUC-1 and/or CEA were observed post vaccination [110]. In a follow-up study involving 14 patients with ovarian cancer, median OS was 15 months in patients receiving the PANVAC vaccine, and those with limited tumor burden and minimal prior chemotherapy seemed to derive the most benefit [111]. Another heterologous prime-boost vaccine regimen was recently examined in a phase II trial in 22 patients with advanced ovarian cancer in clinical remission. Patients received NY-ESO-1-vaccinia as a prime and NY-ESO-1-fowlpox as booster vaccinations. CD4 and CD8 T-cell responses were induced and found to correlate with improved OS. Ovarian cancer patients showed a median PFS and OS of 21 and 48 months, respectively [112].
Immunomodulatory Therapy
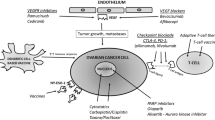
Immunomodulatory therapy aims to tip the balance in the immunosuppressive TME from immune tolerance to immune reactivity. The traditional use of cytokines, including IL-2, IL-12, type I and II IFNs, and tumor necrosis factor α (TNF-α), as immunotherapeutic agents that broadly activate T cells has proven challenging due to systemic toxicity and has met with limited success in ovarian cancer [6, 113]. T-cell activation is regulated not only by costimulatory receptors (including CD28 and CD137) but also by inhibitory receptors or checkpoints, which are induced following TCR stimulation (Fig. 11.1). Therapeutic approaches that block the suppressive signals of checkpoints (checkpoint blockade) or selectively target immunosuppressive cells in the TME (such as Tregs) can sustain the activation and proliferation of tumor-specific T cells and represent one of the most rapidly moving and exciting areas in clinical oncology.

The major T cell-based immunotherapeutic targets and their tumor or antigen-presenting cell ligands. The activation of T cells and their conversion to a cytotoxic phenotype is governed by a network of activating and inhibitory receptors. Using immunotherapeutic agents to increase activation and decrease inhibitory signaling has the potential to generate T cells with enhanced tumor lytic capacity. APC antigen-presenting cell, GAL galectin-9, IDO indoleamine 2,3-dioxygenase, ICOS inducible T-cell costimulator, MHC major histocompatibility complex, PD-1 programmed death 1, PD-L PD-1 ligand, TCR T-cell receptor, TIM-3 T-cell immunoglobulin and mucin domain 3
Depleting Tregs
Targeting CD25: Anti-CD25 (Daclizumab) and Denileukin Diftitox
Tregs constitutively express the IL-2 receptor α chain (CD25). Daclizumab (Zenapax®, F. Hoffmann-La Roche) is an FDA-approved humanized IgG1 mAb that binds to CD25. Traditionally used to inhibit T-cell proliferation in autoimmune disorders, it has recently been used to deplete Tregs in combination with a metastatic breast cancer vaccine. Daclizumab administration led to a marked and prolonged decrease in Tregs and boosted T-cell responses to all vaccine antigens in absence of autoimmunity [114]. Daclizumab is currently being evaluated in combination with a DC-based vaccine in ovarian cancer (NCT01132014). Another approach to targeting CD25 is through denileukin diftitox (Ontak®, Eisai), an FDA-approved engineered fusion protein of IL-2 and diphtheria toxin. In a recent phase II trial of 28 patients with epithelial ovarian cancer, denileukin diftitox administration was well tolerated and significantly depleted functional Tregs from blood and the TME, but showed no significant clinical efficacy [115]. Combination strategies with checkpoint blockade may improve clinical efficacy and have shown promise in preclinical studies [116].
Cyclophosphamide
Cyclophosphamide is a chemotherapeutic agent that has immunomodulatory activity when administered in repeated, low doses (metronomically). It depletes Tregs and restores T-cell function and has been used to augment antitumor immune responses of ACT and vaccination strategies [117]. In a phase I/II trial in 11 patients with recurrent ovarian cancer, a single low dose of cyclophosphamide was explored as an adjuvant to a vaccine regimen of peptide-pulsed DCs. While the 3-year OS was 90 %, the single dose of cyclophosphamide did not reduce the number of circulating Tregs, and no significant survival benefit over controls was observed [118]. In another phase II trial in patients with recurrent ovarian cancer, low-dose cyclophosphamide administered prior to each dose of the p53-SLP vaccine led to p53-specific IFN-γ-producing T cells in 90 % of evaluable patients after two immunizations [119].
Checkpoint Blockade
Immune checkpoints tightly regulate the intensity and duration of the T-cell response and are critical for avoiding autoimmunity. These include the T-cell surface molecules cytotoxic T-lymphocyte antigen 4 (CTLA-4), programmed death 1 (PD-1), T-cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), and lymphocyte activation gene-3 (LAG-3). In addition, the metabolic enzyme indoleamine 2,3-dioxygenase (IDO), which catalyzes the rate-limiting step of the oxidative catabolism of the amino acid tryptophan, regulates T-cell activation. By depleting tryptophan and generating the toxic metabolite kynurenine, IDO can inhibit T-cell proliferation and trigger cell cycle arrest and apoptosis. Kynurenine can also induce naive CD4 T cells into Tregs [120–123].
Activation of immune checkpoint pathways in the TME, however, limits the antitumor immune response. TILs upregulating the expression of checkpoints are hyporesponsive or functionally exhausted [124]. In patients with ovarian cancer, a significant fraction of antigen-specific CD8 TILs co-express LAG-3 and PD-1 and demonstrate impaired effector function [125]. Additionally, Tregs naturally express checkpoints and employ them to suppress effector T cells [124]. Moreover, 56 % of ovarian tumors have demonstrated IDO expression, which correlated with a reduced number of CD8 TILs and with reduced survival in serous (but not other) ovarian cancer histologies [126, 127]. IDO expression was also found to inhibit NK cell intratumoral accumulation and to promote tumor angiogenesis [128]. Reversing TME-mediated immunosuppression via targeting checkpoints with mAbs or inhibitors, an approach coined “checkpoint blockade,” was found to boost immune responses and is becoming increasingly valuable in the clinic.
Anti-CTLA-4 (Ipilimumab)
CTLA-4 (CD152) is an inhibitory co-receptor and member of the B7-CD28 immunoglobulin superfamily. It competes with the costimulatory receptor CD28 for the ligands (B7 molecules) on APCs, leading to downregulation of T-cell activation. Ipilimumab (Yervoy®, BMS) is an FDA-approved human IgG1 mAb that blocks the CTLA-4/B7 interaction to restore CD4 and CD8 effector T-cell activation and can also deplete tumor-infiltrating Tregs [129, 130]. Ipilimumab represents the first standard-of-care immune checkpoint inhibitor, and the majority of clinical experience is derived from studies in patients with melanoma. In a pilot study in two patients with advanced ovarian cancer previously vaccinated with GM-CSF-modified irradiated autologous tumor cells (GVAX), a single dose of ipilimumab triggered a decrease or stabilization of CA-125 levels for several months [131]. In a subsequent study in additional nine patients, three patients had SD, and the extent of therapy-induced tumor necrosis correlated with the intratumoral CD8 T-cell/Treg ratio [132]. An ongoing phase II trial is studying ipilimumab as monotherapy in patients with recurrent platinum-sensitive ovarian cancer (NCT01611558). The primary drawback of ipilimumab is the high frequency of immune-related adverse events (irAEs) like colitis or hypophysitis: approximately 25 % of patients experience an irAE, requiring aggressive management [133].
Anti-PD-1 (Nivolumab and Pembrolizumab) and Anti-PD-L1 (Several mAbs)
PD-1 is another co-inhibitory receptor member of the CD28/B7 immunoglobulin superfamily that binds to its ligands PD-L1 and PD-L2 (mainly expressed by epithelial cells, DCs, and macrophages) to down-modulate the immune response [124]. In addition to the high expression of PD-1 by TILs in ovarian tumors [10, 125], PD-L1 was also found to be highly expressed by ovarian tumor cells and is negatively correlated with CD8 TIL counts and with survival [11]. PD-L1 tumor expression has also been implicated in promoting peritoneal dissemination of ovarian cancer [12]. The blockade of the PD-1 inhibitory pathway is being clinically explored using mAbs targeting either the receptor or its ligands and has so far proven less immunotoxic than ipilimumab.
Nivolumab (Opdivo®, BMS) is an FDA-approved human IgG4 anti-PD-1 mAb that is being investigated in ovarian cancer. In a phase I trial in 15 patients with advanced platinum-resistant ovarian cancer (regardless of PD-L1 expression), nivolumab was well tolerated and led to 3 PR and 4 SD, with an ORR of 17 % [134]. In a recent update, 2 patients with CR survived without disease progression for 17 and 14 months each [135]. Another anti-PD-1 mAb is pembrolizumab (Keytruda®, Merck), an FDA-approved humanized IgG4 mAb. In a recent interim analysis of a phase Ib trial in 26 patients with heavily treated PD-L1+ advanced ovarian cancer, pembrolizumab was well tolerated and achieved 1 CR, 2 PR, and 6 SD with a durable ORR of 11.5 % [136].
Several mAbs that block PD-L1 are being investigated in numerous clinical trials that include ovarian cancer patients. Examples include avelumab (MSB0010718C, human IgG1, Merck Serono), BMS-936559 (human IgG4, BMS-ONO), MPDL3280A (human IgG1, Roche/Genetech), and durvalumab (MEDI4736, human IgG1, MedImmune). In an ongoing phase Ib trial in 75 patients with platinum-resistant or chemotherapy-refractory ovarian cancer (regardless of PD-L1 expression), avelumab demonstrated an acceptable safety profile and had an ORR of 10.7 % in 67 evaluable patients [137]. In a phase I trial involving 17 patients with ovarian cancer, BMS-936559 demonstrated safety and led to 1 PR and 3 SD lasting at least 24 weeks [138].
IDO Inhibitors (Indoximod, GDC-0919, and Epacadostat)
Indoximod (D-1-methyl-tryptophan (D-1-MT), NewLink Genetics) is the first small-molecule IDO inhibitor to enter clinical trials. Preclinical studies in murine models of ovarian cancer have shown that IDO inhibition with the racemic compound 1-MT is synergistic with chemotherapy and that the D (but not the L) racemer is responsible for the majority of antitumor activity [139, 140]. Another IDO inhibitor is the second-generation GDC-0919 (formerly NLG919, NewLink Genetics/Genentech) which specifically inhibits IDO1. Both inhibitors are currently being examined in several clinical trials for solid tumors. A third is the IDO1 inhibitor epacadostat (INCB024360, Incyte Corporation) that is in several clinical trials for ovarian cancer either as monotherapy (NCT01685255 and NCT02042430) or in combination with peptide vaccines (NCT02166905 and NCT02575807) or checkpoint blockade (NCT02178722 and NCT02327078). In a phase I trial in patients with advanced malignancies including ovarian cancer, 90 % inhibition of IDO activity was achieved [141].
Combination Therapies
Combination immunotherapies can synergize to enhance clinical responses by enhancing different stages of the antitumor immune response (antigen uptake and presentation, T-cell activation, and T-cell response maintenance) and modifying various aspects in the TME (angiogenesis and immunosuppression). Combining checkpoint blockade with other immunotherapeutic strategies that have shown limited efficacy as monotherapies, such as vaccination, is an area of intensive research. For example, a phase I trial in six patients with recurrent ovarian cancer examined a DC-based autologous whole tumor lysate vaccine in combination with Treg-depleting metronomic cyclophosphamide, the antiangiogenic mAb bevacizumab, and ACT (vaccine-primed ex vivo CD3/CD28-costimulated peripheral blood autologous T cells). Antitumor immune responses (in the form of increased tumor-reactive T cells and reduced Tregs) and clinical benefit were observed in four patients, including 1 CR, 1 PR, and 2 SD [142]. Checkpoint inhibitors in combination are also being clinically tested. For example, an ongoing phase I trial in patients with advanced solid tumors (including ovarian cancer) is evaluating the combination of tremelimumab (a humanized IgG2 anti-CTLA-4 mAb) with durvalumab (anti-PD-L1) (NCT01975831) [143]. Identification of the optimal immunotherapy combination will require evaluation of not only synergistic mechanisms of action and ideal sequence of dosing but also overlapping toxicities in order to maximize clinical benefit.
Conclusions
Recent advances and rational immunotherapeutic combinations hold immense potential to improve outcomes for patients with ovarian cancer. Nonetheless, a number of challenges remain that are being critically addressed. Immunotherapies are typically administered in conjunction with or post chemotherapy. Several chemotherapeutic drugs utilized in ovarian cancer exert immunostimulatory effects. Examples include platinum compounds (which reduce PD-L2 expression on DCs), trabectedin (which inhibits monocyte differentiation into TAMs), and decitabine (which triggers a type I IFN response) [144–146]. Integrating immunotherapy with chemotherapy requires careful consideration of drugs, dosing, and schedule in order to neutralize the unwarranted immunosuppressive effects and maximize the immunostimulatory effects of chemotherapy [147]. A second challenge is to better characterize the TME not only to identify the rare somatic mutations that are immunogenic but also to understand host-tumor interactions. Like other cancers, ovarian cancer is heterogenic, and recent gene expression profiling studies have recognized several distinct molecular subtypes with markedly different prognoses [148]. Identifying dominant immunosuppressive pathways as well as collateral pathways can improve response rates in immunotherapy by improving the design of combination treatment regimens [149, 150]. Lastly, identification of biomarkers that predict response to immunotherapies will allow patients to be optimally matched with therapies that are expected to deliver the maximum benefit while minimizing unnecessary toxicity.
References
Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348(3):203–13. doi:348/3/203 [pii].
Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102(51):18538–43. doi:10.1073/pnas.0509182102.
Tomsová M, Melichar B, Sedláková I, Steiner I. Prognostic significance of CD3+ tumor-infiltrating lymphocytes in ovarian carcinoma. Gynecol Oncol. 2008;108(2):415–20. doi:10.1016/j.ygyno.2007.10.016.
Hwang WT, Adams SF, Tahirovic E, Hagemann IS, Coukos G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol Oncol. 2012;124(2):192–8. doi:10.1016/j.ygyno.2011.09.039.
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–9. doi:10.1038/nm1093.
Chu CS, Kim SH, June CH, Coukos G. Immunotherapy opportunities in ovarian cancer. Expert Rev Anticancer Ther. 2008;8(2):243–57. doi:10.1586/14737140.8.2.243.
Walters CL, Arend RC, Armstrong DK, Naumann RW, Alvarez RD. Folate and folate receptor alpha antagonists mechanism of action in ovarian cancer. Gynecol Oncol. 2013;131(2):493–8. doi:10.1016/j.ygyno.2013.07.080.
Yakirevich E, Sabo E, Lavie O, Mazareb S, Spagnoli GC, Resnick MB. Expression of the MAGE-A4 and NY-ESO-1 cancer-testis antigens in serous ovarian neoplasms. Clini Cancer Res Off J Am Asso. 2003;9(17):6453–60.
Lin CK, Chao TK, Yu CP, Yu MH, Jin JS. The expression of six biomarkers in the four most common ovarian cancers: correlation with clinicopathological parameters. APMIS Acta Pathologica Micro Immunolo Scandinavica. 2009;117(3):162–75. doi:10.1111/j.1600-0463.2008.00003.x.
Webb JR, Milne K, Nelson BH. PD-1 and CD103 are widely coexpressed on prognostically favorable intraepithelial CD8 T cells in human ovarian cancer. Cancer Immunol Res. 2015;3(8):926–35. doi:10.1158/2326-6066.CIR-14-0239.
Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K, et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A. 2007;104(9):3360–5. doi:10.1073/pnas.0611533104.
Abiko K, Mandai M, Hamanishi J, Yoshioka Y, Matsumura N, Baba T, et al. PD-L1 on tumor cells is induced in ascites and promotes peritoneal dissemination of ovarian cancer through CTL dysfunction. Clini Cancer Res Off J Am Asso. 2013;19(6):1363–74. doi:10.1158/1078-0432.CCR-12-2199.
Makkouk A, Weiner GJ. Cancer immunotherapy and breaking immune tolerance: new approaches to an old challenge. Cancer Res. 2015;75(1):5–10. doi:10.1158/0008-5472.CAN-14-2538.
Martinez Forero I, Okada H, Topalian SL, Gajewski TF, Korman AJ, Melero I. Workshop on immunotherapy combinations. Society for Immunotherapy of Cancer annual meeting Bethesda, November 3, 2011. J Transl Med. 2012;10:108. doi:10.1186/1479-5876-10-108.
Geskin LJ. Monoclonal antibodies. Dermatol Clin. 2015;33(4):777–86. doi:10.1016/j.det.2015.05.015.
Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12(4):237–51. doi:10.1038/nrc3237;10.1038/nrc3237.
Weiner GJ. Building better monoclonal antibody-based therapeutics. Nat Rev Cancer. 2015;15(6):361–70. doi:10.1038/nrc3930.
Siwak DR, Carey M, Hennessy BT, Nguyen CT, McGahren Murray MJ, Nolden L, et al. Targeting the epidermal growth factor receptor in epithelial ovarian cancer: current knowledge and future challenges. J Oncol. 2010;2010:568938. doi:10.1155/2010/568938.
Lu Y, Li X, Liang K, Luwor R, Siddik ZH, Mills GB, et al. Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res. 2007;67(17):8240–7. doi:10.1158/0008-5472.CAN-07-0589.
Patel D, Lahiji A, Patel S, Franklin M, Jimenez X, Hicklin DJ, et al. Monoclonal antibody cetuximab binds to and down-regulates constitutively activated epidermal growth factor receptor vIII on the cell surface. Anticancer Res. 2007;27(5A):3355–66.
Schilder RJ, Pathak HB, Lokshin AE, Holloway RW, Alvarez RD, Aghajanian C, et al. Phase II trial of single agent cetuximab in patients with persistent or recurrent epithelial ovarian or primary peritoneal carcinoma with the potential for dose escalation to rash. Gynecol Oncol. 2009;113(1):21–7. doi:10.1016/j.ygyno.2008.12.003.
Secord AA, Blessing JA, Armstrong DK, Rodgers WH, Miner Z, Barnes MN, et al. Phase II trial of cetuximab and carboplatin in relapsed platinum-sensitive ovarian cancer and evaluation of epidermal growth factor receptor expression: a Gynecologic Oncology Group study. Gynecol Oncol. 2008;108(3):493–9. doi:10.1016/j.ygyno.2007.11.029.
Steffensen KD, Waldstrom M, Pallisgard N, Lund B, Bergfeldt K, Wihl J, et al. Panitumumab and pegylated liposomal doxorubicin in platinum-resistant epithelial ovarian cancer with KRAS wild-type: the PaLiDo study, a phase II nonrandomized multicenter study. Int J Gynecolo Cancer Off J. 2013;23(1):73–80. doi:10.1097/IGC.0b013e3182775fae.
Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ. Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol. 2001;19(14):3312–22.
Tang Z, Qian M, Ho M. The role of mesothelin in tumor progression and targeted therapy. Anticancer Agents Med Chem. 2013;13(2):276–80.
Felder M, Kapur A, Gonzalez-Bosquet J, Horibata S, Heintz J, Albrecht R, et al. MUC16 (CA125): tumor biomarker to cancer therapy, a work in progress. Mol Cancer. 2014;13:129. doi:10.1186/1476-4598-13-129.
Hassan R, Cohen SJ, Phillips M, Pastan I, Sharon E, Kelly RJ, et al. Phase I clinical trial of the chimeric anti-mesothelin monoclonal antibody MORAb-009 in patients with mesothelin-expressing cancers. Clin Cancer Res. 2010;16(24):6132–8. doi:10.1158/1078-0432.CCR-10-2275.
Sabbatini P, Harter P, Scambia G, Sehouli J, Meier W, Wimberger P, et al. Abagovomab as maintenance therapy in patients with epithelial ovarian cancer: a phase III trial of the AGO OVAR, COGI, GINECO, and GEICO--the MIMOSA study. J Clin Oncol. 2013;31(12):1554–61. doi:10.1200/JCO.2012.46.4057.
Berek JS, Taylor PT, Gordon A, Cunningham MJ, Finkler N, Orr J, et al. Randomized, placebo-controlled study of oregovomab for consolidation of clinical remission in patients with advanced ovarian cancer. J Clin Oncol. 2004;22(17):3507–16. doi:10.1200/JCO.2004.09.016.
Berek J, Taylor P, McGuire W, Smith LM, Schultes B, Nicodemus CF. Oregovomab maintenance monoimmunotherapy does not improve outcomes in advanced ovarian cancer. J Clin Oncol. 2009;27(3):418–25. doi:10.1200/JCO.2008.17.8400.
Bagnoli M, Canevari S, Figini M, Mezzanzanica D, Raspagliesi F, Tomassetti A, et al. A step further in understanding the biology of the folate receptor in ovarian carcinoma. Gynecol Oncol. 2003;88(1 Pt 2):S140–4.
Lutz RJ. Targeting the folate receptor for the treatment of ovarian cancer. Transl Cancer Res. 2015;4(1):118–26. doi: 10.3978/j.issr2218-676X.2015.01.04.
Armstrong DK, White AJ, Weil SC, Phillips M, Coleman RL. Farletuzumab (a monoclonal antibody against folate receptor alpha) in relapsed platinum-sensitive ovarian cancer. Gynecol Oncol. 2013;129(3):452–8. doi:10.1016/j.ygyno.2013.03.002.
Drerup JM, Liu Y, Padron AS, Murthy K, Hurez V, Zhang B, et al. Immunotherapy for ovarian cancer. Curr Treat Options Oncol. 2015;16(1):317. doi:10.1007/s11864-014-0317-1.
Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9(6):669–76. doi:nm0603-669 [pii].
Hata K, Watanabe Y, Nakai H, Hata T, Hoshiai H. Expression of the vascular endothelial growth factor (VEGF) gene in epithelial ovarian cancer: an approach to anti-VEGF therapy. Anticancer Res. 2011;31(2):731–7. doi:31/2/731 [pii].
Yang DH, Park JS, Jin CJ, Kang HK, Nam JH, Rhee JH, et al. The dysfunction and abnormal signaling pathway of dendritic cells loaded by tumor antigen can be overcome by neutralizing VEGF in multiple myeloma. Leuk Res. 2009;33(5):665–70. doi:10.1016/j.leukres.2008.09.006.
Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010;70(15):6171–80. doi:10.1158/0008-5472.CAN-10-0153.
Burger RA, Sill MW, Monk BJ, Greer BE, Sorosky JI. Phase II trial of bevacizumab in persistent or recurrent epithelial ovarian cancer or primary peritoneal cancer: a Gynecologic Oncology Group Study. J Clin Oncol. 2007;25(33):5165–71. doi:10.1200/JCO.2007.11.5345.
Cannistra SA, Matulonis UA, Penson RT, Hambleton J, Dupont J, Mackey H, et al. Phase II study of bevacizumab in patients with platinum-resistant ovarian cancer or peritoneal serous cancer. J Clin Oncol. 2007;25(33):5180–6. doi:10.1200/JCO.2007.12.0782.
McGonigle KF, Muntz HG, Vuky J, Paley PJ, Veljovich DS, Greer BE, et al. Combined weekly topotecan and biweekly bevacizumab in women with platinum-resistant ovarian, peritoneal, or fallopian tube cancer: results of a phase 2 study. Cancer. 2011;117(16):3731–40. doi:10.1002/cncr.25967.
Garcia AA, Hirte H, Fleming G, Yang D, Tsao-Wei DD, Roman L, et al. Phase II clinical trial of bevacizumab and low-dose metronomic oral cyclophosphamide in recurrent ovarian cancer: a trial of the California, Chicago, and Princess Margaret Hospital phase II consortia. J Clin Oncol. 2008;26(1):76–82. doi:10.1200/JCO.2007.12.1939.
Pujade-Lauraine E, Hilpert F, Weber B, Reuss A, Poveda A, Kristensen G, et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: the AURELIA open-label randomized phase III trial. J Clini Oncolo Offi J Am Soc. 2014;32(13):1302–8. doi:10.1200/JCO.2013.51.4489.
Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, et al. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008;18(5):349–55. doi:10.1016/j.semcancer.2008.03.004.
Kawamura K, Komohara Y, Takaishi K, Katabuchi H, Takeya M. Detection of M2 macrophages and colony-stimulating factor 1 expression in serous and mucinous ovarian epithelial tumors. Pathol Int. 2009;59(5):300–5. doi:10.1111/j.1440-1827.2009.02369.x.
Takaishi K, Komohara Y, Tashiro H, Ohtake H, Nakagawa T, Katabuchi H, et al. Involvement of M2-polarized macrophages in the ascites from advanced epithelial ovarian carcinoma in tumor progression via Stat3 activation. Cancer Sci. 2010;101(10):2128–36. doi:10.1111/j.1349-7006.2010.01652.x.
Zhang M, He Y, Sun X, Li Q, Wang W, Zhao A, et al. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J Ovarian Res. 2014;7:19. doi:10.1186/1757-2215-7-19.
Hume DA, MacDonald KP. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood. 2012;119(8):1810–20. doi:10.1182/blood-2011-09-379214.
Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25(6):846–59. doi:10.1016/j.ccr.2014.05.016.
Cassier PA, Gomez-Roca CA, Italiano A, Cannarile M, Ries C, Brillouet A, et al. Phase 1 study of RG7155, a novel anti-CSF1R antibody, in patients with locally advanced pigmented villonodular synovitis (PVNS). ASCO annual meeting abstract #10504. 2014. http://meetinglibrary.asco.org/content/131522-144.
Kryczek I, Wei S, Zou L, Zhu G, Mottram P, Xu H, et al. Cutting edge: induction of B7-H4 on APCs through IL-10: novel suppressive mode for regulatory T cells. J Immunol. 2006;177(1):40–4.
Kryczek I, Zou L, Rodriguez P, Zhu G, Wei S, Mottram P, et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. 2006;203(4):871–81. doi:10.1084/jem.20050930.
Kryczek I, Wei S, Zhu G, Myers L, Mottram P, Cheng P, et al. Relationship between B7-H4, regulatory T cells, and patient outcome in human ovarian carcinoma. Cancer Res. 2007;67(18):8900–5. doi:10.1158/0008-5472.CAN-07-1866.
Dangaj D, Lanitis E, Zhao A, Joshi S, Cheng Y, Sandaltzopoulos R, et al. Novel recombinant human b7-h4 antibodies overcome tumoral immune escape to potentiate T-cell antitumor responses. Cancer Res. 2013;73(15):4820–9. doi:10.1158/0008-5472.CAN-12-3457.
Litvinov SV, Bakker HA, Gourevitch MM, Velders MP, Warnaar SO. Evidence for a role of the epithelial glycoprotein 40 (Ep-CAM) in epithelial cell-cell adhesion. Cell Adhes Commun. 1994;2(5):417–28.
Bellone S, Siegel ER, Cocco E, Cargnelutti M, Silasi DA, Azodi M, et al. Overexpression of epithelial cell adhesion molecule in primary, metastatic, and recurrent/chemotherapy-resistant epithelial ovarian cancer: implications for epithelial cell adhesion molecule-specific immunotherapy. Int J Gynecolo Cancer Off J. 2009;19(5):860–6. doi:00009577-200907000-00011 [pii].
Köbel M, Kalloger SE, Boyd N, McKinney S, Mehl E, Palmer C, et al. Ovarian carcinoma subtypes are different diseases: implications for biomarker studies. PLoS Med. 2008;5(12):e232. doi:10.1371/journal.pmed.0050232.
Spizzo G, Went P, Dirnhofer S, Obrist P, Moch H, Baeuerle PA, et al. Overexpression of epithelial cell adhesion molecule (Ep-CAM) is an independent prognostic marker for reduced survival of patients with epithelial ovarian cancer. Gynecol Oncol. 2006;103(2):483–8. doi:10.1016/j.ygyno.2006.03.035.
Heiss MM, Murawa P, Koralewski P, Kutarska E, Kolesnik OO, Ivanchenko VV, et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: results of a prospective randomized phase II/III trial. Int J Cancer. 2010;127(9):2209–21. doi:10.1002/ijc.25423.
Ruf P, Lindhofer H. Induction of a long-lasting antitumor immunity by a trifunctional bispecific antibody. Blood. 2001;98(8):2526–34.
Zeidler R, Mysliwietz J, Csánady M, Walz A, Ziegler I, Schmitt B, et al. The Fc-region of a new class of intact bispecific antibody mediates activation of accessory cells and NK cells and induces direct phagocytosis of tumour cells. Br J Cancer. 2000;83(2):261–6. doi:10.1054/bjoc.2000.1237.
Berek JS, Edwards RP, Parker LP, DeMars LR, Herzog TJ, Lentz SS, et al. Catumaxomab for the treatment of malignant ascites in patients with chemotherapy-refractory ovarian cancer: a phase II study. Int J Gynecol Cancer. 2014;24(9):1583–9. doi:10.1097/IGC.0000000000000286.
Erickson HK, Park PU, Widdison WC, Kovtun YV, Garrett LM, Hoffman K, et al. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006;66(8):4426–33. doi:10.1158/0008-5472.CAN-05-4489.
Golfier S, Kopitz C, Kahnert A, Heisler I, Schatz CA, Stelte-Ludwig B, et al. Anetumab ravtansine: a novel mesothelin-targeting antibody-drug conjugate cures tumors with heterogeneous target expression favored by bystander effect. Mol Cancer Ther. 2014;13(6):1537–48. doi:10.1158/1535-7163.MCT-13-0926.
Liu J, Moore K, Birrer M, Berlin S, Matulonis U, Infante J, et al. Abstract LB-290: Targeting MUC16 with the antibody-drug conjugate (ADC) DMUC5754A in patients with platinum-resistant ovarian cancer: a phase I study of safety and pharmacokinetics. Cancer Res. 2013;73(8 Suppl):LB-290. doi:10.1158/1538-7445.AM2013-LB-290.
Ab O, Whiteman KR, Bartle LM, Sun X, Singh R, Tavares D, et al. IMGN853, a folate receptor-α (FRα)-targeting antibody-drug conjugate, exhibits potent targeted antitumor activity against FRα-expressing tumors. Mol Cancer Ther. 2015;14(7):1605–13. doi:10.1158/1535-7163.MCT-14-1095.
Moore KN, Martin LP, Seward SM, Bauer TM, O’Malley DM, Perez RP, et al. Preliminary single agent activity of IMGN853, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), in platinum-resistant epithelial ovarian cancer (EOC) patients (pts): phase I trial. ASCO annual meeting abstract # 5518. 2015. http://meetinglibrary.asco.org/content/150377-156.
Martin LP, Moore KN, O’Malley DM, Seward SM, Bauer TM, Perez RP, et al. Association of folate receptor alpha (FRα) expression level and clinical activity of IMGN853 (mirvetuximab soravtansine), a FRα-targeting antibody-drug conjugate (ADC), in FRα -expressing platinum-resistant epithelial ovarian cancer (EOC) patients (pts). Proceedings of the 2015 AACR-NCI-EORTC international conference on molecular targets and cancer therapeutics, Nov 5–9. Boston: AACR; 2015.
Gattinoni L, Powell DJ, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6(5):383–93. doi:10.1038/nri1842.
Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12(4):269–81. doi:10.1038/nri3191.
Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–15. doi:10.1038/nm1100.
Freedman RS, Edwards CL, Kavanagh JJ, Kudelka AP, Katz RL, Carrasco CH, et al. Intraperitoneal adoptive immunotherapy of ovarian carcinoma with tumor-infiltrating lymphocytes and low-dose recombinant interleukin-2: a pilot trial. J Immunother Emphasis Tumor Immunol. 1994;16(3):198–210.
Aoki Y, Takakuwa K, Kodama S, Tanaka K, Takahashi M, Tokunaga A, et al. Use of adoptive transfer of tumor-infiltrating lymphocytes alone or in combination with cisplatin-containing chemotherapy in patients with epithelial ovarian cancer. Cancer Res. 1991;51(7):1934–9.
Fujita K, Ikarashi H, Takakuwa K, Kodama S, Tokunaga A, Takahashi T, et al. Prolonged disease-free period in patients with advanced epithelial ovarian cancer after adoptive transfer of tumor-infiltrating lymphocytes. Clin Cancer Res. 1995;1(5):501–7.
Wefers C, Lambert LJ, Torensma R, Hato SV. Cellular immunotherapy in ovarian cancer: targeting the stem of recurrence. Gynecol Oncol. 2015;137(2):335–42. doi:10.1016/j.ygyno.2015.02.019.
Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12(20 Pt 1):6106–15. doi:10.1158/1078-0432.CCR-06-1183.
Song DG, Ye Q, Carpenito C, Poussin M, Wang LP, Ji C, et al. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res. 2011;71(13):4617–27. doi:10.1158/0008-5472.CAN-11-0422.
Kandalaft LE, Powell DJ, Coukos G. A phase I clinical trial of adoptive transfer of folate receptor-alpha redirected autologous T cells for recurrent ovarian cancer. J Transl Med. 2012;10:157. doi:10.1186/1479-5876-10-157.
Tanyi JL, Haas AR, Beatty GL, Morgan MA, Stashwick, Caitlin J, O’Hara MH, et al. Safety and feasibility of chimeric antigen receptor modified T cells directed against mesothelin (CART-meso) in patients with mesothelin expressing cancers. Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; Apr 18–22; Philadelphia: AACR, 2015.
Hung CF, Wu TC, Monie A, Roden R. Antigen-specific immunotherapy of cervical and ovarian cancer. Immunol Rev. 2008;222:43–69. doi:10.1111/j.1600-065X.2008.00622.x.
Melief CJ, van Hall T, Arens R, Ossendorp F, van der Burg SH. Therapeutic cancer vaccines. J Clin Invest. 2015;125(9):3401–12. doi:10.1172/JCI80009.
Odunsi K, Jungbluth AA, Stockert E, Qian F, Gnjatic S, Tammela J, et al. NY-ESO-1 and LAGE-1 cancer-testis antigens are potential targets for immunotherapy in epithelial ovarian cancer. Cancer Res. 2003;63(18):6076–83.
Jäger E, Chen YT, Drijfhout JW, Karbach J, Ringhoffer M, Jäger D, et al. Simultaneous humoral and cellular immune response against cancer-testis antigen NY-ESO-1: definition of human histocompatibility leukocyte antigen (HLA)-A2-binding peptide epitopes. J Exp Med. 1998;187(2):265–70.
Odunsi K, Qian F, Matsuzaki J, Mhawech-Fauceglia P, Andrews C, Hoffman EW, et al. Vaccination with an NY-ESO-1 peptide of HLA class I/II specificities induces integrated humoral and T cell responses in ovarian cancer. Proc Natl Acad Sci U S A. 2007;104(31):12837–42. doi:10.1073/pnas.0703342104.
Diefenbach CS, Gnjatic S, Sabbatini P, Aghajanian C, Hensley ML, Spriggs DR, et al. Safety and immunogenicity study of NY-ESO-1b peptide and montanide ISA-51 vaccination of patients with epithelial ovarian cancer in high-risk first remission. Clin Cancer Res. 2008;14(9):2740–8. doi:10.1158/1078-0432.CCR-07-4619.
Sabbatini P, Tsuji T, Ferran L, Ritter E, Sedrak C, Tuballes K, et al. Phase I trial of overlapping long peptides from a tumor self-antigen and poly-ICLC shows rapid induction of integrated immune response in ovarian cancer patients. Clin Cancer Res. 2012;18(23):6497–508. doi:10.1158/1078-0432.CCR-12-2189.
Tsuji T, Sabbatini P, Jungbluth AA, Ritter E, Pan L, Ritter G, et al. Effect of Montanide and poly-ICLC adjuvant on human self/tumor antigen-specific CD4+ T cells in phase I overlapping long peptide vaccine trial. Cancer Immunol Res. 2013;1(5):340–50. doi:10.1158/2326-6066.CIR-13-0089.
Odunsi K, Matsuzaki J, James SR, Mhawech-Fauceglia P, Tsuji T, Miller A, et al. Epigenetic potentiation of NY-ESO-1 vaccine therapy in human ovarian cancer. Cancer Immunol Res. 2014;2(1):37–49. doi:10.1158/2326-6066.CIR-13-0126.
Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2(8):594–604. doi:10.1038/nrc864.
Nijman HW, Lambeck A, van der Burg SH, van der Zee AG, Daemen T. Immunologic aspect of ovarian cancer and p53 as tumor antigen. J Transl Med. 2005;3:34. doi:10.1186/1479-5876-3-34.
Goodell V, Salazar LG, Urban N, Drescher CW, Gray H, Swensen RE, et al. Antibody immunity to the p53 oncogenic protein is a prognostic indicator in ovarian cancer. J Clin Oncol. 2006;24(5):762–8. doi:10.1200/JCO.2005.03.2813.
Lambeck A, Leffers N, Hoogeboom BN, Sluiter W, Hamming I, Klip H, et al. P53-specific T cell responses in patients with malignant and benign ovarian tumors: implications for p53 based immunotherapy. Int J Cancer. 2007;121(3):606–14. doi:10.1002/ijc.22710.
Rahma OE, Ashtar E, Czystowska M, Szajnik ME, Wieckowski E, Bernstein S, et al. A gynecologic oncology group phase II trial of two p53 peptide vaccine approaches: subcutaneous injection and intravenous pulsed dendritic cells in high recurrence risk ovarian cancer patients. Cancer Immunol Immunother. 2012;61(3):373–84. doi:10.1007/s00262-011-1100-9.
Leffers N, Lambeck AJ, Gooden MJ, Hoogeboom BN, Wolf R, Hamming IE, et al. Immunization with a P53 synthetic long peptide vaccine induces P53-specific immune responses in ovarian cancer patients, a phase II trial. Int J Cancer. 2009;125(9):2104–13. doi:10.1002/ijc.24597.
Leffers N, Vermeij R, Hoogeboom BN, Schulze UR, Wolf R, Hamming IE, et al. Long-term clinical and immunological effects of p53-SLP® vaccine in patients with ovarian cancer. Int J Cancer. 2012;130(1):105–12. doi:10.1002/ijc.25980.
Disis ML, Gooley TA, Rinn K, Davis D, Piepkorn M, Cheever MA, et al. Generation of T-cell immunity to the HER-2/neu protein after active immunization with HER-2/neu peptide-based vaccines. J Clin Oncol. 2002;20(11):2624–32.
Disis ML, Goodell V, Schiffman K, Knutson KL. Humoral epitope-spreading following immunization with a HER-2/neu peptide based vaccine in cancer patients. J Clin Immunol. 2004;24(5):571–8. doi:10.1023/B:JOCI.0000040928.67495.52.
Kandalaft LE, Powell DJ, Singh N, Coukos G. Immunotherapy for ovarian cancer: what’s next? J Clin Oncol. 2011;29(7):925–33. doi:10.1200/JCO.2009.27.2369.
Senzer N, Barve M, Kuhn J, Melnyk A, Beitsch P, Lazar M, et al. Phase I trial of “bi-shRNAi(furin)/GMCSF DNA/autologous tumor cell” vaccine (FANG) in advanced cancer. Mol Ther. 2012;20(3):679–86. doi:10.1038/mt.2011.269.
Oh J, Barve M, Grosen EA, Fine BA, Heffernan TP, Matthews CM, et al. Randomized phase II trial of maintenance autologous tumor cell vaccine (FANG™) following clinical complete response (cCR) in stage III/IV ovarian cancer: preliminary results. Gynecol Oncol. 2015;137(Supplement 1):2–3. doi:10.1016/j.ygyno.2015.01.003.
Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–77. doi:10.1038/nrc3258.
Vlad AM, Diaconu I, Gantt KR. MUC1 in endometriosis and ovarian cancer. Immunol Res. 2006;36(1–3):229–36. doi:10.1385/IR:36:1:229.
Brossart P, Wirths S, Stuhler G, Reichardt VL, Kanz L, Brugger W. Induction of cytotoxic T-lymphocyte responses in vivo after vaccinations with peptide-pulsed dendritic cells. Blood. 2000;96(9):3102–8.
Gray HJ, Gargosky SE, Team C-S. Progression-free survival in ovarian cancer patients in second remission with mucin-1 autologous dendritic cell therapy. ASCO annual meeting abstract #5504. 2014. http://meetinglibrary.asco.org/content/134878-144.
Peethambaram PP, Melisko ME, Rinn KJ, Alberts SR, Provost NM, Jones LA, et al. A phase I trial of immunotherapy with lapuleucel-T (APC8024) in patients with refractory metastatic tumors that express HER-2/neu. Clin Cancer Res. 2009;15(18):5937–44. doi:10.1158/1078-0432.CCR-08-3282.
Hernando JJ, Park TW, Kübler K, Offergeld R, Schlebusch H, Bauknecht T. Vaccination with autologous tumour antigen-pulsed dendritic cells in advanced gynaecological malignancies: clinical and immunological evaluation of a phase I trial. Cancer Immunol Immunother. 2002;51(1):45–52. doi:10.1007/s00262-001-0255-1.
Baek S, Kim YM, Kim SB, Kim CS, Kwon SW, Kim Y, et al. Therapeutic DC vaccination with IL-2 as a consolidation therapy for ovarian cancer patients: a phase I/II trial. Cell Mol Immunol. 2015;12(1):87–95. doi:10.1038/cmi.2014.40.
Chiang CL, Kandalaft LE, Tanyi J, Hagemann AR, Motz GT, Svoronos N, et al. A dendritic cell vaccine pulsed with autologous hypochlorous acid-oxidized ovarian cancer lysate primes effective broad antitumor immunity: from bench to bedside. Clin Cancer Res. 2013;19(17):4801–15. doi:10.1158/1078-0432.CCR-13-1185.
Larocca C, Schlom J. Viral vector-based therapeutic cancer vaccines. Cancer J. 2011;17(5):359–71. doi:10.1097/PPO.0b013e3182325e63.
Gulley JL, Arlen PM, Tsang KY, Yokokawa J, Palena C, Poole DJ, et al. Pilot study of vaccination with recombinant CEA-MUC-1-TRICOM poxviral-based vaccines in patients with metastatic carcinoma. Clin Cancer Res. 2008;14(10):3060–9. doi:10.1158/1078-0432.CCR-08-0126.
Mohebtash M, Tsang KY, Madan RA, Huen NY, Poole DJ, Jochems C, et al. A pilot study of MUC-1/CEA/TRICOM poxviral-based vaccine in patients with metastatic breast and ovarian cancer. Clin Cancer Res. 2011;17(22):7164–73. doi:10.1158/1078-0432.CCR-11-0649.
Odunsi K, Matsuzaki J, Karbach J, Neumann A, Mhawech-Fauceglia P, Miller A, et al. Efficacy of vaccination with recombinant vaccinia and fowlpox vectors expressing NY-ESO-1 antigen in ovarian cancer and melanoma patients. Proc Natl Acad Sci U S A. 2012;109(15):5797–802. doi:10.1073/pnas.1117208109.
Zakharia Y, Rahma O, Khleif SN. Ovarian cancer from an immune perspective. Radiat Res. 2014;182(2):239–51. doi:10.1667/RR13741.1.
Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell Jr DJ, et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med. 2012;4(134):134ra62. doi:10.1126/scitranslmed.3003330;10.1126/scitranslmed.3003330.
Curiel T, Thibodeaux S, Wall S, Pandeswara SL, Daniel B, Drerup J, et al. Denileukin diftitox depletes regulatory T cells without clinical benefit in advanced stage epithelial ovarian carcinoma (VAC3P.945). J Immunol. 2014;192(1 Suppl) 73.7.
Curiel T, Sareddy G, Hurez V, Qin K, Pandeswara SL, Vadlamudi R, et al. B7-H1 blockade improves efficacy of regulatory T cell depletion as cancer immunotherapy by reducing Treg regeneration, possibly through tumor B7-H1 effects (VAC3P.950). J Immunol. 2014;192(1 Suppl) 73.12.
Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, et al. Metronomic cyclophosphamide regimen selectively depletes CD4 + CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007;56(5):641–8. doi:10.1007/s00262-006-0225-8.
Chu CS, Boyer J, Schullery DS, Gimotty PA, Gamerman V, Bender J, et al. Phase I/II randomized trial of dendritic cell vaccination with or without cyclophosphamide for consolidation therapy of advanced ovarian cancer in first or second remission. Cancer Immunol Immunother. 2012;61(5):629–41. doi:10.1007/s00262-011-1081-8.
Vermeij R, Leffers N, Hoogeboom BN, Hamming IL, Wolf R, Reyners AK, et al. Potentiation of a p53-SLP vaccine by cyclophosphamide in ovarian cancer: a single-arm phase II study. Int J Cancer. 2012;131(5):E670–80. doi:10.1002/ijc.27388.
Takikawa O. Biochemical and medical aspects of the indoleamine 2,3-dioxygenase-initiated l-tryptophan metabolism. Biochem Biophys Res Commun. 2005;338(1):12–9. doi:10.1016/j.bbrc.2005.09.032.
Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. 2002;196(4):459–68.
Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189(9):1363–72.
Chen W, Liang X, Peterson AJ, Munn DH, Blazar BR. The indoleamine 2,3-dioxygenase pathway is essential for human plasmacytoid dendritic cell-induced adaptive T regulatory cell generation. J Immunol Baltimore Md 1950. 2008;181(8):5396–404. doi:181/8/5396 [pii].
Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–61. doi:10.1016/j.ccell.2015.03.001.
Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A. 2010;107(17):7875–80. doi:10.1073/pnas.1003345107.
Inaba T, Ino K, Kajiyama H, Yamamoto E, Shibata K, Nawa A, et al. Role of the immunosuppressive enzyme indoleamine 2,3-dioxygenase in the progression of ovarian carcinoma. Gynecol Oncol. 2009;115(2):185–92. doi:10.1016/j.ygyno.2009.07.015.
Takao M, Okamoto A, Nikaido T, Urashima M, Takakura S, Saito M, et al. Increased synthesis of indoleamine-2,3-dioxygenase protein is positively associated with impaired survival in patients with serous-type, but not with other types of, ovarian cancer. Oncol Rep. 2007;17(6):1333–9.
Nonaka H, Saga Y, Fujiwara H, Akimoto H, Yamada A, Kagawa S, et al. Indoleamine 2,3-dioxygenase promotes peritoneal dissemination of ovarian cancer through inhibition of natural killer cell function and angiogenesis promotion. Int J Oncol. 2011;38(1):113–20.
Hoos A, Ibrahim R, Korman A, Abdallah K, Berman D, Shahabi V, et al. Development of ipilimumab: contribution to a new paradigm for cancer immunotherapy. Semin Oncol. 2010;37(5):533–46. doi:10.1053/j.seminoncol.2010.09.015.
Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210(9):1695–710. doi:10.1084/jem.20130579.
Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, Seiden MV, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci U S A. 2003;100(8):4712–7. doi:10.1073/pnas.0830997100.
Hodi FS, Butler M, Oble DA, Seiden MV, Haluska FG, Kruse A, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A. 2008;105(8):3005–10. doi:10.1073/pnas.0712237105.
Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015;33(17):1974–82. doi:10.1200/JCO.2014.59.4358.
Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Matsumura N, et al. Efficacy and safety of anti-PD-1 antibody (Nivolumab: BMS-936558, ONO-4538) in patients with platinum-resistant ovarian cancer. ASCO annual meeting abstract #5570. 2014.
Hamanishi J, Mandai M, Ikeda TI, Minami M, Kawaguchi A, Matsumura N, et al. Durable tumor remission in patients with platinum-resistant ovarian cancer receiving nivolumab. ASCO annual meeting abstract# 5511. 2015.
Varga A, Piha-Paul SA, Ott PA, Mehnert JM, Berton-Rigaud D, Johnson EA, et al. Antitumor activity and safety of pembrolizumab in patients (pts) with PD-L1 positive advanced ovarian cancer: interim results from a phase Ib study. ASCO annual meeting abstract #268. 2015.
Disis ML, Patel MR, Pant S, Infante JR, Lockhart C, Kelly K, et al. Avelumab (MSB0010718C), an anti-PD-L1 antibody, in patients with previously treated, recurrent or refractory ovarian cancer: a phase Ib, open-label expansion trial. ASCO annual meeting abstract #5509. 2015.
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–65. doi:10.1056/NEJMoa1200694.
Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11(3):312–9. doi:10.1038/nm1196.
Hou DY, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, et al. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 2007;67(2):792–801. doi:10.1158/0008-5472.CAN-06-2925.
Newton RC, Scherle PA, Bowman K, Liu X, Beatty GL, O’Dwyer PJ, et al. Pharmacodynamic assessment of INCB024360, an inhibitor of indoleamine 2,3-dioxygenase 1 (IDO1), in advanced cancer patients. ASCO annual meeting abstract #2500. 2012.
Kandalaft LE, Powell DJ, Chiang CL, Tanyi J, Kim S, Bosch M, et al. Autologous lysate-pulsed dendritic cell vaccination followed by adoptive transfer of vaccine-primed ex vivo co-stimulated T cells in recurrent ovarian cancer. Oncoimmunol. 2013;2(1):e22664. doi:10.4161/onci.22664.
Ott PA, Callahan MK, Odunsi K, Park AJ, Pan LS, Venhaus RR, et al. A phase I study to evaluate the safety and tolerability of MEDI4736, an anti-programmed cell death-ligand-1 (PD-L1) antibody, in combination with tremelimumab in patients with advanced solid tumors. ASCO annual meeting abstract #TPS3099. 2015. http://meetinglibrary.asco.org/content/149819-156.
Lesterhuis WJ, Punt CJ, Hato SV, Eleveld-Trancikova D, Jansen BJ, Nierkens S, et al. Platinum-based drugs disrupt STAT6-mediated suppression of immune responses against cancer in humans and mice. J Clin Invest. 2011;121(8):3100–8. doi:10.1172/JCI43656.
Allavena P, Signorelli M, Chieppa M, Erba E, Bianchi G, Marchesi F, et al. Anti-inflammatory properties of the novel antitumor agent yondelis (trabectedin): inhibition of macrophage differentiation and cytokine production. Cancer Res. 2005;65(7):2964–71. doi:10.1158/0008-5472.CAN-04-4037.
Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162(5):974–86. doi:10.1016/j.cell.2015.07.011.
Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. 2015;28(6):690–714. doi:10.1016/j.ccell.2015.10.012.
Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res. 2008;14(16):5198–208. doi:10.1158/1078-0432.CCR-08-0196.
Topalian SL, Wolchok JD, Chan TA, Mellman I, Palucka K, Banchereau J, et al. Immunotherapy: the path to win the war on cancer? Cell. 2015;161(2):185–6.
Chester C, Dorigo O, Berek JS, Kohrt H. Immunotherapeutic approaches to ovarian cancer treatment. J Immunother Cancer. 2015;3:7. doi:10.1186/s40425-015-0051-7.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Makkouk, A., Chester, C., Kohrt, H.E. (2017). The Future in Ovarian Cancer: Advances in Immunotherapies. In: Pujade-Lauraine, E., Ray-Coquard, I., Lécuru, F. (eds) Ovarian Cancers. Springer, Cham. https://doi.org/10.1007/978-3-319-32110-3_11
Download citation
DOI: https://doi.org/10.1007/978-3-319-32110-3_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-32108-0
Online ISBN: 978-3-319-32110-3
eBook Packages: MedicineMedicine (R0)