
Abstract
Calixarenes and resorcinarenes have been bridged at the wider rim with a variety of linking groups for a variety of applications. This chapter will outline some of the strategies used and provide a flavour of the range of linking groups investigated. Bridging calixarenes with four equivalent rings usually results in a mixture of predominantly mono- and di-proximally bridged derivatives. For the synthesis of distally bridged calixarenes, a selectively difunctionalised precursor is usually employed. Commonly used precursors are calixarenes functionalised with hydroxymethyl, halomethyl, formyl, amino or carboxy groups. Linking groups have included polyethers, thioethers, esters, amines, amides, acetals, siloxanes and hydrocarbons.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
The versatile ability of calixarene derivatives to act as hosts for a variety of smaller guest species has been widely demonstrated in the literature [1–6]. This is because calixarenes are relatively large bowl-shaped molecules that are readily synthesised and functionalised. The wider rim of a calixarene bowl may be selectively and variously functionalised to enable the calixarene cavity to engage in a range of binding interactions with smaller guest molecules. This range of interactions can be adjusted to be charged or neutral, and may lead to the cavity being selective for a particular type of guest molecule, depending on the configuration and the type of functional groups on the wider rim. An interesting type of functionalisation would be a bridge that spans over the cavity of the calixarene. Such a bridge could partially enclose the calixarene cavity, and perhaps lead to enhanced containment of guest molecules inside the cavity.
Calixarenes that are distally-bridged on the narrower rim are common in the literature [7–11]. The crown ether bridged calixarene synthesised by Ungaro and co-workers in 1995 is one of many examples [12]. Selective distal functionalisation of a calixarene on the narrower rim was first achieved by treating tetrahydroxycalixarene with 1.1 equivalents of potassium carbonate base with an octyl halide (Scheme 10.1). Simple alkylation of the remaining two distal phenols by pentaethylene glycol ditosylate furnished the crown ether-bridged calixarene in the 1,3-alternate conformation. The calixarene crown ether was then shown to be a membrane transport agent selective for caesium ions over sodium ions. Such a system has been utilised by the U.S. Department of Energy to remove caesium ions from more than 11 million litres of radioactive waste [13, 14].

Selective distal bridging of calixarene at the narrower rim
Distal selectivity is a key requirement for selective distal-bridging of calixarenes. The simple two-step procedure developed by Ungaro and co-workers has been responsible for the many narrow-rim, distally-bridged calixarenes reported in the literature. However, this mechanism of distal selectivity is only applicable to the narrower rim of calixarenes where the hydrogen bonding stabilisation is enabled by the phenols being in proximity to each other. Therefore, the popular method for distal bridging calixarenes does not directly apply to the wider rim of calixarenes. Unfortunately, a bridge at the narrower rim of calixarenes in the cone conformation does not make use of the cavity, which is a key attribute of a calixarene. Generally, the calixarene portion in these narrow-rim bridged calixarenes simply serves as a scaffold. Nevertheless, distal bridging over the wider rim has been accomplished by transferring the distal selectivity from the narrower rim to the wider rim via the stronger para-activating effect of the remaining two phenols [15], as reported in the literature [16, 17].
The first characterisation of calixarenes with a distal bridge at the wider rim was reported by Bӧhmer and co-workers in 1988 [18]. In their pioneering work, distally-bridged calixarenes were synthesised by the TiCl4-catalysed Friedel-Crafts alkylation of α,ω-(p-hydroxypheny1)alkanes with bis(bromomethy1)phenols (Scheme 10.2). TiCl4 was thought to also act as a template for the cyclisation. Calixarenes with various aliphatic bridge lengths were synthesised, but with lower yields. Calixarenes with bridges of eight carbons and longer were observed to be in the cone conformation, but shorter bridges resulted in the cone conformation becoming pinched. The cyclisation reaction with a shorter bridge of four carbons was not successful.

Pioneering synthesis of distally-bridged calixarene
To study the effect of the bridge length and rigidity on the synthesis of a distally-bridged calixarene, Zeng et al. have tried various diacid chlorides to form diester linkages with a distal-diol calixarene (Scheme 10.3) [19]. Each experiment was conducted at dilute concentrations and produced multiple products which were separated by column chromatography into three main fractions. The yields of the three fractions of the various trials are shown in (Table 10.1). At these conditions, it is evident that the longer bridge with an eight-carbon chain gives the highest proportion of distally-bridged calixarene. This could be attributed to the increasing flexibility of the longer chain [19], and perhaps the reduction of strain on the calixarene cone conformation. It is also evident that the distally-bridged calixarene was the predominant product for all tests, except for the teraphthalate, which may have suffered from steric strain/hindrance due to its rigidity. The reactions were also attempted at higher concentrations, producing a greater proportion of calixarene oligomer products, as well as trace amounts of shorter-bridged calixarene dimers. Thus as expected, a higher concentration of the reactants increases the chance of intermolecular reactions between calixarenes at the expense of intramolecular reactions within calixarenes.

Synthesis of distally-bridged calixarenes via diester linkages. Experiments to explore the impact of bridge length and rigidity
In a related reaction, Cacciapaglia et al. constructed a distally-bridged calixarene with acetal linkages (Scheme 10.4) [20]. The outcome of this reaction was also similar to Zheng et al. in that the reaction produced multiple calixarene products that included a distally-bridged calixarene, a calixarene dimer and a mixture of calixarene oligomers. This work was part of an investigation into the synthesis of dynamic ‘living’ polymers which could be manipulated by concentration, to selectively give a particular product or oligomer at equilibrium. This work envisioned the creation of a dynamic family of cyclic calixarene oligomers linked together by formaldehyde acetal linkages, which could be reversibly interchanged by the addition of a catalytic amount of acid. However, when a catalytic amount of trifluoroacetic acid was added to all three acetal calixarene fractions, a distally-bridged ether calixarene formed instead. This was thought to proceed via a benzylic carbocation on the calixarene that could be formed by the acid-catalysed cleavage of one of the acetal bonds. The resultant hemiacetal and stabilised p-propoxy benzyl carbocation would then react in an intramolecular reaction, eliminating formaldehyde and forming the unreactive distal ether bridge. This mechanism would explain the observation that all acetal calixarenes, regardless of the type of acetal linkage, resulted in the same distal ether bridge when treated with acid.

Synthesis of acetal-bridged calixarenes and their decomposition into distally-bridged ether calixarene upon treatment with acid
A very similar calixarene with the same short distal ether bridge has also been synthesised by Arduini et al. Beginning with a distal diol calixarene, but with ethoxyethylether substituents on the narrow rim, reaction with NaH and tosyl chloride gave the distal ether bridged calixarene in 30 % yield [21]. The 1H NMR spectrum of the product provided evidence that the ether-bridged calixarene was rigid, adopting a highly-distorted flattened-cone conformation. The protons of the aromatic rings involved in the bridge appeared at a significantly lower chemical shift (~δ 5.7), which suggested their shielding by the un-bridged proximal aromatic rings. The intermolecular bridging product, a calixarene pair linked by two ether bridges, was also produced, but as a minor product. Interestingly, the intermolecular reaction could be favoured by reacting the distal diol calixarene with a distal dichloromethyl calixarene with caesium hydroxide as base, to give the calixarene pair product in 50 % yield.
The intramolecular bridging on the wider rim of the calixarenes was further studied by Arduini et al. with a McMurry reductive coupling between the formyl groups of various formyl calixarenes (Scheme 10.5) [22]. The reductive coupling of distal diformyl calixarene afforded a distally-bridged calixarene with a diol bridge, after 5 h, which could be further reduced to an alkene after 16 h. A similar result was also reported by Lhoták and Shinkai in separate work [23]. Arduini et al. also explored the possibility for the coupling to occur between formyl groups on proximal aromatic rings of tri and tetra formyl calixarenes. With both these calixarenes, only the distal formyl groups showed coupling, while the remaining formyl groups were reduced to methyl groups.

Distal bridging of formyl calixarenes via intramolecular McMurry reductive coupling
Another calixarene with a short distal bridge is the siloxane-bridged calixarene synthesised by Hudrlik et al. [24] .The synthesis of this calixarene was accomplished in four steps, as described in Scheme 10.6. The distal selectivity on the wider rim of the calixarene was obtained through a selective bromine to lithium exchange, followed by quenching with methanol, as described by Larsen and Jørgensen [25].

Synthesis of distally-bridged siloxane calixarene
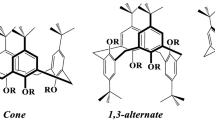
In recent work, Zajícová et al. reported a distally-bridged calixarene obtained from the reductive coupling of a distal dialdoxime calixarene (Scheme 10.7) [16]. The distal selectivity of the starting dialdoxime calixarene was obtained by the stronger activating effect of distal phenols at the narrower rim. The reductive coupling afforded the diamine product in a rather low yield of 26 %, which was attributed to decomposition of the product under aerobic conditions. However, it was discovered that the product easily formed a stable aminal with acetone. Therefore, immediate chromatography of the diamine product with acetone afforded the aminal of the distally-bridged diamine calixarene in 90 % yield. The scope of the reductive coupling was investigated with a number of dialoximine calixarene derivatives with various substituents at both rims of the calixarene, as well as a calixarene in the 1,3-alternate conformation (Fig. 10.1). In all these cases, no distally-bridged calixarene was detected, alluding to the sensitivity of the reductive coupling to the conformation of the starting dialoximine calixarene. A crystal structure of an amide derivative of the distally-bridged calixarene showed that the short bridge, of two carbons, forced the supporting aromatic rings to be bent into the cavity, causing the calixarene to take the pinched cone conformation.

Distal bridging of dialdoxime calixarene via reductive coupling

Reductive coupling with other dialdoxime calixarene derivatives failed to give the distally-bridged product
While investigating the synthesis of phenanthrene calixarenes via photochemical cyclisation, Barton [26] synthesised a distally-bridged calixarene with a cyclobutane bridge (Scheme 10.8). The photolysis of tetra-stilbene calixarene produced a highly complex mixture of cyclisation products including the distally-bridged cyclobutane calixarene, which was isolated in 1 % yield by HPLC. Deducing that the bridging does not occur on the proximal aromatic rings, Hüggenberg et al. attempted the same photochemical cyclisation on proximal di-stilbene calixarene [27]. As anticipated, the proximal di-phenanthrene calixarene was produced as three diastereomers in 67 % yield, without any bridged calixarene. It is interesting that the bridging only occurred on the distal aromatic rings, rather than on proximal.

Photochemical [2 + 2] cycloaddition of tetra-stilbene calixarene to give distal cyclobutane-bridged diphenanthrene calixarene
Liu et al. while exploring the synthesis of cage-like compounds, reported another unexpected distal bridging of calixarenes by intramolecular coupling [28]. The aim was to couple two tetraalkynyl calixarenes via a reversible intermolecular zirconocene coupling to form a cage-like structure. However, the intramolecular coupling was prevalent, and a two-carbon distally bridged calixarene formed instead (Scheme 10.9).

Zirconocene coupling of tetra tetraalkynyl calixarene leads to distal bridging of calixarene
A distally bridged calixarene with the shortest possible bridge has been synthesised by Struck et al. [29]. The distal methylene-bridged calixarene was reported to be exclusively formed by the reaction shown in Scheme 10.10. The ‘collapsed’ cone conformation of the product was evident by signals at δ 5.6 in the 1H NMR spectrum. These signals were attributed to the protons of the bridging aromatic ring, which had become shielded by the neighbouring non-bridging aromatic rings.

Synthesis of distal methylene bridged calixarene
It is noteworthy from the preceding examples that it is possible to span across the wider rim of the calixarene with short bridges. This is possible because the calixarene in the cone conformation is not entirely rigid, despite the steric bulk of the four substituents on the narrower rim. In such a scenario, the cone calixarene oscillates between two pinched-cone conformers, enabling the calixarene to adopt a highly-distorted, pinched cone conformation where the pair of distal aromatic rings involved in bridging are pinched close enough together to form the short bridge. The vibrational motion also allows the other pair of unbridged distal aromatic rings to be forced outwards from the cavity.
Düker and co-workers have synthesised, in good yields, dual tetrathiafulvene proximal bridges across the wider rim of calixarenes for the construction of redox-active molecular architectures (Scheme 10.11) [30]. However bridging the tetrathiafulvene across distal aromatic rings was unsuccessful for calixarene derivatives in both the 1,3-alternate and cone conformations. The lack of success may be due to the conditions used as the bridging reactions were carried out at room temperature. Under these conditions, there may not be sufficient oscillation energy to bring the distal rings close enough to be bridged. Interestingly, distal bridging was possible by using a longer tetrathiafulvene bridging unit, although a calixarene dimer was also produced (Scheme 10.12).

Synthesis of bridged calixarenes with tetrathiafulvene derivatives

Distal bridging of calixarene with tetrathiafulvene
The potential application of calixarenes to function as biomimetic receptors has led Casnati et al. to design distally-bridged calixarenes with peptide bridges. The aim was to use the hydrophobic cavity of the calixarene in conjunction with the hydrogen bonding of the peptide bridge to enable binding of guest molecules. Starting with a distally-functionalised diacid chloride calixarene, the cavity of the calixarene was bridged over by two alanine residues linked together by a nitrogen atom (Scheme 10.13) [31]. The distally-bridged N-linked peptidocalixarene was shown to bind D-Ala-D-Ala (Fig. 10.2), thus mimicking the mode of binding of the vancomycin group of antibiotics. Studies indicated that a proton transfer from the carboxylic acid group of the guest to the amino group of the calixarene pseudopeptide bridge generated a salt which was thought to be the key binding interaction, besides the hydrogen bonding between NH and CO groups. It was speculated that the hydrophobic calixarene cavity may host the methyl group of the non-terminal alanine residue [4].

Synthesis of distally-bridged (L, L) peptidocalixarene

Distally-bridged peptidocalixarene mimics the mode of binding of vancomycin antibiotics by binding D-Ala-D-Ala by the suggested binding mechanism (Image from Casnati et al. [4])
However, some peptidocalixarenes are poor receptors due to intramolecular hydrogen bonding between amino acid residues within the molecule. This is caused by the peptidocalixarene being conformationally flexible. Therefore, to increase conformational rigidity, Sansone et al. have placed a rigid aromatic spacer between the two amino acid residues in a distally-bridged peptidocalixarene that adopted a pinched cone conformation (Scheme 10.14) [32]. This peptidocalixarene was shown to bind anionic guests, having the best affinity for benzoate. From investigations, it appeared again that a proton transfer had occurred, and that the resultant carboxylate anion was electrostatically attracted to the amide protons of the bridge of the peptidocalixarene (Fig. 10.3). π-π Stacking between the aromatic rings also appeared to contribute to the binding [4].

Synthesis of distally-bridged peptidocalixarene linked with a rigid aromatic spacer

Proposed binding of benzoate by distally-bridged peptidocalixarene (Image from Casnati et al. [4])
Distal bridging of calixarenes has also been performed on resorcinarenes. An interesting example is the thiocrown resorcinarenes synthesised by Konishi et al. by the bridging of distal dibromoresorcinarene with 2-mercaptoethyl ether, propane-1,3-dithiol, and ethane-1,2-dithiol (Scheme 10.15) [33]. The octahydroxythiacrown resorcinarene product was reportedly difficult to purify, hence the conversion to octaacetates for characterisation. Despite the various bridge lengths, all the thiocrown resorcinarenes were determined to be in the pinched cone conformation, by analysis of the 1H-NMR chemical shifts and molecular modelling. The bridging was also attempted with rigid dithiols such as 2,6-dimercaptomethylpyridine or m-xylylenedithiol, but to no success. The distal dibromoresorcinarene starting material was obtained by the careful, direct bromination of octahydroxyresorcinarene with limited N-bromosuccinimide [34].

Synthesis of distally-bridged thiacrown resorcinarenes with three bridges of varying length
Other distally-bridged resorcinarenes have been synthesized by Shivanyuk et al. who used aliphatic diamines to bridge distal tetratosylate resorcinarenes by the formation of benzoxazine linkages from a Mannich condensation with formaldehyde (Scheme 10.16) [35]. The cavity of the crown ether-bridged resorcinarene was actually chiral due to the positions of the two benzoxazine linkages. However, no attempt was made to separate the enantiomers. The impact of a shorter bridge on the distal-bridging Mannich condensation was also investigated. Based on the conformation from the crystal structures of the previous chiral benzoxazine resorcinarenes, bridging should not be possible with shorter diamines. Contrary to expectation, the distal bridging was accomplished with ethylene diamine, but the shorter bridge forced the benzoxazine linkages to form on the phenols that were closer together producing the other possible regioisomer which is not chiral. Both these distally-bridged resorcinarene regioisomers took on the pinched cone conformation.

Synthesis of axially-chiral distally-bridged tetratosylate resorcinarene via benzoxazine linkages

Distal bridging of tetratosylate resorcinarene

Studies into the potential enantioselective alkylation of benzaldehyde with diethylzinc in the presence of distally-bridged resorcinarene derivatives
In similar work, the Mannich condensation was also used by Arnott et al. to distally-bridge tetratosylate resorcinarene with various diamines in good yields of 47–80 % (Scheme 10.17) [36]. Various derivatives of these distally-bridged resorcinarenes were then tested for potential to act as enantioselective asymmetric catalysts for the alkylation of benzaldehyde with dimethyl zinc (Scheme 10.18) [37, 38]. Addition of functionality to the bridge, in the form of a dioxane and dimethoxy acetals, was explored for potential coordination to zinc. The different acetals and/or changing the length of the bridge caused a reversal in enantioselectivity. These reversals of enantioselecitivity were indicative that the modifications were causing a significant change in conformation of the bridged resorcinarene. In all these cases, the enantioselectivity of the bridged resorcinarene was limited to about 50 % ee. Arnott et al. have suggested a hypothetic mechanism based on Noyori’s model to explain this limitation [38].
In almost all the examples presented so far in this review, the distal-bridging of calixarenes at the wider rim has been accomplished on calixarenes that have already been distally functionalised. However, if bridging were to be performed on a non-distally-functionalised calixarene with four equally-reactive subunits, multiple bridged-calixarene products are possible. Reinhoudt and coworkers have demonstrated this with flexible crown ether bridges on a cavitand (resorcinarene derivative) [39]. In this work, a cavitand with four equally-reactive phenol subunits was treated with 1.2 equivalents of pentaethyleneglycol ditosylate to give three differently-bridged crowncavitand products (Scheme 10.19). The bridging reaction was performed with various solvents and bases, and the yields of each crowncavitand are shown in Table 10.2. It is evident that the proximally-bridged crowncavitand can be selectively produced in 33 % yield, but with a significant amount of unreacted starting cavitand. The distally-bridged crowncavitand could only be obtained in trace yields of 2–3 % as part of a mixture of all three crowncavitands products. These results show that directly bridging a calixarene with four equally-reactive subunits, with a flexible bridge, will most likely produce either a proximally-bridged calixarene, or a mixture of bridged calixarene products.

Bridging of tetrol cavitand with pentaethyleneglycol ditosylate to form three different crowncavitand products
Bridging calixarenes on the wider rim with crown ethers has also been explored by Nissinen and coworkers in their synthesis of tetramethoxy resorcinarene crown ethers [40]. Tetramethoxy resorcinarene was treated with caesium carbonate base for 15 min, followed by two equivalents of the tetraethylene glycol ditosylate to produce proximally-bridged bis-crown tetramethoxy resorcinarene in 31 % yield (Scheme 10.20). When the deprotonation time with caesium carbonate was extended to 60 min, the bis-crown tetramethoxy resorcinarene was produced in 10 % yield along with the proximally-bridged mono-crown by-product in 13 % yield [41]. Both the mono-crown and this bis-crown tetramethoxy resorcinarenes adopted the flattened cone conformation. The bis-crown tetramethoxy resorcinarene was shown to bind potassium, rubidium, caesium, and silver ions [42–44]. The binding of silver ions was reversible, and was utilised to deliver silver ions to bacteria resulting in cell death and an anti-bacterial effect [43]. The mono-crown tetramethoxy resorcinarene, having a larger cavity, was not able to bind alkali metal cations. Nevertheless, the mono-crown was an optimal host for an acetylcholine (neurotransmitter) guest, binding through interactions between the crown ether with the ammonium portion, as well as hydrogen bonding between the two phenols with the acetate group of the guest.

Synthesis of proximally-bridged crown ether tetramethoxy resorcinarenes
The distal mono-crown tetramethoxy resorcinarene was not reported, signalling a tendency for flexible bridges to form on proximal aromatic rings of a calixarene with equally-reactive subunits.
10.1 Conclusion
As evident from the examples, distally-bridged calixarenes can be obtained from tetra, mono and distally-functionalised calixarenes through a variety of coupling methods. It is also evident that these methods are not applicable to the whole range of different calixarene derivatives, and that achieving selective distal bridging can be challenging. A common issue appears to be the competition between intra and intermolecular coupling. In fact, the examples of distal bridging from tetra and mono functionalised calixarenes were a rather accidental result of dominant intramolecular coupling. The reports of short distal bridged calixarenes demonstrates that the cone conformation of some calixarenes may be more flexible than expected. This flexible cone conformation enables the calixarene to adopt a cone conformation that is pinched, which is characteristic of the distally-bridged calixarenes reported here.
References
Tero, T.-R.; Nissinen, M. Tetrahedron 2014, 70, 1111–1123.
Adhikari, B. B.; Fujii, A.; Schramm, M. P. Eur. J. Org. Chem. 2014, 2972–2979.
Adhikari, B. B.; Roshandel, S.; Fujii, A.; Schramm, M. P. Eur. J. Org. Chem. 2015, 2683–2690.
Casnati, A.; Sansone, F.; Ungaro, R. Acc. Chem. Res. 2003, 36, 246–254.
Mutihac, L.; Lee, J. H.; Kim, J. S.; Vicens, J. Chem. Soc. Rev. 2011, 40, 2777–2796.
Nimse, S. B.; Kim, T. Chem. Soc. Rev. 2013, 42, 366–386.
Arduini, A.; Fabbi, M.; Mantovani, M.; Mirone, L.; Pochini, A.; Secchi, A.; Ungaro, R. J. Org. Chem. 1995, 60, 1454–1457.
Yang, Y.; Cao, X.; Surowiec, M.; Bartsch, R. A. Tetrahedron 2010, 66, 447–454.
Asfari, Z.; Wenger, S.; Vicens, J. Supramol. Sci. 1994, 1, 103–110.
Joseph, R.; Rao, C. P. Chem. Rev. 2011, 111, 4658–4702.
He, Y.; Xiao, Y.; Meng, L.; Zeng, Z.; Wu, X.; Wu, C.-T. Tetrahedron Lett. 2002, 43, 6249–6253.
Casnati, A.; Pochini, A.; Ungaro, R.; Ugozzoli, F.; Arnaud, F.; Fanni, S.; Schwing, M.-J.; Egberink, R. J. M.; de Jong, F.; Reinhoudt, D. N. J. Am. Chem. Soc. 1995, 117, 2767–2777.
Casnati, A. Chem. Commun. 2013, 49, 6827–6830.
Duncan, N. C.; Roach, B. D.; Williams, N. J.; Bonnesen, P. V.; Rajbanshi, A.; Moyer, B. A. Sep. Sci. Technol. 2012, 47, 2074–2087.
Van Loon, J. D.; Arduini, A.; Coppi, L.; Verboom, W.; Pochini, A.; Ungaro, R.; Harkema, S.; Reinhoudt, D. N. J. Org. Chem. 1990, 55, 5639–5646.
Zajícová, M.; Eigner, V.; Budka, J.; Lhoták, P. Tetrahedron Lett. 2015, 56, 5529–5532.
Kanamathareddy, S.; Gutsche, C. D. J. Org. Chem. 1995, 60, 6070–6075.
Goldmann, H.; Vogt, W.; Paulus, E.; Bӧhmer, V. J. Am. Chem. Soc. 1988, 110, 6811–6817.
Zeng, C.-C.; Yuan, H.-S.; Huang, Z.-T. Chin. J. Chem . 2002, 20, 795–802.
Cacciapaglia, R.; Di Stefano, S.; Mandolini, L. J. Phys. Org. Chem. 2008, 21, 688–693.
Arduini, A.; Fanni, S.; Manfredi, G.; Pochini, A.; Ungaro, R.; Sicuri, A. R.; Ugozzoli, F. J. Org. Chem. 1995, 60, 1448–1453.
Arduini, A.; Fanni, S.; Pochini, A.; Sicuri, A. R.; Ungaro, R. Tetrahedron 1995, 51, 7951–7958.
Lhoták, P.; Shinkai, S. Tetrahedron Lett. 1996, 37, 645–648.
Hudrlik, P. F.; Hudrlik, A. M.; Zhang, L.; Arasho, W. D.; Cho, J. J. Org. Chem. 2007, 72, 7858–7862.
Larsen, M.; Jørgensen, M. J. Org. Chem. 1996, 61, 6651–6655.
Barton, O. G., Dissertation, Universitðt Bielefeld, 2008, https://pub.uni-bielefeld.de/publication/2302017 (accessed 16 May 2016).
Hüggenberg, W.; Seper, A.; Oppel, I. M.; Dyker, G. Eur. J. Org. Chem. 2010, 6786–6797.
Liu, F.-Q.; Harder, G.; Tilley, T. D. J. Am. Chem. Soc. 1998, 120, 3271–3272.
Struck, O.; van Duynhoven, J. P. M.; Verboom, W.; Harkema, S.; Reinhoudt, D. N. Chem. Commun. 1996, 1517–1518.
Düker, M. H.; Kutter, F.; Dülcks, T.; Azov, V. A. Supramol. Chem. 2014, 26, 552–560.
Casnati, A.; Fabbi, M.; Pelizzi, N.; Pochini, A.; Sansone, F.; Ungaro, R.; Di Modugno, E.; Tarzia, G. Bioorg. Med. Chem. Lett. 1996, 6, 2699–2704.
Sansone, F.; Baldini, L.; Casnati, A.; Lazzarotto, M.; Ugozzoli, F.; Ungaro, R. PNAS 2002, 99, 4842–4847.
Morikawa, O.; Nakanishi, K.; Miyashiro, M.; Kobayashi, K.; Konishi, H. Synthesis 2000, 233–236.
Konishi, H.; Nakamaru, H.; Nakatani, H.; Ueyama, T.; Kobayashi, K.; Morikawa, O. Chem. Lett. 1997, 26, 185–186.
Shivanyuk, A.; Schmidt, C.; Böhmer, V.; Paulus, E. F.; Lukin, O.; Vogt, W. J. Am. Chem. Soc. 1998, 120, 4319–4326.
Arnott, G.; Bulman Page, P. C.; Heaney, H.; Hunter, R.; Sampler, E. P. Synlett 2001, 412–414.
Arnott, G.; Heaney, H.; Hunter, R.; Page, Philip C. B. Eur. J. Org. Chem. 2004, 5126–5134.
Arnott, G.; Hunter, R. Tetrahedron 2006, 62, 992–1000.
Higler, I.; Boerrigter, H.; Verboom, W.; Kooijman, H.; Spek, A. L.; Reinhoudt, D. N. Eur. J. Org. Chem. 1998, 1998, 1597–1607.
Salorinne, K.; Nissinen, M. Org. Lett. 2006, 8, 5473–5476.
Salorinne, K.; Tero, T.-R.; Riikonen, K.; Nissinen, M. Org. Biomol. Chem. 2009, 7, 4211–4217.
Salorinne, K.; Nissinen, M. Tetrahedron 2008, 64, 1798–1807.
Helttunen, K.; Moridi, N.; Shahgaldian, P.; Nissinen, M. Org. Biomol. Chem. 2012, 10, 2019–2025.
Helttunen, K.; Salorinne, K.; Barboza, T.; Barbosa, H. C.; Suhonen, A.; Nissinen, M. New J. Chem. 2012, 36, 789–795.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Tan, D.A., Mocerino, M. (2016). Calix[4]arenes and Resorcinarenes Bridged at the Wider Rim. In: Neri, P., Sessler, J., Wang, MX. (eds) Calixarenes and Beyond. Springer, Cham. https://doi.org/10.1007/978-3-319-31867-7_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-31867-7_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-31865-3
Online ISBN: 978-3-319-31867-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)