
Abstract
The “Manual of Cardiovascular Proteomics” is the result of the concerted effort of many experts in the field and it addresses the core technologies and approaches that have been implemented since its birth. Although each chapter can be read or studied independently of the others; depending on the level of interest, the whole manual should provide a detailed overview on what is available to the modern scientist who wants to embark on a cardiovascular proteomic expedition. Chapter 1 provides the historical perspective and describes the landmark discoveries that propelled the field forward, along with considerations on how to chose a specific approach and what the first steps to complete a proteomic experiment successfully should be. We hope that you will enjoy the first edition and are looking forward to your feedback in order to improve future editions.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction: The Renaissance of Protein Biochemistry
At the end of the last century, in September 1994 to be exact, a new term was publicly announced during a small meeting titled “2D Electrophoresis: From Protein Maps to Genomes” held in the charming Tuscan hills (Siena, Italy). At this meeting, the term “Proteome” was defined by Marc Wilkins from Sydney as the “protein complement of the genome”, resulting in the birth of the field of “proteomics”. This rather simple moment defined a time in history when the emphasis would slowly but steadily move from the role of genes to that of proteins and their modifications as defining traits of the phenotype, both in health and disease. This “Cultural Revolution” was largely made possible by the remarkable technological advances in the field of protein biochemistry that were achieved in the latter part of the twentieth century. In 1975 Pat O’Farrell and other groups optimized the way to separate and detect over 1100 proteins in a single experiment using two-dimensional polyacrylamide gel electrophoresis (2DE) [1]. Combined with the rapid developments in protein and peptide mass spectrometry (MS), and the creation of protein databases which followed, these combined techniques enabled by the late 1990s the separation, quantification and identification of hundreds to thousands of proteins starting from a single biological sample. Indeed, the study co-Authored by Marc Wilkins the year following the 1994 Siena meeting inaugurated a new era in protein biochemistry [2].
As we write, we have recently celebrated the 21st birthday of the proteome and in retrospect, many things have dramatically improved. To mark this occasion, a Special Issue celebrating the 20th birthday of proteomics, dedicated to the memory of Vitaliano Pallini (the former Supervisor of the Functional Proteomics group at the Molecular Biology Department at the University of Siena, Italy), was recently published in the Journal of Proteomics [3]. We direct the interested reader to this special issue for historical references as several scientists that participated in the birth of proteomics contributed with their personal views and memories.
The “new field” of proteomics (i.e. the complex of technology used to investigate the proteome), which could be also termed advanced protein biochemistry, is the result of the incredible technological advances that have allowed protein biochemists to study the subjects of their investigation in a more efficient way. However, along with the technicalities, as often happens in scientific history, new concepts and ideas arose thanks to the availability of these new technologies. Much like the invention of the microscope allowed van Leeuwenhoek to see animalcules, which led to the discovery of pathogens, proteomic technologies have revealed an almost unimaginable complexity in terms of what are increasingly referred to as proteoforms [4]. Therefore, the impact of proteomics is not only technological, but also highly biological in nature. It is likely that the interest in genes and transcripts that captured the attention of the scientific world (and US Congress) in the 1980s, was also fueled by the relative facility by which nucleic acids can be amplified, a luxury that is still missing in the field of proteomics. However, thanks to the steady advances in protein separation technology, and more recently the implementation of novel MS configurations and protocols, the possibility of mapping the proteome, decorated with its countless post-translational modifications (PTMs), now seems within reach. The outstanding protein scientists who endured long periods of anonymity in the genomic era should not be forgotten as it is thanks to them that we are now capable of achieving all the wonderful things that proteomics is making possible. Thanks to the passion, hard work and dedication of these researchers, the variety and complexity of the proteome has finally emerged. This realization is having and will continue to exert a tremendous impact on modern life sciences and biomedicine and it should be born in mind that, as much as proteomics, more so than other global approaches (or –omics), relies heavily on technologies, the driving force which will propel proteomics in the future will be its impact on biological sciences and biomedicine.
In this chapter, we will highlight the scientific landmarks that made the study of the proteome possible, with specific reference to the cardiovascular system. It is our belief that a thorough understanding of where the proteomic world comes from will highlight the path to a prosperous future.
A Brief History of Proteomics
As mentioned in the introductory paragraph, the “proteomic revolution” could not have been possible without the technique, now 41 years old, which was carefully optimized by Pat O’Farrell in 1975 [1]. Although other groups implemented alternative versions of orthogonal gel separation, the 2DE protocol optimized by O’Farrell was arguably superior at the time it was published [5]. Indeed, by combining the best approaches available at the time and with an eye to the future, he was able to separate and visualize over a thousand proteins from E. Coli, while other groups could “only” see a few hundred proteins. Part of this success was made possible by the choice of isotopic labeling to boost sensitivity by about three orders of magnitude with respect to other groups who used protein staining (such as Coomassie Brilliant Blue) [5]. This technique, which is still in use today, is based on the combination of two independent electrophoretic separation methods applied to the sample in an orthogonal fashion. The first separation, commonly referred to as the first dimension, facilitates the separation of proteins according to their charge properties (isoelectric point, pI) by isoelectric focusing (IEF) under denaturing conditions. This is followed by the second dimension, which exploits the denaturing properties of charged detergents (typically sodium dodecyl sulphate, SDS) in order to separate proteins based on their molecular mass (Mr) (Fig. 1.1, see Chaps. 2 and 7 for details). Both electrophoretic steps are carried out using polyacrylamide gels (PAGE) with different properties that were optimized in the course of the following decade. In fact, another landmark discovery that allowed 2DE to become widely used was the advent of immobilized pH gradients (IPG). The idea, mastered by Pier Giorgio Righetti and colleagues at the University of Milan (Italy) and developed within a large consortium involving several Universities and established scientists as well as an industrial partner (LKB at that time), led to the historical publication by Bjellqvist and colleagues in 1982 [6]. A few years later, based largely on optimization of the IPG 2DE technique in the laboratory of Angelika Gorg, the industrial partner (now Pharmacia) made the technology available at an industrial scale, enabling increased intra- and inter-lab reproducibility of 2DE [7] and the widespread diffusion of 2DE.

Schematic of 2DE-MS. Proteins are first separated by 2DE according to their pI and molecular mass (Mr). The first separation in 2DE (IEF) is commonly referred to as “first dimension” whereas the second step (SDS-PAGE) is commonly referred to as “second dimension”. After in-gel digestion proteins can be identified using MS (see text for details)
Using 2DE technology, Valerie Wasinger and colleagues were able to generate the data disclosed at the’94 Siena meeting and published the following year in the journal Electrophoresis [2]. In what is commonly referred to as the “first proteomic paper”, these new technologies were combined with matrix-assisted laser desorption ionization–time of flight MS (MALDI-TOF MS, see Chap. 2), for the rapid identification of proteins using peptide mass fingerprinting (PMF) [8]. This MS-based approach is based on the digestion of the separated proteins with a protease (e.g. trypsin) and was utilized extensively in the early days of proteomics for the rapid identification of proteins. With the more recent popularity of tandem MS (see Chap. 2), allowing accurate determination of the sequence of peptides of the separated proteins, PMF was almost abandoned, only to make a glorious comeback as a rapid way to identify pathogens in biomedical labs in recent years [9] (see Chap. 18). The last key ingredient that was needed to identify proteins efficiently was the development of databases and algorithms to match the obtained MS spectra with protein names. Among the many contributions made by Norman and Leigh Anderson (father and son), the creation of one of the first successful algorithms to analyze 2DE maps [10] and the first online protein database (the Human Protein Index or HPI) [11] are two advances that accelerated the much needed creation of bioinformatic tools to mine the proteome (see Chaps. 12 and 14).
Operator Independence Days in Separation Techniques
Among the limitations of 2DE, the labor intensive and “artisanal” way they are performed paved the way to the success of more automatized techniques. To anybody who has successfully run a 2D gel, it is obvious how much training and effort is required to optimize both the conditions and the manual skills needed to obtain a well-resolved protein profile. With regard to the visual analysis of 2D gels and considering all of the effort involved in generating them, a parallel with visual arts is perhaps not too much a stretch of the imagination. The love-hate relationship of proteomic scientists with 2DE has made it something of a romantic journey, as testified for instance by the severe problems with the separation of membrane proteins, voiced so passionately by Thierry Rabilloud [12, 13].
Although several protein separation techniques have been implemented over the years, liquid chromatography (LC) became extremely popular for several reasons. Firstly its direct coupling with an MS instrument is relatively straightforward, decreasing the chance of contamination and avoiding a “transfer” step. In the original iteration of what is referred to today as the “shotgun” approach, John Yates and colleagues also applied an “orthogonal” peptide separation approach by combining strong cation exchange (SCX) and reverse phase (RP) chromatography in sequence [14] (Fig. 1.2, see Chap. 4 & 7). This was in the late’90 and the limitations of shotgun approaches, including limited quantitation capabilities, also became evident in the following years. The use of isotopic metabolic or post- labeling, was successfully applied to improve the quantitation capabilities of LC-MS approaches in the decade that followed [15]. Briefly, labeling proteins with isotopic aminoacids (dividing cells [16]) or tags (post-mitotic primary cultures, tissues, etc. [17]), allowed resolution of peaks originating from the same peptide but deriving from different samples in the MS as a mass shift, in a highly quantitative fashion. Several iterations of these reagents resulted in a technique that is now both robust and commercially available (see Chap. 11) [18].

Schematic of LC-MS. In “shotgun” proteomics proteins are first digested into peptides, which are generally more stable and soluble. Peptides can be then separated by liquid chromatographic techniques. Classically, Strong Cation Exchange (SCX) and Reverse Phase (RP) chromatography were combined in series to improve resolution. Liquid chromatography can be physically coupled to MS to identify the resulting peptides (see text and Chap. 4 & 7 for details)
In a similar fashion, at the end the’90s a labeling approach that improved sensitivity, dynamic range and throughput in 2DE was developed by scientists at Carnegie Mellon in Pittsburgh [19]. This technology, currently commercialized by GE Healthcare, involves the pre-electrophoretic fluorescent labeling of protein samples with N-hydroxy-succinimidyl-ester derivatives of fluorescent cyanine (Cy) dyes and is known as two-dimensional difference gel electrophoresis (DIGE). This approach has the advantage that a pair of protein samples can be labeled separately with differently fluorescing Cy3 and Cy5 derivatives. The two samples can be mixed and then separated together on the same 2D gel. The resulting 2D gel is then scanned to acquire the Cy3 and Cy5 images separately using a fluorescent laser scanner. Furthermore, a sample labeled with a third Cy2 dye can be run on each 2D gel and used to normalize the signal among different gels. This DIGE approach dramatically reduces technical variability and exploits the high dynamic range of fluorescent staining for accurate quantitation. The issue of multiple proteins within the same 2DE spot still remains and, therefore, downstream validation of the changes observed in protein levels by other techniques is still essential. In addition, the relatively high price of the dyes and detection systems (a laser scanner able to detect the fluorescent probes) remain limiting factors.
As a result of the enormous advances in MS technology, it is nowadays possible to create an in silico, “unbiased” map of the proteome with little sample manipulation prior to MS [20, 21]. The combination of data independent acquisition (DIA) and targeted methods, such as multiple reaction monitoring (MRM [22]), now facilitate the accurate quantitation of peptides in the absence of isotopic labeling and in a proteome-wide fashion (see Chap. 10 on DIA/SWATH). The higher throughput capabilities and automatization of LC versus 2DE, combined with the increased capacity of detecting membrane and basic proteins further contributed to the popularity of LC-MS in the following decade and through to the present day. However, one limitation of shotgun approaches that remains to this day is that proteins are first digested into peptides, which are more stable, prior to separation by LC. From the standpoint of a biologist it is easy to understand how, by operating at the peptide level from the outset, important information can be lost. Indeed, so-called “top-down” approaches, which allow the analysis of whole proteins by MS (as opposed to “bottom-down” approaches, such as shotgun proteomics), are becoming increasingly popular (see Chap. 8 and [23]). This is also a consequence of the enormous amount of data and information generated by a single shotgun experiment, which can be challenging to store and analyze. The divide between bottom-up and top-down approaches is a blurred one and in fact some argue that 1D (SDS-PAGE or IEF) and 2DE techniques may be right in the middle, offering the opportunity to separate, or at least reduce the complexity of protein samples to a few intact proteins per spot. The capacity to see gross changes in molecular weight (e.g. degradation or proteolysis), or isoelectric point (e.g. phosphorylation) prior to digestion, combined with the natural propensity of the human brain to recognize patterns, suggest that 2DE may still have a role to play as a balance between top-down and bottom-up approaches, while celebrating its forty-first birthday [24].
Over a Century of Mass Spectrometry
Despite MS having a dominant role in proteomics these days, it is not a new technique. Indeed the first reports of the use of a very distant cousin of a modern MS date back to the work of Joseph John Thomson sometime around the turn of the twentieth century. Unbeknown to this young theoretical physicist at Cambridge University, MS would turn into, arguably, the most revolutionary technology in biomedicine over a century later. This embryonic MS, then called a parabola spectrograph, was initially used to investigate the very intimate components of matter, for which Thomson received a Nobel Prize in 1906 for “discovering the electron” [25].
It would take seven decades for MS to be able to be used to sequence proteins thanks to the work and dedication of Klaus Biemann and colleagues, right around the time when protein biochemists were optimizing 2DE [26]. Indeed, the possibility of analyzing large organic molecules using MS was limited by the ability to convert them into ions that would be able to “fly” in the instrument, a requirement for MS analysis. MS was initially applied to smaller molecules such as metabolites or pharmaceuticals, which could be ionized without being fragmented during the process. The way that an MS instrument works can be equated to that of a very precise molecular scale, as we will see in details in the following Chapter. To ionize large organic molecules represented a major challenge in MS history, because the kinetic energy that needs to be transferred to the peptides for them to enter into a gas phase as ions and be separated using magnetic selectors, would cause their fragmentation.
It took the work of several groups and two Nobel prizes to overcome this limitation in the late ‘80s. Two different approaches were pursued starting from either solid (crystallized) or liquid samples. Both proved to be effective in the end and generated the two sources that are still in use today: electrospray ionization (ESI), and matrix-assisted laser desorption ionization (MALDI). As we will learn in the next Chapter an MS instrument can be divided in three sections: a source, which converts peptides into ions in a gas phase; an analyzer (or multiple analyzers in series, as for tandem MS); and a detector to “count” the peptides ions and/or their fragments, separated according to their mass (mass over charge or m/z to be precise) by the analyzer/s (Fig. 1.3).

Anatomy of a mass spectrometer. A mass spectrometer is best described by its “anatomical” components: a source, one or more analyzers and a detector (see text and Chap. 2 for details)
The Nobel prizes were ultimately awarded to John Fenn (ESI) and Koichi Tanaka (MALDI) in 2002, for “their development of soft desorption ionization methods for mass spectrometric analyses of biological macromolecuels [27]”. A little known fact about MALDI, is that although the Nobel prize was awarded to Dr. Tanaka and his group for their pioneering work on ionizing organic macromolecules from solid phase, the MALDI source, as it is currently used, was developed by two German scientists, Michael Karas and Franz Hillenkamp, who also named the technique [25]. This is one of the first examples of MS applied to the cardiovascular realm as the two scientists had a specific interest in mapping Ca2+ stores in cardiac cells [25]. These “soft” ionization methods were first applied to nucleic acids. As we will see in the next paragraph, along with the creation of genomic database, nucleic acid research indirectly contributed to the development of proteomics. Lastly, as we will see in Chapter 2, several advances in MS, including the creation of new analyzers (such as orbital traps [28]) and improved fragmentation approaches [29], have remarkably enhanced the capability of modern instruments to the point that several thousands of proteins and their modifications can be accurately resolved nowadays.
Genomic and Proteomic Databases: From Genes to Proteins, and Back
As mentioned, one of the technologies that was developed during the genomic era and which served the proteomic cause very well was the creation of protein databases. In the first proteomic studies, proteins were identified using chemical Edman degradation and/or genetic tools which allowed scientists to assign a peptide to a protein, by translating it into a genetic sequence and matching it to those available in a particular genome. Since the very beginning of protein sequencing by MS, this new approach helped to identify open reading frames (ORFs) or sequenced genes thus perfecting their publication and annotation [30].
As mentioned above, the creation of protein databases to mine protein sequences was the result of the renaissance mind of the Andersons who created the first online protein database (HPI) [11] and the many implementations which followed. The sequencing of the genome of many organisms, including the human genome, allowed proteomic scientists to build protein databases predicted on the basis of the genetic info and ORFs. On this issue it is interesting to see that proteomic studies helped to find limitations in genomic databases [31]. The quest to match peptide spectra was greatly facilitated by the creation of search and retrieval systems (SRSs), algorithms, such as MASCOT [32], capable of scanning through a large database and returning the likelihood of a true-positive match between the experimental mass spectrum and a protein sequence listed in silico. As can be seen the sequencing of genomes not only allowed proteomics to flourish but for the former to “pay back” by pinpointing limitations in the algorithms used to predict and annotate genes in the respective databases. Indeed, despite genetic information being there it is important to confirm that it is relevant to the phenotype, or expressed. Even when that is the case, the predictive value of genes is highly limited by mRNA stability, alternative splicing and post-translational modifications [33]. Large-scale proteomic studies are inconceivable without the aid of bioinformatics and this finally allowed us to break free from the “one gene to one protein” dogma [34]. It is our hope that, as our technologies and bioinformatic tools perform more accurately and errors are corrected, the detailed picture of the molecular phenotype (aka the proteome) will finally reach a sublime resolution.
Proteomics with a Heart
We already mentioned how, thanks to the work of Michael Karas and Franz Hillenkamp in Frankfurt, cardiovascular research was involved with cutting-edge proteomic research early on in the history of proteomics. There are many unanswered questions in the cardiovascular realm many of which have a deep impact on public health due to the widespread incidence of a multiplicity of cardiovascular diseases [18]. Proteomics would be a natural tool to investigate mechanisms, generate new hypotheses and test the predictive value of novel biomarkers. We are lucky to have participated in the development of cardiovascular proteomics. Indeed, the first studies on the heart using 2DE anticipated the 1995 publication by Valerie Wasinger and colleagues of a much more unpleasant proteome [2]. Mike Dunn and Peter Jungblut were among the first scientists utilizing these methods to study the human heart, and published the first human heart 2D maps the same year the proteome was “born” [35, 36]. Shortly after, the same groups utilized MS to identify numerous myocardial proteins from 2DE [8, 37]. In their seminal report from 1998, Mike Dunn and colleagues compared cardiac biopsies from dilated cardiomyopathy (DCM) and ischemic heart disease (IHD) patients and found several significant changes that were also monitored at the isolated cell level for fibroblasts, mesothelial and endothelial cells, and cardiac myocytes [38]. To our knowledge this is the first report of a proteomic study addressing cardiovascular disease (Fig. 1.4). Interestingly, the changes in desmin and the chaperone alpha-B-crystallin first reported over 25 years ago are still the object of an intense investigation by several scientists (including our group [39]). Specifically, the idea that desmin can form preamyloid oligomers in the heart, similar to those found in Alzheimer’s and Parkinson’s diseases as well as several other proteinopathies, suggest that protein misfolding could play a major role in many diseases affecting the majority of the population in westernized societies, including cardiovascular disease. The role of posttranslational modifications in this process is also rapidly emerging [40]. Therefore, proteomics may also help to generate new views and cutting edge idea in cardiovascular research.

A Timeline for the history of cardiovascular proteomics. See text for details
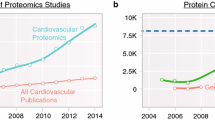
With the new century, proteomics started to feature rapidly in the cardiovascular field. More pioneers of the caliber of Jennifer Van Eyk and Peipei Ping, started exploiting these emerging technologies to address a number of different aspects of cardiovascular disease spanning from signaling [36]. to biomarker discovery [41]. Thanks to the pioneering work of these scientists and others the field grew at an almost exponential pace, scoring a total number of 2011 “cardiovascular proteomics” publications at the time of writing. Although this does not include all of the work done in “specialty areas” and is limited by the searched terms, it does provide a reliable estimate of the upward trend of the field for the last 15 years (Fig. 1.5). The term was used in only five articles in 2000, and in 265 articles in 2014. The appeal of using chromatography over 2DE gels has also rapidly increased and several comparative studies describing the complementary use of the different approaches to achieve more extensive coverage have been published in the last decade. As proteomics evolves, and that is happening very quickly, we are sure that this approach will be increasingly incorporated into studies addressing the main unsolved questions concerning prevention, diagnosis and cure of cardiovascular disease.

Cardiovascular proteomics articles in the last 15 years. Plot displaying the number of articles identified by the search term “cardiovascular proteomics” per year since 2000 (Source, PubMed)
Historical Crossroads: Pick Your New Technologies Wisely
Once proteomics became available to cardiovascular scientists at the beginning of this century, an immediate challenge came with it: what technology should they use to address a specific biomedical question? This is true for both basic and translational studies. The number of technological platforms also exploded. To keep up with a continuously evolving field is not an easy task, especially when it is as technologically based as proteomics. In our opinion, one of the responsibilities of the proteomic community is to explain these aspects to the ever-increasing number of collaborators in a way that is unbiased and comprehensible. Untimely, there is a tendency in some scientific environments to claim potential benefits even if they are only on the horizon of a particular technology. The increasing pressure to obtain research funding at a time when the economy is “breathing slowly” may accentuate this tendency.
There are several examples of why it is best to under-promise and over-deliver than vice versa. Perhaps one of such example that should be born in mind when establishing a collaborative effort or investing in a new technology is the experience with SELDI (surface-enhanced laser desorption ionization). This is a modified and patented version of MALDI that became quite popular in the early days of proteomics. Despite the promise of accelerating the discovery of new biomarkers, when it was first released, the technology was based on a low resolution MALDI-TOF instrument with a proprietary technology to fractionate the samples directly onto the MALDI (or SELDI) plate, the support that allows samples to enter the instrument. Despite the initial “hype” that seems to accompany most new technologies, the use of SELDI has now rapidly declined, probably because the method did not meet initial expectations, possibly due to the high impact of matrix effects, limited reproducibility and limited identification capabilities [42].
This is just one of the many examples that are a feature of the history of proteomics, and probably of many other fields. Another lesson learned is that the SELDI instrument was originally packaged in a “black box” type of format, claiming a simple sample in/data out operation. This is a very important lesson for new and old proteomic scientists alike. The field is complicated, and as much as there is a purpose in trying to simplify technologies in order to allow their broader use, certain aspects simply cannot be simplified. When a technology does not deliver what it had promised, the impact on its future tends to be more dramatic, possibly because there is a collective memory in the scientific community, which tends to adjust to new concepts with a certain delay. It is also possible that this fact alone may limit the chances of “redemption” for technologies that are initially advertised to address more than can be expected from them. The “market” of proteomic technologies is a bit like the stock market in that it is extremely volatile, and at times expensive. For proteomic beginners it may be wise to rely on the advice of more experienced scientists in the field, and since we all have our passions and beliefs, perhaps diversifying the portfolio of “proteomic brokers” could also prove beneficial. Last but not least, proteomic scientists need to make themselves understandable to the general scientific community, avoiding technical jargon when possible and sharing their unbiased knowledge. This book represents an effort in this very direction.
What Do We Leave Behind as We Move Forward: Bringing the Fun Back into Science
This is a tremendously exciting moment in science, including the cardiovascular field. The capabilities offered by the latest generation of proteomic technologies are almost infinite. We can see most of the proteome, quantify it and finally make sense of it. Thinking back on how hard previous generations of scientists had to work in order to optimize these technologies should make us feel extremely lucky. It took decades to couple an LC with an MS, and decades to analyze peptides and proteins using it. It took decades to sequence the genomes that are available today, create the corresponding protein databases and the algorithms to exploit them to generate proteomic data. It almost appears that everything had to be optimized for the current generation of proteomic scientists to “have fun with it”. Proteomics can be painful, as science at large has the tendency to be sometimes. However, it has the potential and capability to bring very diverse expertise around the same table: statisticians, engineers, biochemists and physicians can find some exciting, common ground to move science and medicine forward (hopefully, as is the case with the present book). With this also comes the responsibility to actually change medicine and translate the impact of all these rich technological gifts back into some good for human health. There is an increasing demand for technical expertise and scientists that can translate the difficult languages of math, engineering, protein biochemistry and medicine. It takes time and dedication to have enough knowledge of these languages to be able to contribute something to the table. However, it is not inconceivable that proteomics will become a curriculum in many undergraduate and graduate programs in its own right, as happened previously for biotechnology, molecular biology, etc.. In all, these opportunities for collective growth lie just ahead of us waiting to be picked like ripe fruit.
With such opportunities in prospect, perhaps the best way to move forward is to honor those that took us this far by pursuing the best science possible. Of course quality and integrity are two pillars of science, or at least should be. As we have learned in this chapter, time will tell if what we have fulfilled these two core principles. Moreover, in an era where communication and data access is so rapid it may become increasingly easier to get distracted and lose focus of the scientific goals. In addition, when being exposed to technologies, there is also a risk of falling in love with them. In summary, with all these novelties comes the risk of forgetting the knowledge generated by other means and during earlier times, when perhaps it took longer to separate proteins or to analyze them, but this time could be used to learn about the functional aspects of our discoveries, in-depth. Many proteomic scientists in the cardiovascular field had to write scientific articles explaining why a list of proteins, many of which we had not heard about before, change in a coordinated fashion. What has been published about the role of these mysterious proteins and the biological effect of their known post-translational modifications that we see changing in a particular disease state are very important aspects of our research. To us, another good way to honor the work of the fathers of proteomics would simply be to “do our homework”, try to put our observations in the context of what is already known without the need to rewrite science. Lastly, as we stand on the shoulders of the scientists who made us who we are, we honor their legacy by learning from their mistakes and endeavoring to matchtheir successes.
Funding: American Heart Association 12SDG9210000 and 16IRG27240002; NIH P01 HL107153; RFO University of Bologna to GA
References
O’Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975;250:4007–21.
Wasinger VC, Cordwell SJ, Cerpa-Poljak A, Yan JX, Gooley AA, Wilkins MR, Duncan MW, Harris R, Williams KL, Humphery-Smith I. Progress with gene-product mapping of the Mollicutes: mycoplasma genitalium. Electrophoresis. 1995;16:1090–4.
Bini L, Calvete JJ, Turck N, Hochstrasser D, Sanchez J. Special Issue: “20 years of Proteomics” in memory of Vitaliano Pallini. J Proteomics. 2014;107:1–146.
Smith, L.M., Kelleher, N.L., and Consortium for Top Down Proteomics. Proteoform: a single term describing protein complexity. Nat Methods. 2013;10:186–7.
Righetti PG. The Monkey King: a personal view of the long journey towards a proteomic Nirvana. J Proteomics. 2014;107:39–49.
Bjellqvist B, Ek K, Righetti PG, Gianazza E, Gorg A, Westermeier R, Postel W. Isoelectric focusing in immobilized pH gradients: principle, methodology and some applications. J Biochem Biophys Methods. 1982;6:317–39.
Gorg A, Drews O, Luck C, Weiland F, Weiss W. 2-DE with IPGs. Electrophoresis. 2009;30 Suppl 1:S122–32.
Sutton CW, Pemberton KS, Cottrell JS, Corbett JM, Wheeler CH, Dunn MJ, Pappin DJ. Identification of myocardial proteins from two-dimensional gels by peptide mass fingerprinting. Electrophoresis. 1995;16:308–16.
Randell P. It’s a MALDI but it’s a goodie: MALDI-TOF mass spectrometry for microbial identification. Thorax. 2014;69:776–8.
Anderson NL, Taylor J, Scandora AE, Coulter BP, Anderson NG. The TYCHO system for computer analysis of two-dimensional gel electrophoresis patterns. Clin Chem. 1981;27:1807–20.
Anderson NG, Anderson L. The Human Protein Index. Clin Chem. 1982;28:739–48.
Santoni V, Molloy M, Rabilloud T. Membrane proteins and proteomics: un amour impossible? Electrophoresis. 2000;21:1054–70.
Rabilloud T. Membrane proteins and proteomics: love is possible, but so difficult. Electrophoresis. 2009;30 Suppl 1:S174–80.
Yates 3rd JR, Carmack E, Carmack E, Hays L, Link AJ, Eng JK. Automated protein identification using microcolumn liquid chromatography-tandem mass spectrometry. Methods Mol Biol. 1999;112:553–69.
Patterson SD, Aebersold RH. Proteomics: the first decade and beyond. Nat Genet. 2003;33(Suppl):311–23.
Oda Y, Huang K, Cross FR, Cowburn D, Chait BT. Accurate quantitation of protein expression and site-specific phosphorylation. Proc Natl Acad Sci U S A. 1999;96:6591–6.
Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–9.
Agnetti G, Husberg C, Van Eyk JE. Divide and conquer: the application of organelle proteomics to heart failure. Circ Res. 2011;108:512–26.
Unlu M, Morgan ME, Minden JS. Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis. 1997;18:2071–7.
Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012;11:O111.016717.
Guo T, Kouvonen P, Koh CC, Gillet LC, Wolski WE, Rost HL, Rosenberger G, Collins BC, Blum LC, Gillessen S, Joerger M, Jochum W, Aebersold R. Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps. Nat Med. 2015;21:407–13.
Kuhn E, Wu J, Karl J, Liao H, Zolg W, Guild B. Quantification of C-reactive protein in the serum of patients with rheumatoid arthritis using multiple reaction monitoring mass spectrometry and 13C-labeled peptide standards. Proteomics. 2004;4:1175–86.
Peng Y, Ayaz-Guner S, Yu D, Ge Y. Top-down mass spectrometry of cardiac myofilament proteins in health and disease. Proteomics Clin Appl. 2014;8:554–68.
Oliveira BM, Coorssen JR, Martins-de-Souza D. 2DE: the phoenix of proteomics. J Proteomics. 2014;104:140–50.
Griffiths J. A brief history of mass spectrometry. Anal Chem. 2008;80:5678–83.
Hudson G, Biemann K. Mass spectrometric sequencing of proteins. The structure of subunit I of monellin. Biochem Biophys Res Commun. 1976;71:212–20.
Tabet JC, Rebuffat S. Nobel Prize 2002 for chemistry: mass spectrometry and nuclear magnetic resonance. Med Sci (Paris). 2003;19:865–72.
Makarov A. Electrostatic axially harmonic orbital trapping: a high-performance technique of mass analysis. Anal Chem. 2000;72:1156–62.
Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A. 2004;101:9528–33.
Biemann K. Laying the groundwork for proteomics: mass spectrometry from 1958 to 1988. J Proteomics. 2014;107:62–70.
Pandey A, Mann M. Proteomics to study genes and genomes. Nature. 2000;405:837–46.
Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–67.
Rabilloud T, Hochstrasser D, Simpson RJ. Is a gene-centric human proteome project the best way for proteomics to serve biology? Proteomics. 2010;10:3067–72.
Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–30.
Corbett JM, Wheeler CH, Baker CS, Yacoub MH, Dunn MJ. The human myocardial two-dimensional gel protein database: update 1994. Electrophoresis. 1994;15:1459–65.
Jungblut P, Otto A, Zeindl-Eberhart E, Plessner KP, Knecht M, Regitz-Zagrosek V, Fleck E, Wittmann-Liebold B. Protein composition of the human heart: the construction of a myocardial two-dimensional electrophoresis database. Electrophoresis. 1994;15:685–707.
Thiede B, Otto A, Zimny-Arndt U, Muller EC, Jungblut P. Identification of human myocardial proteins separated by two-dimensional electrophoresis with matrix-assisted laser desorption/ionization mass spectrometry. Electrophoresis. 1996;17:588–99.
Corbett JM, Why HJ, Wheeler CH, Richardson PJ, Archard LC, Yacoub MH, Dunn MJ. Cardiac protein abnormalities in dilated cardiomyopathy detected by two-dimensional polyacrylamide gel electrophoresis. Electrophoresis. 1998;19:2031–42.
Agnetti G, Halperin VL, Kirk JA, Chakir K, Guo Y, Lund L, Nicolini F, Gherli T, Guarnieri C, Caldarera CM, Tomaselli GF, Kass DA, Van Eyk JE. Desmin modifications associate with amyloid-like oligomers deposition in heart failure. Cardiovasc Res. 2014;102:24–34.
Del Monte F, Agnetti G. Protein post-translational modifications and misfolding: new concepts in heart failure. Proteomics Clin Appl. 2014;8:534–42.
Arrell DK, Neverova I, Van Eyk JE. Cardiovascular proteomics: evolution and potential. Circ Res. 2001;88:763–73.
Mischak H, Vlahou A, Ioannidis JP. Technical aspects and inter-laboratory variability in native peptide profiling: the CE-MS experience. Clin Biochem. 2013;46:432–43.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Agnetti, G., Dunn, M.J. (2016). A Historical Perspective on Cardiovascular Proteomics. In: Agnetti, G., Lindsey, M., Foster, D. (eds) Manual of Cardiovascular Proteomics. Springer, Cham. https://doi.org/10.1007/978-3-319-31828-8_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-31828-8_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-31826-4
Online ISBN: 978-3-319-31828-8
eBook Packages: MedicineMedicine (R0)