
Abstract
Ion channels importantly contribute to the function of endothelial cells. They serve as the major source of intracellular Ca2+, which, in turn, controls the production of endothelium-derived vasodilators, the permeability of the endothelium, gene expression, and other properties of endothelial cells. In addition, the activity of ion channels determines the membrane potential of endothelial cells that serves as an important signal for cell-cell communication between endothelial cells and between endothelial cells and overlying smooth muscle cells, and may feed-back to regulate the activity of the ion channels themselves. This review provides an overview of the expression and function of endothelial ion channels that contribute to Ca2+ and membrane potential signaling that is involved in the regulation and modulation of vasomotor tone of resistance arteries and arterioles. Channels discussed include inositol 1,4,5 trisphosphate receptors that mediate agonist-induced Ca2+ release from endoplasmic reticulum stores; members of the transient receptor potential family and other channels that mediate agonist-induced Ca2+ influx through the plasma membrane; Ca2+-activated K+ channels that mediate agonist-induced membrane hyperpolarization; and inward rectifier K+ channels that serve as sensors for changes in extracellular K+ and amplifiers of hyperpolarization induced by the activity of other ion channels. It is emphasized that all of these channels exist as members of macromolecular signaling complexes providing a rich environment for regulation of their activity and the function of endothelial cells in resistance arteries and arterioles.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Vascular endothelium
- Ion channels
- Endothelium-dependent vasodilatation
- Conducted dilatation
- Potassium channels
- Transient receptor potential channels
- Calcium ions
- Inositol 1,4,5 trisphosphate receptors
- Calcium waves
- Calcium sparklets
Introduction
Endothelial cells express a diverse array of ion channels in their plasma membranes and in the membranes of intracellular organelles that contribute to the function of these cells. These channels provide the major source of intracellular Ca2+ that serves as an important second messenger controlling the activity of Ca2+-dependent ion channels and cell membrane potential [40], endothelial cell production of NO, prostaglandins and epoxides of arachidonic acid (EETs) [40] and regulating barrier function of the endothelium [52, 90]. Intracellular Ca2+ is also an important signal controlling gene expression in [117, 130] and proliferation of [120, 123] endothelial cells. Ion channels also participate in cell volume regulation [70]. In addition, plasmalemmal ion channel activity importantly contributes to the membrane potential of endothelial cells that serves as a major signal for cell-cell communication between adjacent endothelial cells and as well as overlying smooth muscle cells due to the expression of homocellular and heterocellular gap junctions in the vascular wall [29]. Membrane potential may also feedback to affect Ca2+ influx through plasmalemmal Ca2+ permeable ion channels by influencing the electrochemical gradient for Ca2+ influx [65, 66, 69, 123], although this topic remains controversial [23, 34, 108, 113, 158]. Thus, ion channels importantly contribute to the function of endothelial cells in health and disease. This review will focus on the expression and function of endothelial ion channels involved in the regulation of vasomotor tone in resistance arteries and arterioles. Because there are considerable changes in ion channel expression and function during proliferation of cells in culture [11, 12, 20, 123, 135], emphasis will be placed on evidence from intact blood vessels and from freshly isolated endothelial cells from the peripheral circulation. The reader is also directed to a number of outstanding earlier reviews of ion channels in endothelial cells for access to earlier literature on this topic [123–125].
Setting the Stage
Most vasodilators that produce endothelium-dependent vasodilatation (see [79]) act on Gαq-protein-coupled receptors which are linked to phospholipase C (PLC)-β producing inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) from membrane phospholipids [13] (Fig. 1.1). The released IP3 activates IP3R in the membranes of the smooth endoplasmic reticulum, releasing stored Ca2+ and increasing cytosolic Ca2+ [13]. The activation of IP3R, loss of Ca2+ from intracellular Ca2+ stores and the DAG produced by PLC-β, activate plasma membrane Ca2+-permeable ion channels [51], allowing Ca2+ to diffuse down its electrochemical gradient into the cells, producing a steady-state increase in intracellular Ca2+ (Figs. 1.1 and 1.2). The sum of these two major events (the release of ER-stored Ca2+ via IP3R and the influx of Ca2+ via plasmalemmal ion channels) produces the well-described, agonist-induced cytosolic Ca2+ transient in endothelial cells (see Fig. 1.2). The increase in intracellular Ca2+ then activates plasmalemmal Ca2+-activated K+ channels to produce hyperpolarization of the endothelial cell membrane as shown in Fig. 1.2c, as well as activation of other Ca2+-dependent processes such as NO production and release of arachidonic acid from membrane phospholipids resulting in increased production of prostacyclin and epoxides of arachidonic acid [40] (Fig. 1.1). The remainder of this review will focus on the expression and function of ion channels responsible for agonist-induced Ca2+ signals and membrane hyperpolarization in endothelial cells as shown in Fig. 1.2, as well as other endothelial cell ion channels that appear important to the regulation of myogenic tone in resistance arteries and arterioles.

Endothelial cell ion channel and Ca2+ signaling overview. Shown is a schematic representation of an endothelial cell and the ion channels and transporters relevant to agonist-induced Ca2+ signaling. Agonists of Gαq-coupled receptors activate PLC-β producing IP3 and DAG . IP3 activates IP3R in the membrane of the endoplasmic reticulum (ER), releasing stored Ca2+ and raising cytosolic Ca2+ as shown. The released Ca2+ and Ca2+ entry through overlying plasma membrane Ca2+ permeable channels further stimulate Ca2+ release via Ca2+-induced-Ca2+-release. The elevated cytosolic Ca2+ will then activate plasma membrane KCa channels to produce membrane hyperpolarization, an important signal for cell-cell communication in resistance arteries and arterioles. This hyperpolarization also has the potential to increase the electrochemical gradient for diffusion of Ca2+ (and other cations) into the endothelial cell counter-acting the depolarizing effect of this cation influx. The DAG produced by the action of PLCs can activate TRPC3 and/or TRPC6 channels in the membrane, contributing to steady-state, agonist-induced Ca2+ influx into the cells. The DAG can also activate PKC, which phosphorylates TRPV4 channels increasing their activity, also contributing to Ca2+ influx. Loss of Ca2+ from the ER is sensed by STIM1, which clusters, interacts with and activates membrane ORAI1, TRPC1 and/or TRPC4 channels. The resultant Ca2+ influx contributes to steady-state, agonist-induced Ca2+ influx. The elevated cytosolic Ca2+ from these processes also activates nitric oxide synthase (eNOS) to stimulate NO production, and phospholipase A2 (PLA2) to produce arachidonic acid (AA) from membrane phospholipids. Arachidonic acid is then converted into vasodilator prostanoids, such as prostacyclin (PGI2), by cyclooxygenase (COX), and epoxides (EETs) by cytochromes P450 (CYP). EETs may contribute to activation of TRPV4. Upon removal of agonist, Ca2+ is pumped back into the ER by the smooth endoplasmic reticulum Ca2+ ATPase (SERCA) and extruded from the cell by the Na+/Ca2+ exchanger (NCX) and the plasma membrane Ca2+ ATPase (PMCA). Sodium that enters the cells via TRP channels is extruded by the Na+/K+ ATPase (NKA) and NCX. CAM calmodulin. PLB phospholamban

Methacholine-induced global Ca2+ transients in arteriolar endothelial cells . Panel a shows an image of an endothelial cell tube enzymatically isolated from a second order hamster cremaster arteriole as described [23]. Panel b shows a representative Ca2+ transient elicited by the muscarinic receptor agonist, methacholine (MCh) from an endothelial cell tube loaded with the ratiometric calcium indicator, Fura-2AM. Panel c shows the MCh-induced membrane hyperpolarization of an endothelial cell tube loaded with the potentiometric indicator, di-8-ANEPPs as described [23]. Panel d shows MCh-induced Ca2+ transients, as in a, under Control conditions, after brief exposure to solutions containing 0 mM Ca2+ demonstrating the loss of the plateau phase of the Ca2+ transient. After return to Ca2+-replete conditions (to obviate depletion of intracellular Ca2+), subsequent exposure to 0 mM Ca2+ and the IP3R-antagonist, xestospongin-D (Xsp-D; 10 μM) abolished the effects of MCh. Panel e shows inhibition of a MCh-induced Ca2+ transient by Xsp-D in the presence of extracellular Ca2+ (2 mM). Data shown in a–e are modified from [23]
What Endoplasmic Reticulum Ion Channels Mediate Agonist-Induced Ca2+ Signals?
Inositol-1,4,5-trisphosphate Receptors
Early studies demonstrated that agonists of Gαq-coupled receptors increased intracellular Ca2+ in endothelial cells that was due to an initial release of Ca2+ from internal stores followed by Ca2+ influx [18, 25, 59, 139, 142, 143] (Figs. 1.1 and 1.2). Pharmacological studies subsequently identified IP3Rs as the primary Ca2+ release channel responsible (see [23, 143] and Fig. 1.2 for examples). Inositol-1,4,5-trisphosphate receptors are large (350 kDa) tetrameric Ca2+ release channels found in the endoplasmic reticulum of all mammalian cells [47, 104]. Each monomer contains an IP3-binding domain that is located in cytoplasmic N-terminus of the proteins [47, 104]. Calcium appears to be the trigger for gating IP3 channels to open (see Table 1.1 for EC50 values for activation of the channels) [47, 104]. However, the response to increases in Ca2+ is biphasic, with higher concentrations of Ca2+ becoming inhibitory (Table 1.1) [47, 104]; Ca2+ activates the channels at low concentrations, but inhibits Ca2+ release at high concentrations (see Fig. 7 in [47] for examples). It has been proposed that the concentration of IP3 determines the affinity of IP3Rs for the inhibitory effects of elevated Ca2+; as the concentration of IP3 increases, higher levels of Ca2+ are required to inhibit the channels (Table 1.1) [47, 104]. Thus, the concentration of IP3 effectively determines the range of cytosolic Ca2+ concentration over which the IP3Rs will be active [47]. However, this has not been observed in all systems [171]. Nonetheless, in the presence of physiologically relevant concentrations of IP3, IP3R can undergo Ca2+-induced-Ca2+ release (CICR ) providing a positive feedback mechanism for release of Ca2+ from adjacent IP3R (as in the case of Ca2+ waves ), as well as amplification, with a limit, of Ca2+ signals originating from Ca2+ influx through overlying plasma membrane ion channels.
There are three isoforms of IP3R, IP3R1, 2 and 3 originating from three genes with modestly different characteristics (see Table 1.1); IP3R2 and 3 appear to have the highest sensitivity for Ca2+-induced activation with IP3R3 having the lowest sensitivity for IP3 [47, 104, 171]. In addition to IP3 and Ca2+, the activity of IP3Rs is also sensitive to cytoplasmic ATP concentrations [47, 104, 171]. In IP3R1 and 3, ATP produces a leftward shift in the Ca2+-activity relationship increasing the affinity of the channels for Ca2+, with little effect on the maximal open-state probability [47, 171]. In distinct contrast, ATP has no effect on Ca2+-sensitivity of IP3R2 receptors, but has large effects on the maximum open-state probability of the channels; low ATP severely reduces the maximal activity of IP3R2 [104, 171]. Given that IP3Rs exist in signaling microdomains adjacent to ATPases in the ER and in the plasma membrane, local ATP concentrations could have a profound effect on IP3R function in an isoform-specific fashion. Finally, IP3R interact with a large number of proteins in the cytosol and in the lumen of the ER that can modulate the activity of these channels by protein-protein interactions, and phosphorylation/dephosphorylation in the case of protein kinases and phosphatases [47, 159].
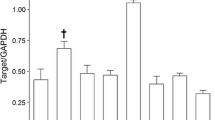
Freshly isolated rat aortic endothelial cells express transcripts for all three isoforms of IP3R with IP3R1 being most highly expressed [118, 119]. Endothelial cells from mouse mesenteric arteries express transcripts for all three isoforms with IP3R2 being most highly expressed [93]. Mouse cremaster arteriolar endothelial cells also express transcripts for all three isoforms, but IP3R3 appears to be most highly expressed (Fig. 1.4a), and protein expression for all three isoforms has been reported [77]. All three IP3R isoforms were detected in the endothelium of Wistar rat basilar and mesenteric arteries by immunocytochemistry [56]. Thus, it appears that there may be regional or species-dependent differences in the expression of IP3R isoforms, and little is known about the localization or function of the individual IP3R isoforms in native endothelial cells.

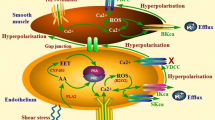
Endothelial cell Ion channels and cell-cell communication in the vessel wall. Shown is a schematic representation of a longitudinal cross section through two endothelial cells and their relationship to overlying smooth muscle cells. Endothelial cells communicate with overlying smooth muscle cells via myoendothelial projections (MEPs ) , that pass through the internal elastic lamina to make contact with overlying smooth muscle cells, as shown. Gap junctions may form at MEPs to yield myoendothelial junctions (MEJs ) allowing endothelial cell hyperpolarization to be conducted to the smooth muscle cells, closing smooth muscle voltage-gated Ca2+ channels (VGCCs) and leading to vasodilatation. Ion channels such as TRPV4, KCa3.1 and IP3R (as shown) may cluster in MEPs to form signaling complexes to direct the endothelial cell responses to vasodilator agonists. Other ion channels such as TRPC3 and KCa2.3 may cluster elsewhere to form other signaling complexes. Abbreviations are as in Fig. 1.1

Expression of transcripts for IP3R and RyR in freshly isolated mouse cremaster arteriolar endothelial cells. Shown are means ± SE (n = 5 cell isolates for IP3R and n = 7 for RyR) abundance of transcripts for IP3R (panel a) and RyR (panel b) isoforms relative to eNOS in endothelial cell tubes isolated from second-order mouse cremaster arterioles. See [23, 178] for methodological details
In co-cultures of smooth muscle and endothelial cells from mouse cremaster arterioles , and in intact vessels, it has been reported that IP3R1 localizes at sites of myoendothelial gap junctions (MEJs ) [77]. Similarly, in intact mouse mesenteric resistance vessels , there are clusters of IP3Rs near holes in the internal elastic lamina [93], sites that have been correlated with projections of endothelial cells (myoendothelial projections, MEPs , Fig. 1.3) towards overlying smooth muscle cells and the localization of MEJs [136]. However, the IP3R isoform expressed in these clusters was not identified. Importantly, these sites were shown to generate localized endothelial cell Ca2+ events that have been termed Ca2+ pulsars [93]. These events are too small and rapid to be detected by global Ca2+ measurements made with Fura 2 (Fig. 1.2, for example), but can be detected using Fluo-4 or genetic Ca2+ sensors such as GCaMP2 and high-speed confocal imaging [83, 93]. Previous studies have shown that KCa3.1 channels also are clustered in the same microdomain [137] providing a means for Ca2+ pulsars to be translated into, for example, changes in membrane potential (see below for more on this topic). In addition, a growing list of proteins congregate in the vicinity of MEJs including TRPA1 channels [37], TRPV4 channels [149, 150], anchoring proteins (e.g., AKAP150 [150]), protein kinases (e.g., PKC [150]), nitric oxide synthase [151], Na+/K+ ATPase [33] and other proteins [152].
Calcium pulsars [83, 93] and Ca2+ waves [36, 83] are present under resting conditions in endothelial cells of pressurized vessels [83, 93] and likely contribute to the resting activity of KCa3.1 channels (in the case of pulsars and waves) and KCa2.3 channels (Ca2+ waves) and endothelial cell membrane potential. Endothelium-dependent vasodilators , such as acetylcholine [93, 147] or adenosine [36], increase the number and frequency of Ca2+ pulsars [93], and also recruit IP3R located throughout endothelial cells to produce asynchronous [93, 147] or synchronous [36, 147] Ca2+ waves and increases in global Ca2+ (Fig. 1.2). Thus, the global Ca2+ signals that have been reported in native microvascular endothelial cells [23, 32, 107, 148] represent a complex mixture of local Ca2+ pulsars and Ca2+ waves in addition to more homogeneous increases in cytosolic Ca2+. Additional studies are needed to define the precise localization of IP3R isoforms and their function related to endothelium-dependent vasomotor activity.
Ryanodine Receptors
Ryanodine receptors (RyR) are composed of very large protein subunits (~500 kDa) that form Ca2+-sensitive-Ca2+-release channels in the endoplasmic reticulum [61]. Similar to IP3R, they are tetramers with three isoforms from three distinct genes: RyR1, RyR2 and RyR3 [61, 62, 185]. Skeletal muscle expresses predominantly RyR1, the heart expresses predominantly RyR2 and RyR3 is expressed in the brain and other tissues [61, 62, 185]. Vascular smooth muscle cells may express all three isoforms, with RyR2 being predominant in resistance artery [166] and arteriolar smooth muscle cells [178]. Immunofluorescence demonstrated RyR expression in guinea pig endocardium and aortic endothelial cells [95], and ryanodine binding sites have been reported in porcine coronary artery endothelial cells [54]. Studies of cultured endothelial cells suggest expression of functional ryanodine receptors [191], and freshly isolated endothelial cells from rabbit aorta display caffeine-induced Ca2+ transients implying the presence of RyR in these cells [132]. In porcine coronary artery endothelial cells, caffeine elicits a Ca2+ transient in only 37 % cells studied suggesting heterogeneity of the distribution and function of endothelial RyR [55]. Transcripts for RyR3, but not RyR1 or RyR2 have been reported in endothelial cells freshly isolated from human mesenteric arteries [89]. Thus, RyR appear to be expressed and functional in macrovascular endothelial cells . However, there is little evidence for expression of ryanodine receptors in microvascular endothelial cells. In mouse mesenteric resistance arteries , where expression of IP3Rs are readily detected, no message for the three RyR isoforms were found, and ryanodine had no effect on basal or acetylcholine-stimulated Ca2+ events [93]. We have also found lack of expression of the three RyR isoforms in endothelial cells from mouse cremaster arterioles (Fig. 1.4b), and caffeine does not elicit a Ca2+ transient in freshly isolated hamster cremaster arteriolar endothelial cells [23] (Fig. 1.5a), although lack of effect of caffeine on global Ca2+ levels does not completely exclude a role for RyR [126]. A lack of effect of the RyR antagonist, ryanodine, on Ca2+ signals in endothelial cells in rat mesenteric arteries also has been observed [83]. Although species and regional heterogeneity in the expression of RyR cannot be excluded, these data suggest that RyR do not play a major role in Ca2+ signaling in endothelial cells of resistance arteries and arterioles.

Failure of caffeine to elicit Ca2+ transients in arteriolar endothelial cells but not smooth muscle cells. Shown are representative responses of an endothelial cell tube (panel a) and a smooth muscle cell (Panel b) isolated by the same method from hamster cremaster arterioles [23], to the RyR agonist, caffeine (10 mM). Consistent with the lack of expression of RyR in arteriolar endothelial cells (see Fig. 1.4b), caffeine failed to elicit a Ca2+ transient above baseline in endothelial cells in endothelial cells (n = 5 isolates from five arterioles), but produced the expected response from smooth muscle cells isolated from the same vessels
What Ion Channels Mediate Agonist-Induced Ca2+ Influx?
TRP Channels
Endothelium-dependent vasodilators not only increase the activity of IP3R, by stimulating the production of IP3, they also result in the activation of ion channels in the plasmalemma of endothelial cells that conduct Ca2+ and are responsible for steady-state increases in intracellular Ca2+ (i.e., the plateau phase of the Ca2+ transient shown in Fig. 1.2). Early studies in cultured endothelial cells provided evidence that agonist-induced Ca2+ entry was electrophysiologically and pharmacologically similar to the Ca2+ entry induced by depletion of intracellular Ca2+ stores [139, 142, 143, 164, 165]. In primary cultures of porcine coronary artery endothelial cells, substance P activates a non-selective, inward whole-cell cation current that can be completely inhibited by blocking IP3-dependent activation of IP3R with heparin [143]. Similarly, block of IP3R with xestospongin-D abolishes methacholine-induced global Ca2+ transients in arteriolar endothelial cells [23] (Fig. 1.2e). These data suggest that, at least under the conditions of these experiments, agonist-induced activation of IP3R , and likely release of Ca2+ from internal stores is required to activate the Ca2+ influx pathway that is responsible for the plateau phase of the agonist-induced global Ca2+ transients, and hence the steady-state phase of agonist-induced endothelial cell hyperpolarization. This does not exclude the activation of receptor, or second-messenger activated Ca2+ influx as the currents activated may be too small to detect by conventional whole-cell methods, or may produce only local changes in Ca2+ that do not influence global Ca2+, particularly as detected by Fura-2.
The ion channels that are responsible for agonist-induced Ca2+ influx in native endothelial cells remain in question. Several members of the transient receptor potential (TRP) family of ion channels, in particular TRPC1, TRPC3, TRPC4, TRPC6 and TRPV4, along with members of the stromal interaction molecule (STIM) and ORAI families appear to be likely candidates, and it is also likely that multiple channels are activated and contribute to the Ca2+ influx activated by endothelium-dependent vasodilators (Fig. 1.1).
The TRP channel family form, in general, cation channels that are weakly Ca2+ selective (permeability for Ca2+/permeability for Na2+ <10) [179]. The channel monomers are assumed to have six membrane spanning domains with the pore between segments 5 and 6, and both the C- and N-termini of the channels located intracellularly, with four monomers forming a functional channel [179].
Endothelial cells express TRPC1 , which may serve as store-operated Ca2+ channels in endothelial cells [124]. However, their function in agonist-induced endothelial hyperpolarization and vasodilatation remains unclear. This may partly be due to the observation that TRPC1 heteromultimerize with other members of the TRPC family (especially TRPC4) as well as STIM/ORAI containing channels [30]. Studies of cultured human pulmonary artery endothelial cells revealed expression of TRPC1, and that antisense oligonucleotide knockdown reduced Ca2+ influx induced by Ca2+ store depletion by about 50 % suggesting that TRPC1 contributes to the Ca2+ influx pathway in these cells [17]. A similar conclusion was drawn in cultured bovine aortic endothelial cells using a TRPC1 antibody to inhibit basic fibroblast growth factor-induced Ca2+ entry [7]. Studies of human and mouse pulmonary microvascular endothelial cells also support a role for TRPC1 in agonist- and Ca2+ store depletion-induced Ca2+ entry and microvascular permeability [5, 92, 114, 155, 156]. However, the roles played by TRPC1 in agonist-induced endothelial cell hyperpolarization and vasodilatation is not as clear. Study of carotid artery endothelial cells from TRPC1 knock-out mice reveal enhanced acetylcholine-induced hyperpolarization, rather than decreased responses predicted based on studies of cultured cells and lung models [140]. Although compensatory upregulation of expression of other channels might explain these results, prior experiments failed to detect upregulation of other TRPC channels [31]. At the least, these results indicate that TRPC1 is not essential for agonist-induce endothelial cell hyperpolarization and that additional channels contribute to the Ca2+ influx induced by agonists in native endothelial cells, although regional heterogeneity in expression and function cannot be excluded. In contrast, studies of native aortic endothelial cells from TRPC1−/− mice demonstrated a small reduction of endothelial cell Ca2+ transients induced by acetylcholine [87], suggesting that TRPC1 channels do participate, to a small extent, in agonist-induced Ca2+ signals. A small reduction in thrombin-induced Ca2+ transients was also observed after siRNA knock down of TRPC1 in cultured pulmonary microvascular endothelial cells [155]. Taken together, these data do not support a major role for TRPC1 in agonist-induced Ca2+ entry into endothelial cells relevant to endothelial cell hyperpolarization and vasodilatation.
TRPC4 is another channel that has been implicated in agonist and store-depletion-induced Ca2+ entry into endothelial cells [48, 155, 162]. In cultured aortic endothelial cells isolated from TRPC4−/− mice, the plateau phase of agonist-induced Ca2+ transients and related endothelial cell hyperpolarization was substantially depressed (but not eliminated) [48], suggesting a major role for TRPC4 in agonist-induced Ca2+ transients in macrovascular endothelial cells. Similarly, use of cultured, pulmonary microvascular endothelial cells isolated from TRPC4−/− mice, as well as siRNA knock down of endogenous TRPC4 from cells isolated from wild-type mice demonstrated a major role for TRPC4 in thrombin- and Ca2+-store-depletion-induced Ca2+ signals [155]. The authors also demonstrated that expression of STIM1 was necessary for normal store-operated Ca2+ entry and showed that STIM1 and TRPC4 interacted and that expression of both proteins was required for normal Ca2+ signaling [155]. Endothelium-dependent vasodilatation is also substantially reduced in vessels from TRPC4−/− mice [48]. These data support a role for TRPC4 as a Ca2+ influx pathway during agonist-induced Ca2+ signaling, with activity triggered by loss of Ca2+ from internal stores as sensed by STIM1 (Fig. 1.1).
In human umbilical vein endothelial cells , the Ca2+ influx induced by agonists or depletion of intracellular stores depends on expression of STIM1 and ORAI1 with STIM1 serving as the sensor of ER Ca2+ and ORAI1 forming the pore of the store-operated channels [1, 155, 190]. Furthermore, in contrast to the studies outlined above, it was shown that effective knock down of TRPC1 or TRPC4 had no effect on Ca2+ store depletion-induced Ca2+ signals in this model [1]. However, studies in murine pulmonary microvascular endothelial cells indicate that expression of ORAI1 is not required for normal endothelial cell Ca2+ signaling [155]. These data suggest that there may be regional or species-dependent differences in the ion channels responsible for agonist-induced Ca2+ signaling in endothelial cells. The role played by STIM1 and ORAI1 in agonist-induced endothelial cell hyperpolarization of endothelial cells in resistance arteries and arterioles has not been reported.
TRPV4 channels also participate in agonist- and shear-stress-induced Ca2+ signals in endothelial cells, and endothelium-dependent vasodilatation in intact vessels [8, 35, 39, 109, 149, 150, 187]. These channels are temperature sensitive [175], stretch-sensitive [170] and can be activated by EETs (Fig. 1.1) and phorbol ester derivatives [170]. Calmodulin interacts with the C-terminal domain and mediates Ca2+-dependent activation of these channels [153]. Activation of TRPV4 channels in endothelial cells increases intracellular Ca2+ [109, 149, 150, 174, 187] and produces vasodilatation [39, 134, 149, 150, 187]. Importantly, the plateau-phase of vasodilator agonist-induced endothelial cell Ca2+ transients are reduced in the endothelium of vessels from TRPV4−/− mice [149, 150, 187]. In mouse mesenteric arteries, TRPV4 channels cluster in the same microdomain as IP3R at MEPs (Fig. 1.3), and endothelium-dependent agonists activate these channels to produce TRPV4-Ca2+ sparklets at the sites of MEPs [149, 150]. Activation of Ca2+ influx through only a few TRPV4 channels per endothelial cell activates endothelial KCa2.3 and KCa3.1 channels and can produce maximal vasodilatation, with activation of KCa3.1 channels, which also cluster at MEJ’s, occurring preferentially at low levels of TRPV4 activation [149]. Block of TRPV4 channels with HC-067047 in mouse mesenteric arteries substantially inhibits the component of acetylcholine-induced vasodilatation that is mediated by activation of endothelial KCa2.3 and KCa3.1 channels, suggesting that TRPV4 channels play a major role in agonist-induced Ca2+ influx that contributes to endothelial cell hyperpolarization and subsequent vasodilatation [149]. In mouse mesenteric arteries, muscarinic receptor agonists activate TRPV4 channels through a signaling pathway involving PLC-β and activation of PKC that is targeted to the channel by the scaffolding protein, AKAP150 [150] (Fig. 1.1). Thus, TRPV4 appears to play a major role in agonist-induced endothelial cell Ca2+ signaling that is related to hyperpolarization and vasodilatation.
Evidence also has been presented suggesting that TRPC3 is involved in agonist-induced Ca2+ signaling in endothelial cells [75, 86, 87, 98, 141, 184]. These TRP channels can be activated by DAG formed through the action of PLCs on membrane phospholipids [71] (Fig. 1.1). In cerebral vascular smooth muscle, activation of IP3R1, independent from release of Ca2+, results in activation of TRPC3 [4]. It is not known if a similar interaction occurs in endothelial cells. TRPC3 is involved in flow- and bradykinin-induced vasodilatation in rat small mesenteric arteries, but not dilation induced by histamine, ATP or cyclopiazonic acid [98]. As with TRPV4, TRPC3 appears to cluster near MEPs in rat mesenteric resistance artery endothelial cells [141]. In rat mesenteric arteries, the TRPC3 blocker, Pyr3, inhibited endothelial cell hyperpolarization and the portion of acetylcholine-induced endothelium-dependent vasodilatation mediated by activation of KCa2.3 and KCa3.1 channels and the consequent endothelial cell hyperpolarization [141]. These data suggest a close physical and functional relationship between TRPC3 and the KCa channels that mediate agonist-induced endothelial cell hyperpolarization. In contrast, in porcine coronary arteries, the TRPC3 antagonist Pyr3 inhibited the portion of bradykinin-induced vasodilatation that is mediated by NO, NO production in isolated endothelial cells and bradykinin-induced endothelial cell Ca2+ transients suggesting that TRPC3 channels contributed to Ca2+ signaling directed at NO production in this system [75]. In murine cerebral arteries, ATP-induced endothelial cell Ca2+ transients are reduced by Pyr3 or in cells isolated from TRPC3-/- mice, and Ca2+ entry through TRPC3 appears to selectively activate KCa2.3 channels during the plateau-phase of agonist-induced endothelial cell hyperpolarization [86]. Thus, there appear to be regional and likely species-dependent differences in the roles played by TRPC3 in endothelial cell Ca2+ signaling and hyperpolarization. This likely represents differences in the localization of the channels and the signaling microdomains in which they are expressed.
Endothelial cells in cerebral arteries also express TRPA1 channels that, when activated, produce endothelial cell hyperpolarization and endothelium-dependent vasodilatation [37, 154]; the endothelium of mouse, rat or human coronary, renal or mesenteric arteries do not express transcripts or protein for TRPA1 [154]. The TRPA1 subunits have 14–18 ankyrin repeats at their amino-terminal that give the channels their name, are activated by a diverse array of chemicals including pungent substances found in mustards and garlic and are heavily expressed in sensory nerves [9]. In endothelial cells of cerebral arteries, these channels cluster at MEPs and co-localize with KCa3.1 channels [37, 38, 154] (Fig. 1.3). Activation of TRPA1 leads to TRPA1-Ca2+ sparklets [154]. Vasodilatation induced by activation of TRPA1 channels with allyl isothiocyanate (AITC) in substantially inhibited by the KCa3.1 blocker, TRAM34 and abolished by the combination of TRAM34 and the KCa2.3 blocker, apamin, but is unaffected by blockade of nitric oxide synthase or cyclooxygenase [37, 154]. These data indicate that Ca2+ influx through TRPA1 primarily activates endothelial cell KCa channels to produce vasodilatation. Furthermore, as with TRPV4 channels [149], only a small number of active TRPA1 channel clusters per endothelial cell are required for maximal vasodilatation [154]. It was also shown that vasodilatation induced by AITC could be inhibited by Ba2+ suggesting that inward rectifier K+ channels either amplify the hyperpolarization induced by activation of endothelial cell KCa channels, or transduce the endothelial cell KCa channel activation by detecting the K+ released through the KCa channels [37] (Fig. 1.1). In the endothelium of cerebral arteries, TRPA1 co-localizes with NADPH oxidase (NOX) isoform 2, and lipid peroxides produced by NOX2 activate these channels to produce vasodilatation [154].
Expression and function of TRPV3 channels in rat cerebral artery endothelial cells also has been reported [38]. These channels appear to be more uniformly expressed than TRPV4 or TRPA1 [38]. Activation of TRPV3 with agents such as the oregano monoterpenoid phenol, carvacrol, increases endothelial cell Ca2+ and activates endothelial cell KCa channels to produce endothelial cell hyperpolarization and vasodilatation [38].
Finally, endothelial cells also express TRPC6 that appears to contribute to Ca2+ signaling [21, 60, 100, 127, 145]. As for TRPC3, TRPC6 is activated by DAG produced simultaneously with the formation of IP3 by PLCs [71] (Fig. 1.1 and Table 1.1). Human pulmonary artery endothelial cells express TRPC6, and siRNA knockdown of this channel impairs Ca2+ signaling and increases monolayer permeability induced by thrombin [145]. Human dermal microvascular endothelial cells in culture express TRPC6, but not TRPC3, and the plateau phase of the histamine-induced increase in Ca2+ is reduced by SKF96365 in these cells. Furthermore, histamine-induced increases in microvascular permeability are abolished in TRPC6−/− mice supporting a role for this channel in regulation of microvascular Ca2+ signaling and permeability [21]. Increased microvascular permeability induced by vascular endothelial growth factor (VEGF) appears to be mediated by TRPC6 [127], and VEGF-induced Ca2+ transients are reduced in cultured human microvascular endothelial cells expressing a dominant-negative form of TRPC6 [60]. Mouse aortic endothelial cells express TRPC6 and TRPC3, but carbachol-induced Ca2+ transients in isolated endothelial cells as well as carbachol-induced endothelium-dependent relaxation of aortas are reduced only in vessels isolated from TRPC6−/− mice [100]. The role of TRPC6 in agonist-induced Ca2+ signaling related to vasodilatation of arterioles and resistance arteries remains to be established.
Taken together, these data suggest that it is likely that there are multiple ion channels in the plasma membrane of endothelial cells that conduct Ca2+ into the cells to activate endothelial cell KCa channels (and other processes) to produce endothelial cell hyperpolarization (Fig. 1.1). Agonists that activate Gαq-coupled receptors appear to activate multiple channels including TRPC3, TRPC4, TRPV4 and possibly TRPC6 (Fig. 1.1). However, the roles played by each of these channels, together, in concert, remains to be established. Regional and species dependent heterogeneity in expression and function of these channels cloud our view of the larger picture. The use of isolated, intact blood vessels using multiple approaches (as for studies of TRPV4 [149, 150]) appears to be a step in the right direction in solving this puzzle.
Voltage-Gated Ca2+ Channels
The expression of voltage-gated Ca+ channels (VGCC ) in endothelial cells is controversial. Capillary endothelial cells from bovine adrenal medullas were reported to express both L-type and T-type VGCC as assessed by patch clamp techniques [167]. However, the function of these channels was not defined. More recently, mouse and rat pulmonary microvascular endothelial cells have been shown to express CaV3.1-based T-type VGCC that appear to be involved in thrombin-induced Ca2+ signals, cell adhesion, exocytosis of von Willebrand’s Factor and depolarization-induced Ca2+ signaling [177, 180, 189]. Endothelial cells of mesenteric arterioles label with antibodies for CaV3.2, but not CaV1.2 or CaV3.1 and these channels were proposed to participate in conducted vasomotor responses [16]. Immunofluorescence identified CaV3.1 and CaV3.2 in endothelial cells in rat middle cerebral artery and its branches, although their function was not studied [91]. In mouse mesenteric arteries, CaV3.1 has been detected in the endothelium by immunofluorescence, was colocalized with eNOS and appeared to play a role in depolarization-induced NO synthesis based on studies employing CaV3.1 knockout mice [157]. In mouse cremaster muscle arterioles, CaV3.2 has been implicated in electrically-induced, conducted vasodilatation based on the pharmacology of this response [45]. However, the location of the channel was not established. Thus, there appears to be some evidence for expression of T-type channels in endothelial cells of resistance arteries and arterioles. However, their electrophysiological function has not been adequately explored in other than cultured cells, particularly in the peripheral microcirculation. Whether hyperpolarization of endothelial cells induced by vasodilators can activate current through T-type channels by reducing voltage-dependent inactivation and recruiting a window-current has not been established. Depolarization of freshly isolated hamster arteriolar endothelial cells does not, in and of itself, elicit a global change in intracellular Ca2+ [23]. These data do not support a major role for VGCC in these endothelial cells.
Which Ion Channels Mediate Agonist-Induced Endothelial Cell Hyperpolarization?
Early studies in endothelial cells from conduit arteries and in cultured cells showed that agonists of Gαq-protein-coupled receptors resulted in Ca2+ signals in endothelial cells [18, 25, 142], activation of endothelial cell Ca2+-activated K+ channels [18, 24, 25, 53, 142] and endothelial cell hyperpolarization [18, 142].
KCa2.3 and KCa3.1 Channels
Native endothelial cells in resistance arteries primarily express two types of KCa channels: KCa2.3, the small-conductance, KCa (SKCa) channel and KCa3.1, the intermediate-conductance KCa (IKCa) channel [41, 57, 88, 137, 144, 160]. The channels are the products of distinct genes (KCNN3 and KCNN4, respectively [176]) (Table 1.1). Both channels are voltage insensitive and use calmodulin as the Ca2+ sensor, which interacts with the intracellular C-terminus of both channels to gate channel opening [44, 182]. The concentration of free Ca2+ required for 50 % of maximal activation of both KCa2.3 [182] and KCa3.1 [78] is on the order of 300 nM, with the threshold for activity at approximately 100 nM and maximal activity at 1 μM [78, 182]. These channels display distinct pharmacology that has aided in elucidating their function in intact vessels (Table 1.1).
Both KCa2.3 and KCa3.1 can contribute to endothelial cell hyperpolarization induced by agonists of Gαq-protein coupled receptors , such as acetylcholine acting at M3-muscarinic receptors: for example, in guinea-pig carotid artery [26], rat mesenteric arteries preconstricted with phenylephrine [27] and porcine coronary arteries [19], inhibition of agonist-induced hyperpolarization requires block of both KCa2.3 and KCa3.1. However, in several systems, agonist-induced hyperpolarization of endothelial cells appears to be mediated by KCa3.1 alone. In rat middle cerebral arteries , block of KCa3.1 alone abolishes endothelial cell hyperpolarization and subsequent vasodilatation induced by UTP [108]. Similarly, in mouse carotid arteries and cremaster muscle microcirculation , KCa3.1 appear to dominate acetylcholine-induced vasodilatation based on studies using knockouts of KCa2.3 or KCa3.1 [15].
The distribution of KCa2.3 and KCa3.1 in the plasma membrane of endothelial cells is neither random nor uniform. In the endothelium of mouse [93] and rat [137] mesenteric resistance arteries and rat cerebral arteries [37], KCa3.1 appear to cluster at MEPs (Fig. 1.3), with some of these MEPs containing gap junction proteins (Connexin 40 and 37) and forming gap junctions with overlying vascular smooth muscle cells (MEJs ) (see [136] and references therein). At MEPs and MEJ’s, KCa3.1 likely exist in macromolecular signaling complexes with IP3Rs [93], TRPA1 channels [37], TRPV4 channels [149, 150], anchoring proteins (e.g., AKAP150 [150]), protein kinases (e.g., PKC [150]) and likely G-protein coupled receptors [150]. This localization appears to facilitate activation of KCa3.1 channels by local Ca2+ signals [37, 93, 149, 150] and transmission of KCa3.1-mediated hyperpolarization from endothelial cells to overlying smooth muscle cells via MEJs (Fig. 1.3). In addition, it has been proposed that activation of KCa3.1 channels by Ca2+ influx through TRPV4 channels has a positive-feedback effect on Ca2+ influx through TRPV4 [128] (Fig. 1.1).
In contrast, KCa2.3 channels appear to localize on the periphery of endothelial cells [137]. These channels also likely exist in signaling microdomains. In rat mesenteric arteries KCa2.3 localizes in cholesterol-rich areas (caveolae or lipid rafts) and colocalizes with caveolin-1 [2]. In rat cerebral arteries, Ca2+ influx through TRPC3 channels selectively activates KCa2.3 [86], suggesting co-localization of the two channels. The localization of KCa2.3 in caveolae adjacent to gap junction plaques between endothelial cells also likely explains why shear-stress-induced dilation is severely attenuated in carotid arteries from mice with conditional knockout of KCa2.3 [15]. The differential cellular distribution of KCa2.3 and KCa3.1 and their respective signaling microdomains likely explain how these channels contribute to different aspects of agonist-induced endothelial cell hyperpolarization [27, 144].
In addition to agonist-mediated events , endothelial cell KCa2.3 and KCa3.1 appear to be active at rest contributing to endothelial cell membrane potential and the regulation of myogenic tone particularly in small resistance arteries and arterioles. For example, overexpression of KCa2.3 hyperpolarizes mouse mesenteric artery endothelial cells and reduces myogenic tone, whereas conditional knockout depolarizes the cells and increases tone [160]. Similarly, blockade of either KCa2.3 or KCa3.1, or both channels increases myogenic tone in rat cerebral parenchymal arterioles [22, 63]. It is noteworthy that in upstream middle cerebral arteries, under similar conditions, inhibition of KCa2.3 and KCa3.1 had a smaller effect on myogenic tone than was observed in downstream parenchymal arterioles, despite their importance in agonist-induced vasodilatation [22]. These data suggest that there are regional differences in the roles played by these channels with increased function in the microcirculation.
KCa1.1 Channels
The expression and function of Large-conductance Ca2+-activated K+ (KCa1.1) channels in endothelial cells remains controversial [135]. These voltage and Ca2+-sensitive K+ channels have a much larger conductance (~250 pS) than KCa2.3 or KCa3.1, are intrinsically sensitive to Ca2+ (i.e., they do not require association with calmodulin), and display distinct pharmacology [73] (Table 1.1). These channels are highly expressed in vascular smooth muscle cells [80, 122]. Studies of cultured endothelial cells have repeatedly demonstrated expression and function of KCa1.1 channels (see [135] for references). However, the function and expression of KCa1.1 in native and freshly isolated endothelial cells is not clear. For example, in patch-clamp studies of freshly isolated endothelial cells from bovine coronary arteries [50], mouse carotid arteries [15], and rat cerebral parenchymal arterioles [63] no currents characteristic of KCa1.1 are detected and essentially all Ca2+-activated currents are inhibited by the combination of a KCa2.3 and a KCa3.1 blocker. However, currents through KCa1.1 have been recorded in endothelial cells isolated from rats exposed to chronic hypoxia [76, 131] or from endothelial cells acutely exposed to β-methylcyclodextrin to deplete membrane cholesterol [131], effects that could be reversed by exposure of cells to a membrane permeant version of caveolin-1 [131]. These data are consistent with an earlier study in cultured endothelial cells indicating that KCa1.1 are targeted to caveolae and that caveolin-1 inhibited the function of KCa1.1 in that microdomain [173]. These data suggest that, in some endothelial cells, KCa1.1 channels may be silent under normal conditions, but can be upregulated by conditions, such as chronic hypoxia, that disrupt membrane microdomains. This idea is consistent with the hypothesis that KCa1.1 expression and function in endothelial cells only occurs during stress or pathological conditions [135]. Immunohistochemical localization of KCa1.1 has been reported in rat cremaster arterioles [163]. There must be regional or species differences in expression of KCa1.1 because no transcripts or protein for KCa1.1 were detected in bovine coronary artery endothelial cells [50] excluding the possibility that there are silent KCa1.1 present in these cells. We also have not detected transcripts for KCa1.1 channels in endothelial cells isolated from mouse cremaster arterioles or upstream feed arteries (n = 5 endothelial cell isolates from five mice, Jackson, unpublished observations). Additional studies where message, protein expression, protein localization and electrophysiology are performed will be required to resolve this issue.
KIR2.X Channels
Endothelial cells also express inward rectifier K+ (KIR) channels [123]. In resistance arteries and arterioles , endothelial cells appear to express two to four isoforms of KIR channels: strong inward rectifiers, KIR2.1, KIR2.2, and KIR2.3 (Table 1.1) and weak inward rectifiers KIR6.1 and KIR6.2 that form ATP-sensitive K+ (KATP) channels. The function of KATP channels will not be addressed. In general, KIR channels are tetramers of KIR subunits, with each subunit having two membrane-spanning domains with a pore-forming loop in between [68]. As shown in Fig. 1.6, at potentials more negative than the K+ equilibrium potential, KIR channels have a higher conductance and pass current in the inward direction, whereas at more positive potentials, channel conductance is reduced. This inward-rectification gives KIR channels their name and results from voltage-dependent block of the channel pore by intracellular Mg2+ and polyamines [68]. The physiological function of KIR channels is predominantly mediated by the outward “hump” in the current-voltage relationship (Fig. 1.6). Over membrane potentials that accompany this portion of the current-voltage relationship for KIR, outward currents through these channels will tend to hyperpolarize cells, driving the membrane potential towards the K+ equilibrium potential. It is this characteristic of the strong inward rectifiers like KIR2.1 and 2.2 that sets the resting membrane potential of cardiac muscle, skeletal muscle and nerves to be quite negative and near the K+ equilibrium potential [68]. Elevation of extracellular K+ increases the conductance of KIR channels (Fig. 1.6) and shifts the outward hump to more positive membrane potentials which allows these channels to transduce changes in extracellular K+ concentration into hyperpolarizing outward K+ currents (more on this below). The strong inward rectifier KIR channels can be blocked by micromolar concentration of Ba2+ (Table 1.1), which has been the primary pharmacological tool used to study the physiological function of these channels.

KIR channel currents in arteriolar endothelial cells . Shown are mean ± SE (n = 5) current-voltage relationships recorded from freshly isolated hamster cremaster arteriolar endothelial cells (see Fig. 1.2 and [23, 81] for method details) with 5 or 15 mM K+ in the extracellular solution as indicated. At rest, these cells have a resting membrane potential of ~−30 mV [23], and KIR channels contribute little to the resting membrane potential. However, note that membrane hyperpolarization from this potential, due to opening of KCa channels, for example, will recruit current through KIR channels, amplifying the original hyperpolarization. KIR channels are also sensitive to increases in extracellular [K+] as shown, which increases the conductance of these channels and shifts the reversal potential and the outward “hump” to more positive potentials as shown. Thus, increases in extracellular K+, due to release of K+ from active skeletal muscle, or from release of K+ from endothelial KCa channels, for example, will also recruit outward current through KIR channels that will hyperpolarize endothelial cells. Figure modified from [81]
In cultured endothelial cells, currents through strong inward rectifier KIR channels dominate the whole-cell, current-voltage relationship [3, 123], and from studies of cultured macrovascular and microvascular endothelial cells it was proposed that macrovascular, but not microvascular endothelial cells express KIR channels [123]. However, studies of freshly isolated endothelial cells from porcine brain capillaries [74], guinea pig coronary capillaries [169], rat mesenteric resistance arteries [28], rat cerebral resistance arteries [86], mouse cerebral penetrating arterioles [101] and hamster cremaster arterioles [81] (Fig. 1.6) all display Ba2+-sensitive KIR currents . Expression of message for KIR2.1, 2.2 and possibly 2.3 has been reported in guinea pig coronary capillary endothelial cells [99]. Similarly, in primary cultures of rat brain capillary endothelial cells KIR2.1, 2.2 and a much lower level of KIR2.3 have been detected [115]. In mouse and rat skeletal muscle arteriolar endothelial cells, we have detected the expression of KIR2.1, but not KIR2.2 at the protein level (Fig. 1.7).

Expression of KIR protein in skeletal muscle arterioles . Top panels show western blots for KIR2.1 and KIR2.2 in mouse and rat skeletal muscle arteriolar whole homogenates demonstrating expression of both proteins. In both panels each lane is as follows: Lane 1 = 1° + 2° antibody for indicated protein, Lane 2 = 2° antibody only, Lane 3 = 1° antibody only, Lane 4 = 1° + 2° antibody for indicated protein and Lane 5 = 2° antibody only. Lanes 1–3 contain mouse abdominal arteriolar whole homogenate and Lanes 4 and 5 contain rat cremaster arteriolar whole homogenate. Representative of three experiments. Antibodies: 1° Kir2.1 (1:200) (Alomone) + 2° HRP-conjugated mouse anti-rabbit, light chain specific (1:25,000) (Jackson Immuno Research)1° Kir2.2 (1:400) (Alomone) + 2° HRP-conjugated goat anti-rabbit (1:2000) (Cell Signaling). Fig. 1.7 (continued) Bottom panels show immunofluorescence localization of KIR2.1 and KIR2.2 in mouse abdominal arteriolar smooth muscle cells and endothelial cells. Vessels were enzymatically dissociated to yield single smooth muscle cells and endothelial cell tubes (labeled ECs) as described [23], the cells were fixed with 4 % paraformaldehyde and stained with the same antibodies as in top panels. Smooth muscle cells were identified by staining for α-smooth muscle actin (Green in middle panels). Note that endothelial cells expressed only KIR2.1, whereas smooth muscle cells expressed both KIR2.1 and 2.2 (Red in right panels). Data are representative of three isolates. Similar results were obtained in cells isolated from rat cremaster arterioles
The function of KIR2.X channels in endothelial cells of resistance arteries has been understudied. Theoretically, the expression of these channels in endothelial cells could serve to amplify endothelial cell hyperpolarization induced by the activation of other K+ channels and also serve to transduce small increases in extracellular K+ into cell hyperpolarization. In blood vessels where KIR2.X channels are expressed in the vascular smooth muscle, evidence for both of these functions has been demonstrated: smooth muscle KIR channels serve to amplify smooth muscle hyperpolarization induced by other means [82, 146] and it is well established that elevated extracellular K+ causes vasodilatation that is mediated, at least in part, by activation of smooth muscle KIR2.X channels [85, 101, 122, 186]. It has been argued that endothelial KIR channels do not play similar roles, primarily based on studies of rat mesenteric arteries in which the endothelial cells, but not the smooth muscle cells express functional KIR channels [146]. However, this hypothesis has not been adequately tested, particularly in arterioles with only a single layer of smooth muscle and strong expression of KIR channels. Endothelial and smooth muscle-selective knockdown, as proposed by Longden and Nelson [101], would provide an appropriate system in which to better evaluate the roles played by endothelial KIR channels.
Concluding Remarks
Endothelial cells importantly contribute to the regulation of the vasomotor function of resistance arteries and arterioles, and it is clear that a number of ion channels in these cells contribute to this function. As noted throughout this chapter, there appears to be substantial regional and species differences in the expression and function of endothelial cell ion channels that clouds a clear view of the function of individual channels and the interplay among and between specific channels. It appears that many, if not all of these channels exist in macromolecular signaling complexes. This further complicates analysis of the individual roles played by specific channels. While knockout approaches are certainly an important tool in studying the function of specific channels, given these macromolecular complexes, the likelihood of off target effects in disrupting other functions of such complexes is significant, but usually overlooked, and may explain why different answers are achieved using knockout approaches vs. pharmacology. The use of cell-specific conditional knockouts, as well as knockins of dominant negative channels coupled with careful pharmacological approaches may help to resolve the questions that still remain. Because of the inherent heterogeneity of expression and function of ion channels in different vascular beds, the detailed study of individual beds among species and particularly in humans is to be encouraged rather than stifled.
References
Abdullaev IF, Bisaillon JM, Potier M, Gonzalez JC, Motiani RK, Trebak M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ Res. 2008;103:1289–99.
Absi M, Burnham MP, Weston AH, Harno E, Rogers M, Edwards G. Effects of methyl beta-cyclodextrin on EDHF responses in pig and rat arteries; association between SK(Ca) channels and caveolin-rich domains. Br J Pharmacol. 2007;151:332–40.
Adams DJ, Hill MA. Potassium channels and membrane potential in the modulation of intracellular calcium in vascular endothelial cells. J Cardiovasc Electrophysiol. 2004;15:598–610.
Adebiyi A, Zhao G, Narayanan D, Thomas-Gatewood CM, Bannister JP, Jaggar JH. Isoform-selective physical coupling of TRPC3 channels to IP3 receptors in smooth muscle cells regulates arterial contractility. Circ Res. 2010;106:1603–12.
Ahmmed GU, Mehta D, Vogel S, Holinstat M, Paria BC, Tiruppathi C, Malik AB. Protein kinase Calpha phosphorylates the TRPC1 channel and regulates store-operated Ca2+ entry in endothelial cells. J Biol Chem. 2004;279:20941–9.
Alagem N, Dvir M, Reuveny E. Mechanism of Ba2+ block of a mouse inwardly rectifying K+ channel: differential contribution by two discrete residues. J Physiol Lond. 2001;534:381–93.
Antoniotti S, Lovisolo D, Fiorio Pla A, Munaron L. Expression and functional role of bTRPC1 channels in native endothelial cells. FEBS Lett. 2002;510:189–95.
Bagher P, Beleznai T, Kansui Y, Mitchell R, Garland CJ, Dora KA. Low intravascular pressure activates endothelial cell TRPV4 channels, local Ca2+ events, and IKCa channels, reducing arteriolar tone. Proc Natl Acad Sci U S A. 2012;109:18174–9.
Baraldi PG, Preti D, Materazzi S, Geppetti P. Transient receptor potential ankyrin 1 (TRPA1) channel as emerging target for novel analgesics and anti-inflammatory agents. J Med Chem. 2010;53:5085–107.
Beech DJ, Xu SZ, McHugh D, Flemming R. TRPC1 store-operated cationic channel subunit. Cell Calcium. 2003;33:433–40.
Bergdahl A, Gomez MF, Wihlborg A-K, Erlinge D, Eyjolfson A, Xu S-Z, Beech DJ, Dreja K, Hellstrand P. Plasticity of TRPC expression in arterial smooth muscle: correlation with store-operated Ca2+ entry. Am J Physiol Cell Physiol. 2005;288:C872–80.
Berra-Romani R, Mazzocco-Spezzia A, Pulina MV, Golovina VA. Ca2+ handling is altered when arterial myocytes progress from a contractile to a proliferative phenotype in culture. Am J Physiol Cell Physiol. 2008;295:C779–90.
Berridge M. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–25.
Boulay G, Zhu X, Peyton M, Jiang M, Hurst R, Stefani E, Birnbaumer L. Cloning and expression of a novel mammalian homolog of Drosophila Transient Receptor Potential (Trp) involved in calcium entry secondary to activation of receptors coupled by the Gq class of G protein. J Biol Chem. 1997;272:29672–80.
Brahler S, Kaistha A, Schmidt VJ, Wolfle SE, Busch C, Kaistha BP, Kacik M, Hasenau AL, Grgic I, Si H, Bond CT, Adelman JP, Wulff H, de Wit C, Hoyer J, Kohler R. Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation. 2009;119:2323–32.
Braunstein TH, Inoue R, Cribbs L, Oike M, Ito Y, Holstein-Rathlou NH, Jensen LJ. The role of L- and T-type calcium channels in local and remote calcium responses in rat mesenteric terminal arterioles. J Vasc Res. 2009;46:138–51.
Brough GH, Wu S, Cioffi D, Moore TM, Li M, Dean N, Stevens T. Contribution of endogenously expressed Trp1 to a Ca2+-selective, store-operated Ca2+ entry pathway. FASEB J. 2001;15:1727–38.
Busse R, Fichtner H, Luckhoff A, Kohlhardt M. Hyperpolarization and increased free calcium in acetylcholine-stimulated endothelial cells. Am J Physiol. 1988;255:H965–9.
Bychkov R, Burnham MP, Richards GR, Edwards G, Weston AH, Feletou M, Vanhoutte PM. Characterization of a charybdotoxin-sensitive intermediate conductance Ca2+-activated K+ channel in porcine coronary endothelium: relevance to EDHF. Br J Pharmacol. 2002;137:1346–54.
Chatterjee S, Al-Mehdi AB, Levitan I, Stevens T, Fisher AB. Shear stress increases expression of a KATP channel in rat and bovine pulmonary vascular endothelial cells. Am J Physiol Cell Physiol. 2003;285:C959–67.
Chen W, Oberwinkler H, Werner F, Gassner B, Nakagawa H, Feil R, Hofmann F, Schlossmann J, Dietrich A, Gudermann T, Nishida M, Del Galdo S, Wieland T, Kuhn M. Atrial natriuretic peptide-mediated inhibition of microcirculatory endothelial Ca2+ and permeability response to histamine involves cGMP-dependent protein kinase I and TRPC6 channels. Arterioscler Thromb Vasc Biol. 2013;33:2121–9.
Cipolla MJ, Smith J, Kohlmeyer MM, Godfrey JA. SKCa and IKCa Channels, myogenic tone, and vasodilator responses in middle cerebral arteries and parenchymal arterioles: effect of ischemia and reperfusion. Stroke. 2009;40:1451–7.
Cohen KD, Jackson WF. Membrane hyperpolarization is not required for sustained muscarinic agonist-induced increases in intracellular Ca2+ in arteriolar endothelial cells. Microcirculation. 2005;12:169–82.
Colden-Stanfield M, Schilling WP, Possani LD, Kunze DL. Bradykinin-induced potassium current in cultured bovine aortic endothelial cells. J Membr Biol. 1990;116:227–38.
Colden-Stanfield M, Schilling WP, Ritchie AK, Eskin SG, Navarro LT, Kunze DL. Bradykinin-induced increases in cytosolic calcium and ionic currents in cultured bovine aortic endothelial cells. Circ Res. 1987;61:632–40.
Corriu C, Feletou M, Canet E, Vanhoutte PM. Endothelium-derived factors and hyperpolarization of the carotid artery of the guinea-pig. Br J Pharmacol. 1996;119:959–64.
Crane GJ, Gallagher N, Dora KA, Garland CJ. Small- and intermediate-conductance calcium-activated K+ channels provide different facets of endothelium-dependent hyperpolarization in rat mesenteric artery. J Physiol. 2003;553:183–9.
Crane GJ, Walker SD, Dora KA, Garland CJ. Evidence for a differential cellular distribution of inward rectifier K channels in the rat isolated mesenteric artery. J Vasc Res. 2003;40:159–68.
de Wit C, Griffith TM. Connexins and gap junctions in the EDHF phenomenon and conducted vasomotor responses. Pflugers Arch. 2010;459:897–914.
Dietrich A, Fahlbusch M, Gudermann T. Classical Transient Receptor Potential 1 (TRPC1): channel or channel regulator? Cells. 2014;3:939–62.
Dietrich A, Kalwa H, Storch U, Mederos Y, Schnitzler M, Salanova B, Pinkenburg O, Dubrovska G, Essin K, Gollasch M, Birnbaumer L, Gudermann T. Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflugers Arch. 2007;455:465–77.
Dora KA, Doyle MP, Duling BR. Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles. Proc Natl Acad Sci U S A. 1997;94:6529–34.
Dora KA, Gallagher NT, McNeish A, Garland CJ. Modulation of endothelial cell KCa3.1 channels during endothelium-derived hyperpolarizing factor signaling in mesenteric resistance arteries. Circ Res. 2008;102:1247–55.
Dora KA, Garland CJ. Linking hyperpolarization to endothelial cell calcium events in arterioles. Microcirculation. 2013;20:248–56.
Du J, Ma X, Shen B, Huang Y, Birnbaumer L, Yao X. TRPV4, TRPC1, and TRPP2 assemble to form a flow-sensitive heteromeric channel. FASEB J. 2014;28:4677–85.
Duza T, Sarelius IH. Localized transient increases in endothelial cell Ca2+ in arterioles in situ: implications for the coordination of vascular function. Am J Physiol Heart Circ Physiol. 2004;286:H2322–31.
Earley S, Gonzales AL, Crnich R. Endothelium-dependent cerebral artery dilation mediated by TRPA1 and Ca2+-activated K+ channels. Circ Res. 2009;104:987–94.
Earley S, Gonzales AL, Garcia ZI. A dietary agonist of transient receptor potential cation channel V3 elicits endothelium-dependent vasodilation. Mol Pharmacol. 2010;77:612–20.
Earley S, Pauyo T, Drapp R, Tavares MJ, Liedtke W, Brayden JE. TRPV4-dependent dilation of peripheral resistance arteries influences arterial pressure. Am J Physiol Heart Circ Physiol. 2009;297:H1096–102.
Edwards G, Feletou M, Weston AH. Endothelium-derived hyperpolarising factors and associated pathways: a synopsis. Pflugers Arch. 2010;459:863–79.
Eichler I, Wibawa J, Grgic I, Knorr A, Brakemeier S, Pries AR, Hoyer J, Kohler R. Selective blockade of endothelial Ca2+-activated small- and intermediate-conductance K+-channels suppresses EDHF-mediated vasodilation. Br J Pharmacol. 2003;138:594–601.
Estacion M, Li S, Sinkins WG, Gosling M, Bahra P, Poll C, Westwick J, Schilling WP. Activation of human TRPC6 channels by receptor stimulation. J Biol Chem. 2004;279:22047–56.
Everaerts W, Zhen X, Ghosh D, Vriens J, Gevaert T, Gilbert JP, Hayward NJ, McNamara CR, Xue F, Moran MM, Strassmaier T, Uykal E, Owsianik G, Vennekens R, De Ridder D, Nilius B, Fanger CM, Voets T. Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. Proc Natl Acad Sci U S A. 2010;107:19084–9.
Fanger CM, Ghanshani S, Logsdon NJ, Rauer H, Kalman K, Zhou J, Beckingham K, Chandy KG, Cahalan MD, Aiyar J. Calmodulin mediates calcium-dependent activation of the intermediate conductance KCa channel, IKCa1. J Biol Chem. 1999;274:5746–54.
Figueroa XF, Chen CC, Campbell KP, Damon DN, Day KH, Ramos S, Duling BR. Are voltage-dependent ion channels involved in the endothelial cell control of vasomotor tone? Am J Physiol Heart Circ Physiol. 2007;293:H1371–83.
Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002;82:893–922.
Foskett J, White C, Cheung K, Mak D. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007;87:593–658.
Freichel M, Suh SH, Pfeifer A, Schweig U, Trost C, Weissgerber P, Biel M, Philipp S, Freise D, Droogmans G, Hofmann F, Flockerzi V, Nilius B. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4−/− mice. Nat Cell Biol. 2001;3:121–7.
Gafni J, Munsch JA, Lam TH, Catlin MC, Costa LG, Molinski TF, Pessah IN. Xestospongins: potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron. 1997;19:723.
Gauthier KM, Liu C, Popovic A, Albarwani S, Rusch NJ. Freshly isolated bovine coronary endothelial cells do not express the BK(Ca) channel gene. J Physiol. 2002;545:829–36.
Gees M, Colsoul B, Nilius B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb Perspect Biol. 2010;2:a003962.
Goddard LM, Iruela-Arispe ML. Cellular and molecular regulation of vascular permeability. Thromb Haemost. 2013;109:407–15.
Gordon JL, Martin W. Endothelium-dependent relaxation of the pig aorta: relationship to stimulation of 86Rb efflux from isolated endothelial cells. Br J Pharmacol. 1983;79:531–41.
Graier WF, Paltauf-Doburzynska J, Hill BJ, Fleischhacker E, Hoebel BG, Kostner GM, Sturek M. Submaximal stimulation of porcine endothelial cells causes focal Ca2+ elevation beneath the cell membrane. J Physiol. 1998;506(Pt 1):109–25.
Graier WF, Simecek S, Bowles DK, Sturek M. Heterogeneity of caffeine- and bradykinin-sensitive Ca2+ stores in vascular endothelial cells. Biochem J. 1994;300:637–41.
Grayson TH, Haddock RE, Murray TP, Wojcikiewicz RJH, Hill CE. Inositol 1,4,5-trisphosphate receptor subtypes are differentially distributed between smooth muscle and endothelial layers of rat arteries. Cell Calcium. 2004;36:447.
Grgic I, Kaistha BP, Hoyer J, Kohler R. Endothelial Ca+-activated K+ channels in normal and impaired EDHF-dilator responses—relevance to cardiovascular pathologies and drug discovery. Br J Pharmacol. 2009;157:509–26.
Halaszovich CR, Zitt C, Jungling E, Luckhoff A. Inhibition of TRP3 channels by lanthanides. Block from the cytosolic side of the plasma membrane. J Biol Chem. 2000;275:37423–8.
Hallam TJ, Pearson JD. Exogenous ATP raises cytoplasmic free calcium in fura-2 loaded piglet aortic endothelial cells. FEBS Lett. 1986;207:95–9.
Hamdollah Zadeh MA, Glass CA, Magnussen A, Hancox JC, Bates DO. VEGF-mediated elevated intracellular calcium and angiogenesis in human microvascular endothelial cells in vitro are inhibited by dominant negative TRPC6. Microcirculation. 2008;15:605–14.
Hamilton S. Ryanodine receptors. Cell Calcium. 2005;38:253–60.
Hamilton S, Serysheva I. Ryanodine receptor structure: progress and challenges. J Biol Chem. 2009;284:4047–51.
Hannah RM, Dunn KM, Bonev AD, Nelson MT. Endothelial SK(Ca) and IK(Ca) channels regulate brain parenchymal arteriolar diameter and cortical cerebral blood flow. J Cereb Blood Flow Metab. 2011;31:1175–86.
Harteneck C, Gollasch M. Pharmacological modulation of diacylglycerol-sensitive TRPC3/6/7 channels. Curr Pharm Biotechnol. 2011;12:35–41.
He P, Curry FE. Depolarization modulates endothelial cell calcium influx and microvessel permeability. Am J Physiol Heart Circ Physiol. 1991;261:H1246–54.
He P, Curry FE. Endothelial cell hyperpolarization increases [Ca2+]i and venular microvessel permeability. J Appl Physiol. 1994;76:2288–97.
Heady TN, Gomora JC, Macdonald TL, Perez-Reyes E. Molecular pharmacology of T-type Ca < sup > 2 + </sup > channels. Jpn J Pharmacol. 2001;85:339–50.
Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90:291–366.
Himmel HM, Whorton AR, Strauss HC. Intracellular calcium, currents, and stimulus-response coupling in endothelial cells. Hypertension. 1993;21:112–27.
Hoffmann EK, Lambert IH, Pedersen SF. Physiology of cell volume regulation in vertebrates. Physiol Rev. 2009;89:193–277.
Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–63.
Hondeghem LM, Ayad MJ, Robertson RM. Verapamil, diltiazem and nifedipine block the depolarization-induced potentiation of norepinephrine contractions in rabbit aorta and porcine coronary arteries. J Pharmacol Exp Ther. 1986;239:808–13.
Hoshi T, Pantazis A, Olcese R. Transduction of voltage and Ca2+ signals by Slo1 BK channels. Physiology (Bethesda). 2013;28:172–89.
Hoyer J, Popp R, Meyer J, Galla HJ, Gogelein H. Angiotensin II, vasopressin and GTP[gamma-S] inhibit inward-rectifying K+ channels in porcine cerebral capillary endothelial cells. J Membr Biol. 1991;123:55–62.
Huang JH, He GW, Xue HM, Yao XQ, Liu XC, Underwood MJ, Yang Q. TRPC3 channel contributes to nitric oxide release: significance during normoxia and hypoxia-reoxygenation. Cardiovasc Res. 2011;91:472–82.
Hughes JM, Riddle MA, Paffett ML, Gonzalez Bosc LV, Walker BR. Novel role of endothelial BKCa channels in altered vasoreactivity following hypoxia. Am J Physiol Heart Circ Physiol. 2010;299:H1439–50.
Isakson BE. Localized expression of an Ins(1,4,5)P3 receptor at the myoendothelial junction selectively regulates heterocellular Ca2+ communication. J Cell Sci. 2008;121:3664–73.
Ishii TM, Silvia C, Hirschberg B, Bond CT, Adelman JP, Maylie J. A human intermediate conductance calcium-activated potassium channel. Proc Natl Acad Sci U S A. 1997;94:11651–6.
Jackson WF. The endothelium-derived relaxing factor. J Reconstr Microsurg. 1989;5(3):263–71.
Jackson WF. Ion channels and vascular tone. Hypertension. 2000;35(1 Pt 2):173–8.
Jackson WF. Potassium channels in the peripheral microcirculation. Microcirculation. 2005;12:113–27.
Jantzi MC, Brett SE, Jackson WF, Corteling RL, Vigmond EJ, Welsh DG. Inward rectifying potassium channels facilitate cell-to-cell communication in hamster retractor muscle feed arteries. Am J Physiol Heart Circ Physiol. 2006;291:H1319–28.
Kansui Y, Garland CJ, Dora KA. Enhanced spontaneous Ca2+ events in endothelial cells reflect signalling through myoendothelial gap junctions in pressurized mesenteric arteries. Cell Calcium. 2008;44:135–46.
Kiyonaka S, Kato K, Nishida M, Mio K, Numaga T, Sawaguchi Y, Yoshida T, Wakamori M, Mori E, Numata T, Ishii M, Takemoto H, Ojida A, Watanabe K, Uemura A, Kurose H, Morii T, Kobayashi T, Sato Y, Sato C, Hamachi I, Mori Y. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc Natl Acad Sci U S A. 2009;106:5400–5.
Knot HJ, Zimmermann PA, Nelson MT. Extracellular K(+)-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K(+) channels. J Physiol. 1996;492(Pt 2):419–30.
Kochukov MY, Balasubramanian A, Abramowitz J, Birnbaumer L, Marrelli SP. Activation of endothelial transient receptor potential C3 channel is required for small conductance calcium-activated potassium channel activation and sustained endothelial hyperpolarization and vasodilation of cerebral artery. J Am Heart Assoc. 2014;3.
Kochukov MY, Balasubramanian A, Noel RC, Marrelli SP. Role of TRPC1 and TRPC3 channels in contraction and relaxation of mouse thoracic aorta. J Vasc Res. 2013;50:11–20.
Kohler R, Brakemeier S, Kuhn M, Behrens C, Real R, Degenhardt C, Orzechowski HD, Pries AR, Paul M, Hoyer J. Impaired hyperpolarization in regenerated endothelium after balloon catheter injury. Circ Res. 2001;89:174–9.
Kohler R, Brakemeier S, Kuhn M, Degenhardt C, Buhr H, Pries A, Hoyer J. Expression of ryanodine receptor type 3 and TRP channels in endothelial cells: comparison of in situ and cultured human endothelial cells. Cardiovasc Res. 2001;51:160.
Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol. 2010;72:463–93.
Kuo IY, Ellis A, Seymour VA, Sandow SL, Hill CE. Dihydropyridine-insensitive calcium currents contribute to function of small cerebral arteries. J Cereb Blood Flow Metab. 2010;30:1226–39.
Kwiatek AM, Minshall RD, Cool DR, Skidgel RA, Malik AB, Tiruppathi C. Caveolin-1 regulates store-operated Ca2+ influx by binding of its scaffolding domain to transient receptor potential channel-1 in endothelial cells. Mol Pharmacol. 2006;70:1174–83.
Ledoux J, Taylor MS, Bonev AD, Hannah RM, Solodushko V, Shui B, Tallini Y, Kotlikoff MI, Nelson MT. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci U S A. 2008;105:9627–32.
Lee JH, Gomora JC, Cribbs LL, Perez-Reyes E. Nickel block of three cloned T-type calcium channels: low concentrations selectively block alpha1H. Biophys J. 1999;77:3034–42.
Lesh RE, Marks AR, Somlyo AV, Fleischer S, Somlyo AP. Anti-ryanodine receptor antibody binding sites in vascular and endocardial endothelium. Circ Res. 1993;72:481–8.
Leuner K, Heiser JH, Derksen S, Mladenov MI, Fehske CJ, Schubert R, Gollasch M, Schneider G, Harteneck C, Chatterjee SS, Muller WE. Simple 2,4-diacylphloroglucinols as classic transient receptor potential-6 activators--identification of a novel pharmacophore. Mol Pharmacol. 2010;77:368–77.
Liao P, Yu D, Li G, Yong TF, Soon JL, Chua YL, Soong TW. A smooth muscle Cav1.2 calcium channel splice variant underlies hyperpolarized window current and enhanced state-dependent inhibition by nifedipine. J Biol Chem. 2007;282:35133–42.
Liu CL, Huang Y, Ngai CY, Leung YK, Yao XQ. TRPC3 is involved in flow- and bradykinin-induced vasodilation in rat small mesenteric arteries. Acta Pharmacol Sin. 2006;27:981–90.
Liu GX, Derst C, Schlichthorl G, Heinen S, Seebohm G, Bruggemann A, Kummer W, Veh RW, Daut J, Preisig-Muller R. Comparison of cloned Kir2 channels with native inward rectifier K+ channels from guinea-pig cardiomyocytes. J Physiol. 2001;532:115–26.
Loga F, Domes K, Freichel M, Flockerzi V, Dietrich A, Birnbaumer L, Hofmann F, Wegener JW. The role of cGMP/cGKI signalling and Trpc channels in regulation of vascular tone. Cardiovasc Res. 2013;100:280–7.
Longden TA, Nelson MT. Vascular inward rectifier k(+) channels as external k(+) sensors in the control of cerebral blood flow. Microcirculation. 2015;22:183–96.
Mackrill JJ. Ryanodine receptor calcium channels and their partners as drug targets. Biochem Pharmacol. 2010;79:1535–43.
Mak D, McBride S, Foskett J. Regulation by Ca2+ and inositol 1,4,5-trisphosphate (InsP3) of single recombinant type 3 InsP3 receptor channels. Ca2+ activation uniquely distinguishes types 1 and 3 insp3 receptors. J Gen Physiol. 2001;117:435–46.
Mak DD, Foskett JK. Inositol 1,4,5-trisphosphate receptors in the endoplasmic reticulum: a single-channel point of view. Cell Calcium. 2014;58:67–78.
Mak DO, McBride S, Foskett JK. Inositol 1,4,5-trisphosphate [correction of tris-phosphate] activation of inositol trisphosphate [correction of tris-phosphate] receptor Ca2+ channel by ligand tuning of Ca2+ inhibition. Proc Natl Acad Sci U S A. 1998;95:15821–5.
Mannhold R. KATP channel openers: structure-activity relationships and therapeutic potential. Med Res Rev. 2004;24:213–66.
Marrelli SP. Selective measurement of endothelial or smooth muscle [Ca(2+)](i) in pressurized/perfused cerebral arteries with fura-2. J Neurosci Methods. 2000;97:145–55.
Marrelli SP, Eckmann MS, Hunte MS. Role of endothelial intermediate conductance KCa channels in cerebral EDHF-mediated dilations. Am J Physiol Heart Circ Physiol. 2003;285:H1590–9.
Marrelli SP, O’Neil RG, Brown RC, Bryan Jr RM. PLA2 and TRPV4 channels regulate endothelial calcium in cerebral arteries. Am J Physiol Heart Circ Physiol. 2007;292:H1390–7.
Martin RL, Lee JH, Cribbs LL, Perez-Reyes E, Hanck DA. Mibefradil block of cloned T-type calcium channels. J Pharmacol Exp Ther. 2000;295:302–8.
Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J Biochem. 1997;122:498–505.
McNamara CR, Mandel-Brehm J, Bautista DM, Siemens J, Deranian KL, Zhao M, Hayward NJ, Chong JA, Julius D, Moran MM, Fanger CM. TRPA1 mediates formalin-induced pain. Proc Natl Acad Sci U S A. 2007;104:13525–30.
McSherry IN, Spitaler MM, Takano H, Dora KA. Endothelial cell Ca2+ increases are independent of membrane potential in pressurized rat mesenteric arteries. Cell Calcium. 2005;38:23–33.
Mehta D, Ahmmed GU, Paria BC, Holinstat M, Voyno-Yasenetskaya T, Tiruppathi C, Minshall RD, Malik AB. RhoA interaction with inositol 1,4,5-trisphosphate receptor and transient receptor potential channel-1 regulates Ca2+ entry. Role in signaling increased endothelial permeability. J Biol Chem. 2003;278:33492–500.
Millar ID, Wang S, Brown PD, Barrand MA, Hladky SB. Kv1 and Kir2 potassium channels are expressed in rat brain endothelial cells. Pflugers Arch. 2008;456:379–91.
Miller M, Shi J, Zhu Y, Kustov M, Tian JB, Stevens A, Wu M, Xu J, Long S, Yang P, Zholos AV, Salovich JM, Weaver CD, Hopkins CR, Lindsley CW, McManus O, Li M, Zhu MX. Identification of ML204, a novel potent antagonist that selectively modulates native TRPC4/C5 ion channels. J Biol Chem. 2011;286:33436–46.
Minami T. Calcineurin-NFAT activation and DSCR-1 auto-inhibitory loop: how is homoeostasis regulated? J Biochem. 2014;155:217–26.
Mountian I, Manolopoulos VG, De Smedt H, Parys JB, Missiaen L, Wuytack F. Expression patterns of sarco/endoplasmic reticulum Ca(2+)-ATPase and inositol 1,4,5-trisphosphate receptor isoforms in vascular endothelial cells. Cell Calcium. 1999;25:371–80.
Mountian II, Baba-Aissa F, Jonas JC, De Humbert S, Wuytack F, Parys JB. Expression of Ca(2+) transport genes in platelets and endothelial cells in hypertension. Hypertension. 2001;37:135–41.
Munaron L. Intracellular calcium, endothelial cells and angiogenesis. Recent Pat Anticancer Drug Discov. 2006;1:105–19.
Narahashi T, Tsunoo A, Yoshii M. Characterization of two types of calcium channels in mouse neuroblastoma cells. J Physiol. 1987;383:231–49.
Nelson M, Quayle J. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. 1995;268:C799–822.
Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev. 2001;81:1415–59.
Nilius B, Droogmans G, Wondergem R. Transient receptor potential channels in endothelium: solving the calcium entry puzzle? Endothelium. 2003;10:5–15.
Nilius B, Viana F, Droogmans G. Ion channels in vascular endothelium. Annu Rev Physiol. 1997;59:145–70.
Paltauf-Doburzynska J, Posch K, Paltauf G, Graier WF. Stealth ryanodine-sensitive Ca2+ release contributes to activity of capacitative Ca2+ entry and nitric oxide synthase in bovine endothelial cells. J Physiol. 1998;513(Pt 2):369–79.
Pocock TM, Foster RR, Bates DO. Evidence of a role for TRPC channels in VEGF-mediated increased vascular permeability in vivo. Am J Physiol Heart Circ Physiol. 2004;286:H1015–26.
Qian X, Francis M, Kohler R, Solodushko V, Lin M, Taylor MS. Positive feedback regulation of agonist-stimulated endothelial Ca2+ dynamics by KCa3.1 channels in mouse mesenteric arteries. Arterioscler Thromb Vasc Biol. 2014;34:127–35.
Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev. 1997;77:1165–232.
Quinlan KL, Naik SM, Cannon G, Armstrong CA, Bunnett NW, Ansel JC, Caughman SW. Substance P activates coincident NF-AT- and NF-kappa B-dependent adhesion molecule gene expression in microvascular endothelial cells through intracellular calcium mobilization. J Immunol. 1999;163:5656–65.
Riddle MA, Hughes JM, Walker BR. Role of caveolin-1 in endothelial BKCa channel regulation of vasoreactivity. Am J Physiol Cell Physiol. 2011;301:C1404–14.
Rusko J, Van Slooten G, Adams DJ. Caffeine-evoked, calcium-sensitive membrane currents in rabbit aortic endothelial cells. Br J Pharmacol. 1995;115:133–41.
Saleem H, Tovey SC, Molinski TF, Taylor CW. Interactions of antagonists with subtypes of inositol 1,4,5-trisphosphate (IP3) receptor. Br J Pharmacol. 2014;171:3298–312.
Saliez J, Bouzin C, Rath G, Ghisdal P, Desjardins F, Rezzani R, Rodella LF, Vriens J, Nilius B, Feron O, Balligand JL, Dessy C. Role of caveolar compartmentation in endothelium-derived hyperpolarizing factor-mediated relaxation: Ca2+ signals and gap junction function are regulated by caveolin in endothelial cells. Circulation. 2008;117:1065–74.
Sandow SL, Grayson TH. Limits of isolation and culture: intact vascular endothelium and BKCa. Am J Physiol Heart Circ Physiol. 2009;297:H1–7.
Sandow SL, Hill CE. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circ Res. 2000;86:341–6.
Sandow SL, Neylon CB, Chen MX, Garland CJ. Spatial separation of endothelial small- and intermediate-conductance calcium-activated potassium channels (K(Ca)) and connexins: possible relationship to vasodilator function? J Anat. 2006;209:689–98.
Schaefer M, Plant TD, Obukhov AG, Hofmann T, Gudermann T, Schultz G. Receptor-mediated regulation of the nonselective cation channels TRPC4 and TRPC5. J Biol Chem. 2000;275:17517–26.
Schilling WP, Cabello OA, Rajan L. Depletion of the inositol 1,4,5-trisphosphate-sensitive intracellular Ca2+ store in vascular endothelial-cells activates the agonist-sensitive Ca2+-influx pathway. Biochem J. 1992;284:521–30.
Schmidt K, Dubrovska G, Nielsen G, Fesus G, Uhrenholt TR, Hansen PB, Gudermann T, Dietrich A, Gollasch M, de Wit C, Kohler R. Amplification of EDHF-type vasodilatations in TRPC1-deficient mice. Br J Pharmacol. 2010;161:1722–33.
Senadheera S, Kim Y, Grayson TH, Toemoe S, Kochukov MY, Abramowitz J, Housley GD, Bertrand RL, Chadha PS, Bertrand PP, Murphy TV, Tare M, Birnbaumer L, Marrelli SP, Sandow SL. Transient receptor potential canonical type 3 channels facilitate endothelium-derived hyperpolarization-mediated resistance artery vasodilator activity. Cardiovasc Res. 2012;95:439–47.
Sharma NR, Davis MJ. Mechanism of substance P-induced hyperpolarization of porcine coronary artery endothelial cells. Am J Physiol. 1994;266:H156–64.
Sharma NR, Davis MJ. Substance-P-induced calcium-entry in endothelial-cells is secondary to depletion of intracellular stores. Am J Physiol Heart Circ Physiol. 1995;268:H962–73.
Si H, Heyken WT, Wolfle SE, Tysiac M, Schubert R, Grgic I, Vilianovich L, Giebing G, Maier T, Gross V, Bader M, de Wit C, Hoyer J, Kohler R. Impaired endothelium-derived hyperpolarizing factor-mediated dilations and increased blood pressure in mice deficient of the intermediate-conductance Ca2+-activated K+ channel. Circ Res. 2006;99:537–44.
Singh I, Knezevic N, Ahmmed GU, Kini V, Malik AB, Mehta D. Galphaq-TRPC6-mediated Ca2+ entry induces RhoA activation and resultant endothelial cell shape change in response to thrombin. J Biol Chem. 2007;282:7833–43.
Smith PD, Brett SE, Luykenaar KD, Sandow SL, Marrelli SP, Vigmond EJ, Welsh DG. KIR channels function as electrical amplifiers in rat vascular smooth muscle. J Physiol. 2008;586:1147–60.
Socha MJ, Domeier TL, Behringer EJ, Segal SS. Coordination of intercellular Ca2+ signaling in endothelial cell tubes of mouse resistance arteries. Microcirculation. 2012;19:757–70.
Socha MJ, Hakim CH, Jackson WF, Segal SS. Temperature effects on morphological integrity and Ca2+ signaling in freshly isolated murine feed artery endothelial cell tubes. Am J Physiol Heart Circ Physiol. 2011;301:H773–83.
Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill-Eubanks DC, Nelson MT. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science. 2012;336:597–601.
Sonkusare SK, Dalsgaard T, Bonev AD, Hill-Eubanks DC, Kotlikoff MI, Scott JD, Santana LF, Nelson MT. AKAP150-dependent cooperative TRPV4 channel gating is central to endothelium-dependent vasodilation and is disrupted in hypertension. Sci Signal. 2014;7:ra66.
Straub AC, Billaud M, Johnstone SR, Best AK, Yemen S, Dwyer ST, Looft-Wilson R, Lysiak JJ, Gaston B, Palmer L, Isakson BE. Compartmentalized connexin 43S-nitrosylation/denitrosylation regulates heterocellular communication in the vessel wall. Arterioscler Thromb Vasc Biol. 2011;31:399–407.
Straub AC, Zeigler AC, Isakson BE. The myoendothelial junction: connections that deliver the message. Physiology (Bethesda). 2014;29:242–9.
Strotmann R, Schultz G, Plant TD. Ca2+-dependent potentiation of the nonselective cation channel TRPV4 is mediated by a C-terminal calmodulin binding site. J Biol Chem. 2003;278:26541–9.
Sullivan MN, Gonzales AL, Pires PW, Bruhl A, Leo MD, Li W, Oulidi A, Boop FA, Feng Y, Jaggar JH, Welsh DG, Earley S. Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci Signal. 2015;8:ra2.
Sundivakkam PC, Freichel M, Singh V, Yuan JP, Vogel SM, Flockerzi V, Malik AB, Tiruppathi C. The Ca(2+) sensor stromal interaction molecule 1 (STIM1) is necessary and sufficient for the store-operated Ca(2+) entry function of transient receptor potential canonical (TRPC) 1 and 4 channels in endothelial cells. Mol Pharmacol. 2012;81:510–26.
Sundivakkam PC, Kwiatek AM, Sharma TT, Minshall RD, Malik AB, Tiruppathi C. Caveolin-1 scaffold domain interacts with TRPC1 and IP3R3 to regulate Ca2+ store release-induced Ca2+ entry in endothelial cells. Am J Physiol Cell Physiol. 2009;296:C403–13.
Svenningsen P, Andersen K, Thuesen A, Shin H-S, Vanhoutte P, Skøtt O, Jensen B, Hill C, Hansen PL. T-type Ca2+ channels facilitate NO-formation, vasodilatation and NO-mediated modulation of blood pressure. Pflügers Arch. 2014;466:2205–14.
Takano H, Dora KA, Spitaler MM, Garland CJ. Spreading dilatation in rat mesenteric arteries associated with calcium-independent endothelial cell hyperpolarization. J Physiol. 2004;556(pt.3):887–903.
Taylor CW, Tovey SC, Rossi AM, Lopez Sanjurjo CI, Prole DL, Rahman T. Structural organization of signalling to and from IP3 receptors. Biochem Soc Trans. 2014;42:63–70.
Taylor MS, Bonev AD, Gross TP, Eckman DM, Brayden JE, Bond CT, Adelman JP, Nelson MT. Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ Res. 2003;93:124–31.
Thorneloe KS, Sulpizio AC, Lin Z, Figueroa DJ, Clouse AK, McCafferty GP, Chendrimada TP, Lashinger ES, Gordon E, Evans L, Misajet BA, Demarini DJ, Nation JH, Casillas LN, Marquis RW, Votta BJ, Sheardown SA, Xu X, Brooks DP, Laping NJ, Westfall TD. N-((1S)-1-{[4-((2S)-2-{[(2,4-dichlorophenyl)sulfonyl]amino}-3-hydroxypropanoyl)-1-piperazinyl]carbonyl}-3-methylbutyl)-1-benzothiophene-2-carboxamide (GSK1016790A), a novel and potent transient receptor potential vanilloid 4 channel agonist induces urinary bladder contraction and hyperactivity: part I. J Pharmacol Exp Ther. 2008;326:432–42.
Tiruppathi C, Freichel M, Vogel SM, Paria BC, Mehta D, Flockerzi V, Malik AB. Impairment of store-operated Ca2+ entry in TRPC4(−/−) mice interferes with increase in lung microvascular permeability. Circ Res. 2002;91:70–6.
Ungvari Z, Csiszar A, Koller A. Increases in endothelial Ca(2+) activate K(Ca) channels and elicit EDHF-type arteriolar dilation via gap junctions. Am J Physiol Heart Circ Physiol. 2002;282:H1760–7.
Vaca L, Kunze DL. Depletion and refilling of intracellular Ca2+ stores induce oscillations of Ca2+ current. Am J Physiol. 1993;264:H1319–22.
Vaca L, Kunze DL. Depletion of intracellular Ca2+ stores activates a Ca2+-selective channel in vascular endothelium. Am J Physiol Cell Physiol. 1994;267:C920–5.
Vaithianathan T, Narayanan D, Asuncion-Chin MT, Jeyakumar LH, Liu J, Fleischer S, Jaggar JH, Dopico AM. Subtype identification and functional characterization of ryanodine receptors in rat cerebral artery myocytes. Am J Physiol Cell Physiol. 2010;299:C264–78.
Vinet R, Vargas FF. L- and T-type voltage-gated Ca2+ currents in adrenal medulla endothelial cells. Am J Physiol. 1999;276:H1313–22.
Vogt-Eisele AK, Weber K, Sherkheli MA, Vielhaber G, Panten J, Gisselmann G, Hatt H. Monoterpenoid agonists of TRPV3. Br J Pharmacol. 2007;151:530–40.
von Beckerath N, Dittrich M, Klieber HG, Daut J. Inwardly rectifying K+ channels in freshly dissociated coronary endothelial cells from guinea-pig heart. J Physiol. 1996;491(Pt 2):357–65.
Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B. Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Natl Acad Sci U S A. 2004;101:396–401.
Wagner II LE, Yule DI. Differential regulation of the InsP(3) receptor type-1 and -2 single channel properties by InsP(3), Ca(2)(+) and ATP. J Physiol. 2012;590:3245–59.
Wang H-R, Wu M, Yu H, Long S, Stevens A, Engers DW, Sackin H, Daniels JS, Dawson ES, Hopkins CR, Lindsley CW, Li M, McManus OB. Selective inhibition of the Kir2 family of inward rectifier potassium channels by a small molecule probe: the discovery, SAR, and pharmacological characterization of ML133. ACS Chem Biol. 2011;6:845–56.
Wang XL, Ye D, Peterson TE, Cao S, Shah VH, Katusic ZS, Sieck GC, Lee HC. Caveolae targeting and regulation of large conductance Ca(2+)-activated K+ channels in vascular endothelial cells. J Biol Chem. 2005;280:11656–64.
Watanabe H, Davis JB, Smart D, Jerman JC, Smith GD, Hayes P, Vriens J, Cairns W, Wissenbach U, Prenen J, Flockerzi V, Droogmans G, Benham CD, Nilius B. Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. J Biol Chem. 2002;277:13569–77.
Watanabe H, Vriens J, Suh SH, Benham CD, Droogmans G, Nilius B. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J Biol Chem. 2002;277:47044–51.
Wei AD, Gutman GA, Aldrich R, Chandy KG, Grissmer S, Wulff H. International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol Rev. 2005;57:463–72.
Wei Z, Manevich Y, Al-Mehdi AB, Chatterjee S, Fisher AB. Ca2+ flux through voltage-gated channels with flow cessation in pulmonary microvascular endothelial cells. Microcirculation. 2004;11:517–26.
Westcott EB, Goodwin EL, Segal SS, Jackson WF. Function and expression of ryanodine receptors and inositol 1,4,5-trisphosphate receptors in smooth muscle cells of murine feed arteries and arterioles. J Physiol. 2012;590:1849–69.
Wu LJ, Sweet TB, Clapham DE. International Union of Basic and Clinical Pharmacology. LXXVI. Current progress in the mammalian TRP ion channel family. Pharmacol Rev. 2010;62:381–404.
Wu S, Haynes Jr J, Taylor JT, Obiako BO, Stubbs JR, Li M, Stevens T. Cav3.1 (alpha1G) T-type Ca2+ channels mediate vaso-occlusion of sickled erythrocytes in lung microcirculation. Circ Res. 2003;93:346–53.
Wulff H, Kohler R. Endothelial small-conductance and intermediate-conductance KCa channels: an update on their pharmacology and usefulness as cardiovascular targets. J Cardiovasc Pharmacol. 2013;61:102–12.
Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, Maylie J, Adelman JP. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395:503–7.
Xu W, Lipscombe D. Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci. 2001;21:5944–51.
Yeon SI, Kim JY, Yeon DS, Abramowitz J, Birnbaumer L, Muallem S, Lee YH. Transient receptor potential canonical type 3 channels control the vascular contractility of mouse mesenteric arteries. PLoS One. 2014;9:e110413.
Zalk R, Lehnart S, Marks A. Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem. 2007;76:367–85.
Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL. Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K(+) current in K(+)-mediated vasodilation. Circ Res. 2000;87:160–6.
Zhang DX, Mendoza SA, Bubolz AH, Mizuno A, Ge ZD, Li R, Warltier DC, Suzuki M, Gutterman DD. Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension. 2009;53:532–8.
Zheng W, Rampe D, Triggle DJ. Pharmacological, radioligand binding, and electrophysiological characteristics of FPL 64176, a novel nondihydropyridine Ca2+ channel activator, in cardiac and vascular preparations. Mol Pharmacol. 1991;40:734–41.
Zhou C, Chen H, Lu F, Sellak H, Daigle JA, Alexeyev MF, Xi Y, Ju J, van Mourik JA, Wu S. Cav3.1 (alpha1G) controls von Willebrand factor secretion in rat pulmonary microvascular endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2007;292:L833–44.
Zhou M-H, Zheng H, Si H, Jin Y, Peng JM, He L, Zhou Y, Muñoz-Garay C, Zawieja DC, Kuo L, Peng X, Zhang SL. Stromal Interaction Molecule 1 (STIM1) and Orai1 mediate histamine-evoked calcium entry and Nuclear Factor of Activated T-cells (NFAT) signaling in human umbilical vein endothelial cells. J Biol Chem. 2014;289:29446–56.
Ziegelstein RC, Spurgeon HA, Pili R, Passaniti A, Cheng L, Corda S, Lakatta EG, Capogrossi MC. A functional ryanodine-sensitive intracellular Ca2+ store is present in vascular endothelial cells. Circ Res. 1994;74:151–6.
Zucchi R, Ronca-Testoni S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol Rev. 1997;49:1–51.
Acknowledgements
Supported NIH grants RO1-HL32469, RO1-HL086483 and PO1-HL070687.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Jackson, W.F. (2016). Endothelial Cell Ion Channel Expression and Function in Arterioles and Resistance Arteries. In: Levitan, PhD, I., Dopico, MD, PhD, A. (eds) Vascular Ion Channels in Physiology and Disease. Springer, Cham. https://doi.org/10.1007/978-3-319-29635-7_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-29635-7_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-29633-3
Online ISBN: 978-3-319-29635-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)