
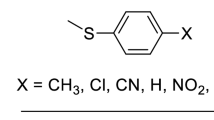
Abstract
In this chapter, we put forward an emerging concept on which the therapeutic action of some Mn porphyrins (MnPs) used in vivo may not be restricted to an SOD-type antioxidant catalytic role per se, but involve P450-type oxidation catalysis. ROS/RNS-type oxidants, such as hydrogen peroxide, hydroperoxides, and hypochlorite, are all classic oxygen donors in the chemistry literature of metalloporphyrin-based systems, mostly unavailable in the Pubmed database. Whereas the association of MnPs to a pro-oxidant therapeutic role is rather recent, MnPs have been explored as biomimetic oxidant catalysts for more than three decades in the fields of chemistry and catalysis. Merging the biological new discoveries and the typical oxidation chemistry findings would bring a breath of fresh air to both chemistry and biology with very positive prospects to the medicinal development of MnP-based therapeutics. The chapter includes, thus, a brief description of the general mechanism of the cytochromes P450 and related heme-containing enzymes, such as peroxidases (e.g., horseradish peroxidase) followed by the presentation of selected aspects of the 30+ years of biomimetic chemistry of MnPs as P450 models. It is not the goal of this chapter to provide a full account on the MnP-based biomimetic oxidation systems, but highlight some of the main features of P450-type MnP reactions that may become relevant on interpreting some of the biological results on the use of MnPs as redox-active therapeutics.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
9.1 Highlights on Metalloporphyrin-Based Biomimetic Chemistry
Iron porphyrins (FePs) are ubiquitous in living organisms. Naturally occurring porphyrins, such as protoporphyrin IX (Fig. 9.1) or a closely related tetrapyrrole ligand, are able to bind Fe(II) or Fe(III) ions very tightly, yielding the well-known “heme” groups [1–3]. Heme is rarely free in biological systems [4, 5], being often bound to or associated with specific proteins collectively known as hemeproteins (or hemoproteins) [1–3]. These resulting metalloproteins play a variety of diverse and critical roles in many biological processes, in which the heme unit acts as the prosthetic group (e.g., hemoglobin, myoglobin, and cytochrome c) [1–3, 6–8]. Many important redox-based metalloenzymes, such as cytochromes P450, nitric oxide synthases, cytochrome c oxidase, and some peroxidases and catalases, are also hemeproteins [2, 3, 8–13]. The reactivity of the heme group is, thus, largely controlled by the apoprotein moiety, which ultimately dictates the heme biological function [6–11].

Structure of metalloporphyrins relevant to this chapter
These hemeproteins have fascinated chemists and biochemists for more than a century [14]. The attempts to model their structural features, their electronic structure and corresponding spectroscopic signatures, and their reactivity have been under the scrutiny of biomimetic chemists before biomimetic chemistry was even termed as such. The term “biomimetic chemistry ” was coined by Breslow [15, 16] in the early 1970s to describe a scientific investigation strategy that uses Nature as inspiration either to develop a chemical process related to those found in the natural systems, or to prepare a compound that imitates a biological material in its structure and/or function. The goal is not to merely reproduce Nature, but, nevertheless, to draw inspiration from it, which allows experimentations with ligands and metals not associated with biology, including non-naturally occurring ligands, and a full range of different metals, including those biologically uncommon ones (e.g., Ru [17–19]).
During the past 50 years or so, chemists have been quite busy using metalloporphyrins as biomimetic models of metalloproteins and/or metalloenzymes [6–14, 17–24]. Historically, most of the initial studies were devoted to the modeling of dioxygen transport and storage by hemoglobin and myoglobin, respectively [6, 7, 14]. In the 1970s, many groups (e.g., Collman, Baldwin, Momenteau, James, Dolphin, Traylor, and Basolo) carried out elegant studies on synthetic metalloporphyrins as models for describing the reversible coordination of O2 to hemoglobin and myoglobin [6–8, 25–27]. The most promising systems as O2 carriers were those of Fe(II); some success was also achieved with Co(II), but Mn(II) and Mn(III) were ineffective as O2 carriers (of note, reversible O2-coordination to Mn(II) porphyrins could be achieved under unsuitable conditions, i.e., in toluene at −78 °C [28]). The main challenges in these systems, in particular the Fe(II) ones, were [6, 7]: (a) to avoid the irreversible O2-oxidation of FeIIP to FeIIIP, which may be achieved by preventing the approximation of water to the metal site; and (b) to prevent the irreversible formation of μ-oxo complexes, i.e., PFeIII–O–FeIIIP, which resulted from the reaction between the (O2)FeIIP adduct and a second molecule of FeIIP. In the hemeproteins, the bulky protein moiety, which is not present in the model systems, creates not only a hydrophobic region around the metal center but also prohibits the formation of μ-oxo species by sterically hindering the approximation of two heme groups [6, 7]. In this regard, the search for architecturally structured porphyrin macrocycles bearing bulky substituents with hydrophobic cavities around the metal center originated the classic “picket fence” [25], “capped” [26], and “basket handle” [27] porphyrins. Eventually, FePs were able to model hemoglobin and myoglobin with respect to reversible O2-coordination, provided a good description of the spectroscopic characteristics of these systems, and revealed subtle details on the role played by the protein on modulating the reactivity of heme to yield reversible and selective O2-coordination [6, 7]. The technological applications of these complexes as O2-carriers or air fractioning devices, however, have been hindered by the complexity and fragility of the systems. Whereas the development of FePs as O2-carriers for artificial blood formulations is still pursued [29], the drawbacks that need to be solved include concepts such as oxidative and nitrosative stress [30, 31]. Looking at these systems retrospectively, the early attempts to use simple FeP complexes as O2-carriers in vivo in the 1970s were bound to fail, given that these “naked” systems are prone to side-reactions and side-effect complications associated with the implications of some reactivity patterns unforeseen then and still much of a challenge now: the reactivity of FePs toward NO [32], a molecule whose biology and biologically relevant chemistry blossomed from the 1980s on [33]. Coordination of O2 to ferrous FePs yields the adduct (O2)FeIIP, which, by internal electron transfer, exists as a ferric superoxide-bound complex, (O2 −)FeIIIP [6]; superoxide may react with NO at diffusion limiting rates to yield the powerful oxidant peroxynitrite [34]. Despite the stress associated with peroxynitrite on its own [34], a high concentration, bolus dose of FePs (as artificial red blood cell substituent) would scavenge NO directly (via coordination to FePs) or indirectly (via FeP-derived superoxide pathways), leading to cardiovascular alterations, to the least.
The 1979 seminal paper by Groves and coworkers [35] on the use of a synthetic FeP as a biomimetic model of the cytochromes P450, which is a ferric heme-containing metalloenzyme , revealed that the same (or even simpler) FePs that used to be studied as hemoglobin/myoglobin models could also be investigated as P450 models [20]. The mechanism of the cytochromes P450 is rather complex [36] (see Sect. 9.2.1) and the use of model compounds appeared as an alternative to dissect the structural aspects, electronic features, and reactivity behavior of these enzymes [11, 37]. These possibilities were quickly taken by many groups worldwide and the results on these P450 model systems inaugurated the chemistry of metalloporphyrins as biomimetic oxidant catalysts, which remains quite an active and dynamic field of research since then [37]. Among the most studied metalloporphyrin-based catalysts are those of Mn(III), Fe(III), and Ru(VI) porphyrins [17, 20, 21, 38–40]. Upon the success in exploring metalloporphyrin complexes as biomimetic P450 models [37], the studies expanded rapidly to include other redox-active heme-based proteins, such as peroxidases, catalase, and nitric oxide synthase [11, 24, 38, 41].
Another very active area of research on using metalloporphyrins as biomimetic models is that of modeling of the superoxide dismutase (SOD) enzymes [22, 23, 42] (see Chap. 8 by Batinić-Haberle et al.), even though none of the four known isoforms of SOD (i.e., Cu,ZnSOD, MnSOD, FeSOD, and NiSOD) contains the porphyrin ring in their structure nor uses porphyrin as cofactor at all (see Chap. 7 by Policar). Despite SOD and metalloporphyrins being structurally unrelated, FePs and Mn porphyrins (MnPs) are among the most active models of SOD [22, 23, 43, 44], i.e., FePs and MnPs can model the SOD function, but, obviously, cannot account for any of the enzyme spectroscopic features. At this point, it is worth pointing out that along this chapter, the word “mimic” will be reserved for a model compound that is able to perform at rates meaningfully comparable to that of the modeled enzyme. For example, cationic MnPs, such as MnTE-2-PyP5+ (Fig. 9.1), are SOD mimics, as their catalytic rate constant (log k cat) are close to that of SOD; additionally, MnTE-2-PyP5+ is able to substitute for the SOD enzyme in vivo in SOD-deficient microorganisms such as bacteria (Escherichia coli) and yeast (Saccharomyces cerevisiae) [23, 45]. Conversely, whereas FePs and MnPs are able to emulate the reactions of catalase, their catalase activities are rather low (as low as 0.006 % of that of the enzyme, in the best case scenario) [41]; these metalloporphyrins may, thus, be suitable biomimetic models of catalase, but are not catalase mimics [41].
The first studies on the SOD activity of metalloporphyrins appeared also in 1979 with the work by Pasternack and Halliwell [46], who showed that simple water-soluble metalloporphyrins could model the reactions of SOD on dismuting the superoxide radical ion to H2O2 and O2. The metalloporphyrin-based SOD mimic field remained relatively dormant [47–52] as compared to the developments witnessed at P450 biomimetic systems. Of note, Meunier published in 1992 a classical review with already more than 600 references on P450-type reactions associated with metalloporphyrins [20]. The breakthrough in developing potent porphyrin-based SOD mimics appeared in 1998 with the work by Batinić-Haberle et al. [53] where the ortho substituted Mn(III) meso-tetrakis(N-methylpyridinium-2-yl)porphyrin, MnTM-2-PyP5+ (Fig. 9.1), was found able to dismutate superoxide at a, then, unusually high rate constant (log k cat = 7.72). The SOD-based biomimetic chemistry of metalloporphyrins flourished and soon became clear that a good SOD model should emulate the reduction potential of SOD enzymes (ca. +300 mV vs. NHE) [54–57] and bear positive charges in close vicinity to the metal center, in order to guide the superoxide anion to the reactive site via electrostatic facilitation [58, 59], in a similar fashion to the role that cationic amino acids residues play on SOD enzymes themselves. The first empirical structure–activity relationships on Mn-porphyrin-based SOD mimics accounting for both thermodynamic (reduction potential, E ½ [54]) and electrostatic facilitation (i.e., the impact the charges of cationic, anionic, and neutral porphyrins exert on SOD activity, log k cat [58]) emerged [60] and have recently been extended to include many non-porphyrin complex models [42, 61]. Some MnP complexes are as SOD active as the SOD enzymes themselves [43], which represents a successful achievement in biomimetic chemistry: designed, nonstructurally related, small-molecule complexes mimicking closely the reactivity patterns and rates of Nature’s own SOD enzymes.
Nowadays, water-soluble Mn porphyrins comprise the class of metalloporphyrins most investigated in biological systems [22, 23, 42, 45, 62, 63]. Their use in vivo spams a wide range of organisms, from microorganisms (e.g., bacteria, yeast) to animals, such as rodents, dogs, and primates, including humans [22, 23, 42, 45, 62, 63]. Historically, the development of MnPs for medicinal applications was initiated and had relied considerably on exploring the SOD mimic properties of the complexes. Their in vivo efficiency in a variety of oxidative stress-based injuries and pathological states has usually been ascribed to the SOD activity of the mimic [22, 23, 61–63]. As SOD enzymes are within the first lines of defense among the endogenous antioxidant enzymes against oxidative damage (see Chap. 8 by Batinić-Haberle et al.), these MnPs have, thus, been often referred to as catalytic antioxidants [64, 65].
In this chapter, we put forward an emerging concept on which the therapeutic action of some MnPs used in vivo might not be restricted to an SOD-type antioxidant catalytic role per se, but involve P450-type oxidation catalysis. In the next sections, a brief description of the general mechanism of the cytochromes P450 and related enzyme systems will be followed by the presentation of selected aspects of the 30+ years of biomimetic chemistry of MnPs as P450 models. It is not the goal of this chapter to provide a full account on the MnP-based biomimetic oxidation systems, but highlight some of the main features of P450-type MnP reactions that may become relevant on interpreting some of the biological results on the use of MnPs as redox-active therapeutics.
9.2 A Brief Overview on Cytochrome P450-Catalyzed Oxidations and Related Systems
9.2.1 Cytochrome P450 Family
The search for efficient catalysts that are able to promote the selective insertion of oxygen atoms (oxygenation reactions) in many organic substrates under mild conditions of temperature and pressure remains a challenge in chemical catalysis. This is particularly relevant when developing synthetic routes to the hydroxylation of the inert C–H bonds of alkanes or the selective oxidation of aromatic compounds [20, 21, 38–40, 66].
The direct conversion of an alkane into an alcohol using cheap and environmentally friendly oxidants, such as O2, has been intensely sought after, as this would allow the development of novel synthetic routes to convert hydrocarbons, easily available and of low cost, in products to fine chemistry and pharmaceuticals [24, 67, 68]. On industrial settings, C–H bond activation is usually achieved using heterogeneous catalysis at high temperature, but polyoxygenation is often observed, which renders the systems of compromised selectivity [67].
In biological systems, however, the family of cytochrome P450 enzymes are able to catalyze the monooxygenation of a large variety of organic substrates (RH) , under ambient temperature and pressure, using atmospheric O2 and a biological reductant, such as the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) , as represented in (9.1) [9, 36]. The cytochromes P450 belong to a superfamily of heme-containing monooxygenase enzymes with similar spectral properties and that share a distinct spectroscopic feature: they exhibit an intense absorption band (Soret band) at ~450 nm in the reduced form in the presence of CO [13, 36, 37, 69]. They all contain an Fe(III) protoporphyrin IX (Fig. 9.1) in the active site, whose binding to the protein is achieved by coordination of the thiolate moiety of a residual cysteine (the proximal ligand) to the heme Fe(III) center [11, 36]. The heme coordination site trans to the cysteine proximal ligand comprises the distal site, where binding of O2 takes place. The distal site is often occupied by water molecules, which are easily replaced by O2 in the presence of a suitable substrate [13, 36]. The polypeptide protein part has molecular weight of ca. 45,000 Da and is responsible for both rendering the catalytic site hydrophilic and directing the substrate toward the active metal site [20, 36, 37, 39].
The cytochromes P450 are membrane bound enzymes [70] widely spread among the living organisms, including unicellular microorganisms (e.g., bacteria and fungi) and complex organisms (such as humans) [71]. They exert important roles in vital processes of the organisms, being involved as catalysts for the biosynthesis of prostaglandins and steroids and for the oxidative biotransformation of xenobiotics, such as drugs, pesticides, carcinogens, polycyclic aromatic hydrocarbons, alkanes, and alkenes [12, 71].
The catalytic cycle of P450 is still a matter of intense research, with particular focus on the elucidation of the electronic structure and reactivity pattern of the high-valent intermediate species [12, 13, 72, 73]. Given the high molecular weight of the cytochromes P450, it is rather difficult to determine with accuracy the mechanistic details of the oxidation reactions, as well as the molecular structure of all intermediates involved in the processes. In this context, the use of biomimetic systems helped a great deal in shedding some light on the transformations associated with the enzymatic processes [37]. Even in simple model systems, the characterization of many short-lived intermediates is challenging. Two main issues remain of keen interest in the P450 reactions: the mechanism by which O2 is activated to yield the active intermediate species and the mechanism by which the oxygen atom is transferred to the substrate [72, 74–78].
A proposed mechanism that accounts for the most likely intermediate species during P450-catalyzed oxidation reactions is illustrated in Fig. 9.2 [12, 36, 74–78]. The P450 resting state (A) is a hexacoordinate low-spin Fe(III) species with a water molecule as distal ligand. Upon the approximation of the substrate RH, the protein changes its conformation, releasing the axial water molecule to yield a pentacoordinate high-spin Fe(III) species (B), which accommodates the substrate in a pocket in the vicinity of the active site. The degree of low-to-high spin-state transition that accompanies iron coordination-sphere change may lead to ca. 80–130 mV increase in the P450 Fe(III)/Fe(II) reduction potential [79–82] (see Sect. 9.4), which is crucial for the Fe(III) → Fe(II) reduction that follows. Reduction of (B) by one electron leads to a pentacoordinate Fe(II) species (C) with the Fe atom slightly shifted toward the proximal cysteine ligand; coordination of O2 to the pentacoordinate ferrous species (C) yields the hexacoordinate adduct (O2)FeIIP (D). This species can also be described as a superoxide-bound ferric species (D′) resulting from an internal electron transfer from the ferrous iron to the coordinated O2 in (D) [80, 81]. A second 1-electron reduction yields a Fe(III) species containing a peroxide moiety coordinated on an end-on (η1) fashion (E) [77]. Protonation of the peroxide moiety leads to species (F, where X = H). Heterolytic cleavage of the O–OH bond followed by protonation of the resulting hydroxide leads to the release of water and concomitant formation of the high-valent Fe(IV)-oxo porphyrin π-cation species (Compound I), formally a Fe(V)-oxo species. Compound I, then, abstracts a hydrogen atom from the nearby substrate, yielding an organic radical R• and Fe(IV)-hydroxo species (G). As the substrate-derived radical R• is kept within the active site of the P450 enzyme, this allows a recombination of R• with the coordinated hydroxide moiety and an internal electron transfer to yield the Fe(III) species (H) with formation of the oxygenated product ROH. Release of the product and coordination of water restores the resting state species (A). The transformations from Compound I to species (H) are also referred to as the “oxygen rebound mechanism” [83, 84]. Of note, the reduction of the cytochromes P450 from the ferric to the ferrous form in the presence of CO leads to a catalytically dead carbonyl species (P450-CO) [13, 20, 36, 37, 69]. As noted previously, this species is the one responsible for the characteristic 450 nm absorption band of the cytochromes P450.

Catalytic cycle of the cytochromes P450 . The “long cycle” is associated with the following general steps, starting from the resting state species (A): exclusion of water from the active site upon substrate (RH) approach and accommodation to substrate pocket (A → B), iron reduction and O2 coordination (B → D), heterolytic O–OH bond cleavage via proton-dependent reductive electron transfers to yield the high-valent Fe(IV)-oxo porphyrin π-cation-based active species, compound I (D → Compound I), substrate oxidation via a “oxygen rebound mechanism” (Compound I → H), followed by product release and water coordination restoring the enzyme resting state (H → A). The “short P450 cycles” are brought upon the use of peroxides XOOH, such as hydrogen peroxide (X = H) or alkyl hydroperoxides (X = alkyl), or active-oxygen donors, such as iodosylbenzene (PhIO), and involve the so-called peroxide shunt or iodosylbenzene shunt pathways, respectively. See text for additional details on the species along the cycle. Adapted from [2, 24, 77]
Shorter P450 catalytic cycles are available by using some oxygen donors other than O2. The use of peroxides, such as hydrogen peroxide (XOOH, X = H) or alkyl hydroperoxides (XOOH, X = alkyl), shortcuts the cycle from species (B) into species (F) [77]. If oxidation is carried out with iodosylbenzene (PhIO) as oxygen donor, the cycle becomes even shorter and species (B) is converted directly into the high-valent active species (Compound I) [24]. These shortcuts are named “peroxide shunt” or “iodosylbenzene shunt” pathways, after the oxygen donor used, and have been the basis for justifying and validating the studies with such oxidants in biomimetic model systems [14].
This mechanism describes roughly most of the cytochromes P450 oxidation reactions, such as hydroxylations, epoxidations, oxidative dehydrogenation, sulfoxidations, N-oxidations, and N-demethylations [13, 24, 36]. These P450 reactions are usually 2-electron oxidation processes: the oxygen transfer from the Fe(IV)-oxo porphyrin π-cation radical active species Compound I (formally an Fe(V)-oxo species) to the substrate is accompanied by the reduction of the Fe center from a formal +5 oxidation state to a +3 oxidation state. This is however, not a universal behavior, as P450 enzymes may also be involved in two consecutive 1-electron oxidations in a similar fashion as peroxidases. Indeed, Hrycay and Bandiera [12] have recently reviewed the peroxidase-like activity of cytochromes P450.
9.2.2 Single-Electron Oxidation Systems
Heme-containing peroxidases, such as horseradish peroxidase (HRP) , are usually associated with 1-electron oxidation of organic substrates (AH) using hydrogen peroxide as final electron acceptor (9.2) [85–87].
Analogously to the P450 systems, HRP also recruits a high-valent Fe(IV)-oxo porphyrin π-cation radical (Compound I) intermediate as active species. HRP-Compound I reactivity, however, is very different from that of P450-Compound I: HRP uses preferentially H2O2 as final electron acceptor for the oxidation of two equivalents of substrate by two independent 1-electron oxidations [86, 87]. The features that render the oxidation mode of HRP rather distinct from that of P450 start with the different amino acid residue used as heme proximal ligand: heme-Fe(III) in HRP is coordinated to the protein via an imidazole N-atom of a residual histidine [87], whereas P450s are Cys thiolate bound hemeproteins [11].
A simplified catalytic cycle for heme-containing peroxidase, such as HRP, is depicted in Fig. 9.3 [87]. The resting state in these systems is a pentacoordinate Fe(III) species that, upon coordination of a deprotonated hydrogen peroxide anion, yields a hexacoordinate Fe(III) species (Compound 0). Heterolytic cleavage of the coordinated hydroperoxide moiety in the presence of a proton yields the high-valent Fe(IV)-oxo porphyrin π-cation species (Compound I) and water. Compound I, reacts via a 1-electron process with a substrate AH (equivalent to A− H+) via a 1-electron reduction at the porphyrin π-cation site, to yield a Fe(IV)-oxo porphyrin species (Compound II) and a 1-electron oxidized species A− → A•. Compound II reacts further with another AH molecule to restore the resting Fe(III) state via a 1-electron reduction at the meal site, producing a second equivalent of 1-eletron-oxidized A− → A•.

Catalytic cycle of heme-containing peroxidases , such as horseradish peroxidase (HRP) . Adapted from [87]
9.3 Selected Aspects of Metalloporphyrin-Based Biomimetic Oxidations
9.3.1 General Features
The development of synthetic porphyrin-based oxidation catalysts that are able to model the reactivity of cytochromes P450 has been the focus of many researchers for the past three decades [20, 24, 37, 38, 40, 66, 88–90]. During this period of time, the oxidation of a large variety of organic substrates (Fig. 9.4) using many oxygen donors (such as iodosylbenzene, hypochlorite, hydrogen peroxide, organic peracids, monopersulfate, amine N-oxides, and O2) have been accomplished [17, 20, 37]. Among the metalloporphyrins tested, those of Mn(III) and Fe(III) have yielded the most efficient oxidation catalysts [20, 24, 37, 40]. The mechanisms of such reactions are usually ascribed to a putative high-valent metal-oxo species, in close analogy with Compound I in the hemeprotein systems [21, 90]. The results accumulated on the use of metalloporphyrins as oxidation catalysts have been the subject of many reviews, books chapters, or entire books over those years. We highlight below a few selected aspects of these systems that could be useful for interpreting the biological effects observed with Mn porphyrins in vivo.

Typical oxidation reactions catalyzed by metalloporphyrins
In natural enzyme systems, the wide range of reactivity, quimioselectivity, regioselectivity, stereoselectivity, oxidant preference (e.g., O2 vs. H2O2), oxidant and substrate activation mode, and overall oxidation rate of enzymes that share the exact same heme cofactor, such as cytochromes P450 [12, 36, 74–78] and horseradish peroxidases [9, 85–87], cannot, thus, depend on the heme moiety by itself. In fact, the reactivity of the heme group in these systems is modulated by the proximal ligand (e.g., Cys vs. His on P450 vs. HRP, respectively), by the distal site amino acid envelope, and by the overall protein shape and charge distribution [11, 13, 36]. Conversely, biomimetic models lack the protein, which implies that the substrate and oxidant accessibility and the reactivity control must be modulated by the design of the metalloporphyrin itself, i.e., the metal selection, the nature of the porphyrin ring substituents, the choice of axial ligand and, finally, the microenvironment around the macrocycle [17, 20, 24, 37, 88, 91, 92].
Although O2 would be the oxidant of choice for catalytic oxidations [24, 67], the activation of O2 by the model systems poses some complications. Under aerobic conditions, most common Fe and Mn porphyrins exist in the +3 oxidation state. The coordination of O2 to these metals is only favored, however, in lower oxidation states, such as Fe(II) and Mn(II) [6, 28]. Therefore, Fe(III) and Mn(III) porphyrins should first be reduced by some sacrificial reductant to yield their Fe(II) and Mn(II) analogues [17, 20]. Upon O2-coordination, an internal 1-electron transfer from the Fe(II) center to O2 yields a Fe(III)-superoxo complex. A further 1-electron reduction at the superoxo site is required to yield a peroxo species, which upon O–O bond heterolytic cleavage oxidizes the metal(III) center by 2-electrons to give rise to the active high-valent Fe(V)-oxo or Mn(V)-oxo species. That is, in order to reach the formation of the putative metal-oxo active species, the complex needs to undergo two consecutive 1-electron reductions [17]. The highly oxidizing metal-oxo species needs, thus, to be formed in the presence of an excess of a sacrificial reductant. If the substrate is somewhat unreactive, such as alkanes, some significant O2-oxidizing equivalents may be lost during the catalysis, by oxidation of the sacrificial reductant in detriment of substrate oxidation. Nevertheless, some O2-oxidations have been accomplished by metalloporphyrins using various sacrificial reductants [93], such as aldehydes [20], Zn metal [20], and ascorbate [94–97]. The O2-oxidation of a cancer pro-drug under biologically relevant conditions [97] is described below (see Sect. 9.3.2).
In most model systems, given the inherent impossibility to control the orchestrated access of oxidants and reductants to the metal center [17, 93], the majority of the biomimetic studies are carried out with oxygen donors, such as PhIO, sodium hypochlorite, alkyl hydroperoxides (LOOH), or hydrogen peroxide, to emulate either the peroxide-shunt or the PhIO-shunt pathways of P450 (Fig. 9.2) and yield the metal-oxo species equivalent to Compound I [14, 17, 20, 24, 90]. In the enzyme systems, the reactivity of Compound I is controlled by the protein moiety: whereas P450-Compound I usually carries out formally a single 2-electron substrate oxidation process to return to the resting state, HRP-Compound I carries out two 1-electron oxidations to reach the resting state [12]. It is worth noting that in the model systems, as model-Compound I lacks the protein moiety, its selectivity and mode of substrate oxidation cannot be accurately defined a priori. The prevalence of the 2-electron oxidation pathway over the two 1-electron oxidation pathway in the model-Compound I depends, thus, on a series of factor [20, 24, 90, 93], such as the chosen metal center, the porphyrin substituents, the nature of the oxygen donor system, and the medium conditions (e.g., pH [98]). It is worth noting that the organic transformations carried out by P450-type model reactions are typically of overall non-radical nature, whereas HRP-type model reactions are usually associated with radical species/products.
Although hemeproteins are naturally Fe-containing biomolecules , Mn(III) porphyrins have been considerably versatile oxidation catalysts [20, 37]. The hydroxylation of saturated hydrocarbons using the system Mn(III)-porphyrin/PhIO was simultaneously described by Groves and coworkers [99] and Hill and Schardt [100]. The advantages of the Mn(III) porphyrin systems over the corresponding Fe(III) counterparts are: longer life-time of the catalyst, and higher catalytic efficiency [20]. Mn porphyrin-based systems are, however, more prone to be involved in 1-electron processes (radical-type reactions) which compromise the selectivity of the Mn systems as compared to the corresponding Fe analogues [90, 101, 102]. Whereas the oxidation of alkanes by Fe(III) porphyrin/PhIO systems usually results in the formation of alcohol with little or no amount of the corresponding ketone, in the Mn(III) systems ketone is regularly found along with the desired alcohol product [20, 90]. In some systems, even alkyl halides are found as products resulting from the abstraction of halide of the MnP counter-ion, the porphyrin ring, or the solvent [101, 103]. The increased radical character of Mn(III) porphyrin systems is usually invoked to explain, for example, the decreased selectivity toward the alcohol [90]. A general reaction mechanism scheme for Mn(III) porphyrin systems is presented in Fig. 9.5.

Hypothetical mechanism scheme for the oxidations catalyzed by Mn(III) porphyrins using an oxygen donor AO. The box in the left highlights the major oxygenation pathways leading to the oxidized substrate ROH. The right top box presents the major catalyst deactivation pathway, which is associated with catalyst oxidative destruction. Other competing pathways, including those of radical nature, are presented in the right bottom box. Adapted from [100]
A fundamental problem associated with metalloporphyrin catalyzed oxidations is the vulnerability of the macrocycle toward bimolecular oxidative destruction [24, 38, 104], in which a metal-oxo species generated in situ attacks another metalloporphyrin molecule. This is typically a situation where the macrocycle competes with the substrate for the active intermediate and is, therefore, accentuated when substrates of low reactivity are used [91, 103, 105, 106]. For example, the oxidation of alkanes catalyzed by MnPs is usually accompanied by partial-to-total destruction of the porphyrin, whereas if the substrate is an olefin instead, little destruction may be noticed under the same oxidizing conditions [105]. In the presence of an active oxidant, such as PhIO, ClO−, or H2O2, but in the absence of suitable substrates, FePs and MnPs are prone to oxidative destruction [41, 103, 107]. The attempts to develop MnP- and FeP-based catalase models are particularly hindered by the destruction of the metalloporphyrins by H2O2 [41]. Of note, while H2O2 is a good oxygen donor to FePs and MnPs, leading to the corresponding metal-oxo species, H2O2 itself is not easily oxidized, being, thus, a very poor substrate to the metal-oxo species; in absence of a good substrate, the other metalloporphyrin molecules in the system become a suitable substrate to the electrophilic metal-oxo species [41, 104, 107]. The vulnerability of the metal-free macrocycle toward oxidation is increased with complexation of redox-active metals, such as Fe(III) and Mn(III): whereas bleaching of MnPs and FePs in the absence of a substrate is rapidly observed upon incubation with H2O2 [41], for example, the metal-free porphyrin is much less susceptible to destruction under the same conditions. In fact, incubation of free-base porphyrin mixtures with H2O2 [108] or other strong quinone-based oxidants [109, 110] is a common procedure in porphyrin synthesis.
A great deal of efforts has been dedicated toward the synthesis of new metalloporphyrins, introducing bulky and/or electron-withdrawing groups on the porphyrin ring, in an attempt to make the catalyst more efficient, selective, and, especially, more stable against oxidative destruction [24, 38, 111]. Another way to decrease the likelihood of the bimolecular oxidative destruction processes has been the immobilization of FePs and MnPs onto a solid surface or micelle interface [39, 88, 95, 96]. A variety of supports have been studied, such as silica gel (functionalized or not), ion-exchange resins, polymers, clays, and zeolite [20, 88]. MnPs immobilized in vesicles and micelles have also been studied as oxidation catalysts in liquid-liquid microheterogeneous systems exhibiting great regioselectivity, efficiency, and stability [95, 102, 112]. Of note, the immobilization of metalloporphyrins may reveal other beneficial outcomes induced by the steric and/or electronic effects associated with the support, in analogy to the protein moiety of the hemeproteins [39, 88].
9.3.2 Systems of Biological Interest
The P450-based biomimetic models have been increasingly explored for the oxidation of biomolecules or substrates of biological relevance, such as drugs, pro-drugs, dyes, pesticides, and many other xenobiotics, as reviewed elsewhere [20, 37, 40, 89, 113, 114]. Given that synthetic metalloporphyrins are usually more easily accessible than purified P450 enzymes, the reactions may be carried out in preparative scale, allowing isolation and/or full characterization of products. Commonly, the goal is to guide the identification of likely metabolites and/or putative intermediates in the more analytically demanding biological P450 oxidation systems.
The first studies on the oxidation of drugs under porphyrin-based biomimetic conditions were reported in the late 1980s and early 1990s, using the simplest, water-insoluble Fe(III) or Mn(III) porphyrins, such as FeTPP+ and MnTPP+ [115–118]. The oxidation of nicotine by MnTPP+ using PhIO as oxidant yielded two products, 3-hydroxynicotine and cotinine, that were identical to the P450-oxidation metabolites observed in vivo [118]. The variety of drugs studied thus far increased considerably and included, for example, caffeine (the legal drug most universally consumed [119]), anti-inflammatory drugs, anesthetics, analgesics, steroids, natural products, antibiotics, anticancer drugs, anti-epileptic drugs, anti-psychotic drugs, antipyretic drugs, antiarrhythmic drugs, antihistaminic drugs, among others [113–130]. These studies are generally carried out under simple reaction conditions: the substrate (drug), the oxygen donor (e.g., H2O2, t-BuOOH, NaOCl, PhIO), with FePs or MnPs as catalyst, in a suitable solvent.
Vitamin K3 was prepared in a single step via the oxidation of 2-methylnaphthalene with hydrogen persulfate (HSO5 −) in aqueous solution at room temperature (Fig. 9.6) [131]. The reaction was catalyzed by anionic or cationic water-soluble metalloporphyrins (Fe and Mn). Although the substrate is not biologically available, it is worth noting that this demonstrates the potential of MnPs and FePs for the oxidation of aromatic compounds to biologically relevant quinones under ambient conditions. The biomimetic system makes use of HSO5 −, which is prepared from sulfate and H2O2.

MnP-based biomimetic oxidation of 2-methylnaphthalene to yield vitamin K3. Adapted from [131]
The rich chemistry of metalloporphyrin-based biomimetic catalysis on a naturally occurring substrate using a biologically ubiquitous oxidant can be illustrated by the H2O2-oxidation of all-(E)-retinol, a natural form of vitamin A, using the simplest Fe(III) porphyrin, FeTPP+ under ambient conditions [132]. This biomimetic approach subjected the substrate to a variety of transformations, such as epoxidations, allylic hydroxylations, oxidative dehydrogenation, C–C bond cleavage, and isomerizations (Fig. 9.7). This is in direct contrast with the P450-catalyzed all-(E)-retinol oxidation, which yields primarily the alcohol dehydrogenation product, retinal. The unrelated nature of the products of the biomimetic versus the enzymatic systems is worth noting while using synthetic metalloporphyrin in vitro or in vivo: in the presence of both a suitable substrate and an active oxidant, metalloporphyrin-based oxidations could give rise to products of completely distinct nature from those usually expected as naturally occurring metabolites.

H2O2-oxidation of naturally occurring all-(E)-retinol using FeP as biomimetic catalyst. Adapted from [132]
Naturally occurring reactive oxygen/nitrogen species, such as lipid hydroperoxides (LOOH) and peroxynitrite (ONOO−), are suitable oxygen donors to MnPs under biological oxidative-stress conditions [133, 134]. Both oxidants can convert cationic MnPs (e.g., MnTE-2-PyP5+) into the oxidizing Mn(IV)-oxo porphyrin species in aqueous solution. The high-valent species was further demonstrated to be able to carry out the oxidation of glutathione, ascorbate, and urate under biologically relevant conditions [134].
MnP-based biomimetic systems using O2 to carry out oxidation reactions of biologically relevant substrates is uncommon. The activation of O2 usually requires a sacrificial reductant. A biologically available reductant, present in cells at relative high concentrations (millimolar), is ascorbate. Mansuy and coworkers [94] used the O2/ascorbate system for the MnTPP+-catalyzed oxidations, epoxidations, and dehydrogenations of alkanes and olefins under biphasic (water/benzene) conditions. Groves and Neumann [95, 96] incorporated steroidal-appended MnPs and FePs into phospholipid bilayer vesicles (Fig. 9.8) and showed that the MnP-based catalytic constructs were able to carry out the selective hydroxylation of cholesterol at carbon C-25 using the O2/ascorbate oxidant system under mild conditions in Tris buffer at pH 8.6; the FeP-based catalytic construct catalyzed the O2/ascorbate-oxidation of desmosterol (Fig. 9.8). Of note, the corresponding homogeneous (non-vesicle) systems gave rise to small amounts of many products.

More recently, Spasojević and coworkers [97] showed that water-soluble Fe and Mn porphyrins are able to catalyze the hydroxylation of the anticancer pro-drug cyclophosphamide to active metabolite 4-hydroxycyclophosphamide using the O2/ascorbate system (Fig. 9.9) in yields similar or higher than those typically obtained by the action of microsomal P450 enzymes in vivo. The cationic Mn porphyrins MnTM-2-PyP5+ and MnTM-3-PyP5+ used for the cyclophosphamide oxidation by the O2/ascorbate system have regularly been investigated as SOD mimics for the development of redox-based experimental therapeutics. Of note, the O2/ascorbate/MnP systems have recently been forwarded to in vivo studies as a prospective anticancer treatment [107].

The hydroxylation of the anticancer pro-drug cyclophosphamide to active metabolite 4-hydroxycyclophosphamide using the O2/ascorbate system catalyzed by the MnP- and FeP-based biomimetic models. Adapted from [97]
9.4 Pro-oxidative Role of Mn-Porphyrins Under Oxidative Stress Conditions: Biomimetic Oxidation Catalysis in Biological Milieu?
The inorganic medicinal c hemistry of MnPs is in its infancy as compared to classic metal-containing drugs, such as cisplatin. The design of MnP-based therapeutics did not follow the standard medicinal chemistry approach of defining a specific biomacromolecule target (e.g., protein, enzyme, nucleic acids), but aimed at ubiquitous reactive oxygen/nitrogen species (ROS/RNS) [22, 23, 135] and, thus, at the cell Redoxome [136].
The early studies on MnP in vivo were carried out in microorganism (e.g., E. coli) to verify their SOD activity in a complex biological matrix or to test them as an indirect mechanistic probe for ROS/RNS [45, 53, 137]. The reports on in vivo animal studies of MnPs as prospective experimental therapeutics began in the 2000s [22, 23]. Despite the short period of time, two of the designed and tested compounds (i.e., MnTE-2-PyP5+ [54] and MnTnBuOE-2-PyP5+ [138]) reached translational studies and are now entering phase I/II Clinical Trials [42, 62] (see also Chap. 8 by Batinić-Haberle et al.). The MnPs of medicinal interest were primarily designed as SOD mimics and/or peroxynitrite scavengers exploiting the radioprotector or catalytic antioxidant properties of the compounds [22, 23].
The overall effect commonly observed in the biological systems was regularly equivalent to that expected from the role played by an endogenous/exogenous antioxidant [22]. Because of these overall antioxidant effects, MnPs themselves were usually referred to as antioxidant catalyst [64, 65]. However, recent studies challenged this view by providing solid evidences that the mode of action of SOD mimic compounds is frequently pro-oxidative while the effects observed appear antioxidative [62, 139–146] (see also Chap. 8 by Batinić-Haberle et al.).
Early aqueous solution studies revealed that the SOD activity and the peroxynitrite decomposition efficacy of MnPs are linearly correlated, and established that powerful SOD mimics are also potent peroxynitrite scavengers [22, 23]. Thus, good SOD mimics would provide good drug candidates for treating ailments and diseases of oxidative-stress nature. Of course, the in vivo efficiency of any particular drug depends on its bioavailability, subcellular/cellular/tissue distribution, pharmacokinetics, dosing regimen, toxicity, etc. A correlation of animal in vivo efficacy and SOD activity of MnPs is emerging (see Chap. 8 by Batinić-Haberle et al.). A rough picture in SOD-specific microorganism-based models or in vitro systems indicates, however, that little amounts of powerful SOD mimics are needed for protection against superoxide-driven oxidative stress [22, 23, 45]. Conversely, the fair-to-low SOD activity of some compounds may be compensated by a great accumulation within the cell/tissue of interest [147–149]. A puzzling question that arises, however, is how to explain the overwhelming amount of biological data on MnPs that are not SOD mimics at all, such as MnTBAP3− [150]. Evidences associating MnTBAP3− with an oxidant role in biological systems have been recently reported [146].
The rational development of MnP-based SOD mimics have most commonly been guided by designing the porphyrin ligand so that the resulting MnP complex would exhibit Mn(III)/Mn(II) reduction potential close to that of the SOD enzymes [22, 23, 54, 55, 59, 60, 145, 151]. Although electrostatics plays also a major role in modulating the approach of superoxide to the metal center [58–60], the use of the enzyme reduction potential as a guide has been a rule of thumb. Indeed, cationic MnPs whose metal-centered reduction potentials are near ca. +300 mV vs. NHE have high SOD activity [22, 23, 42, 138] (Table 9.1). Conversely, MnPs (regardless of their overall charge) of moderate to negative reduction potential, such as MnTM-4-PyP5+ [53] and MnTBAP3− [150], have moderate to negligible SOD activity, respectively [22, 23] (Table 9.1).
Cytochromes P450 exhibit a very rich electrochemical behavior [70, 79–82, 152]. Their Fe(III)/Fe(II) reduction potential show a remarkable substrate dependency giving that the substrate modulates the high- versus low-spin contributions on the Fe center spin state [80]. Table 9.2 exemplifies the dramatic changes in both human and bacterial P450 reduction potential with respect to the nature of the substrate [82, 152]. The Fe(III)/Fe(II) reduction potential of the resting state complex, a water-coordinated Fe(III) porphyrin thiolate species (Fig. 9.2, A) is very negative, e.g., −220 mV vs. NHE for human cytochrome P450 CYP3A4 [81, 82]. This low potential is easily achieved even with the simplest meso-tetraphenylporphyrin derivatives [22, 23], such as MnTPP+ [153] or MnTBAP3− [150]. The reaction of MnP-based model compounds with active oxygen donors (e.g., PhIO, H2O2) resembles, thus, that of the resting state of P450 to yield the active metal-oxo active species.
Although it is tempting to use the same reduction potential strategy used in the development of SOD mimics to guide the design of P450-model design, care must be exercised. In the SOD case, the Mn(III)/Mn(II) redox couple represents the exact Mn(III)P and Mn(II)P species involved in the dismutation process. In the P450 case, the rate-determining step is usually ascribed to the oxygen transfer from the formal Mn(V)-oxo species to the substrate. If the oxidation takes place via a typical P450-type reaction, the oxo-Mn(V)P species would be reduced directly to the Mn(III)P resting state in a single step. This implies, thus, that the redox couple of interest to describe this 2-electron process would be the Mn(V)/Mn(III) one, which is available for a few MnP systems only [154]. As the model compounds lack the protein moiety to favor a P450-type vs. peroxidase-type reaction, it is likely that 1-electron transfer processes may be also operative, depending on the nature of the substrate available, the porphyrin substituents, and the overall conditions. Irrespective of the mechanism being predominately either P450-like or peroxidase-like, if the catalytically relevant reactions are significantly dependent on the 2-electron cycling of the Mn(V)P/Mn(III)P species or the Mn(IV)P/Mn(II)P species, i.e., the rate determining step being associated with Mn(V)-oxo/Mn(III) or Mn(IV)-oxo/Mn(II) couples, then the electrochemically available Mn(III)/Mn(II) reduction potentials could be eventually a surrogate descriptor for the MnP reactivity under catalytic conditions. Comprehensive studies on correlating P450-type catalysis and Mn(III)/Mn(II) reduction potentials are still limited [105, 106, and references therein], but a bell-shape behavior is likely. However, recent data from our laboratories (Falcão, Pinto, Reboucas, unpublished) on the hydroxylation of cyclohexane catalyzed by isomeric ortho, meta, and para Mn(III) N-methylpyridinium porphyrins (MnTM-2-PyP5+, MnTM-3-PyP5+, and MnTM-4-PyP5+, respectively, Fig. 9.1) under conditions of low catalyst degradation, resulted in approximately the same total yield of oxygenated products (~91 %) regardless of the isomer used. This suggests that the efficiency of the isomers in this particular oxidation system is essentially independent of the Mn(III)/Mn(II) reduction potential, which ranges from +52 mV (meta isomer [53]) to +220 mV vs. NHE (ortho isomer [53]): a 168 mV spam (Table 9.1). It is worth noting, however, that a more direct correlation between Mn(III)/Mn(II) reduction potential and the catalytic activity for metalloporphyrin-catalyzed oxidations should be likely found for radical O2-autoxidation reactions in which the metal center cycles indeed between +3 and +2 oxidation states [67, 68, 155], resembling the behavior observed in the SOD systems.
Whether or not the Mn(III)/Mn(II) reduction potentials (or any another redox couple for that matter) would ever be a good descriptor for biomimetic oxidation catalysts is still uncertain. What is worth noting, however, is that even MnPs of very low Mn(III)/Mn(II) reduction potentials, which are unable to carry out SOD-like reactions in vivo, may give a suitable entry to Mn-oxo species in the presence of an oxidative stress-derived oxidant, such as H2O2 or ClO− commonly used in the chemical biomimetic systems [20]. In MnP-loaded cells under oxidative stress, all ingredients needed for MnP-based biomimetic oxidation catalysis are present in the cell milieu: a good source of oxygen donor (ROS/RNS), the catalyst (MnP), and a plethora of substrates (e.g., proteins, lipids, nucleic acids). In such conditions of high substrate availability, vulnerability of the MnP toward bimolecular destruction should, thus, be minimized, preserving catalytic function [105]. An antioxidant overall effect could still be observed via biomimetic oxidation catalysis if ROS/RNS is intercepted by MnP species, which would then undergo oxidation to the corresponding Mn-oxo porphyrin species. Either Mn(V)-oxo or Mn(IV)-oxo porphyrin species are still very potent oxidants, being able to oxidize even inert aliphatic C-H bonds if better (more reducing) biomolecules are not present. Contrary to regular ROS/RNS (e.g., H2O2, ONOO−, LOOH, etc) Mn-oxo porphyrin species are bulky oxidants. If the ROS/RNS-sensitive oxidation sites are either cofactors or active centers buried within biomolecules such as enzymes, proteins, and transcription factors, the formation of the Mn-oxo porphyrin species would protect these ROS/RNS-sensitive sites against oxidation under oxidative stress conditions. Again, since Mn-oxo species are potent oxidants [154], they are likely to react within the cell milieu regenerating the MnP resting state. Given the size of the MnPs, the MnP-catalyzed oxidations in the cells is most likely to take place at sacrificial amino acid residues (e.g., Met or Cys) on protein surfaces [156–160], or other endogenous small molecular weight reductant, such as glutathione, ascorbate, or urate [107, 134, 161]. This mode of action where MnPs act as biomimetic oxidation catalysts of overall protective role is illustrated in Fig. 9.10.

Schematic cartoon illustrating the putative role of MnP-based biomimetic oxidation catalysts in protecting the active site (central circle) of a generic enzyme against oxidation by ROS/RNS-based oxygen donors (OD). MnP would intercept OD to form Mn-oxo porphyrin species, which could then direct the oxidation to sites of less physiological consequences, such as sacrificial residues [156–160] at protein surface (triangles). Oxidation of the active site by an OD-generated Mn-oxo porphyrin species would be hampered by steric demands. Yellow color indicates reduced state, whereas oxidizes sites are colored in red
9.5 Concluding Remarks
Manganese and iron porphyrins are well-established biomimetic oxidation catalysts under purely chemical settings. They have been able to provide an entry to single compounds or materials that model remarkably well the electronic structure and reactivity profile of many heme-containing oxidation enzymes, such as cytochromes P450 and peroxidases. These studies contributed to both unravel the underlying species involved in the enzymatic systems and develop attractive oxidation catalysts to the functionalization of a number of organic substrates, including the efficient activation of inert C–H bonds under mild conditions.
Mn porphyrins are among the metalloporphyrins most studied in vivo. Their design and medicinal applications have been largely driven by exploiting the ability of MnPs to mimic the activity of the SOD enzymes. This has been proven a remarkably efficient strategy to yield potent redox-active therapeutics, in particular those derived from cationic porphyrins. However, the large number of studies on the in vivo efficacy of MnPs of low SOD activity, such as neutral porphyrins and MnTBAP3−, toward ameliorating many oxidative stress-related conditions is unsettling; when biological effects do not arise from sample impurities [150, 162, 163], the mechanistic association with SOD activity needs, obviously, further revision [137]. Catalase-like activity of MnPs in general cannot account for the therapeutic effects of MnPs [41].
MnPs uptaken by cell systems under oxidative stress conditions are subjected to oxidative pressures similar to those found in standard chemical systems. In MnP-treated tissues, all components for biomimetic oxidation catalysis are available in the cell milieu: ROS/RNS-based oxidants (hydrogen peroxide, hydroperoxides, hypochlorite, etc.), the MnP-based catalyst, and many biomolecules as substrates.
The transformation of ROS/RNS-derived oxygen donor to high-valent Mn-oxo species switches the oxidant size from a small reactive species into a bulkier and sterically demanding metal-oxo species of equally high oxidation power; such larger metal-based oxidant is most likely to react, however, with (sacrificial) surface protein-residues, while preserving untouched the buried, active sites of large biomolecules. The formation of MnP-based metal-oxo species may render the oxidant also less susceptible to diffusion, helping containing the oxidative burst to a restricted region of the cell. Conversely, as Mn-oxo porphyrin species react readily with thiol and thioether moieties [134, 161, 164], such as those of surface-active Cys and Met residues of transcription factors, Mn porphyrin action as biomimetic oxidation catalysts may play a significant role in affecting major cell signaling pathways (see also Chap. 8 by Batinić-Haberle et al.).
MnPs of low Mn(III)/Mn(II) reduction potential can barely work as SOD mimics (if at all) [150, 165], which increases the likelihood of the compounds being biologically active via a biomimetic oxidation catalysis pathway. This may be of particular relevance to negatively charged and/or neutral porphyrin-based compounds [166, 167], or other metal-based complexes of low reduction potential, such as Mn salens, whose biological data as redox-active therapeutics are clearly of undeniable significance [168] (see also Chap. 11 by Doctrow and coworkers). On the other hand, for cationic MnPs of high Mn(III)/Mn(II) reduction potentials, the contribution of the biomimetic oxidation catalysis versus the standard antioxidant catalysis to the overall biological effects remains uncertain, given that such compounds are also potent SOD mimics on their own, and should be of particular interest to research in the near future.
Abbreviations
- AH:
-
A generic 1-electron donor (AH → A• + H+ + 1e−)
- AO:
-
A generic oxygen donor such as PhIO, ClO−, and H2O2 etc.
- FeP:
-
Iron porphyrin
- FeTPP+:
-
Fe(III) meso-tetraphenylporphyrin
- HRP:
-
Horseradish peroxidase
- LOOH:
-
Lipid hydroperoxide or an alkyl hydroperoxide
- MnP:
-
Manganese porphyrin
- MnTBAP3− :
-
Mn(III) meso-tetrakis(benzoic acid)porphyrin or Mn(III) meso-tetrakis(carboxyphenyl)porphyrin
- MnTE-2-PyP5+ :
-
Mn(III) meso-tetrakis(N-ethylpyridinium-2-yl)porphyrin
- MnTM-2-PyP5+ :
-
Mn(III) meso-tetrakis(N-methylpyridinium-2-yl)porphyrin
- MnTM-3-PyP5+ :
-
Mn(III) meso-tetrakis(N-methylpyridinium-3-yl)porphyrin
- MnTM-4-PyP5+ :
-
Mn(III) meso-tetrakis(N-methylpyridinium-4-yl)porphyrin
- MnTnBuOE-2-PyP5+ :
-
Mn(III) meso-tetrakis(N-n-butoxyethylpyridinium-2-yl)porphyrin
- MnTnHex-2-PyP5+ :
-
Mn(III) meso-tetrakis(N-n-hexylpyridinium-2-yl)porphyrin
- MnTPP+:
-
Mn(III) meso-tetraphenylporphyrin
- NADP+ :
-
Nicotinamide adenine dinucleotide phosphate
- NADPH:
-
Reduced form of nicotinamide adenine dinucleotide phosphate
- P450:
-
Cytochrome P450
- PhIO:
-
Iodosylbenzene
- RH:
-
A generic organic substrate usually an alkane
- SOD:
-
Superoxide dismutase
References
Milgrom LR. The colours of life: an introduction to the chemistry of porphyrins and related compounds. Oxford: Oxford University Press; 1997.
Lippard SJ, Berg JM. Principles of bioinorganic chemistry. Mill Valley: University Science; 1994.
da Silva JJRF, Williams RJP. The biological chemistry of the elements: the inorganic chemistry of life. New York: Oxford University Press; 2001. p. 343–69.
Kumar S, Bandyopadhyay U. Free heme toxicity and its detoxification systems in human. Toxicol Lett. 2005;157:175–88.
Aich A, Freundlich M, Vekilov PG. The free heme concentration in healthy human erythrocytes. Blood Cells Mol Dis. 2015;55:402–9.
Jones RD, Summerville DA, Basolo F. Synthetic oxygen carriers related to biological systems. Chem Rev. 1979;79:139–79.
James BR. Interaction of dioxygen with metalloporphyrins. In: Dolphin D, editor. The porphyrins, vol. V. New York: Academic Press; 1977. p. 205–302.
Traylor TG. Synthetic model compounds for hemoproteins. Acc Chem Res. 1981;14:102–9.
de Montellano PRO. Catalytic sites of hemoprotein peroxidases. Annu Rev Pharmacol Toxicol. 1992;32:89–107.
Collman JP, Boulatov R, Sunderland CJ, Fu L. Functional analogues of cytochrome c oxidase, myoglobin, and hemoglobin. Chem Rev. 2004;104:561–88.
Mansuy D, Battioni P. Biochemistry and binding: activation of small molecules. In: Kadish KM, Smith KM, Guilard R, editors. The porphyrin handbook, vol. 4. New York: Academic Press; 2000. p. 1–15.
Hrycay EG, Bandiera SM. The monooxygenase, peroxidase, and peroxygenase properties of cytochrome P450. Archiv Biochem Biophys. 2012;522:71–89.
Meunier B, Visser SP, Shaik S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem Rev. 2004;104:3947–80.
Sheldon RA. Oxidation catalysis by metalloporphyrins: a historical perspective. In: Sheldon RA, editor. Metalloporphyrins in catalytic oxidations. New York: Marcel Dekker; 1994. p. 5–27.
Breslow R. Centenary lecture. Biomimetic chemistry. Chem Soc Rev. 1972;1:553–80.
Breslow R. Preface. In: Breslow R, editor. Artificial enzymes. Weinheim: Wiley-VCH; 2014. p. 9–10.
Ezhova MB, James BR. Catalytic oxidations using ruthenium porphyrins. In: Simándi LI, editor. Advances in catalytic activation of dioxygen by metal complexes. Dordrecht: Kluwer Academic; 2003. p. 1–77.
Rebouças JS, Patrick BO, James BR. Thiol, disulfide, and trisulfide complexes of Ru porphyrins: potential models for Iron-Sulfur bonds in heme-proteins. J Am Chem Soc. 2012;134:3555–70.
Rebouças JS, James BR. Molecular recognition using Ruthenium(II) porphyrin thiol complexes as probes. Inorg Chem. 2013;52:1084–98.
Meunier B. Metalloporphyrins as versatile catalysts for oxidation reactions and oxidative DNA cleavage. Chem Rev. 1992;92:1411–56.
Groves JT, Shalyaev K, Lee J. Oxometalloporphyrins in oxidative catalysis. In: Kadish KM, Smith KM, Guilard R, editors. The porphyrin handbook, vol. 4. San Diego: Academic Press; 2000. p. 17–40.
Batinić-Haberle I, Rebouças JS, Spasojević I. Superoxide dismutase mimics: chemistry, pharmacology and therapeutic potential. Antioxid Redox Signal. 2010;13:877–918.
Batinić-Haberle I, Rebouças JS, Benov B, Spasojević I. Chemistry, biology and medical effects of water-soluble metalloporphyrins. In: Kadish KM, Smith KM, Guilard R, editors. Handbook of porphyrin science, vol. 11. New York: World Scientific; 2011. p. 291–394.
Dolphin D, Traylor TG, Xie LY. Polyhaloporphyrins: unusual ligands for metals and metal-catalyzed oxidations. Acc Chem Res. 1997;30:251–9.
Collman JP, Gagne RR, Halbert TR, Marchon JC, Reed CA. Reversible oxygen adduct formation in ferrous complexes derived from a picket fence porphyrin. Model for oxymyoglobin. J Am Chem Soc. 1973;95:7868–70.
Almog J, Baldwin JE, Huff J. Reversible oxygenation and autoxidation of a capped porphyrin iron(II) complex. J Am Chem Soc. 1975;97:227–8.
Momenteau M, Loock B, Mispelter J, Bisagni E. “Basket handle” porphyrins and their ferrous complexes as stables oxygen carriers. Nouv J Chim. 1979;3:77–9.
Jones RD, Summerville DA, Basolo F. Manganese(II) porphyrin oxygen carriers. Equilibrium constants for the reaction of dioxygen with para-substituted meso-tetraphenylporphinatomanganese(II) complexes. J Am Chem Soc. 1978;100:4416–24.
Tsuchida E, Sou K, Nakagawa A, Sakai H, Komatsu T, Kobayashi K. Artificial oxygen carriers, hemoglobin vesicles and albumin-hemes, based on bioconjugate chemistry. Bioconjug Chem. 2009;20:1419–40.
Alayash AI. Blood substitutes: why haven’t we been more successful? Trends Biotechnol. 2014;32:177–85.
Riess JG. Oxygen carriers (“blood substitutes”)—raison d’etre, chemistry, and some physiology. Chem Rev. 2001;101:2797–920.
Cheng L, Richter-Addo GB. Binding and activation of nitric oxide by metalloporphyrins and heme. In: Kadish KM, Smith KM, Guilard R, editors. The porphyrin handbook, vol. 4. New York: Academic Press; 2000. p. 219–91.
Koshland Jr DE. The molecule of the year. Science. 1992;258:1861.
Szabó C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–80.
Groves JT, Nemo TE, Myers RS. Hydroxylation and epoxidation catalyzed by iron-porphine complexes. Oxygen transfer from iodosylbenzene. J Am Chem Soc. 1979;101:1032–3.
de Montellano PRO. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem Rev. 2010;110:932–48.
Mansuy D. A brief history of the contribution of metalloporphyrin models to cytochrome P450 chemistry and oxidation catalysis. C R Chimie. 2007;10:392–413.
Wijesekera TP, Dolphin D. Synthetic aspects of porphyrin and metalloporphyrin chemistry. In: Sheldon RA, editor. Metalloporphyrin in catalytic oxidations. New York: Marcel Dekker; 1994. p. 193–239.
Suslick KS. Shape-selective oxidations by metalloporphyrins. In: Kadish KM, Smith KM, Guilard R, editors. The porphyrin handbook, vol. 4. New York: Academic Press; 2000. p. 41–63.
Che CM, Lo VK, Zhou CY, Huang JS. Selective functionalisation of saturated C–H bonds with metalloporphyrin catalysts. Chem Soc Rev. 2011;40:1950–75.
Tovmasyan A, Maia CGC, Weitner T, Carballal S, Sampaio RS, Lieb D, Ghazaryan R, Ivanovic-Burmazovic I, Ferrer-Sueta G, Radi R, Rebouças JS, Spasojević I, Benov L, Batinić-Haberle I. A comprehensive evaluation of catalase-like activity of different classes of redox-active therapeutics. Free Radic Biol Med. 2015;86:308–21.
Batinić-Haberle I, Tovmasyan A, Roberts ER, Vujaskovic Z, Leong KW, Spasojević I. SOD therapeutics: latest insights into their structure-activity relationships and impact on the cellular redox-based signaling pathways. Antioxid Redox Signal. 2014;20:2372–415.
DeFreitas-Silva G, Rebouças JS, Spasojević I, Benov L, Idemori YM, Batinić-Haberle I. SOD-like activity of Mn(II) β-octabromo-meso-tetrakis(N-methylpyridinium-3-yl)porphyrin equals that of the enzyme itself. Arch Biochem Biophys. 2008;477:105–12.
Tovmasyan A, Weitner T, Sheng H, Lu M, Rajic Z, Warner DS, Spasojević I, Rebouças JS, Benov L, Batinić-Haberle I. Differential coordination demands in Fe versus Mn water-soluble cationic metalloporphyrins translate into remarkably different aqueous redox chemistry and biology. Inorg Chem. 2013;52:5677–91.
Tovmasyan A, Rebouças JS, Benov L. Simple biological systems for assessing the activity of superoxide dismutase mimics. Antioxid Redox Signal. 2014;20:2416–36.
Pasternack RF, Halliwell B. Superoxide dismutase activities of an iron porphyrin and other iron complexes. J Am Chem Soc. 1979;101:1026–31.
Faraggi M, Peretz P, Weinraub D. Chemical properties of water-soluble porphyrins. 4. The reaction of a ‘picket-fence-like’ iron (III) complex with the superoxide oxygen couple. Int J Radiat Biol Relat Stud Phys Chem Med. 1986;49:951–68.
Weinraub D, Peretz P, Faraggi M. Chemical properties of water-soluble porphyrins. 1. Equilibriums between some ligands and iron(III) tetrakis(4-N-methylpyridyl)porphyrin. J Phys Chem. 1982;86:1839–42.
Ilan Y, Rabani J, Fridovich I, Pasternack RF. Superoxide dismuting activity of an iron porphyrin. Inorg Nucl Chem Lett. 1981;17:93–6.
Pasternack RF, Banth A, Pasternack JM, Johnson CS. Catalysis of the disproportionation of superoxide by metalloporphyrins. III. J Inorg Biochem. 1981;15:261–7.
Peretz P, Solomon D, Weinraub D, Faraggi M. Chemical properties of water-soluble porphyrins 3. The reaction of superoxide radicals with some metalloporphyrins. Int J Radiat Biol Relat Stud Phys Chem Med. 1982;42:449–56.
Weinraub D, Levy P, Faraggi M. Chemical properties of water-soluble porphyrins. 5. Reactions of some manganese (III) porphyrins with the superoxide and other reducing radicals. Int J Radiat Biol Relat Stud Phys Chem Med. 1986;50:649–58.
Batinić-Haberle I, Benov L, Spasojević I, Fridovich I. The ortho effect makes manganese(III) meso-tetrakis(N-methylpyridinium-2-yl)porphyrin a powerful and potentially useful superoxide dismutase mimic. J Biol Chem. 1998;273:24521–8.
Batinić-Haberle I, Spasojević I, Hambright P, Benov L, Crumbliss AL, Fridovich I. Relationship among redox potentials, proton dissociation constants of pyrrolic nitrogens, and in vivo and in vitro superoxide dismutating activities of Manganese(III) and Iron(III) water-soluble porphyrins. Inorg Chem. 1999;38:4011–22.
Batinić-Haberle I, Liochev SI, Spasojević I, Fridovich I. A potent superoxide dismutase mimic: manganese beta-octabromo-meso-tetrakis-(N-methylpyridinium-4-yl)porphyrin. Arch Biochem Biophys. 1997;343:225–33.
Batinić-Haberle I, Spasojević I, Stevens RD, Hambright P, Fridovich I. Manganese(III) meso-tetrakis(ortho-N-alkylpyridyl)porphyrins. Synthesis, characterization, and catalysis of O2 ∙− dismutation. J Chem Soc Dalton Trans. 2002:2689–96.
Batinić-Haberle I, Spasojević I, Stevens RD, Hambright P, Neta P, Okado-Matsumoto A, Fridovich I. New class of potent catalysts of O2-dismutation. Mn(III) ortho-methoxyethylpyridyl- and di-ortho-methoxyethylimidazolylporphyrins. Dalton Trans. 2004:1696–702.
Spasojević I, Batinić-Haberle I, Rebouças JS, Idemori YM, Fridovich I. Electrostatic contribution in the catalysis of O2 •– dismutation by superoxide dismutase mimics. MnIIITE-2-PyP5+ versus MnIIIBr8T-2-PyP+. J Biol Chem. 2003;278:6831–7.
Rebouças JS, Spasojević I, Tjahjono DH, Richaud A, Méndez F, Benov L, Batinić-Haberle I. Redox modulation of oxidative stress by Mn porphyrin-based therapeutics: the effect of charge distribution. Dalton Trans. 2008:1233–42.
Rebouças JS, DeFreitas-Silva G, Spasojević I, Idemori YM, Benov L, Batinić-Haberle I. Impact of electrostatics in redox modulation of oxidative stress by Mn porphyrins: protection of SOD-deficient Escherichia coli via alternative mechanism where Mn porphyrin acts as a Mn carrier. Free Radic Biol Med. 2008;45:201–10.
Batinić-Haberle I, Tovmasyan A, Spasojević I. The complex mechanistic aspects of redox-active compounds, commonly regarded as SOD mimics. BioInorg React Mech. 2013;9:35–58.
Batinić-Haberle I, Rajic Z, Tovmasyan A, Rebouças JS, Ye X, Leong KW, Dewhirst MW, Vujaskovic Z, Benov L, Spasojević I. Diverse functions of cationic Mn(III) N-substituted pyridyl porphyrins, recognized as SOD mimics. Free Radic Biol Med. 2011;51:1035–53.
Tovmasyan A, Sheng H, Weitner T, Arulpragasam A, Lu M, Warner DS, Vujaskovic Z, Spasojević I, Batinić-Haberle I. Design, mechanism of action, bioavailability and therapeutic effects of Mn porphyrin-based redox modulators. Med Princ Pract. 2013;22:103–30.
Sheng H, Spasojević I, Warner DS, Batinić-Haberle I. Mouse spinal cord compression injury is ameliorated by intrathecal cationic manganese(III) porphyrin catalytic antioxidant therapy. Neurosci Lett. 2004;366:220–5.
Mackensen GB, Patel M, Sheng H, Calvi CL, Batinić-Haberle I, Day BJ, Liang LP, Fridovich I, Crapo JD, Pearlstein RD, Warner DS. Neuroprotection from delayed post ischemic administration of a metalloporphyrin catalytic antioxidant. J Neurosci. 2001;21:4582–92.
Feiters MC, Rowan AE, Nolte RJM. From simple to supramolecular cytochrome P450 mimics. Chem Soc Rev. 2000;29:375–84.
Ellis PE, Lyons JE. Selective air oxidation of light alkanes catalyzed by activated metalloporphyrins—the search for a suprabiotic system. Coord Chem Rev. 1990;105:181–93.
Grinstaff MW, Hill MG, Labinger JA, Gray HB. Mechanism of catalytic oxygenation of alkanes by halogenated iron porphyrins. Science. 1994;264:1311–3.
Holm RH. Metal-centered oxygen atom transfer reactions. Chem Rev. 1987;87:1401–49.
Baylon JL, Lenov IL, Sligar SG, Tajkhorshid E. Characterizing the membrane-bound state of cytochrome P450 3A4: structure, depth of insertion, and orientation. J Am Chem Soc. 2013;135:8542–51.
Pochapsky TC, Kazanis S, Dang M. Conformational plasticity and structure/function relationships in cytochromes P450. Antioxid Redox Signal. 2010;13:1273–96.
Shaik S, Kumar D, Visser SP, Altun A, Thiel W. Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem Rev. 2005;105:2279–328.
Nam W. High-valent iron(IV)—oxo complexes of heme and non-heme ligands in oxygenation reactions. Acc Chem Res. 2007;40:522–31.
Rittle J, Green MT. Cytochrome P450 compound I: capture, characterization, and C–H bond activation kinetics. Science. 2010;330:933–7.
Jung C. The mystery of cytochrome P450 Compound I: a mini-review dedicated to Klaus Ruckpaul. Biochim Biophys Acta. 1814;2011:46–57.
Krest CM, Onderko EL, Yosca TH, Calixto JC, Karp RF, Livada J, Rittle J, Green MT. Reactive intermediates in cytochrome p450 catalysis. J Biol Chem. 2013;288:17074–81.
Denisov IG, Mak PJ, Makris TM, Sligar SG, Kincaid JR. Resonance Raman characterization of the peroxo and hydroperoxo intermediates in cytochrome P450. J Phys Chem A. 2008;112:13172–9.
Cojocaru V, Balali-Mood K, Sansom MSP, Wade RC. Structure and dynamics of the membrane-bound cytochrome P450 2C9. PLoS Comput Biol. 2011;7, e1002152.
Das A, Grinkova YV, Sligar SG. Redox potential control by drug binding to cytochrome P450 3A4. J Am Chem Soc. 2007;129:13778–9.
Lewis DF, Hlavica P. Interactions between redox partners in various cytochrome P450 systems: functional and structural aspects. Biochim Biophys Acta. 2000;1460:353–74.
Fisher MT, Sligar SG. Control of heme protein redox potential and reduction rate: linear free energy relation between potential and ferric spin state equilibrium. J Am Chem Soc. 1985;107:5018–9.
Sligar SG. Coupling of spin, substrate, and redox equilibria in cytochrome P450. Biochemistry. 1976;15:5399–406.
Groves JT, McClusky GA. Aliphatic hydroxylation via oxygen rebound. Oxygen transfer catalyzed by iron. J Am Chem Soc. 1976;98:859–61.
Groves JT, McClusky GA. Aliphatic hydroxylation by highly purified liver microsomal cytochrome P-450. Evidence for a carbon radical intermediate. Biochem Biophys Res Commun. 1978;81:154–60.
Veitch NC. Horseradish peroxidase: a modern view of a classic enzyme. Phytochemistry. 2004;65:249–59.
Kobayashi S, Nakano M, Kimura T, Schaap AP. On the mechanism of the peroxidase-catalyzed oxygen-transfer reaction. Biochemistry. 1987;26:5019–22.
Fujii H. Model complexes of heme peroxidases. In: Raven E, Dunford B, editors. Heme peroxidases. Cambridge: RSC Metallobiology; 2016. p. 183–217.
Nakagaki S, Ferreira GKB, Marcalb AL, Ciuffi KJ. Metalloporphyrins immobilized on silica and modified silica as catalysts in heterogeneous processes. Curr Org Synth. 2014;11:67–88.
Hongjian L, Zhang XP. Catalytic C–H functionalization by metalloporphyrins: recent developments and future directions. Chem Soc Rev. 2011;40:1899–909.
Gunter MJ, Turner P. Metalloporphyrins as models for the cytochromes p-450. Coord Chem Rev. 1991;108:115–61.
Silva VS, Meireles AM, Martins DCS, Rebouças JS, DeFreitas-Silva G, Idemori YM. Effect of imidazole on biomimetic cyclohexane oxidation by first-, second-, and third-generation manganese porphyrins using PhIO and PhI(OAc)2 as oxidants. Appl Catal A-Gen. 2015;491:17–27.
Iamamoto Y, Serra OA, Idemori YM. Iron(III) porphyrins atropisomers as catalysts for cyclohexane hydroxylations. A biomimetical system. J Inorg Biochem. 1994;54:55–66.
Tabushi I. Reductive dioxygen activation by use of artificial P-450 systems. Coord Chem Rev. 1988;86:1–42.
Mansuy D, Fontecave M, Bartoli JF. Mono-oxygenase-like dioxygen activation leading to alkane hydroxylation and olefin epoxidation by an Mn(porphyrin)-ascorbate biphasic system. J Chem Soc Chem Commun. 1983:253–4.
Groves JT, Neumann R. Enzymic regioselectivity in the hydroxylation of cholesterol catalyzed by a membrane-spanning metalloporphyrin. J Org Chem. 1988;53:3891–3.
Groves JT, Neumann R. Regioselective oxidation catalysis in synthetic phospholipid vesicles. Membrane-spanning steroidal metalloporphyrins. J Am Chem Soc. 1989;111:2900–9.
Spasojević I, Colvin OM, Warshany KR, Batinić-Haberle I. New approach to the activation of anti-cancer pro-drugs by metalloporphyrin-based cytochrome P450 mimics in all-aqueous biologically relevant system. J Inorg Biochem. 2006;100:1897–902.
Nam W, Choi HJ, Han HJ, Cho SH, Lee HJ, Han SY. Use of 2-methyl-1-phenylpropan-2-yl hydroperoxide (MPPH) as a mechanistic probe for the heterolytic versus homolytic O-O bond cleavage of tert-alkyl hydroperoxide by iron(III) porphyrin complex. Chem Commun. 1999;4:387–8.
Groves JT, Kruper Jr WJ, Haushalter RC. Hydrocarbon oxidations with oxometalloporphinates. Isolation and reactions of a (porphinato)manganese(V) complex. J Am Chem Soc. 1980;102:6375–7.
Hill CL, Schardt BC. Alkane activation and functionalization under mild conditions by a homogeneous manganese(III)porphyrin-iodosylbenzene oxidizing system. J Am Chem Soc. 1980;102:6374–5.
Smegal JA, Hill CL. Hydrocarbon functionalization by the (iodosylbenzene)manganese(IV) porphyrin complexes from the (tetraphenylporphinato)manganese(III)-iodosylbenzene catalytic hydrocarbon oxidation system. Mechanism and reaction chemistry. J Am Chem Soc. 1983;105:3515–21.
Suslick KS, van Deusen-Jeffries S. Shape selective biomimetic oxidation catalysis. In: Atwood JL, Davies JED, MacNicol DD, Vögtle F, Lehn J-M, editors. Comprehensive supramolecular chemistry, vol. 5. Oxford: Elsevier; 1996. p. 141–70.
Rebouças JS, Carvalho MEMD, Idemori YM. Perhalogenated 2-pyridylporphyrin complexes: synthesis, self-coordinating aggregation properties, and catalytic studies. J Porphyrins Phthalocyanines. 2002;6:50–7.
Serra AC, Marçalo EC, Gonsalves AMd’AR. A view on the mechanism of metalloporphyrin degradation in hydrogen peroxide epoxidation reactions. J Mol Catal A-Chem. 2004;215:17–21.
do Nascimento E, de F Silva G, Caetano FA, Fernandes MA, da Silva DC, de Carvalho ME, Pernaut JM, Rebouças JS, Idemori YM. Partially and fully beta-brominated Mn-porphyrins in P450 biomimetic systems: effects of the degree of bromination on electrochemical and catalytic properties. J Inorg Biochem. 2005;99:1193–204.
da Silva DC, DeFreitas-Silva G, do Nascimento E, Rebouças JS, Barbeira PJ, de Carvalho ME, Idemori YM. Spectral, electrochemical, and catalytic properties of a homologous series of manganese porphyrins as cytochrome P450 model: the effect of the degree of beta-bromination. J Inorg Biochem. 2008;102:1932–41.
Tovmasyan A, Sampaio RS, Boss MK, Bueno-Janice JC, Bader BH, Thomas M, Rebouças JS, Orr M, Chandler JD, Go YM, Jones DP, Venkatraman TN, Haberle S, Kyui N, Lascola CD, Dewhirst MW, Spasojević I, Benov L, Batinić-Haberle I. Anticancer therapeutic potential of Mn porphyrin/ascorbate system. Free Radic Biol Med. 2015;89:1231–47.
Johnstone RAW, Nunes MLPG, Pereira MM, Gonsalves AMd’AR, Serra AC. Improved syntheses of 5, 10, 15, 20-tetrakisaryl- and tetrakisalkylporphyrins. Heterocycles. 1996;43:1423–37.
Lindsey JS, Schreiman IC, Hsu HC, Kearney PC, Marguerettaz AM. Rothemund and Adler-Longo reactions revisited: synthesis of tetraphenylporphyrins under equilibrium conditions. J Org Chem. 1987;52:827–36.
Lindsey JS, Wagner RW. Investigation of the synthesis of ortho-substituted tetraphenylporphyrins. J Org Chem. 1989;54:828–36.
Atkinson ST, Brady SP, James JP, Nolan KB. Synthetic haems as mimics for high valent intermediates in haemoprotein catalysed oxidations. Synthesis and oxidation of chloro-7,8,17,18-tetracyano-5,10,15,20-tetraphenylporphyrinatoiron(III), a haem which contains strongly electron-withdrawing groups in the β-pyrrole positions. Pure Appl Chem. 1995;67:1109–16.
Borocci S, Marotti F, Mancini G, Monti D, Pastorini A. Selectivity in the oxidation of limonene by amphiphilized metalloporphyrins in micellar media. Langmuir. 2001;17:7198–203.
Lohmann W, Karst U. Biomimetic modeling of oxidative drug metabolism: strategies, advantages and limitations. Anal Bioanal Chem. 2008;391:79–96.
Bernadou J, Meunier B. Biomimetic chemical catalysts in the oxidative activation of drugs. Adv Synth Catal. 2004;346:171–84.
Masumoto H, Takeuchi K, Ohta S, Hirobe M. Application of chemical P450 model systems to studies on drug metabolism. I. Phencyclidine: a multi-functional model substrate. Chem Pharm Bull. 1989;37:1788–94.
Pautet F, Barret R, Daudon M. Optimization of biomimetic oxidation reactions achieved with the aid of an iodosylbenzene and meso-tetraphenylporphinatoiron (III): application to antergan. Pharm Acta Helv. 1988;63:140–4.
Salmeen IT, Foxall-VanAken S, Ball JC. A preliminary study of an iron porphyrin-iodosylbenzene system for activation of mutagens in the Ames assay. Mutat Res Lett. 1988;207:111–5.
Chauncey MA, Ninomiya SI. Metabolic studies with model cytochrome p-450 systems. Tetrahedron Lett. 1990;31:5901–4.
Neves CMB, Simões MMQ, Santos ICMS, Domingues FMJ, Neves MGPMS, Paz FAA, Silva AMS, Cavaleiro JAS. Oxidation of caffeine with hydrogen peroxide catalyzed by metalloporphyrins. Tetrahedron Lett. 2011;52:2898–902.
Schaab EH, Crotti AEM, Iamamoto Y, Kato MJ, Lotufo LVC, Lopes NP. Biomimetic oxidation of piperine and piplartine catalyzed by iron(III) and manganese(III) porphyrins. Biol Pharm Bull. 2010;33:912–6.
Breslow R, Zhang X, Huang Y. Selective catalytic hydroxylation of a steroid by an artificial cytochrome P-450 enzyme. J Am Chem Soc. 1997;119:4535–6.
Fang Z, Breslow R. A thiolate ligand on a cytochrome P-450 mimic permits the use of simple environmentally benign oxidants for biomimetic steroid hydroxylation in water. Bioorg Med Chem Lett. 2005;15:5463–6.
Melo AJB, Iamamoto Y, Maestrin APJ, Smith JRL, Santos MD, Lopes NP, Bonato PS. Biomimetic oxidation of praziquantel catalysed by metalloporphyrins. J Mol Catal A-Chem. 2005;226:23–31.
Hartmann J, Bartels P, Mau U, Witter M, Tümpling WV, Hofmann J, Nietzschmann E. Degradation of the drug diclofenac in water by sonolysis in presence of catalysts. Chemosphere. 2008;70:453–61.
Santos MD, Martins PR, Santos PA, Bortocan R, Iamamoto Y, Lopes NP. Oxidative metabolism of 5-o-caffeoylquinic acid (chlorogenic acid), a bioactive natural product, by metalloporphyrin and rat liver mitochondria. Eur J Pharm Sci. 2005;26:62–70.
Mac Leod TCO, Faria AL, Barros VP, Queiroz MEC, Assis MD. Primidone oxidation catalyzed by metalloporphyrins and Jacobsen catalyst. J Mol Catal A-Chem. 2008;296:54–60.
Neves CMB, Simões MMQ, Domíngues MRM, Santos ICMS, Neves MGPMS, Paz FAA, Silva AMS, Cavaleiro JAS. Oxidation of diclofenac catalyzed by manganese porphyrins: synthesis of novel diclofenac derivatives. RSC Adv. 2012;2:7427–38.
Simões MMQ, Neves CMB, Pires SMG, Neves MGPMS, Cavaleiro JAS. Mimicking P450 processes and the use of metalloporphyrins. Pure Appl Chem. 2013;85:1671–81.
Faria AL, Mac Leod TCO, Assis MD. Carbamazepine oxidation catalyzed by iron and manganese porphyrins supported on aminofunctionalized matrices. Catal Today. 2008;133–135:863–9.
CarvalhoDa-Silva D, Mac Leod TCO, Faria AL, Santos JS, Carvalho MEMD, Rebouças JS, Idemori YM, Assis MD. Carbamazepine oxidation catalyzed by manganese porphyrins: effects of the β-bromination of the macrocycle and the choice of oxidant. Appl Catal A-Gen. 2011;408:25–30.
Song R, Sorokin A, Bernadou J, Meunier B. Metalloporphyrin-catalyzed oxidation of 2-methylnaphthalene to vitamin K3 and 6-methyl-1,4-naphthoquinone by potassium monopersulfate in aqueous solution. J Org Chem. 1997;62:673–8.
Waldmann D, König T, Schreier P. Iron(III)porphinate/H2O2-mediated conversion of all-(E)-retinol. Z Naturforsch B. 1995;50:589–94.
Bloodsworth A, O’Donnell VB, Batinić-Haberle I, Chumley PH, Hurt JB, Day BJ, Crow JP, Freeman BA. Manganese-porphyrin reactions with lipids and lipoproteins. Free Radic Biol Med. 2000;28:1017–29.
Ferrer-Sueta G, Batinić-Haberle I, Spasojević I, Fridovich I, Radi R. Catalytic scavenging of peroxynitrite by isomeric Mn(III) N-methylpyridylporphyrins in the presence of reductants. Chem Res Toxicol. 1999;12:442–9.
Batinić-Haberle I, Tovmasyan A, Spasojević I. An educational overview of the chemistry, biochemistry and therapeutic aspects of Mn porphyrins—from superoxide dismutation to H2O2-driven pathways. Redox Biol. 2015;5:43–65.
Buettner GR, Wagner BA, Rodgers VG. Quantitative redox biology: an approach to understand the role of reactive species in defining the cellular redox environment. Cell Biochem Biophys. 2013;67:477–83.
Batinić-Haberle I, Cuzzocrea S, Rebouças JS, Ferrer-Sueta G, Mazzon E, Di Paola R, Radi R, Spasojević I, Benov L, Salvemini D. Pure MnTBAP selectively scavenges peroxynitrite over superoxide: comparison of pure and commercial MnTBAP samples to MnTE-2-PyP in two models of oxidative stress injury, an SOD-specific Escherichia coli model and carrageenan-induced pleurisy. Free Radic Biol Med. 2009;46:192–201.
Rajic Z, Tovmasyan A, Spasojević I, Sheng H, Lu M, Li AM, Gralla EB, Warner DS, Benov L, Batinić-Haberle I. A new SOD mimic, Mn(III) ortho N-butoxyethylpyridylporphyrin, combines superb potency and lipophilicity with low toxicity. Free Radic Biol Med. 2012;52:1828–34.
Jaramillo MC, Frye JB, Crapo JD, Briehl MM, Tome ME. Increased manganese superoxide dismutase expression or treatment with manganese porphyrin potentiates dexamethasone-induced apoptosis in lymphoma cells. Cancer Res. 2009;69:5450–7.
Jaramillo MC, Briehl MM, Crapo JD, Batinić-Haberle I, Tome ME. Manganese porphyrin, MnTE-2-PyP5+, acts as a pro-oxidant to potentiate glucocorticoid-induced apoptosis in lymphoma cells. Free Radic Biol Med. 2012;52:1272–84.
Lee K, Briehl MM, Mazar AP, Batinić-Haberle I, Rebouças JS, Glinsmann-Gibson B, Rimsza LM, Tome ME. The copper chelator ATN-224 induces peroxynitrite-dependent cell death in hematological malignancies. Free Radic Biol Med. 2013;60:157–67.
Jaramillo MC, Briehl MM, Batinić-Haberle I, Tome ME. Manganese (III) meso-tetrakis-N-ethylpyridinium-2-yl porphyrin acts as a pro-oxidant to inhibit electron transport chain proteins, modulate bioenergetics, and enhance the response to chemotherapy in lymphoma cells. Free Radic Biol Med. 2015;83:89–100.
Tse HM, Milton MJ, Piganelli JD. Mechanistic analysis of the immunomodulatory effects of a catalytic antioxidant on antigen-presenting cells: implication for their use in targeting oxidation-reduction reactions in innate immunity. Free Radic Biol Med. 2004;36:233–47.
Delmastro-Greenwood MM, Tse HM, Piganelli JD. Effects of metalloporphyrins on reducing inflammation and autoimmunity. Antioxid Redox Signal. 2014;20:2465–77.
Batinić-Haberle I, Spasojević I, Tse HM, Tovmasyan A, Rajic Z, St Clair DK, Vujaskovic Z, Dewhirst MW, Piganelli JD. Design of Mn porphyrins for treating oxidative stress injuries and their redox-based regulation of cellular transcriptional activities. Amino Acids. 2012;42:95–113.
Celic T, Španjol J, Bobinac M, Tovmasyan A, Vukelic I, Rebouças JS, Batinić-Haberle I, Bobinac D. Mn porphyrin-based SOD mimic, MnTnHex-2-PyP5+, and non-SOD mimic, MnTBAP3−, suppressed rat spinal cord ischemia/reperfusion injury via NF-κB pathways. Free Radic Res. 2014;48:1426–42.
Kos I, Benov L, Spasojević I, Rebouças JS, Batinić-Haberle I. High lipophilicity of meta Mn(III) N-alkylpyridylporphyrin-based superoxide dismutase mimics compensates for their lower antioxidant potency and makes them as effective as ortho analogues in protecting superoxide dismutase-deficient Escherichia coli. J Med Chem. 2009;52:7868–72.
Kos I, Rebouças JS, DeFreitas-Silva G, Salvemini D, Vujaskovic Z, Dewhirst MW, Spasojević I, Batinić-Haberle I. Lipophilicity of potent porphyrin-based antioxidants: comparison of ortho and meta isomers of Mn(III) N-alkylpyridylporphyrins. Free Radic Biol Med. 2009;47:72–8.
Spasojević I, Kos I, Benov LT, Rajic Z, Fels D, Dedeugd C, Ye X, Vujaskovic Z, Rebouças JS, Leong KW, Dewhirst MW, Batinić-Haberle I. Bioavailability of metalloporphyrin-based SOD mimics is greatly influenced by a single charge residing on a Mn site. Free Radic Res. 2011;45:188–200.
Rebouças JS, Spasojević I, Batinić-Haberle I. Pure manganese(III) 5,10,15,20-tetrakis(4-benzoic acid)porphyrin (MnTBAP) is not a superoxide dismutase mimic in aqueous systems: a case of structure-activity relationship as a watchdog mechanism in experimental therapeutics and biology. J Biol Inorg Chem. 2008;13:289–302.
Tovmasyan A, Carballal S, Ghazaryan R, Melikyan L, Weitner T, Maia CGC, Rebouças JS, Radi R, Spasojević I, Benov L, Batinić-Haberle I. Rational design of superoxide dismutase (SOD) mimics: the evaluation of the therapeutic potential of new cationic Mn porphyrins with linear and cyclic substituents. Inorg Chem. 2014;53:11467–83.
Lewis DFV. The P450 catalytic cycle. In: Lewis DFV, editor. Guide to cytochromes P450: structure and function. New York: Taylor & Francis; 2001. p. 49–68.
Spasojević I, Batinić-Haberle I. Manganese(III) complexes with porphyrins and related compounds as catalytic scavengers of superoxide. Inorg Chim Acta. 2001;317:230–42.
Lahaye D, Groves JT. Modeling the haloperoxidases: reversible oxygen atom transfer between bromide ion and an oxo-Mn(V) porphyrin. J Inorg Biochem. 2007;101:1786–97.
Lyons JE, Ellis Jr PE. Reactions of alkanes with dioxygen: toward suprabiotic systems. In: Sheldon RA, editor. Metalloporphyrins in catalytic oxidations. New York: Marcel Dekker; 1994. p. 297–324.
Levine RL, Mosoni L, Berlett BS, Stadtman ER. Methionine residues as endogenous antioxidants in proteins. Proc Natl Acad Sci U S A. 1996;93:15036–40.
Stadtman ER, Moskovitz J, Levine RL. Oxidation of methionine residues of proteins: biological consequences. Antioxid Redox Signal. 2003;5:577–82.
Requejo R, Hurd TR, Costa NJ, Murphy MP. Cysteine residues exposed on protein surfaces are the dominant intramitochondrial thiol and may protect against oxidative damage. FEBS J. 2010;277:1465–80.
Pamplona R, Costantini D. Molecular and structural antioxidant defenses against oxidative stress in animals. Am J Physiol Regul Integr Comp Physiol. 2011;301:R843–63.
Bender A, Hajieva P, Moosmann B. Adaptive antioxidant methionine accumulation in respiratory chain complexes explains the use of a deviant genetic code in mitochondria. Proc Natl Acad Sci U S A. 2008;105:16496–501.
Bueno-Janice JC, Tovmasyan A, Batinić-Haberle I. Comprehensive study of GPx activity of different classes of redox-active therapeutics—implications for their therapeutic actions. Free Radic Biol Med. 2015;87:S86–7.
Rebouças JS, Spasojević I, Batinić-Haberle I. Quality of potent Mn porphyrin-based SOD mimics and peroxynitrite scavengers for pre-clinical mechanistic/therapeutic purposes. J Pharm Biomed Anal. 2008;48:1046–9.
Rebouças JS, Kos I, Vujasković Z, Batinić-Haberle I. Determination of residual manganese in Mn porphyrin-based superoxide dismutase (SOD) and peroxynitrite reductase mimics. J Pharm Biomed Anal. 2009;50:1088–91.
Marques A, Marin M, Ruasse MF. Hydrogen peroxide oxidation of mustard-model sulfides catalyzed by iron and manganese tetraarylporphyrins. Oxygen transfer to sulfides versus H2O2 dismutation and catalyst breakdown. J Org Chem. 2001;66:7588–95.
Lahaye D, Muthukumaran K, Hung CH, Gryko D, Rebouças JS, Spasojević I, Batinić-Haberle I, Lindsey JS. Design and synthesis of manganese porphyrins with tailored lipophilicity: investigation of redox properties and superoxide dismutase activity. Bioorg Med Chem. 2007;15:7066–86.
Rosenthal RA, Huffman KD, Fisette LW, Damphousse CA, Callaway WB, Malfroy B, Doctrow SR. Orally available Mn porphyrins with superoxide dismutase and catalase activities. J Biol Inorg Chem. 2009;14:979–91.
Rausaria S, Ghaffari MM, Kamadulski A, Rodgers K, Bryant L, Chen Z, Doyle T, Shaw MJ, Salvemini D, Neumann WL. Retooling manganese(III) porphyrin-based peroxynitrite decomposition catalysts for selectivity and oral activity: a potential new strategy for treating chronic pain. J Med Chem. 2011;54:8658–69.
Rosenthal RA, Fish B, Hill RP, Huffman KD, Lazarova Z, Mahmood J, Medhora M, Molthen R, Moulder JE, Sonis ST, Tofilon PJ, Doctrow SR. Salen Mn complexes mitigate radiation injury in normal tissues. Anticancer Agents Med Chem. 2011;11:359–72.
Acknowledgments
The authors thank Drs. Ynara M. Idemori, Brian R. James, Artak Tovmasyan, Ludmil Benov, Margaret E. Tome, Dayse C. S. Martins, Gilson DeFreitas-Silva, and Shirley Nakagaki, for many helpful scientific discussions throughout these years. Financial support by The Brazilian Research Council (CNPq, Brazil), CAPES Foundation (Ministry of Education, Brazil), Financiadora de Estudos e Projetos (FINEP, Brazil), and the National Institutes of Health (NIH, USA) are greatly acknowledged.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Pinto, V.H.A., Falcão, N.K.S.M., Bueno-Janice, J.C., Spasojević, I., Batinić-Haberle, I., Rebouças, J.S. (2016). Cytochrome P450-Like Biomimetic Oxidation Catalysts Based on Mn Porphyrins as Redox Modulators. In: Batinić-Haberle, I., Rebouças, J., Spasojević, I. (eds) Redox-Active Therapeutics. Oxidative Stress in Applied Basic Research and Clinical Practice. Springer, Cham. https://doi.org/10.1007/978-3-319-30705-3_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-30705-3_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-30703-9
Online ISBN: 978-3-319-30705-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)